

Abstract
The preparation of natural rubber (NR)–silica (SiO2) elastomeric composites with excellent mechanical properties along with better self-healing ability remains a key challenge. Inspired by the energy dissipation and repairability of sacrificial bonds in biomaterials, a strategy for combining covalent and noncovalent sacrificial networks is engineered to construct a dual hybrid network. Here, the approach used to fabricate the composites was self-assembly of NR, bearing proteins and phospholipids on its outer bioshell, with SiO2 via metal-ion-mediated heteroaggregation effected by reversible electrostatic and H-bonds. Further, covalent cross-links were incorporated by a silane coupling agent, bis [3-(triethoxysilyl) propyl] tetrasulfide. The intrinsic self-healing ability of the composite at the molecular level was studied by broadband dielectric spectroscopy that unraveled the mechanism of the healing process. The synergistic effect between the molecular interdiffusion of the cross-linked NR chains and the electrostatic and H-bonding interactions imparted an exceptional self-healing characteristic to the liquid–liquid-mixing-prepared NR–SiO2 composites with improved mechanical performance. Specifically, the segmental relaxation dynamics of the healed composite was largely restricted due to increased number of ion–dipole interactions and S–S cross-links at the junction of the cut surface. We envisage that this extraordinary healing property, unreported yet, would be of great importance toward the design of novel NR–SiO2 elastomeric hybrids with superior mechanical properties.
1. Introduction
Reinforcement of polymers by fillers such as carbon black (CB) and silica (SiO2) is vital because almost all kinds of neat rubber suffer from poor mechanical strength.1−6 CB as a versatile reinforcing filler has greatly enhanced the dynamic and mechanical properties of elastomeric composites.7−11 However, the source of CB is petroleum-based, whereas the fossil reserve is inadequate. Moreover, CB generates numerous pollutants, thereby triggering serious ecological hazards.12 SiO2, which is independent of oil resource, has gained importance in the manufacture of “green tires”, as it offers significant advantages over CB in terms of low rolling resistance and significantly improved performance of the tire tread component.13−15 However, the silanol groups on the SiO2 surface, the poor dispersion of polar SiO2 in the nonpolar rubber, and weak polymer–filler interactions pose persistent complications for the use of SiO2 in the tire industry. To maximize the usage of SiO2 in rubber composites, especially in tires, exhaustive efforts have been focused on the techniques to combine SiO2 and polymers into a nanocomposite. Strategies such as shear mixing, surface modification of SiO2, and adsorption of dispersants on the filler surface have been validated to be effective in improving the dispersion of SiO2.16−22 For instance, sulfide-containing silanes, such as [3-(triethoxysilyl) propyl] tetrasulfide (TESPT), have been commonly used in the tire industry.23 The formation of an interphase in the composite has been influenced by the accessibility of filler surfaces, determined by the dispersion state of the filler in the polymer matrix.24−27 Consequently, the ultimate performance of elastomeric composites has been determined by the dispersion state of the filler and the polymer–filler interactions, effectively enhancing their mechanical strength. However, the crack propagation in elastomeric products during the service period has led to mechanical failures. Thus, self-healing rubbers have been developed and have attracted significant attention in the recent years to address this problem.28−37 A sacrificial network has been seen to be a vital factor in determining such properties in biological materials, such as spider silk, mussel, and bones.38,39 Accordingly, sacrificial bonds have been assimilated into rubbers by various authors40−45 for imparting super strength and self-healing. Specifically, noncovalent interactions, such as H-bonding, metal–ligand coordination, and π–π stacking, and hydrophobic interactions have been exploited to construct such supramolecular assemblies so that they break first upon being damaged and re-form after application of external stimuli. However, compared to the strong covalent bonds, the weak noncovalent bonds are generally stimuli-responsive and exhibit a reversible association–dissociation mechanism on exposure to the external stimulus. Consequently, such reversible behavior bestows the supramolecular composites with ensured self-healing capability but results in poor mechanical performance. Even though silica technology is effectively being practiced in tire industries,46−48 the above discussed problems are still encountered while using silica as fillers especially in NR. Therefore, the fabrication of an elastomeric composite with excellent mechanical and self-healing properties remains a crucial task for engineering high-performance composites.
Inspired by the energy dissipation mechanism and the reparability of sacrificial bonds, we engineered here a dual hybrid network to exploit the contribution of sacrificial and covalent bonds in enhancing self-healing and mechanical performance of the composite simultaneously. Here, we first incorporated weaker electrostatic and H-bonds via metal-ion-mediated heteroaggregation between NR (bearing proteins and phospholipids) and SiO2.49 Further, stronger covalent bonds were established via the bis [3-(triethoxysilyl) propyl] tetrasulfide (TESPT) silane coupling agent. This approach resolved two fundamental issues discussed above: (i) first, it allowed us to improve the dispersion of SiO2 particles in the NR matrix by avoiding their random aggregation and (ii) it enabled the incorporation of reversible sacrificial bonds. However, these kinds of ionic cross-links have not yet been realized in self-healing composites. For example, an epoxy-functionalized NR with low degree of cross-links has been reported to display self-healing ability at higher temperature via interdiffusion of NR chains along with stronger epoxy interactions. Differing from the previous reports,50,51 we effectively synthesized NR–SiO2 composites via Mg2+-induced heteroaggregation to produce a reversible supramolecular network mainly constructed by ion–dipole, electrostatic, and H-bonding interactions at room temperature. Further, the covalent network between SiO2 and NR was established by adding TESPT to the resultant composite. As anticipated, this supramolecular network imparted an exceptional self-healing characteristic to the NR with simultaneously improved mechanical performance than that of the conventionally prepared composites. Scheme 1 depicts the sequence of events leading to the desired cross-linked network of the composite.
Scheme 1. Schematic of the Synthetic Pathway for the Formation of NR–SiO2 Composite Illustrating the Supramolecular and Covalent Cross-Link Networks in the Composite Material.

2. Results and Discussion
2.1. Key Strategy in the Making of NR–SiO2 Composites
The essential feature of our approach in the fabrication of self-healing and high-mechanical-strength composites was to introduce H-bonds and ionic cross-links between NR and SiO2 prior to covalent bond formation. Our strategy was based on a controlled metal-ion-induced heteroaggregation that allowed generation of massive ion–dipole and H-bond cross-links. The proteins of NR could be ionized in three different ways depending on the pH of the system (Scheme 2). At a pH less than the isoelectric point (4.5–5.0), the NR particles are positively charged and remain neutral at isoelectric conditions.
Scheme 2. Effect of pH on the Net Charge on the Linked Protein (P) Component of NR.
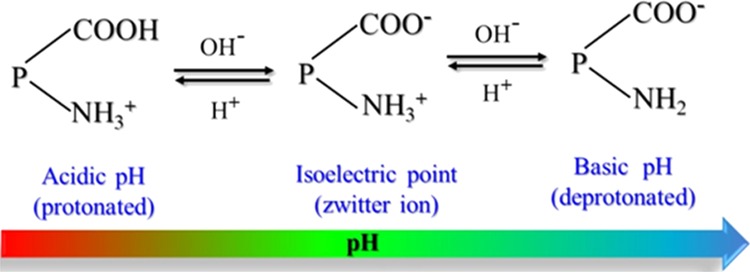
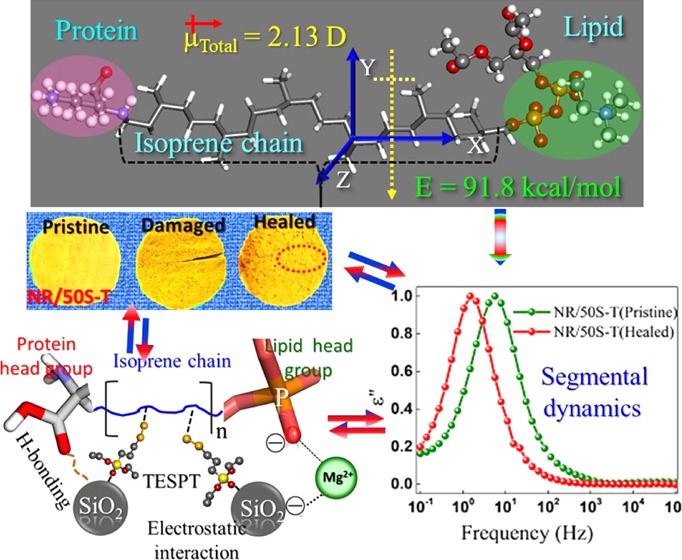
At a basic pH (pH ∼ 10) higher than the isoelectric point, acidic ionization takes place and NR particles would thus be negatively charged. Besides, lipid molecules in natural rubber carry permanent negative charge. Therefore, the electrostatic forces exerted by Mg2+ ions in a binary suspension of repulsive charged colloids of NR and SiO2 particles would drive the mutual assembly via reversible electrostatic and H-bonds. Simultaneously, the buffering ability of the amino acid on NR would trigger self-assembly of SiO2 by preventing their random aggregation. Thus, the resultant composites were stabilized via the formation of supramolecular networks, initiated from ion–dipole and H-bonding interactions, as confirmed by Fourier transform infrared (FTIR) analysis (Figure S1, Supporting Information (SI)). For specific nomenclature of the composites, for example NR/50S-T, etc., refer to the Experimental Section.
2.2. Molecular Dynamics in NR and NR–SiO2 Composites
2.2.1. Segmental Dynamics in NR–SiO2 Composites as Revealed by Broadband Dielectric Spectroscopy (BDS)
Natural rubber comprises low-polarity cis-polyisoprene chains. Because of the asymmetry in its molecular structure, NR has components of dipole moment both parallel and perpendicular to the chain contour and therefore exhibits two relaxation modes: (i) a segmental mode at temperatures above the glass-transition temperature (Tg ∼ −65 °C) with its origin in local motions of the perpendicular dipole moment component of the segmental NR chain motion and (ii) a dielectric normal mode caused by the parallel dipole moment component along the chain contour at temperatures well beyond Tg.52−54 It has been reported that the presence of cross-links slows down the segmental dynamics of polymer chains and lowers the frequency shift of the segmental mode due to the formation of polar functionalities such as (i) mono, di, or polysulfide cross-links, (ii) carbonyl groups, and (iii) S–C bonds in vulcanized composites.55−58 Therefore, cross-linking of NR with sulfur and TESPT will increase the total number of polar groups, which restricts the mobility of polymer chains and would cause the segmental mode to shift toward lower frequencies. As shown in Figure 1a, the molecular mobility of the polymer segments in pristine NR is influenced by the presence of filler and by cross-linking, with the position of the maxima shifting to lower frequencies in both the composites. However, it is interesting to note that the lower frequency shifts in NR/50S-T are much more predominant compared with DNR/50S-T. Such a variation in the NR chain dynamics could be attributed to enhanced dipolar interactions caused by Mg2+ present in NR50S-T that had strongly restricted the segmental dynamics of the neighboring NR chains in the composite. In essence, Mg2+ established a molecular bridge (salt bridge) that clamped the negatively charged NR and SiO2 colloidal particles and formed a supramolecular network in the composite. These structural developments contributed to the overall cross-linked network, serving as a new kind of ionic cross-linker along with the disulfide (S–S) cross-links. Such a network structure was missing in DNR/50S-T that was fabricated using a dry mixing procedure. Consequently, the cross-linking of NR chains in NR/50S-T could effectively take place with increasing number of dipoles involved in the segmental relaxation process, causing the segmental mode to shift to lower frequencies. The presence of disulfide and C–S cross-links was further confirmed by Raman spectra, vide Figure 1b. Prominent Raman bands at around 500 and 700 cm–1 appeared due to S–S and C–S linkages in the composite.59 The more predominant S–S and C–S peak intensities in NR/50S-T, compared to the other composite, implied an increased fraction of disulfide bonds created between the NR chains, in accordance with the bound rubber content, as discussed in Section 2.3.
Figure 1.

(a) Normalized dielectric loss ε″ vs frequency spectra for pristine NR and DNR/50S-T and NR/50S-T NR–SiO2 composites in the segmental mode region at temperature T = −40 °C. (b) Raman spectra of pristine NR and composites depicting the disulfide (S–S) cross-links.
2.2.2. Molecular Dipole Oscillations in Natural Rubber (NR)
Molecular oscillation of dipoles that are related to the molecular mobility of side groups, segments, or whole polymer chains undergoes different relaxation processes, which can be studied experimentally via BDS. However, molecular-level understanding is necessary to probe the mechanism of such dipole oscillations and their response to the applied electric field. Consequently, molecular mechanics (MM+) simulations were performed to shed light on the oscillation of molecular dipoles under an external electric field. The molecular structure for NR was modeled via MM+ simulations, where the main approach was to obtain the minimum-energy configuration of the potential energy surface of the studied molecular system using equations of classical mechanics that describe the structure and physical properties of molecules. The representative model is shown in Figure 2, depicting the orientation of the molecular dipole moment perpendicular to the chain contour (segmental mode) adopted from our previous studies.49
Figure 2.
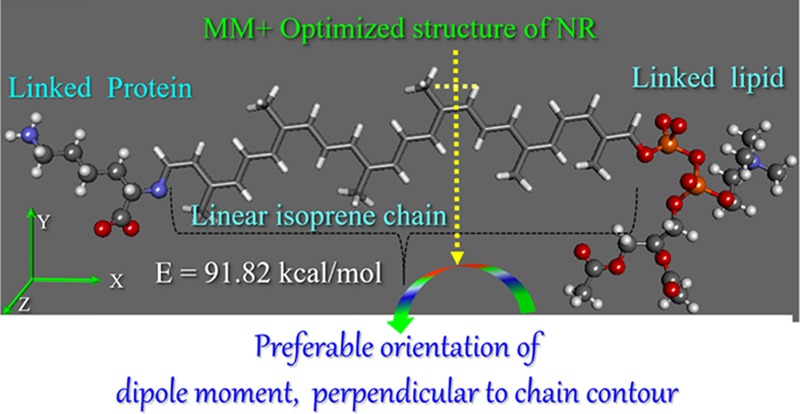
Representative optimized structure of natural rubber (NR) with five repeating isoprene chains bifunctionalized with lysine and phospholipids.
To study the effect of applied electric filed on the oscillation of molecular dipoles, an electric field interaction term was introduced into the Hamiltonian of the modeled NR system and the corresponding equations were solved numerically. The dipolar oscillation in NR was calculated using a consequential set-by-step procedure with varying electric field, using single-point calculations. Figure 3a presents the possible orientation of molecular dipoles in the model NR chain in the absence of an external electric field (E = 0). Our calculation shows that the total dipole moment (μTotal = 2.132 D) of the NR chain is oriented perpendicular to the molecular chain axis/along the chain contour. However, interestingly, the component of the dipole moment along the z direction, μz = 1.955 D, perpendicular to the molecular axis was found higher than the molecular dipole in x direction, μx = 0.177 D, parallel to the molecular axis, with μy = 0. Therefore, as shown in Figure 2, NR molecular chains prefer to lie with the orientation of their dipole perpendicular to the molecular axis in the absence of electric field (E = 0). As discussed above, the μz and μx components of the dipole moment for the modeled NR chain can be related to the experimentally observed segmental and normal modes, respectively, via BDS. As shown in Figure 3b, the application of external electric field E along the direction of average dipole moment causes reorientation of the dipole and leads to switching of the molecular dipole at E = ∼2 MV/cm. The driving force for reorientation could be the minimization of the total energy of the modeled NR chain (66.79 vs 91.82 kcal/mol at E = 0, vide Figure 2), which changes for different dipole configurations. Therefore, the mechanism of molecular switching in the NR chain by a simple rotation of molecular dipole along the applied electric field can also be a very useful parameter to unravel the polymer chain dynamics near the filler surface, for which we intended to carry out further BDS studies.
Figure 3.
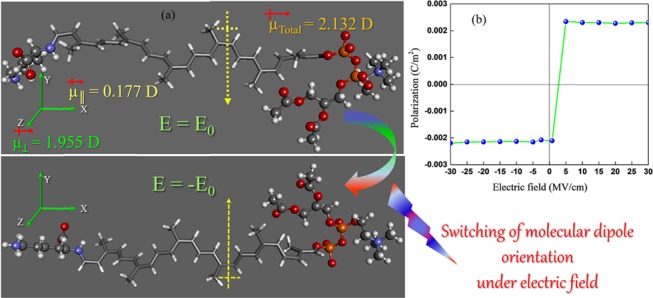
(a) Variation of the dipole orientation for the NR molecular chain upon application of electric field. (b) Depiction of polarization switching in the model NR chain under an electric field (E ∼ 2 MV/cm).
2.3. Characteristic Swelling Behavior of the Composites
Bound rubber (BR) is defined as the rubber that cannot be dissolved by a good solvent. Toluene is an excellent solvent for NR, while it is a bad solvent for silica. Figure 4a shows the swelling behavior of the prepared NR–SiO2 composites. In this process, neat NR dissolved in toluene within 1 h, while NR–SiO2 composites could not be fully swollen in toluene even after 7 days. It is obvious that the upper part of the vial is the clear toluene phase, while the lower part is the NR–SiO2 composite. The inclusion of SiO2 into the NR matrix could enhance the toluene resistance of the composites, which could result from two factors:60 (i) tortuosity effect and (ii) decreased NR polymer free volume fraction. Further, the mesoporous structure of silica can strongly enable the physical and mechanical entrapment of NR chains by interlocking, thereby acting as a driving force to improve the mechanical reinforcement. As shown in Figure 4b, the bound rubber content of NR/50S-0T with no TESPT was the lowest due to weak physical cross-links between SiO2 and NR. However, the BR contents in NR/50S-T (5 phr TESPT), NR/50S-RT (R: reduced, 2.5 phr TESPT), and DNR/50S-T (5 phr TESPT under dry condition) composites were higher compared to those in the composite without TESPT.
Figure 4.

(A) Photographs of the swelling behavior of NR–SiO2 composites in toluene at specific time intervals: (a) NR, (b) NR/75S-T, (c) NR/50S-T, (d) NR/25S-T, (e) NR/50S-0T, (f) NR/50S-RT, and (g) DNR/50S-T. (B) Comparison of bound rubber contents in NR–SiO2 composites.
Addition of TESPT (silane coupling agent) established an improved coupling between NR and SiO2 that further cross-linked the NR chains during vulcanization. Interestingly, the BR content of NR/50S-RT (reduced TESPT) is almost similar to that of NR/50S-T and higher than that of DNR/50S-T, which could be due to controlled dispersion of the filler and enhanced reactivity of TESPT even at low levels.
2.4. Filler Networking and Mechanical Behavior of NR–SiO2 Composites
The storage modulus (G′) of the NR–SiO2 composites in the rubbery state can reveal the extent of SiO2–SiO2 interactions. Figure 5a clearly shows that the G′ of all of the composites decreases significantly upon increasing strain. This phenomenon is called “Payne effect”,61 expressed by the difference between G′ values at small and large strains (ΔG′), and occurs due to breakdown of the filler network that follows release of the trapped NR under oscillatory shear.62,63 As expected, the NR/75S-T (75 phr SiO2) and NR/25S-T (25 phr SiO2) composites exhibited highest and lowest ΔG′ values. In the former case, the high concentration of SiO2 particles led to a significantly higher SiO2–SiO2 interaction than in the later. On the other hand, the higher ΔG′ in the case of the DNR/50S-T composite can be attributed to poor dispersion of filler particles in the NR matrix. Here, severe agglomeration among the filler particles even in the presence of TESPT could be observed, as evident from transmission electron microscopy (TEM) analysis (Figure S3, SI). Further, the NR/50S-T and NR/50S-RT composites (prepared via liquid–liquid mixing) exhibited lower magnitude (ΔG′) of Payne effect, suggesting a more uniform dispersion of the filler and a weaker filler network in the composites. It is in correspondence with the conclusion that the liquid mixing approach mediated by Mg2+ ions has a positive effect on the dispersion of silica by preventing their random aggregation. A close correlation between the Si–O–Si peak maximum and the Payne effect was validated via Figure S4, SI. With increasing silica loading, the SiO2–SiO2 interaction enhanced, resulting in Si–O–Si frequency shifts to lower wavenumbers.
Figure 5.

(a) Payne effect illustrating the filler networking in the NR–SiO2 composites. (b) Representative stress–strain curves of the NR–SiO2 composites depicting their mechanical performance.
The stress–strain behavior straightly reflects the mechanical performance of NR–SiO2 composites. Representative stress–strain curves for the composites are plotted in Figure 5b. The NR/50S-0T composite has relatively poor mechanical performance with a lower modulus (stress at 300% strain) as 6.96 MPa. Further, the elongation at break was as high as 495% for this composite. Here, Mg2+ could only facilitate the dispersion of silica in the NR matrix by establishing a salt bridge between NR and SiO2 colloidal particles. In spite of this, the interfacial interaction was still weak, as Mg2+ could not covalently bridge silica and the NR chains. The remarkable enhancement in the extensibility of the NR/50S-0T composite is due to slippage of the entangled NR chains that are noncovalently linked to SiO2 due to the absence of the cross-linker (TESPT). A noteworthy scenario was that the addition of TESPT significantly improved the modulus and tensile strength of NR–SiO2 composites, as given in Table 1. For instance, the calculated modulus and tensile strength of NR/50S-T of 17.55 and 18.5 MPa, respectively, were much better than those of the DNR/50S-T composite (16.94 and 17.95 MPa, respectively). The enhanced mechanical performance of NR/50S-T could be due to the highly controlled dispersion of silica particles attained via liquid–liquid mixing. Simultaneously, the incorporation of covalent cross-links between NR and SiO2 further enhanced the filler reinforcing properties. However, such improvement in the reinforcing effect (tensile strength) reduced the material’s extensibility, and as a result, the stretching of NR rubber chains that are anchored and entrapped on the SiO2 surface was effectively arrested. Consequently, the elongation at break decreased. Notably, the reduced elongation at break for NR/50S-T and NR/50S-RT (312 and 345%, respectively) as compared to that for NR/50S-0T (495% with no TESPT) indicated the NR chain extensibility to be affected by TESPT due to strong covalent cross-links. Moreover, the increasing amount of NR molecular domain that was trapped inside the silica pores partially behaved like “dead” polymer and might have increased the modulus and strength of the composite. Altogether, our present work has the following improvements: (1) the TESPT dosage could be significantly reduced by adopting the liquid mixing approach, where controlled silica dispersion was attained via Mg2+ ions, following which TESPT was added, and (2) the processing and performance characteristics became superior to those of the traditional composite.
Table 1. Mechanical Properties of NR–SiO2 Compositesa.
| sample | modulus (MPa) | tensile strength (MPa) | elongation at break (%) | toughness (MJ/m3) |
|---|---|---|---|---|
| NR | 4.39 (0. 4) | 7.96 (0. 6) | 580 (9.0) | 8.34 (0.5) |
| NR/25S-T | 15.32 (0.5) | 20.2 (0.4) | 358 (11.5) | 10.14 (0.6) |
| NR/50S-T | 17.55 (0. 6) | 18.5 (0. 4) | 312 (10.3) | 7. 72 (0.7) |
| NR/75S-T | 18.97 (0.7) | 20.4 (0.3) | 315 (10.1) | 8.97 (0.2) |
| NR/50S-0T | 6.91 (0.4) | 14.3 (0.3) | 495 (7.3) | 12.46 (0.8) |
| NR/50S-RT | 16.12 (0.6) | 19.9 (0.4) | 345 (13) | 9.64 (0.2) |
| DNR/50S-T | 16.94 (0.2) | 17.95 (0.3) | 313 (11.5) | 7.94 (0.4) |
The standard error is presented in parentheses
2.5. Monitoring Self-Healing Characteristics of the NR–SiO2 Composites
2.5.1. Macroscale Mechanical Testing Analysis
The self-healing behavior of NR–SiO2 composites was evaluated by cutting the sample at the middle part using a clean surgical blade. The cut surfaces were brought together immediately with a slight pressure to assure the docking of the cut surfaces. The samples were allowed to heal upon heating them from room temperature (22 °C) to 60 °C for 24 h, and the healing performance was noted by acquiring optical micrographs of the specimen as well as manual stretching (Figure S5, SI). Better healing was observed at 50 °C after 24 h of healing time. According to Tee et al.,64 even though the NR chain mobility at higher healing temperatures evidently facilitates the healing efficiency, the extreme deterioration of electrostatic and ion pair interactions could decline the ionic bond strength and disturb the reconstruction ability of the supramolecular networks.65 Hence, in the present investigation, the healing performance was evaluated at 50 °C. The healing integrity of the composite is clearly observed in Figure 6a,b. Interestingly, the healed sample, particularly NR/50S-T, did not fracture at the cut even by manual stretching. However, the healing efficiency of DNR/50S-T, the covalently cross-linked composite, was poor, as shown in Figure S6, SI. Such a response can be attributed to the presence of sacrificial networks in NR/50S-T that exchanged the formed ionic species near the cut surface and reconstructed the supramolecular network during healing. Thus, the self-healing ability of NR/50S-T was endorsed to the reversible ionic and H-bonding interactions that increased the adhesion and repaired the cut surfaces. The meticulous self-healing behavior at the junction of the cut position was evaluated via optical microscopy, as shown in Figure 6a. Even though the crack between the two cut surfaces remained apparent, a clear shrinkage was noticed after healing by self-fusion of the cut surfaces. Markedly, the crack between the two cut surfaces for NR/50S-T and NR/50S-0T composites was almost unnoticeable, confirming the effective self-healing effect. However, the crack between the cut surfaces of the DNR/50S-T composite could be markedly seen owing to partial healing. The self-healing capability was also evidenced in the stress–strain measurements of the healed samples at 50 °C for various healing times. The tensile behavior of the original and self-healed NR/50S-T and DNR/50S-T composites at 50 °C for 24 h exhibited best healing efficiencies, as shown in Figure 7a,b. The stress–strain curves of healed composites closely followed the shapes of the original composites. The area under the curve (tensile energy) before and after healing was used to calculate the healing performance of the composites, and the values are tabulated in Table 2. It is worth mentioning that the self-healing ability of the NR/50S-T composite originated from the effective reversible ion–dipole and H-bond sacrificial networks in addition to the S–S cross-links.
Figure 6.
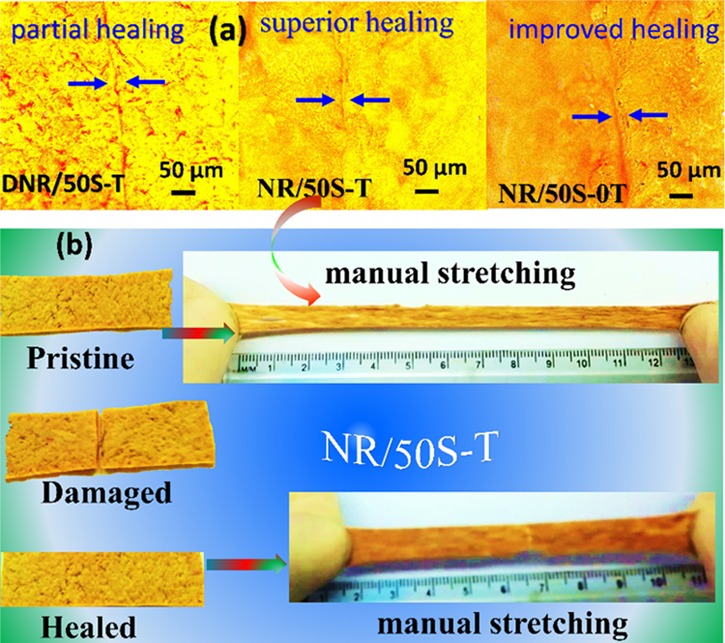
(a) Optical microscopy images of self-healing behavior of DNR/50S-T, NR/50S-T, and NR/50S-0T composites and (b) photographs of the NR/50S-T composite illustrating self-healing behavior at 50 °C and 24 h healing time.
Figure 7.

Stress–strain curves of the original and self-healed composites at 50 °C at various healing times, (a) NR/50S-T and (b) DNR/50S-T, and (c) comparison of mechanical performance of the healed samples at 50 °C after 24 h healing time. (d) Healing efficiencies of NR/50S-T and DNR/50S-T composites at various healing times.
Table 2. Comparison of Mechanical Performance of NR/50S-T and DNR/50S-T Composites before and after Healinga.
| sample | tensile strength (MPa) | toughness (MJ/m3) | healing efficiency (%) |
|---|---|---|---|
| NR/50S-T (original) | 18.5 (0.4) | 7.72 (0.7) | |
| NR/50S-T (healed) | 17.1 (0.3) | 6.1 (0.5) | 79.01 (1.5) |
| DNR/50S-T (original) | 17.94 (0.3) | 7.94 (0.4) | |
| DNR/50S-T (healed) | 14.2 (0.2) | 4.1 (0.2) | 51.6 (1.8) |
The standard error is presented in parentheses
The slightly higher modulus in self-healed composites has resulted from the labile polysulfide and disulfide bridges, allowing for the rearrangement of the broken bonds at the healed interface, in addition to the supramolecular network. Thus, the total number of polar groups involved has increased during the healing reaction, thereby changing the molecular mobility due to the reconnection of the chains. This redistribution of the structure led to the formation of a new network with different architectures with respect to the pristine samples. As a result, the total cross-links across the cut surface increased after the healing reaction. This phenomenon is further supported by the frequency shifts in the dielectric spectra, as shown in Figure 8. Similar results were reported by Hernández et al.55,66 in natural rubber networks with multiple reformable bonds. Their findings were remarkable in terms of understanding the formation of new heterogeneous networks during the healing process, especially during the restoration of macroscopic damages due to increased number of ion–dipole interactions in the healed composite. Similarly, Li et al.67 demonstrated the effects of thermal aging on the tensile behavior of natural rubber composites. Here, the observed increase in the modulus was attributed to the increased number of cross-links upon aging the composite. In the present case, the damaged samples were exposed to a certain temperature for a particular time for the healing reaction to proceed, leading to an increasing modulus.
Figure 8.

Normalized dielectric loss ε′ vs frequency spectra for the composites in the segmental mode region at a selected temperature (T = −40 °C): (a) pristine and healed NR/50S-T composite, (b) pristine and healed DNR/50S-T composite, healed for 24 h at 50 °C.
Having determined the mechanical performance and macroscopic healing ability of the NR–SiO2 composites, the following section is devoted to unravel the mechanism of molecular processes involved in the healing behavior by means of dielectric relaxation spectroscopy.
2.5.2. Mechanism of Self-Healing: A Broadband Dielectric Perspective
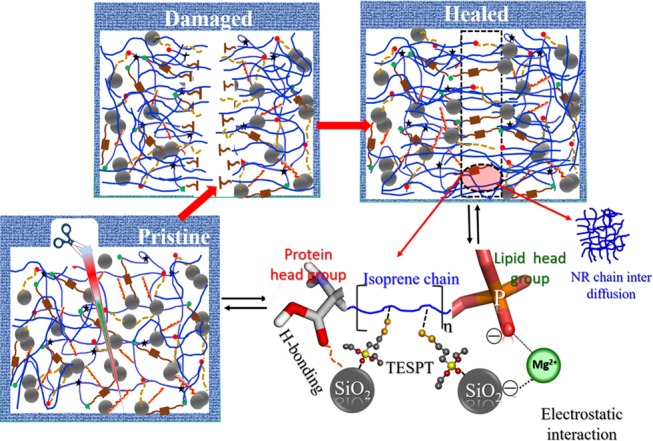
Despite the fact that healing of macroscopic and optically measureable damage is important, the healing of unseen internal damage (chain scission) to the polymer network is of equal significance. Dielectric spectroscopy is a sensitive method for probing such multilevel molecular relaxations over a broad frequency range of 10–1 < f (Hz) < 107. Molecular oscillations of dipoles that are related to the molecular mobility of side groups, segments, or whole polymer chains, which show up as different relaxation processes, are highly sensitive to BDS. Accordingly, the evolution of the segmental dynamics during the healing of internal damage in the NR–SiO2 composite was studied by BDS.68−78 In this section, we do not emphasize on the quantification of the damage and healing as such by BDS but monitor the molecular processes and the state of the material at the end of the healing treatment. Information on the structural state of the material after healing was followed by taking the molecular mobility spectrum to probe the segmental relaxation process over a wide frequency range and at a selected temperature (T = −40 °C) for pristine NR and the composites, as shown in Figure 8. This temperature, just above the Tg of NR, was chosen since the segmental relaxation was well resolved within the studied frequency window.79Figure 8 shows the normalized dielectric loss spectra of pristine and healed NR/50S-T (fabricated via liquid–liquid mixing) and DNR/50S-T (prepared by the dry compounding procedure) composites. It is interesting to note that the segmental mode of the healed composites was shifted to lower frequencies compared with the pristine composites. This effect was more predominant in the NR/50S-T composite, as shown in Figure 8a, than in DNR/50-T, as shown in Figure 8b. Such a behavior can be explained by the state of the polymer network on account of healing. Schönhals and Schlosser80 proposed a phenomenological model for amorphous polymers in which the shape of the dielectric loss peak was related to the behavior at low and high frequencies, controlled by specific inter- and intramolecular interactions, respectively. Accordingly, the self-healing process in NR/50S-T was mainly attributed to changes in the NR chain molecular mobility in addition to its reorganized structure. The main contribution to the segmental dynamics came from large-scale motions at low frequencies from the respective ion–dipole, H-bonding, and electrostatic interactions including the S–S cross-links. However, at higher frequencies, intrachain mobility/diffusion contributed to the observed segmental relaxation. Scheme 3 shows the schematic depiction of the healing process in NR/50S-T. The plausible mechanism of the self-healing process could therefore be interdiffusion of mobile NR chains across the boundary faces in addition to the supramolecular networks that exchange the newly formed dangling free groups near the cut surface within the fracture region, enabling reconstruction.
Scheme 3. Self-Healing Mechanism in the NR–SiO2 Composite Illustrating the Reconstruction of Supramolecular Networks in the Composite Material and Interpenetration of Mobile NR Chains.
3. Conclusions
We have engineered high-mechanical-strength, yet self-healable, dual hybrid network NR–SiO2 composites. The reversible ionic and H-bonding supramolecular network between the NR and SiO2 facilitated the self-healing capability. The covalent cross-links, physical entanglements, and interlocks improved the mechanical and thermal stability of the composites. The tensile tests clearly indicated that the composite without TESPT has self-healing ability but inferior mechanical performance. The addition of Mg2+ could only facilitate the dispersion of silica by disturbing the H-bonding among silica particles, thereby establishing a molecular bridge between NR and SiO2 via ion–dipole and H-bonding interactions. Further, the incorporation of TESPT endowed the composites with superior mechanical, self-healing, and processing characteristics simultaneously. Specifically, the segmental relaxation dynamics studied via BDS unraveled the characteristic molecular motions in the composite, leading to the mechanism of self-healing. Collectively, the present methodology promises (a) an improved silica dispersion in NR, (b) a greatly reduced dosage of TESPT; (c) a possible way to save mixing energy due to easy incorporation of SiO2, and (d) fabrication of industrially feasible composites with superior mechanical and effective self-healing properties and opens up a promising pathway to engineer smart multifunctional composites with versatile functions.
4. Experimental Section
4.1. Materials
Natural rubber latex (NR) with a total solid content of 60% was purchased from Mardec R.K. Latex, Kerala, India. Magnesium sulfate (MgSO4) was purchased from Sigma-Aldrich, and silica and bis [3-(triethoxysilyl) propyl] tetrasulfide (TESPT) were provided by MRF Ltd. All reagents were of analytical grade and used without further purification. The solutions used were made in non-ionic Milli-Q water with 18.2 MΩ cm resistivity. The salt concentrations were appropriately adjusted to obtain an ionic strength of 100 mM.
4.2. Synthesis of Self-Assembled NR–SiO2 Composites
NR–SiO2 master batches were prepared by wet compounding and self-assembling techniques. Briefly, SiO2 particles of 25, 50, and 75 phr (phr = weight of filler per 100 g of NR) were dispersed in 200 mL of Milli-Q water and sonicated for ∼10 min; subsequently, SiO2 dispersion was added to NR (100 phr) suspension followed by MgSO4 addition so as to reach an ionic strength of 100 mM. The samples were left for 1 h at room temperature. After removing the supernatant, the samples were dried in a vacuum oven at 50 °C for 72 h.
4.3. Preparation of Compounded Rubber Composites
To prepare the NR–SiO2 rubber composites, NR–SiO2 master batches were compounded with rubber ingredients with a basic formulation as listed in Table 3 on a two-roll mill according to the processes described in the Supporting Information. Scheme 1 collates the sequence of processes toward the synthesis of the composites. Additionally, composites with no TESPT and with 2.5 phr of reduced TESPT and a conventional composite via dry mixing were also synthesized. Henceforth, the samples are abbreviated NR/25S-T, NR/50S-T, NR/75S-T, NR/50S-0T, NR/50S-RT, and DNR/5S0-T in further discussions.
Table 3. Composition of the Composite Materialsa.
| composition
(phr) | |||||||
|---|---|---|---|---|---|---|---|
| sample | NR | SiO2 | SA | ZnO | T | CBS | S |
| NR/25S-T | 100 | 25 | 2 | 3 | 2.5 | 1 | 1.5 |
| NR/50S-T | 100 | 50 | 2 | 3 | 5 | 1 | 1.5 |
| NR/75S-T | 100 | 75 | 2 | 3 | 7.5 | 1 | 1.5 |
| NR/50S-0T | 100 | 50 | 2 | 3 | 0 | 1 | 1.5 |
| NR/50S-RT | 100 | 50 | 2 | 3 | 2.5 | 1 | 1.5 |
| DNR/50S-T | 100 | 50 | 2 | 3 | 5 | 1 | 1.5 |
Rubber ingredients: zinc oxide (ZnO), stearic acid (SA), TESPT (T), N-cyclohexyl-2-benzothiazole sulfonamide (CBS), and sulfur (S). NR/25S-T should be read as 100 phr NR; 25 phr SiO2 in the presence of TESPT (T). NR/50S-0T is the composite without TESPT. NR/50S-RT is the composite with reduced TESPT (T). DNR/50S-T is the composite prepared under dry mixing.
4.4. Characterization of NR–SiO2 Composites
The surface area and porosity of the SiO2 filler were measured by the Brunauer–Emmett–Teller (BET) technique using a Quantachrome Nova2000e series surface area analyzer. The structural morphology of the composites was unraveled with transmission electron microscopy (TEM) with a JEM-1010, JEOL instrument at an acceleration voltage of 80 kV. Thin slices of the samples with a thickness of ∼120 nm were obtained by cryo-microtoming at −120 °C using a Leica EM FC6 instrument and were transferred onto carbon-coated copper grids. Following this, the interaction between SiO2 and NR in the nanocomposites was studied with a Spectrum Two PerkinElmer FTIR spectrophotometer by acquiring spectra at 4 cm–1 resolutions, averaged over 32 scans. An FT-Raman spectrometer (Bruker RAMII) working in a confocal mode, connected to a Leica microscope, was used for the measurement of the Raman spectra. The laser beam was focused by a 100× magnification objective of a confocal microscope. Each spectrum was collected in the frequency range 100–3500 cm–1 over 60 s and with 10 accumulations to avoid electronic peaks and average background. Thermogravimetric analyses (TGA) were performed with a TGA Discovery series, TA Instruments, New Castle, DE machine. Samples were heated over a temperature range of 22 °C (room temperature) to 900 °C at a heating rate of 20 °C/min under a N2 atmosphere.
4.5. Broadband Dielectric Spectroscopy (BDS)
BDS measurements were done on an α high-resolution dielectric analyzer (Novocontrol Technologies GmbH, Hundsangen, Germany). The complex permittivity ε*(ω) = ε′(ω) – iε″(ω) was measured by carrying out isothermal frequency sweeps over a wide frequency range of 10–1 < f (Hz) < 107 (where f = ω/2π is the frequency of the applied electric field and ω is the angular frequency). The circular-shaped samples (pristine and healed composites) were placed in the dielectric cell between two parallel gold-plated electrodes for measuring the permittivity. The thickness and diameter of each sample were about 1.5 and 20 mm, respectively. The temperature during the measurement was controlled by a nitrogen jet; a temperature error of 0.1 K during every single frequency sweep was observed.
Swelling study was performed as
follows: the sediments of NR and NR–SiO2 composites
were cut into small pieces and dipped in toluene for 1 week at room
temperature with 0.5 g NR content in all of the samples. To ensure
sufficient swelling, the solvent was renewed every 24 h. Optical images
of the samples at specific time intervals were taken with a camera.
The bound rubber content (BR) was measured by extracting the dissolved
NR after 7 days at room temperature. The remnant was then dried at
50 °C in a vacuum oven to a constant weight. The BR was calculated
as 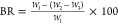 , where W1 is the weight of the rubber
component in the
compound and W2 and W3 are the weights of the compound before and after extraction,
respectively.
, where W1 is the weight of the rubber
component in the
compound and W2 and W3 are the weights of the compound before and after extraction,
respectively.
Tensile tests were conducted on a dumbbell specimen
using a Zwick/Roell-Z010 testing machine with an optical elongation
sensor at a cross-head speed rate of 200 mm/min at room temperature;
five measurements were carried out for each sample. The dynamic rheological
properties of the rubber compounds were measured by a RPA2000 rubber
processing analyzer (RPA, α Technologies Co.) with strain amplitudes
varying from 0.28 to 400%, at a frequency of 1 Hz. For self-healing
tests, dumbbell-shaped samples were cut in the middle with a surgical
blade and the two cut surfaces were brought back in contact by hand
and then allowed to heal at 50 °C. After healing for 24 h, the
samples were consequently tested on a tensile tester. Subsequently,
the healing efficiency “η” was calculated as 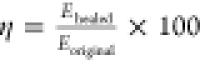 , where Eoriginal and Ehealed correspond to the respective tensile
energy (E)
for the original and healed samples.
, where Eoriginal and Ehealed correspond to the respective tensile
energy (E)
for the original and healed samples.
4.6. Computational Methodology
Molecular simulations were performed using MM+ molecular mechanics force field with Hyperchem version 8.0, Hypercube Inc. Here, geometry optimization was pursued under the Polak–Ribiere conjugate gradient method. Single-point calculations were carried out to determine the final optimized values of the physical properties, such as dipole moment, volume, polarizability, etc. This procedure was repeated under every value of electric field applied for a correlation with the results obtained from BDS.
Acknowledgments
Thanks are due to the Department of Physics, IIT-Madras, for making available the Broad Band Dielectric Spectroscopy facility. Financial support from MRF Ltd. Chennai, India, is gratefully acknowledged.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01243.
Compounding procedure and supramolecular interactions via spectral characterization using FTIR, TEM images, optical photographs of composites illustrating self-healing behavior, BET isotherm, TGA, and the swelling behavior of the composites (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tang Z.; Huang J.; Wu X.; Guo B.; Zhang L.; Liu F. Interface Engineering toward Promoting Silanization by Ionic Liquid for High-Performance Rubber/Silica Composites. Ind. Eng. Chem. Res. 2015, 54, 10747–10756. 10.1021/acs.iecr.5b03146. [DOI] [Google Scholar]
- Baeza G. P.; Genix A.-C.; Degrandcourt C.; Petitjean L.; Gummel J.; Schweins R.; Couty M.; Oberdisse J. Effect of Grafting on Rheology and Structure of a Simplified Industrial Nanocomposite Silica/SBR. Macromolecules 2013, 46, 6621–6633. 10.1021/ma401016d. [DOI] [Google Scholar]
- Morozov I. A. Structural-Mechanical AFM Study of Surface Defects in Natural Rubber Vulcanizates. Macromolecules 2016, 49, 5985–5992. 10.1021/acs.macromol.6b01309. [DOI] [Google Scholar]
- Mujtaba A.; Keller M.; Ilisch S.; Radusch H. J.; Thurn-Albrecht T.; Saalwächter K.; Beiner M. Mechanical Properties and Cross-Link Density of Styrene-Butadiene Model Composites Containing Fillers with Bimodal Particle Size Distribution. Macromolecules 2012, 45, 6504. 10.1021/ma300925p. [DOI] [Google Scholar]
- Tang Z.; Zhang L.; Feng W.; Guo B.; Liu F.; Jia D. Rational Design of Graphene Surface Chemistry for High-Performance Rubber/Graphene Composites. Macromolecules 2014, 47, 8663–8673. 10.1021/ma502201e. [DOI] [Google Scholar]
- Bokobza L. Multiwall carbon nanotube elastomeric composites: A review. Polymer 2007, 48, 4907–4920. 10.1016/j.polymer.2007.06.046. [DOI] [Google Scholar]
- a Dannenberg E. M.Carbon Black, Physics, Chemistry, and Elastomer Reinforcement; Jean-Baptiste D.; Andries V., Eds.; Dekker: New York, 1976; p 362. [Google Scholar]; b Dannenberg E. M. Carbon black, physics, chemistry, and elastomer reinforcement, Jean-Baptiste Donnet and Andries Voet, Dekker, New York, 1976, 362 pp. $39.50. J. Polym. Sci., Polym. Lett. Ed. 1977, 15, 631–632. 10.1002/pol.1977.130151013. [DOI] [Google Scholar]
- Klüppel M.; Heinrich G. Fractal Structures in Carbon Black Reinforced Rubbers. Rubber Chem. Technol. 1995, 68, 623–651. 10.5254/1.3538763. [DOI] [Google Scholar]
- Kraus G. Reinforcement of Elastomers by Carbon Black. Rubber Chem. Technol. 1978, 51, 297–321. 10.5254/1.3545836. [DOI] [Google Scholar]
- Vleugels N.; Pille-Wolf W.; Dierkes W. K.; Noordermeer J. W. M. Understanding The Influence Of Oligomeric Resins On Traction And Rolling Resistance Of Silica-Reinforced Tire Treads. Rubber Chem. Technol. 2015, 88, 65–79. 10.5254/rct.14.86947. [DOI] [Google Scholar]
- Miloskovska E.; Friedrichs C.; Hristova-Bogaerds D.; Persenair O.; van Duin M.; Hansen M. R.; de With G. Chemical Mapping of Silica Prepared via Sol-Gel Reaction in Rubber Nanocomposites. Macromolecules 2015, 48, 1093–1103. 10.1021/ma5020929. [DOI] [Google Scholar]
- Angellier H.; Molina-Boisseau S.; Lebrun L.; Dufresne A. Processing and Structural Properties of Waxy Maize Starch Nanocrystals Reinforced Natural Rubber. Macromolecules 2005, 38, 3783–3792. 10.1021/ma050054z. [DOI] [Google Scholar]
- Stöckelhuber K. W.; Svistkov A. S.; Pelevin A. G.; Heinrich G. Impact of Filler Surface Modification on Large Scale Mechanics of Styrene Butadiene/Silica Rubber Composites. Macromolecules 2011, 44, 4366–4381. 10.1021/ma1026077. [DOI] [Google Scholar]
- Goerl U.; Hunsche A.; Mueller A.; Koban H. G. Investigations into the Silica/Silane Reaction System. Rubber Chem. Technol. 1997, 70, 608–623. 10.5254/1.3538447. [DOI] [Google Scholar]
- Wolff S. Chemical Aspects of Rubber Reinforcement by Fillers. Rubber Chem. Technol. 1996, 69, 325–346. 10.5254/1.3538376. [DOI] [Google Scholar]
- Chen L.; Jia Z.; Tang Y.; Wu L.; Luo Y.; Jia D. Novel functional silica nanoparticles for rubber vulcanization and reinforcement. Compos. Sci. Technol. 2017, 144, 11–17. 10.1016/j.compscitech.2016.11.005. [DOI] [Google Scholar]
- Liu X.; Kuang W.; Guo B. Preparation of rubber/graphene oxide composites with in-situ interfacial design. Polymer 2015, 56, 553–562. 10.1016/j.polymer.2014.11.048. [DOI] [Google Scholar]
- Coran A. Y.; Donnet J.-B. The Dispersion of Carbon Black in Rubber Part II. The Kinetics of Dispersion in Natural Rubber. Rubber Chem. Technol. 1992, 65, 998–1015. 10.5254/1.3538656. [DOI] [Google Scholar]
- Kuang W.; Yang Z.; Tang Z.; Guo B. Wrapping of polyrhodanine onto tubular clay and its prominent effects on the reinforcement of the clay for rubber. Composites, Part A 2016, 84, 344–353. 10.1016/j.compositesa.2016.02.015. [DOI] [Google Scholar]
- Nah C.; Huh M.-Y.; Rhee J. M.; Yoon T.-H. Plasma surface modification of silica and its effect on properties of styrene-butadiene rubber compound. Polym. Int. 2002, 51, 510–518. 10.1002/pi.897. [DOI] [Google Scholar]
- Lee J.-y.; Lee T.; Kim K.; Kim B.; Kwag G.; Kim J.-y.; Ji S.; Kim W.; Paik H.-j. Poly(styrene-r-butadiene)-b-poly(poly(ethylene glycol) methyl ether methacrylate) as a silica dispersant in rubber compounds. Polym. Int. 2014, 63, 908–914. 10.1002/pi.4644. [DOI] [Google Scholar]
- Takei T.; Oda R.; Miura A.; Kumada N.; Kinomura N.; Ohki R.; Koshiyama H. Effect of dispersion of sepiolite in sepiolite-NBR composite on the tensile strength. Composites, Part B 2013, 44, 260–265. 10.1016/j.compositesb.2012.05.034. [DOI] [Google Scholar]
- Kaewsakul W.; Sahakaro K.; Dierkes W. K.; Noordermeer J. W. M. Mechanistic aspects of silane coupling agents with different functionalities on reinforcement of silica-filled natural rubber compounds. Polym. Eng. Sci. 2015, 55, 836–842. 10.1002/pen.23949. [DOI] [Google Scholar]
- Liu J.; Gao Y.; Cao D.; Zhang L.; Guo Z. Nanoparticle Dispersion and Aggregation in Polymer Nanocomposites: Insights from Molecular Dynamics Simulation. Langmuir 2011, 27, 7926–7933. 10.1021/la201073m. [DOI] [PubMed] [Google Scholar]
- Wang M.-J. The Role of Filler Networking in Dynamic Properties of Filled Rubber. Rubber Chem. Technol. 1999, 72, 430–448. 10.5254/1.3538812. [DOI] [Google Scholar]
- Majesté J.-C.; Vincent F. A kinetic model for silica-filled rubber reinforcement. J. Rheol. 2015, 59, 405–427. 10.1122/1.4906621. [DOI] [Google Scholar]
- Huang Y.; Schadler L. S. Understanding the strain-dependent dielectric behavior of carbon black reinforced natural rubber-An interfacial or bulk phenomenon?. Compos. Sci. Technol. 2017, 142, 91–97. 10.1016/j.compscitech.2017.02.003. [DOI] [Google Scholar]
- Zhang X.; Tang Z.; Guo B.; Zhang L. Enabling Design of Advanced Elastomer with Bioinspired Metal-Oxygen Coordination. ACS Appl. Mater. Interfaces 2016, 8, 32520–32527. 10.1021/acsami.6b10881. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Huang J.; Guo B.; Zhang L.; Liu F. Bioinspired Engineering of Sacrificial Metal-Ligand Bonds into Elastomers with Supramechanical Performance and Adaptive Recovery. Macromolecules 2016, 49, 1781–1789. 10.1021/acs.macromol.5b02756. [DOI] [Google Scholar]
- Zhang X.; Tang Z.; Huang J.; Lin T.; Guo B. Strikingly improved toughness of nonpolar rubber by incorporating sacrificial network at small fraction. J. Polym. Sci., Part B: Polym. Phys. 2016, 54, 781–786. 10.1002/polb.23980. [DOI] [Google Scholar]
- Huang J.; Tang Z.; Yang Z.; Guo B. Bioinspired Interface Engineering in Elastomer/Graphene Composites by Constructing Sacrificial Metal-Ligand Bonds. Macromol. Rapid Commun. 2016, 37, 1040–1045. 10.1002/marc.201600226. [DOI] [PubMed] [Google Scholar]
- Liu J.; Wang S.; Tang Z.; Huang J.; Guo B.; Huang G. Bioinspired Engineering of Two Different Types of Sacrificial Bonds into Chemically Cross-Linked cis-1,4-Polyisoprene toward a High-Performance Elastomer. Macromolecules 2016, 49, 8593–8604. 10.1021/acs.macromol.6b01576. [DOI] [Google Scholar]
- Xu C.; Nie J.; Wu W.; Fu L.; Lin B. Design of self-healable supramolecular hybrid network based on carboxylated styrene butadiene rubber and nano-chitosan. Carbohydr. Polym. 2019, 410–419. 10.1016/j.carbpol.2018.10.080. [DOI] [PubMed] [Google Scholar]
- Wu W.; Xu C.; Zheng Z.; Lin B.; Fu L. Strengthened, recyclable shape memory rubber films with a rigid filler nano-capillary network. J. Mater. Chem. A 2019, 7, 6901–6910. 10.1039/C9TA01266D. [DOI] [Google Scholar]
- Ge S.; Liu Q.; Li M.; Liu J.; Lu A.; Li F.; Zhang S.; Sun Q.; Xiong L. Enhanced mechanical properties and gelling ability of gelatin hydrogels reinforced with chitin whiskers. Food Hydrocolloids 2018, 1–12. 10.1016/j.foodhyd.2017.09.023. [DOI] [Google Scholar]
- Niu H.; Xia X.; Wang C.; Kong B.; Liu Q. Thermal stability and gel quality of myofibrillar protein as affected by soy protein isolates subjected to an acidic pH and mild heating. Food Chem. 2018, 188–195. 10.1016/j.foodchem.2017.09.055. [DOI] [PubMed] [Google Scholar]
- Rajisha K. R.; Maria H. J.; Pothan L. A.; Ahmad Z.; Thomas S. Preparation and characterization of potato starch nanocrystal reinforced natural rubber nanocomposites. Int. J. Biol. Macromol. 2014, 147–153. 10.1016/j.ijbiomac.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Harrington M. J.; Masic A.; Holten-Andersen N.; Waite J. H.; Fratzl P. Iron-Clad Fibers: A Metal-Based Biological Strategy for Hard Flexible Coatings. Science 2010, 328, 216–220. 10.1126/science.1181044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball P. Does nature know best?. Nat. Mater. 2003, 2, 510. 10.1038/nmat952. [DOI] [Google Scholar]
- Becker N.; Oroudjev E.; Mutz S.; Cleveland J. P.; Hansma P. K.; Hayashi C. Y.; Makarov D. E.; Hansma H. G. Molecular nanosprings in spider capture-silk threads. Nat. Mater. 2003, 2, 278–283. 10.1038/nmat858. [DOI] [PubMed] [Google Scholar]
- Liu J.; Liu J.; Wang S.; Huang J.; Wu S.; Tang Z.; Guo B.; Zhang L. An advanced elastomer with an unprecedented combination of excellent mechanical properties and high self-healing capability. J. Mater. Chem. A 2017, 5, 25660–25671. 10.1039/C7TA08255J. [DOI] [Google Scholar]
- Wang D.; Guo J.; Zhang H.; Cheng B.; Shen H.; Zhao N.; Xu J. Intelligent rubber with tailored properties for self-healing and shape memory. J. Mater. Chem. A 2015, 3, 12864–12872. 10.1039/C5TA01915J. [DOI] [Google Scholar]
- Xu C.; Cao L.; Huang X.; Chen Y.; Lin B.; Fu L. Self-Healing Natural Rubber with Tailorable Mechanical Properties Based on Ionic Supramolecular Hybrid Network. ACS Appl. Mater. Interfaces 2017, 9, 29363–29373. 10.1021/acsami.7b09997. [DOI] [PubMed] [Google Scholar]
- Cao L.; Yuan D.; Xu C.; Chen Y. Biobased, self-healable, high strength rubber with tunicate cellulose nanocrystals. Nanoscale 2017, 9, 15696–15706. 10.1039/C7NR05011A. [DOI] [PubMed] [Google Scholar]
- Xu C.; Cao L.; Lin B.; Liang X.; Chen Y. Design of Self-Healing Supramolecular Rubbers by Introducing Ionic Cross-Links into Natural Rubber via a Controlled Vulcanization. ACS Appl. Mater. Interfaces 2016, 8, 17728–17737. 10.1021/acsami.6b05941. [DOI] [PubMed] [Google Scholar]
- Ramier J.; Gauthier C.; Chazeau L.; Stelandre L.; Guy L. Payne effect in silica-filled styrene–butadiene rubber: Influence of surface treatment. J. Polym. Sci., Part B: Polym. Phys. 2007, 45, 286–298. 10.1002/polb.21033. [DOI] [Google Scholar]
- Lung C. Y. K.; Matinlinna J. P. Aspects of silane coupling agents and surface conditioning in dentistry: An overview. Dent. Mater. 2012, 28, 467–477. 10.1016/j.dental.2012.02.009. [DOI] [PubMed] [Google Scholar]
- Castellano M.; Conzatti L.; Turturro A.; Costa G.; Busca G. Influence of the Silane Modifiers on the Surface Thermodynamic Characteristics and Dispersion of the Silica into Elastomer Compounds. J. Phys. Chem. B 2007, 111, 4495–4502. 10.1021/jp0702144. [DOI] [PubMed] [Google Scholar]
- Sattar M. A.; Nair A. S.; Xavier P. J.; Patnaik A. Natural rubber–SiO2 nanohybrids: interface structures and dynamics. Soft Matter 2019, 15, 2826–2837. 10.1039/C9SM00254E. [DOI] [PubMed] [Google Scholar]
- Rahman M. A.; Sartore L.; Bignotti F.; Di Landro L. Autonomic Self-Healing in Epoxidized Natural Rubber. ACS Appl. Mater. Interfaces 2013, 5, 1494–1502. 10.1021/am303015e. [DOI] [PubMed] [Google Scholar]
- Rahman M. A.; Penco M.; Peroni I.; Ramorino G.; Grande A. M.; Di Landro L. Self-Repairing Systems Based on Ionomers and Epoxidized Natural Rubber Blends. ACS Appl. Mater. Interfaces 2011, 3, 4865–4874. 10.1021/am201417h. [DOI] [PubMed] [Google Scholar]
- Carretero-González J.; Ezquerra T. A.; Amnuaypornsri S.; Toki S.; Verdejo R.; Sanz A.; Sakdapipanich J.; Hsiao B. S.; López-Manchado M. A. Molecular dynamics of natural rubber as revealed by dielectric spectroscopy: The role of natural cross-linking. Soft Matter 2010, 6, 3636–3642. 10.1039/c003087b. [DOI] [Google Scholar]
- Boese D.; Kremer F. Molecular dynamics in bulk cis-polyisoprene as studied by dielectric spectroscopy. Macromolecules 1990, 23, 829–835. 10.1021/ma00205a023. [DOI] [Google Scholar]
- Hernández M.; Carretero-González J.; Verdejo R.; Ezquerra T. A.; López-Manchado M. A. Molecular Dynamics of Natural Rubber/Layered Silicate Nanocomposites As Studied by Dielectric Relaxation Spectroscopy. Macromolecules 2010, 43, 643–651. 10.1021/ma902379t. [DOI] [Google Scholar]
- Hernández M.; Grande A. M.; van der Zwaag S.; García S. J. Monitoring Network and Interfacial Healing Processes by Broadband Dielectric Spectroscopy: A Case Study on Natural Rubber. ACS Appl. Mater. Interfaces 2016, 8, 10647–10656. 10.1021/acsami.6b02259. [DOI] [PubMed] [Google Scholar]
- Hernández M.; Ezquerra T. A.; Verdejo R.; López-Manchado M. A. Role of Vulcanizing Additives on the Segmental Dynamics of Natural Rubber. Macromolecules 2012, 45, 1070–1075. 10.1021/ma202325k. [DOI] [Google Scholar]
- Calderwood J. H. Dielectric Spectroscopy of Polymers. Phys. Bull. 1977, 28, 572. 10.1088/0031-9112/28/12/036. [DOI] [Google Scholar]
- Hernández M.; Valentín J. L.; López-Manchado M. A.; Ezquerra T. A. Influence of the vulcanization system on the dynamics and structure of natural rubber: Comparative study by means of broadband dielectric spectroscopy and solid-state NMR spectroscopy. Eur. Polym. J. 2015, 68, 90–103. 10.1016/j.eurpolymj.2015.04.021. [DOI] [Google Scholar]
- Campbell D. S. Structural characterization of vulcanizates part X. Thiol-disulfide interchange for cleaving disulfide crosslinks in natural rubber vulcanizates. J. Appl. Polym. Sci. 1969, 13, 1201–1214. 10.1002/app.1969.070130609. [DOI] [Google Scholar]
- Yu P.; He H.; Luo Y.; Jia D.; Dufresne A. Elastomer Reinforced with Regenerated Chitin from Alkaline/Urea Aqueous System. ACS Appl. Mater. Interfaces 2017, 9, 26460–26467. 10.1021/acsami.7b08294. [DOI] [PubMed] [Google Scholar]
- Payne A. R. The Dynamic Properties of Carbon Black-Loaded Natural Rubber Vulcanizates. Part I. J. Appl. Polym. Sci. 1962, 6, 57. 10.1002/app.1962.070061906. [DOI] [Google Scholar]
- Kotal M.; Srivastava S. K.; Bhowmick A. K. Thermoplastic polyurethane and nitrile butadiene rubber blends with layered double hydroxide nanocomposites by solution blending. Polym. Int. 2010, 59, 2–10. 10.1002/pi.2686. [DOI] [Google Scholar]
- Kong L.; Li F.; Wang F.; Miao Y.; Huang X.; Zhu H.; Lu Y. In situ assembly of SiO2 nanodots/layered double hydroxide nanocomposite for the reinforcement of solution-polymerized butadiene styrene rubber/butadiene rubber. Compos. Sci. Technol. 2018, 158, 9–18. 10.1016/j.compscitech.2018.01.036. [DOI] [Google Scholar]
- Tee B. C. K.; Wang C.; Allen R.; Bao Z. An electrically and mechanically self-healing composite with pressure- and flexion-sensitive properties for electronic skin applications. Nat. Nanotechnol. 2012, 7, 825. 10.1038/nnano.2012.192. [DOI] [PubMed] [Google Scholar]
- Xu C.; Chen Y.; Wang Y.; Zeng X. Temperature dependence of the mechanical properties and the inner structures of natural rubber reinforced by in situ polymerization of zinc dimethacrylate. J. Appl. Polym. Sci. 2013, 128, 2350–2357. 10.1002/app.38263. [DOI] [Google Scholar]
- Hernández M.; Grande A. M.; van der Zwaag S.; García S. J.; Dierkes W.; Bijleveld J. Turning Vulcanized Natural Rubber into a Self-Healing Polymer: Effect of the Disulfide/Polysulfide Ratio. ACS Sustainable Chem. Eng. 2016, 4, 5776–5784. 10.1021/acssuschemeng.6b01760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Liu X.; Hu X.; Luo W. Changes in tensile and tearing fracture properties of carbon-black filled rubber vulcanizates by thermal aging. Polym. Adv. Technol. 2015, 26, 1331–1335. 10.1002/pat.3683. [DOI] [Google Scholar]
- Carroll B.; Cheng S.; Sokolov A. P. Analyzing the Interfacial Layer Properties in Polymer Nanocomposites by Broadband Dielectric Spectroscopy. Macromolecules 2017, 50, 6149–6163. 10.1021/acs.macromol.7b00825. [DOI] [Google Scholar]
- Holt A. P.; Sangoro J. R.; Wang Y.; Agapov A. L.; Sokolov A. P. Chain and Segmental Dynamics of Poly(2-vinylpyridine) Nanocomposites. Macromolecules 2013, 46, 4168–4173. 10.1021/ma400418b. [DOI] [Google Scholar]
- Holt A. P.; Griffin P. J.; Bocharova V.; Agapov A. L.; Imel A. E.; Dadmun M. D.; Sangoro J. R.; Sokolov A. P. Dynamics at the Polymer/Nanoparticle Interface in Poly(2-vinylpyridine)/Silica Nanocomposites. Macromolecules 2014, 47, 1837–1843. 10.1021/ma5000317. [DOI] [Google Scholar]
- Baeza G. P.; Dessi C.; Costanzo S.; Zhao D.; Gong S.; Alegria A.; Colby R. H.; Rubinstein M.; Vlassopoulos D.; Kumar S. K. Network dynamics in nanofilled polymers. Nat. Commun. 2016, 7, 11368 10.1038/ncomms11368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jouault N.; Moll J. F.; Meng D.; Windsor K.; Ramcharan S.; Kearney C.; Kumar S. K. Bound Polymer Layer in Nanocomposites. ACS Macro Lett. 2013, 2, 371–374. 10.1021/mz300646a. [DOI] [PubMed] [Google Scholar]
- Trinh G. H.; Desloir M.; Dutertre F.; Majesté J.-C.; Dalmas F.; Baeza G. P. Isostructural softening of the filler network in SBR/silica nanocomposites. Soft Matter 2019, 3122–3132. 10.1039/C8SM02592D. [DOI] [PubMed] [Google Scholar]
- Chen Q.; Gong S.; Moll J.; Zhao D.; Kumar S. K.; Colby R. H. Mechanical Reinforcement of Polymer Nanocomposites from Percolation of a Nanoparticle Network. ACS Macro Lett. 2015, 4, 398–402. 10.1021/acsmacrolett.5b00002. [DOI] [PubMed] [Google Scholar]
- Harton S. E.; Kumar S. K.; Yang H.; Koga T.; Hicks K.; Lee H.; Mijovic J.; Liu M.; Vallery R. S.; Gidley D. W. Immobilized Polymer Layers on Spherical Nanoparticles. Macromolecules 2010, 43, 3415–3421. 10.1021/ma902484d. [DOI] [Google Scholar]
- Cui G.; Boudara V. A. H.; Huang Q.; Baeza G.; J. Wilson A.; Hassager O.; J. Read D.; Mattsson J. Linear shear and nonlinear extensional rheology of unentangled supramolecular side-chain polymers. J. Rheol. 2018, 62, 1155–1174. 10.1122/1.5012349. [DOI] [Google Scholar]
- Voylov D. N.; Holt A. P.; Doughty B.; Bocharova V.; Meyer H. M.; Cheng S.; Martin H.; Dadmun M.; Kisliuk A.; Sokolov A. P. Unraveling the Molecular Weight Dependence of Interfacial Interactions in Poly(2-vinylpyridine)/Silica Nanocomposites. ACS Macro Lett. 2017, 6, 68–72. 10.1021/acsmacrolett.6b00915. [DOI] [PubMed] [Google Scholar]
- Holt A. P.; Bocharova V.; Cheng S.; Kisliuk A. M.; White B. T.; Saito T.; Uhrig D.; Mahalik J. P.; Kumar R.; Imel A. E.; Etampawala T.; Martin H.; Sikes N.; Sumpter B. G.; Dadmun M. D.; Sokolov A. P. Controlling Interfacial Dynamics: Covalent Bonding versus Physical Adsorption in Polymer Nanocomposites. ACS Nano 2016, 10, 6843–6852. 10.1021/acsnano.6b02501. [DOI] [PubMed] [Google Scholar]
- Kremer F.; Schönhals A.. Broadband Dielectric Spectroscopy; Springer: New York, 2003; p 721. [Google Scholar]
- Schönhals A.; Schlosser E. Dielectric-Relaxation in Polymeric Solids 0.1. A New Model for the Interpretation of the Shape of the Dielectric-Relaxation Function. Colloid Polym. Sci. 1989, 267, 125–132. 10.1007/BF01410350. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.