

Abstract

The direct synthesis of complex chemicals from simple precursors (such as syngas) is one of the main objectives of current research in heterogeneous catalysis. To rationally design catalytic materials for this purpose, it is essential to identify the critical elementary reaction steps that ultimately determine a catalyst’s activity and selectivity with respect to a desired product. Unfortunately, the number of potentially relevant elementary steps is in the thousands, even for relatively simple target species like ethanol. The challenge of identifying the critical steps is thus akin to finding the proverbial needle in a haystack. Recently, a model-reduction scheme has been proposed, which tackles this problem by prescreening the barriers of all potential reactions with computationally inexpensive approximations. Although this route appears highly promising, it raises the question of how the starting point of the model-reduction process can be determined. In this contribution, we present a systematic method for enumerating all intermediates and elementary reactions relevant to a chemical process of interest. Using this approach, we construct reaction networks for C,H,O-containing systems consisting of up to four non-hydrogen atoms (more than 1 million reactions). Importantly, the scheme goes beyond simple bond-breaking reactions and allows considering rearrangement and transfer reactions as well. The presented reaction networks thus cover the chemistry of syngas-based processes (and beyond) to an unprecedented scale.
Introduction
Microscopic understanding of the mechanisms of chemical processes in terms of elementary reaction steps and intermediates is a prerequisite for the rational design of heterogeneous catalysts. Although the experimental and theoretical study of real catalysts in situ is still in its infancy, much progress has been made by studying model catalysts, such as single crystal surfaces. The state-of-the-art computational approach to this problem is the use of DFT calculations for determining the kinetics of elementary reactions, which are then fed into microkinetic models (e.g., mean field or kinetic Monte Carlo, kMC, simulations).1 In particular, great effort has been devoted to the study of heterogeneous transition metal (TM) catalysts for such essential processes as ammonia synthesis, CO methanation, or the water–gas shift reaction.2−13
Catalyst material design is a highly complex optimization problem.14 On the one hand, the search space of potential catalysts is huge. Even when only considering TMs, numerous surface terminations and orientations, active sites, and alloy compositions need to be taken into account.15,16 On the other hand, there are multiple relevant objectives to aim for, ranging from catalyst turnover frequency and selectivity to other economic considerations such as material cost, processability, and stability.17 Finding the optimal catalyst is therefore only possible if thousands of potential candidates can be screened effectively.
The first-principles (1p) microkinetic approach mentioned above is, unfortunately, far too computationally demanding to be applied in this context. This is mainly due to the large computational effort required for determining activation barriers (and consequently reaction rates) using chain-of-states methods such as the nudged elastic band.18−20 Following the pioneering work by Nørskov and co-workers, it has therefore become common to focus on a single rate-limiting step in screening studies, greatly reducing the number of barriers that need to be calculated (and assuming that the mechanism of the process does not change for different catalysts or conditions).11,21,22
Even for this elementary step of interest, the barrier is typically not calculated explicitly for each catalyst in a screening study. Instead, the Brønsted–Evans–Polanyi principle is used, which postulates a linear relationship between the reaction and activation energies.23,24 Furthermore, similar linear scaling relationships exist between atomic and molecular adsorption energies and between atomic adsorption energies and electronic properties of the metal surface (most prominently, the d-band center of TMs).2,25−28 When combining these scaling relations, the turnover frequency of a complex process can often be related to one or two molecular adsorption energies. This makes the large-scale screening of catalysts possible.5,6,21,26,29
The strategies outlined above can be summarized under the label “model reduction”. In principle, the kinetics of a chemical process depends on a number of coupled elementary steps (the reaction network), but the full complexity does not need to be taken into account to make predictions. Model reduction generally becomes possible if a process is simple enough so that the full model can be studied on (at least) one representative catalyst. The essential features of the full model can then be extracted using sensitivity analysis or rate limitation arguments.30−34
For more complex processes (e.g., the synthesis of higher alcohols from syngas), solving the full model, however, becomes prohibitively expensive. Consequently, model reduction is even more desirable for these cases, but there is no clear prescription on how it can be achieved systematically. Most commonly, a fairly large but still tractable reaction network is postulated as a starting point.35,36 This is somewhat unsatisfactory, however, as the initial reaction network is then biased by the chemical intuition of the researcher and important steps may be missing.
It should be noted that, although surface catalysis is our main motivation, the above discussion also pertains to other fields where kinetic modeling is performed, for example, in combustion or solution chemistry.37,38 The main difference is that gas-phase chemistry tends to be more complex (i.e., the reaction networks are larger) than surface chemistry, whereas the individual calculations of reaction energetics are significantly less expensive. Meanwhile, the basic workflow of identifying a reaction network, determining rates, and running microkinetic simulations is the same.
In principle, an unbiased and automatic construction of reaction networks is possible by exploring a high-dimensional potential energy surface using the electronic structure theory. In the gas phase, this has, for example, been applied for predicting fragmentation in electron-impact mass spectrometry.39,40 Similarly, reaction mechanisms in solution have been explored, often using implicit solvation models for a simplified treatment of the solvent environment.41−43 At the core, all of these methods rely on a large number of electronic structure calculations, either to drive (biased) molecular dynamics simulations or to find minimum energy paths. As an example, the “context-driven” exploration algorithm of Simm and Reiher required more than 80000 geometry optimizations and ca. 10000 transition-state searches to unravel the first steps of the formose reaction.41 This is clearly prohibitive for calculations on extended surfaces.
To deal with more complex chemistries and environments, several knowledge-driven approaches have been proposed in the chemical engineering literature, such as the reaction mechanism generator and the reaction description language.38,44−49 Here, all elementary reaction types that are expected to occur under the given conditions are encoded into heuristics, so-called “reaction rules”. Such approaches are extremely powerful, because they allow constructing chemically meaningful reaction networks without running expensive quantum mechanical calculations a priori. Furthermore, the differentiation of elementary steps into reaction types allows the use of reasonably accurate empirical expressions for reaction rates and thermochemistry.44,50 In this way, a starting point for microkinetic simulations can be obtained, and the most important rates can subsequently be refined with electronic structure calculations.37
Both the electronic structure and rule-based approaches have something in common that the reaction network is generated in a stepwise fashion, focusing on the most important steps between the educt and product. In a recent paper, Ulissi et al. took an inverse approach to this problem by starting from a large set of possible elementary reaction steps.51 The key feature of their method is that reaction energies and rates are estimated with inexpensive approximations using machine learning and scaling relations, whereas the most important steps are initially identified via simple rate limitation arguments. For these steps, rates are subsequently determined through DFT calculations, whose results are in turn used to refine the machine-learning model. In this way, the size of the model is continually reduced, whereas the accuracy for the most relevant steps is increased. Once the most important subnetwork is found with the desired accuracy, a full (mean-field) microkinetic simulation can be performed.
In our view, this is an extremely promising route for understanding complex chemical reactions on catalyst surfaces. There remains, however, some bias in how the set of possible fragments and elementary reactions is initially defined. In the case of ref (51), only simple bond-breaking reactions were considered. Rule-based approaches can include more complex reactions, but this still leads to bias in the selection of the reaction rules that are to be included.
The goal of this publication is therefore twofold. First, an algorithm for the systematic and unbiased enumeration of elementary reaction steps is provided, focusing on reactions of organic molecules. Second, this method is used to generate reaction networks for carbon-, hydrogen-, and oxygen-containing molecules. The largest data set reported contains more than 1 million reactions and thus represents an unprecedentedly complete view of the fundamental building blocks of reaction mechanisms in heterogeneous catalysis. A particularly interesting aspect is the richness of more complex elementary reactions (e.g., rearrangements or transfer reactions) that emerges from this analysis.
In contrast to most previous approaches to the automatic generation of reaction networks, we do not focus on a given set of educts (or a desired product). Instead, our objective is to construct a reaction network connecting all fragments within a given subset of chemical space (bounded, e.g., by molecular size and composition). Consequently, the presented algorithm consists of (1) the enumeration of all fragments within a chemical subspace and (2) finding all elementary reactions that connect them. In this sense, our work is more in the spirit of previous effort directed at enumerating molecules in chemical space (see, e.g., the GDB databases of Reymond and co-workers).52,53 We hope that the fragment and reaction databases presented in this paper can become similarly useful to researchers in both chemical and data-driven research, as the GDB data sets of closed-shell molecules already are.54−56
Results and Discussion
Enumeration of Complete Chemical Subspaces
We begin with some definitions. The “size” of a molecule is designated by the number of non-hydrogen atoms it contains (i.e., its heavy atom count). Chemical subspaces are defined by the elements included and the size of the largest molecules. [CHO]4 thus refers to the set of all molecules containing at least one C, H, or O atom and at most four C and/or O atoms. A complete chemical subspace is defined as a set of molecules, where breaking any bond in any molecule creates fragments that are also part of the subspace (see Figure 1). Note that this definition of completeness does not preclude additional boundary conditions, for example, regarding the number of heteroatoms in a molecule or the number of (fused) rings (see below). In the following, we will use [CHO]n subspaces to illustrate the concept. The generalization to more elements and larger subspaces (within the realm of organic chemistry) is trivial.
Figure 1.

(a) Algorithm for the generation of complete chemical subspaces. The numbering of the sections refers to the explanations in full text. In the “Sanitize” step, implicit hydrogen atoms are added or removed where necessary and duplicate molecules are removed from the list. The “Found new?” step checks whether new structures have been added to the database in the previous iteration of the current section. (b) The complete [CHO]2 subspace. Breaking any bond in any of the depicted molecules creates fragments that are also part of the subspace.
A molecule is defined by its connectivity, that is, by the number and types of atoms it contains and whether they are connected by a chemical bond (regardless of bond order). As a consequence, different resonance structures or stereoisomers are counted as a single species (see below). Throughout, simple valence rules are enforced, for example, that all carbon atoms are at most connected to four other atoms. In other words, bond orders are used as an auxiliary quantity to enforce valence rules, but they are disregarded when comparing whether two molecules are identical. Molecular information (e.g., composition, connectivity, and bond order) is stored and manipulated using the RDKit package.57 A flowchart depicting the enumeration algorithm is shown in Figure 1a.
The algorithm starts with a list including the simplest closed-shell molecules within the targeted chemical space (i.e., H2, CH4, and H2O in [CHO]n spaces). This list is iteratively enlarged. In step 1, all saturated molecules up to a given size are constructed. This is achieved by incrementally replacing all hydrogen atoms with −CH3 and −OH groups until the maximum size is reached. In step 2, rings are introduced by connecting atoms that are separated by at least one other atom. In step 3, bond orders are incremented wherever the valence of the adjacent atoms allows it. Up to this point, the canonical SMILES format in RDKit is used to avoid duplication of molecules due to, for example, atom ordering or resonance structures. Furthermore, implicit hydrogen atoms are assumed to saturate all free valences. In step 4, open-shell (poly)radicals are then introduced by iteratively removing these hydrogen atoms. Exemplary structures added at each step are shown in Figure 2a.
Figure 2.
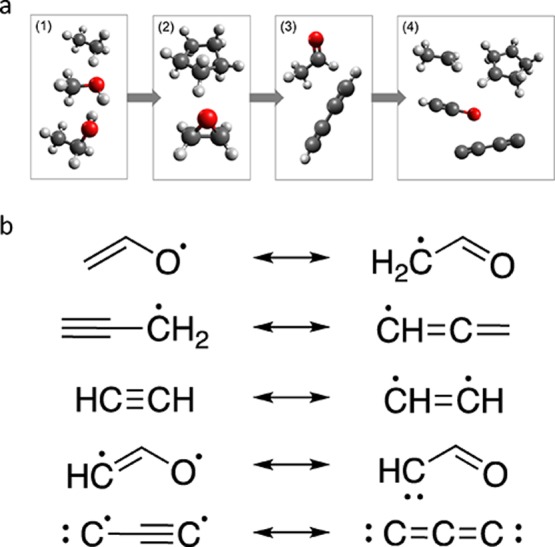
(a) Illustration of exemplary structures added at each step of the algorithm in Figure 1. After step 1, the set consists of acyclic saturated molecules. In step 2, intramolecular bonds are added to form cyclic saturated structures. In step 3, bond orders are incremented to form unsaturated molecules. In step 4, hydrogen atoms are removed to form open-shell systems. (b) Examples of redundant structures filtered by the algorithm in step 5.
Because of the presence of open-shell systems, additional precaution needs to be taken to avoid duplication. For example, neighboring radicals connected by single or double bonds are equivalent to the corresponding closed-shell species with the next higher bond order and therefore excluded. There are also more subtle equivalencies, in particular resonance structures. Consequently, in the final step 5, all such redundant molecules are excluded, so that only molecules with unique graphs remain in the list. Examples of redundant structures are shown in Figure 2b. The canonical SMILES format in RDKit does not map such structures to the same string and consequently cannot be used to check for isomorphism in these cases. A Python function that can be used to this end is provided in the Supporting Information.
The subspaces generated in this manner can be subjected to additional boundary conditions (filters), regarding composition or structural motives. It is important to reemphasize that this does not compromise the completeness of the space as defined above. Although not strictly necessary, filtering the chemical space is very beneficial for excluding uninteresting but combinatorically frequent structural motifs.53 What is deemed interesting obviously depends on the application.
In the present work, only systems with a single ring are considered, and systems with more than two heteroatoms (i.e., oxygen) are excluded. Additionally, triple bonds in rings are excluded. For the chemical spaces considered (up to n = 4), these are reasonable choices, as this allows excluding frameworks with fused three-membered rings and oxygen chains, which are almost universally unstable. However, such boundary conditions should only be adopted after careful consideration. Note, for example, that through the present choices ozone is excluded, which could be a relevant chemical under certain conditions. Also, for larger molecules, multiple rings and heteroatoms are certainly reasonable, and the filters should accordingly be adapted. In the following, we always refer to chemical subspaces subject to the above filters. These are designated as [CHO]nF, to distinguish them from the unfiltered sets. Note that the filters do not apply to the smaller spaces (n = 1, 2), as there are no two-membered rings and there cannot be more than two heteroatoms in these spaces.
Throughout this manuscript, all molecules are depicted in their charge neutral state. This corresponds to the situation expected on a TM surface (where additionally any spin polarization is generally assumed to be quenched) or in the gas phase but certainly not in (polar) solution or on an oxide surface. For our purposes, the distinction is irrelevant, as the elementary reactions for charged and uncharged species are identical. Similarly, the possible elementary reactions for different stereoisomers are the same (though the reaction energetics will in general be different). This also applies to the equivalencies shown in Figure 2b. The “correct” Lewis structure is to some extent a matter of perspective, and we do not intend to judge which representation is more physically meaningful. Although it can be interesting to, for example, debate the correct bond order of C2, this question is not relevant for the present work.58,59
Enumeration of Elementary Reactions
Mathematically, a reaction network can be seen as a directed bipartite graph G with two types of nodes, representing molecules and reactions (see Figure 3a).60−62 Molecule nodes are connected to reaction nodes via directed edges, but molecules are not directly connected to each other. This is similar to the “virtual flask” concept used by Simm and Reiher.41 The directions of the edges in Figure 3 should not be misunderstood as imposing a directionality on the reaction. Indeed, all reactions are assumed to be reversible. The directed edges instead differentiate the molecule nodes connected to a given reaction node into two sets. The molecules belonging to each set can be both products and educts in the reaction, but molecules from the two sets cannot react with each other. Consequently, the directions shown in the figures are arbitrary, in the sense that the graph is equally valid if all edges connected to a reaction node are reversed. This representation can in principle treat reactions with an arbitrary number of educts and products.
Figure 3.
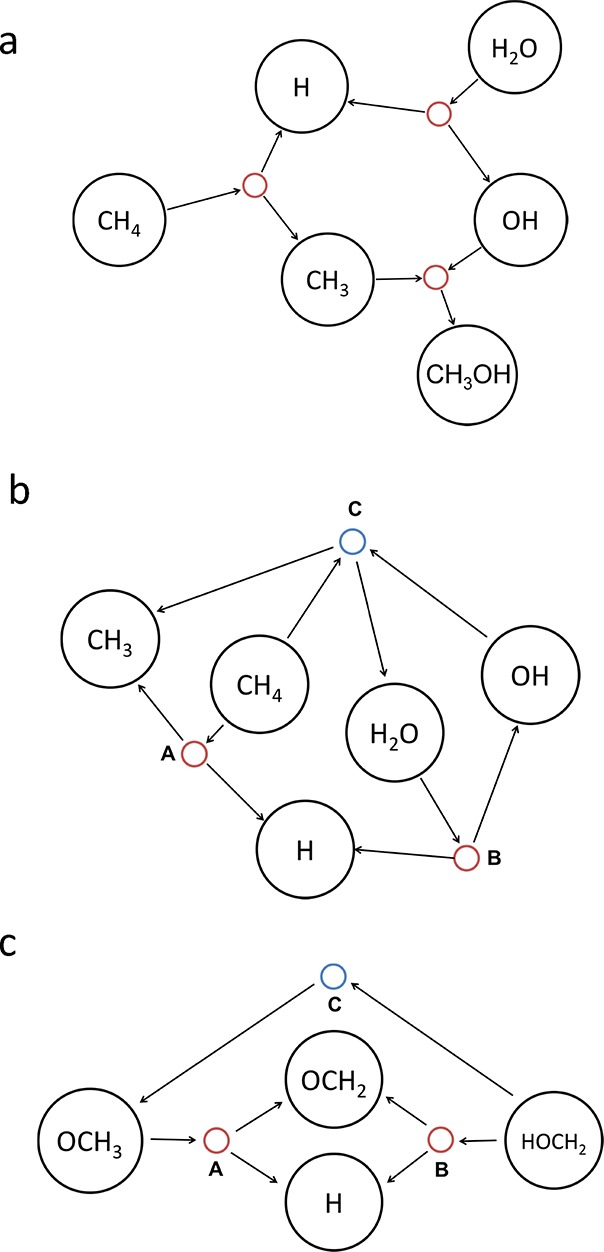
(a) Simple reaction network as a directed bipartite graph. Large nodes represent molecules, and small nodes represent reactions. (b) Transfer reactions can be derived from combinations of bond-breaking reactions. In this case, the dissociation of methane into the methyl radical and atomic hydrogen (A) and the dissociation of water into the hydroxyl radical and atomic hydrogen (B) are combined to define a new reaction (C), in which methane reacts with the hydroxyl radical to form the methyl radical and water. (c) Derivation of rearrangement reactions from G0. Both the oxygen-centered methoxy radical and the carbon-centered methanol radical can dissociate into formaldehyde and atomic hydrogen (reactions A and B, respectively). As reactions A and B share both products, a new reaction (C) can be defined, in which the methoxy radical rearranges to the (carbon-centered) methanol radical.
The goal of this work is the enumeration of the elementary reactions in a chemical subspace, that is, all reactions that can take place in a single step. However, there is no rigorous definition of what makes a specific reaction elementary, based solely on structural information. To circumvent this problem, we propose a hierarchical method of deriving increasingly complex reactions.
We begin with the simplest possible reaction type, namely, the dissociation of a bond in a molecule:
| 1 |
Thanks to the completeness of the subspaces considered herein, all nodes of this simple reaction network are known from the outset. To construct the corresponding graph G0, we simply need to break each bond in each molecule and connect the original molecule and its fragments via a reaction node. We refer to this graph as the “Generation 0” (G0) network in the following.
From G0, we can derive the next more complex set of reactions. This is done by analyzing which of the reactions in G0 share one or more products. In general, it holds that given two reactions
| 2 |
| 3 |
a new bimolecular reaction can be defined as:
| 4 |
This new reaction is characterized by the simultaneous dissociation of one bond and the formation of another. Such reactions, for example, include transfer reactions, as shown in Figure 3b.
Similarly, given the reactions
| 5 |
| 6 |
a new reaction can be defined as:
| 7 |
Again, a bond is dissociated and another is formed, with the difference that the reaction is a unimolecular rearrangement (see Figure 3c).
In this manner, a new “Generation 1” graph G1 that contains all nodes and edges of G0 and the new nodes and edges corresponding to these rearrangement and transfer reactions can be defined. The same procedure can now be applied to G1, to form the next more complex graph G2, and so forth.
It should be noted that this procedure increases the complexity of reactions both in terms of the number of bonds that are broken/formed and of their molecularity. As an example, consider two reactions in G1:
| 8 |
| 9 |
The reactions share a reactant (OH) and can therefore be combined to a new reaction:
| 10 |
Reaction 10 is now trimolecular and can be interpreted as a hydrogen-mediated version of reaction 9. In principle, G2 will contain a very large number of such reactions, which would not in general be considered elementary. Indeed, higher than bimolecular reactions are usually excluded on entropic grounds in microkinetic studies of surface reactions.63
In the present work, we therefore only consider uni- or bimolecular reactions. Still, it is important to note that the framework is general enough to derive reactions with higher molecularity. A special case where this becomes relevant is reactions in which solvent molecules (e.g., water) participate, for example, in electrochemical processes. As solvent molecules are available in extremely high concentration, the entropic argument does not hold in this case. It could then be advisable to include trimolecular reactions with water. Similarly, effects of acid or base catalysis (assuming high concentrations of H+ or OH–) can be included in this way.
Properties of [CHO]nF Subspaces
Using the above method, [CHO]nF subspaces with n = 1–4 were constructed. The corresponding data sets (SMILES strings) are hosted online at the reaction network repository (http://rnet.theo.ch.tum.de).64 Unsurprisingly, the complete spaces are much larger than the corresponding sets of closed-shell molecules (see Table 1). For example, [CHO]4F contains 679 species, of which only 99 are simple closed-shell molecules as included in other enumerated databases, such as the GDB-17 database or the derived QM-7 set.53,54 The data set presented here therefore includes parts of chemical space that are unexplored to date. Beyond the application at hand (constructing chemical reaction networks for surface catalysis), we therefore expect the presented data to be highly valuable for training and validating machine-learning methods.
Table 1. Statistics of Chemical Subspace Generation.
| subspace | acyclic, saturated backbones | all saturated backbones | closed-shell molecules | all fragmentsa | redundant structures |
|---|---|---|---|---|---|
| [CHO]1F | 2 | 2 | 2 | 9 | 0 |
| [CHO]2F | 5 | 5 | 9 | 31 | 0 |
| [CHO]3F | 10 | 13 | 28 | 131 | 23 |
| [CHO]4F | 20 | 32 | 99 | 679 | 236 |
Redundant (e.g., resonance) structures are not included in this number.
It also becomes clear from the table that filtering redundant molecules is absolutely essential, as these structures become very abundant for larger subspaces (e.g., 236 for [CHO]4F). In Figure 4, the molecular weight distribution of the chemical subspaces is shown. This reflects the exponential growth of chemical subspaces with increasing molecular size.
Figure 4.
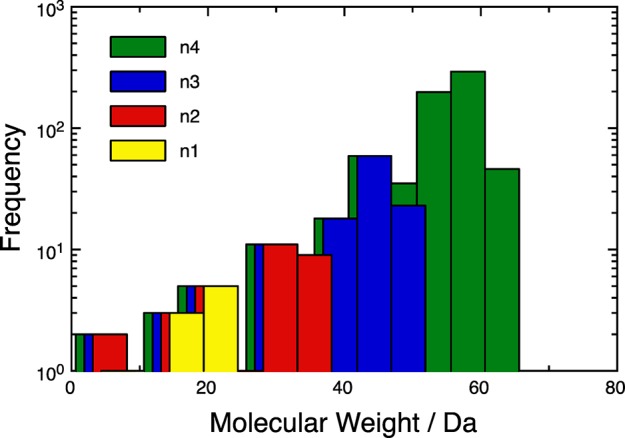
Molecular weight distributions in the [CHO]nF subspaces. The histograms use a bin size of 5 Da.
An interesting feature of the [CHO]nF subspaces is that they contain a large number of constitutional isomers for some compositions. For example, there are 48 isomers with the composition C3H4O in [CHO]4F. Of these, 12 are common closed-shell molecules (e.g., acrolein and cyclopropanone), whereas the rest are open-shell species (see Figure 5). Although many of the latter may seem rather exotic, they are absolutely essential for our purposes. On the one hand, they can represent important (even if short-lived) intermediates in a reaction network. On the other hand, even if that is not the case, a molecule can be part of an elementary dissociation reaction that is used to construct an important higher-level rearrangement or transfer reaction (see above), without itself being included in the final reaction network.
Figure 5.

Isomeric structures with the composition C3H4O contained in the [CHO]4F subspace.
Compositions with high isomeric abundance may play an important role in the selectivity of complex processes. All the isomers in Figure 5 can in principle rearrange into each other (though not all such rearrangements are kinetically feasible). Specifically, this will be the case if the internal rearrangement kinetics of the transiently formed species can compete with the kinetics of its bimolecular decay reaction. Among other things, this depends on the concentration of reaction partners. If the coreactant is abundant, the rearrangement kinetics may be too slow to influence the outcome of the reaction, whereas the (unimolecular) rearrangement will dominate if the coreactant concentration is low. Where these regimes actually lie in terms of realistic reaction conditions is obviously dependent on the process. As microkinetic models of surface reactions generally do not consider rearrangement reactions at all (or assume that they are equilibrated), their role may have been overlooked in some cases (see below).
Properties of [CHO]nF Reaction Networks
The sizes of the zeroth and first generation reaction networks derived for [CHO]1–4F are shown in Table 2. The number of reactions for both G0 and G1 increases exponentially with the size of the largest molecule in the subspace. Even for relatively small sets, the number of reactions in G1 is very large. Specifically, there are more than 20000 reactions for [CHO]3F and more than 1 million for [CHO]4F.
Table 2. Sizes of Zeroth- and First-Generation Reaction Networks Derived for [CHO]nF Subspaces.
| n | G0 reaction | G1 reaction |
|---|---|---|
| 1 | 6 | 15 |
| 2 | 52 | 555 |
| 3 | 387 | 21006 |
| 4 | 3149 | 1134592 |
To understand the structure and composition of these networks, it is instructive to consider the simplest case of the [CHO]1 graphs. The corresponding G0 graph is shown in Figure 6a. As the subspace only contains methane, water, and the corresponding radicals, the only elementary reactions in G0 are dehydrogenations. Accordingly, H is at the center of the graph and connected to reactions with all other species arranged at the periphery. Furthermore, there is no reaction connecting carbon- and oxygen-containing molecules, effectively splitting the network into two sides.
Figure 6.

(a) Generation 0 (G0) reaction network for [CHO]1. (b) Generation 1 (G1) reaction network for [CHO]1. (c) Generation 0 (G0) reaction network for [CHO]3F. (d) Exemplary reactions from G0, G1, and G2 for [CHO]3F, where curved arrows on the educt side indicate bond formation and bold red bonds indicate bond breaking.
This changes for the G1 network (see Figure 6b). Here, multiple new transfer reactions are included both among and between species from both sides. Because of the simplicity of the subspace, these are all hydrogen transfer reactions. For example, there are a number of disproportionation reactions (e.g., OH + OH → H2O + O), as well as regular transfer reactions (e.g., H2O + CH3 → OH + CH4).
For larger subspaces, such graph representations become increasingly complex, and their visual inspection has few benefits (beyond aesthetics). It is however interesting to note that the qualitative topology of the G0 network for [CHO]3F (see Figure 6c) resembles the one for [CHO]1. In both cases, the hydrogen atom is at the center and connected to reactions with almost all species, whereas some molecules are at the periphery and only connect to very few reactions.
Going beyond the first generation of derived reactions, the complexity of the individual reactions further increases but not necessarily their number. This is because the reaction derivation scheme is self-terminating. There are a finite number of combinatorically feasible reactions of the type A + B → C + D for each subspace. For [CHO]1, these are completely enumerated within two generations (with only four additional reactions in G2).
However, enumeration for the next larger subspace ([CHO]2) only terminates after four generations, with more than 600 reactions in G2. This trend continues for larger subspaces, so that complete enumeration of all reactions quickly becomes unfeasible. Of course, the point of the hierarchical enumeration scheme is not to enumerate all combinatorically possible reactions66 but to exhaustively enumerate only the ones that are simple enough to occur in a single step. To this end, it is instructive to look at the chemistry of the respective reactions.
The G0 reactions by construction only consist of simple bond-breaking steps, whereas later generations can be decomposed into combinations of bond-breaking and formation steps. Examples of reactions from G0, G1, and G2 are shown in Figure 6d. It becomes clear that G2 reactions are already quite complex. In the example shown, two bonds are broken and two are formed. It can be assumed that the reaction is very unlikely to occur in a single step. Here, it is important to reemphasize that throughout this manuscript, “bond breaking” refers to changes in connectivity, not to changes in the bond order or hybridization. Accordingly, concerted shifts of electron pairs, as they are commonly postulated in physical organic chemistry, do not affect the complexity of reactions in our scheme.
The purpose of this paper is to define reaction networks for surface reactions, particularly on TM catalysts. From this perspective, we argue that reactions in G2 and beyond should not be considered elementary. Indeed, reaction networks postulated in the literature typically almost exclusively consist of G0 reactions, in addition to surface adsorption/desorption and (if explicitly resolved, for instance, in kMC simulations)1 diffusion steps.
The comparatively large reaction network [for acetaldehyde and ethanol synthesis on Rh(111)] used as the starting point by Ulissi et al. consists, for instance, of 249 bond-breaking reactions and 8 adsorption/desorption reactions (for H2, CO, H2O, CO2, methane, methanol, ethanol, and acetaldehyde).51 This network was constructed from initially 99 intermediates with up to 4 non-hydrogen atoms. In contrast, the [CHO]3F subspace (which is the smallest complete set that includes ethanol) is significantly larger, containing 131 intermediates and 387 bond-breaking reactions. Additionally, several OCCO species are considered in ref (51), though they were not found to be relevant in the final mechanism. The [CHO]4F subspace (which includes these species) contains 679 intermediates and 3149 bond-breaking reactions.
These G0 reaction networks thus represent an unprecedentedly complete starting point for model reduction studies of this type. Beyond this, the [CHO]3F G1 reaction network also contains more than 20000 transfer (Figure 3b) and rearrangement reactions (Figure 3c), which are entirely absent in most literature-reported reaction networks. This absence can be attributed to several factors.
First, reaction networks are often based on the (implicit or explicit) assumption that most intermediates are only formed transiently and do not persist long enough to react with each other. This assumption would exclude a large share of the above transfer reactions. However, some intermediates must react with each other to form more complex chemical structures, so a part of the 20000 reactions could still be a part of these reaction networks. Furthermore, this argument does not apply to the rearrangement reactions, which are all unimolecular.
Second, it could be argued that the barriers of these reactions are simply too high for them to play a role, although transfer and rearrangement reactions do frequently occur in solution and gas-phase chemistry. To the best of our knowledge, there is little data on how TM surfaces affect the barriers of these reactions. This is currently being investigated by our group.
Third, there is a technical reason why such reactions are typically disregarded, namely, their sheer number. As long as there is little data on which reactions are likely and cannot be disregarded, it is hard to justify why some reactions should be included and thousands of others should be excluded. Ideally, a quantitative study of the reaction energies and barriers of G1 reactions on TM surfaces can uncover trends that allow defining a useful rule of thumb with respect to which reactions are important.
Conclusions
In this paper, algorithms that allow the systematic enumeration of reaction intermediates and elementary reaction steps likely to occur in surface catalysis were presented. Some formal aspects of the graph theoretical nature of reaction networks were discussed, from which a hierarchy of increasingly complex reactions was derived. We believe that the presented data sets of fragments and elementary reactions (available at http://rnet.theo.tum.de) will prove to be a valuable resource for the catalysis community.
It should be emphasized that the focus of this work is on the enumeration of all elementary reactions within a chemical subspace, not on the elucidation of the reaction mechanism of a specific process (which is the goal of most approaches cited in the Introduction). Furthermore, the elementary reactions are derived exclusively from considering how complex a chemical transformation is in terms of changing the molecular graph. In this sense, the approach is neither biased by (nor does it profit from) chemical knowledge in the traditional sense.
The presented work provides a solid basis for future model reduction studies, aiming to extract the most relevant elementary steps that contribute to a process of interest. In this context, it will be interesting to explore how path-search algorithms can be used to help in preselecting relevant subsets. Predicting the selectivity of a catalyst toward a given product from 1p studies is proving very challenging for complex processes. It is likely that this is not just due to inadequacies of the theoretical description (e.g., errors in density functional approximations) but rather because important elementary steps are missing from the reaction network.65
Acknowledgments
Funding by the TUM University Foundation and the International Graduate School for Science and Engineering (IGSEE) is gratefully acknowledged. This work was supported by the German Research Foundation (DFG) and the Technical University of Munich within the funding programme Open Access Publishing.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03200.
Description of reported data sets and Python code for determining isomorphism of SMILES strings (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Reuter K. Ab Initio Thermodynamics and First-Principles Microkinetics for Surface Catalysis. Catal. Lett. 2016, 146, 541–563. 10.1007/s10562-015-1684-3. [DOI] [Google Scholar]
- Nørskov J. K.; Abild-Pedersen F.; Studt F.; Bligaard T. Density Functional Theory in Surface Chemistry and Catalysis. Proc. Natl. Acad. Sci. 2011, 108, 937–943. 10.1073/pnas.1006652108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T.; Reuter K. Structure Sensitivity in Oxide Catalysis: First-Principles Kinetic Monte Carlo Simulations for CO Oxidation at RuO2(111). J. Chem. Phys. 2015, 143, 204702. 10.1063/1.4936354. [DOI] [PubMed] [Google Scholar]
- Matera S.; Meskine H.; Reuter K. Adlayer Inhomogeneity without Lateral Interactions: Rationalizing Correlation Effects in CO Oxidation at RuO2(110) with First-Principles Kinetic Monte Carlo. J. Chem. Phys. 2011, 134, 064713. 10.1063/1.3553258. [DOI] [PubMed] [Google Scholar]
- Sehested J.; Larsen K. E.; Kustov A. L.; Frey A. M.; Johannessen T.; Bligaard T.; Andersson M. P.; Nørskov J. K.; Christensen C. H. Discovery of Technical Methanation Catalysts Based on Computational Screening. Top. Catal. 2007, 45, 9–13. 10.1007/s11244-007-0232-9. [DOI] [Google Scholar]
- Liu P. Water-Gas Shift Reaction on oxide/Cu(111): Rational Catalyst Screening from Density Functional Theory. J. Chem. Phys. 2010, 133, 204705. 10.1063/1.3506897. [DOI] [PubMed] [Google Scholar]
- Grabow L. C.; Gokhale A. A.; Evans S. T.; Dumesic J. A.; Mavrikakis M. Mechanism of the Water Gas Shift Reaction on Pt: First Principles, Experiments, and Microkinetic Modeling. J. Phys. Chem. C 2008, 112, 4608–4617. 10.1021/jp7099702. [DOI] [Google Scholar]
- Callaghan C.; Fishtik I.; Datta R.; Carpenter M.; Chmielewski M.; Lugo A. An Improved Microkinetic Model for the Water Gas Shift Reaction on Copper. Surf. Sci. 2003, 541, 21–30. 10.1016/S0039-6028(03)00953-1. [DOI] [Google Scholar]
- Gokhale A. A.; Dumesic J. A.; Mavrikakis M. On the Mechanism of Low-Temperature Water Gas Shift Reaction on Copper. J. Am. Chem. Soc. 2008, 130, 1402–1414. 10.1021/ja0768237. [DOI] [PubMed] [Google Scholar]
- Mhadeshwar A. B.; Vlachos D. G. Microkinetic Modeling for Water-Promoted CO Oxidation, Water-Gas Shift, and Preferential Oxidation of CO on Pt. J. Phys. Chem. B 2004, 108, 15246–15258. 10.1021/jp048698g. [DOI] [Google Scholar]
- Honkala K.; Hellman A.; Remediakis I. N.; Logadottir A.; Carlsson A.; Dahl S.; Christensen C. H.; Nørskov J. K. Ammonia Synthesis from First-Principles Calculations. Science 2005, 307, 555–558. 10.1126/science.1106435. [DOI] [PubMed] [Google Scholar]
- Medford A. J.; Wellendorff J.; Vojvodic A.; Studt F.; Abild-Pedersen F.; Jacobsen K. W.; Bligaard T.; Nørskov J. K. Assessing the Reliability of Calculated Catalytic Ammonia Synthesis Rates. Science 2014, 345, 197–200. 10.1126/science.1253486. [DOI] [PubMed] [Google Scholar]
- Hellman A.; Honkala K.; Remediakis I. N.; Logadóttir Á.; Carlsson A.; Dahl S.; Christensen C. H.; Nørskov J. K. Ammonia Synthesis and Decomposition on a Ru-Based Catalyst Modeled by First-Principles. Surf. Sci. 2009, 603, 1731–1739. 10.1016/j.susc.2008.10.059. [DOI] [Google Scholar]
- Nørskov J. K.; Bligaard T.; Rossmeisl J.; Christensen C. H. Towards the Computational Design of Solid Catalysts. Nat. Chem. 2009, 1, 37–46. 10.1038/nchem.121. [DOI] [PubMed] [Google Scholar]
- Reuter K.; Plaisance C. P.; Oberhofer H.; Andersen M. Perspective: On the Active Site Model in Computational Catalyst Screening. J. Chem. Phys. 2017, 146, 040901. 10.1063/1.4974931. [DOI] [PubMed] [Google Scholar]
- Singh A. R.; Montoya J. H.; Rohr B. A.; Tsai C.; Vojvodic A.; Nørskov J. K. Computational Design of Active Site Structures with Improved Transition-State Scaling for Ammonia Synthesis. ACS Catal. 2018, 8, 4017–4024. 10.1021/acscatal.8b00106. [DOI] [Google Scholar]
- Reuter K.; Metiu H.. A Decade of Computational Surface Catalysis. In Handbook of Materials Modeling: Applications: Current and Emerging Materials; Andreoni W., Yip S., Eds.; Springer International Publishing: Cham, 2018; pp 1–11. [Google Scholar]
- Smidstrup S.; Pedersen A.; Stokbro K.; Jónsson H. Improved Initial Guess for Minimum Energy Path Calculations. J. Chem. Phys. 2014, 140, 214106. 10.1063/1.4878664. [DOI] [PubMed] [Google Scholar]
- Henkelman G.; Uberuaga B. P.; Jónsson H. Climbing Image Nudged Elastic Band Method for Finding Saddle Points and Minimum Energy Paths. J. Chem. Phys. 2000, 113, 9901–9904. 10.1063/1.1329672. [DOI] [Google Scholar]
- Weinan E.; Ren W.; Vanden-Eijnden E. Simplified and Improved String Method for Computing the Minimum Energy Paths in Barrier-Crossing Events. J. Chem. Phys. 2007, 126, 164103. 10.1063/1.2720838. [DOI] [PubMed] [Google Scholar]
- Jacobsen C. J. H.; Dahl S.; Clausen B. S.; Bahn S.; Logadottir A.; Nørskov J. K. Catalyst Design by Interpolation in the Periodic Table: Bimetallic Ammonia Synthesis Catalysts. J. Am. Chem. Soc. 2001, 123, 8404–8405. 10.1021/ja010963d. [DOI] [PubMed] [Google Scholar]
- Besenbacher F.; Chorkendorff I.; Clausen B. S.; Hammer B.; Molenbroek A. M.; Nørskov J. K.; Stensgaard I. Design of a Surface Alloy Catalyst for Steam Reforming. Science 1998, 279, 1913–1915. 10.1126/science.279.5358.1913. [DOI] [PubMed] [Google Scholar]
- Wang S.; Temel B.; Shen J.; Jones G.; Grabow L. C.; Studt F.; Bligaard T.; Abild-Pedersen F.; Christensen C. H.; Nørskov J. K. Universal Brønsted-Evans-Polanyi Relations for C–C, C–O, C–N, N–O, N–N, and O–O Dissociation Reactions. Catal. Lett. 2011, 141, 370–373. 10.1007/s10562-010-0477-y. [DOI] [Google Scholar]
- Bligaard T.; Nørskov J. K.; Dahl S.; Matthiesen J.; Christensen C. H.; Sehested J. The Brønsted-Evans-Polanyi Relation and the Volcano Curve in Heterogeneous Catalysis. J. Catal. 2004, 224, 206–217. 10.1016/j.jcat.2004.02.034. [DOI] [Google Scholar]
- Montemore M. M.; Medlin J. W. Scaling Relations between Adsorption Energies for Computational Screening and Design of Catalysts. Catal. Sci. Technol. 2014, 4, 3748–3761. 10.1039/C4CY00335G. [DOI] [Google Scholar]
- Jones G.; Bligaard T.; Abild-Pedersen F.; Nørskov J. K. Using Scaling Relations to Understand Trends in the Catalytic Activity of Transition Metals. J. Phys. Condens. Matter 2008, 20, 064239. 10.1088/0953-8984/20/6/064239. [DOI] [PubMed] [Google Scholar]
- Abild-Pedersen F.; Greeley J.; Studt F.; Rossmeisl J.; Munter T. R.; Moses P. G.; Skúlason E.; Bligaard T.; Nørskov J. K. Scaling Properties of Adsorption Energies for Hydrogen-Containing Molecules on Transition-Metal Surfaces. Phys. Rev. Lett. 2007, 99, 016105. 10.1103/PhysRevLett.99.016105. [DOI] [PubMed] [Google Scholar]
- Hammer B.; Norskov J. K. Why Gold Is the Noblest of All the Metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]
- Andersen M.; Medford A. J.; Nørskov J. K.; Reuter K. Analyzing the Case for Bifunctional Catalysis. Angew. Chem. Int. Ed. 2016, 55, 5210–5214. 10.1002/anie.201601049. [DOI] [PubMed] [Google Scholar]
- Meskine H.; Matera S.; Scheffler M.; Reuter K.; Metiu H. Examination of the Concept of Degree of Rate Control by First-Principles Kinetic Monte Carlo Simulations. Surf. Sci. 2009, 603, 1724–1730. 10.1016/j.susc.2008.08.036. [DOI] [Google Scholar]
- Campbell C. T. The Degree of Rate Control: A Powerful Tool for Catalysis Research. ACS Catal. 2017, 2770–2779. 10.1021/acscatal.7b00115. [DOI] [Google Scholar]
- Sutton J. E.; Lorenzi J. M.; Krogel J. T.; Xiong Q.; Pannala S.; Matera S.; Savara A. Electrons to Reactors Multiscale Modeling: Catalytic CO Oxidation over RuO2. ACS Catal. 2018, 8, 5002–5016. 10.1021/acscatal.8b00713. [DOI] [Google Scholar]
- Döpking S.; Plaisance C. P.; Strobusch D.; Reuter K.; Scheurer C.; Matera S. Addressing Global Uncertainty and Sensitivity in First-Principles Based Microkinetic Models by an Adaptive Sparse Grid Approach. J. Chem. Phys. 2018, 148, 034102. 10.1063/1.5004770. [DOI] [PubMed] [Google Scholar]
- Hoffmann M. J.; Engelmann F.; Matera S. A Practical Approach to the Sensitivity Analysis for Kinetic Monte Carlo Simulation of Heterogeneous Catalysis. J. Chem. Phys. 2017, 146, 044118. 10.1063/1.4974261. [DOI] [PubMed] [Google Scholar]
- Choi Y. M.; Liu P. Mechanism of Ethanol Synthesis from Syngas on Rh(111). J. Am. Chem. Soc. 2009, 131, 13054–13061. 10.1021/ja903013x. [DOI] [PubMed] [Google Scholar]
- Mei D.; Rousseau R.; Kathmann S. M.; Glezakou V. A.; Engelhard M. H.; Jiang W.; Wang C.; Gerber M. A.; White J. F.; Stevens D. J. Ethanol Synthesis from Syngas over Rh-based/SiO2 Catalysts: A Combined Experimental and Theoretical Modeling Study. J. Catal. 2010, 271, 325–342. 10.1016/j.jcat.2010.02.020. [DOI] [Google Scholar]
- Broadbelt L. J.; Stark S. M.; Klein M. T. Computer Generated Pyrolysis Modeling: On-the-Fly Generation of Species, Reactions, and Rates. Ind. Eng. Chem. Res. 1994, 33, 790–799. 10.1021/ie00028a003. [DOI] [Google Scholar]
- Gao C. W.; Allen J. W.; Green W. H.; West R. H. Reaction Mechanism Generator: Automatic Construction of Chemical Kinetic Mechanisms. Comput. Phys. Commun. 2016, 203, 212–225. 10.1016/j.cpc.2016.02.013. [DOI] [Google Scholar]
- Grimme S. Towards First Principles Calculation of Electron Impact Mass Spectra of Molecules. Angew. Chem. Int. Ed. 2013, 52, 6306–6312. 10.1002/anie.201300158. [DOI] [PubMed] [Google Scholar]
- Maeda S.; Ohno K.; Morokuma K. Systematic Exploration of the Mechanism of Chemical Reactions: The Global Reaction Route Mapping (GRRM) Strategy Using the ADDF and AFIR Methods. Phys. Chem. Chem. Phys. 2013, 15, 3683–3701. 10.1039/c3cp44063j. [DOI] [PubMed] [Google Scholar]
- Simm G. N.; Reiher M. Context-Driven Exploration of Complex Chemical Reaction Networks. J. Chem. Theory Comput. 2017, 13, 6108–6119. 10.1021/acs.jctc.7b00945. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Kim J. W.; Kim Z.; Kim W. Y. Efficient Prediction of Reaction Paths through Molecular Graph and Reaction Network Analysis. Chem. Sci. 2018, 9, 825–835. 10.1039/C7SC03628K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L.-P.; Titov A.; McGibbon R.; Liu F.; Pande V. S.; Martínez T. J. Discovering Chemistry with an Ab Initio Nanoreactor. Nat. Chem. 2014, 6, 1044–1048. 10.1038/nchem.2099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper M. R.; Van Geem K. M.; Pyl S. P.; Marin G. B.; Green W. H. Comprehensive Reaction Mechanism for N-Butanol Pyrolysis and Combustion. Combust. Flame 2011, 158, 16–41. 10.1016/j.combustflame.2010.06.002. [DOI] [Google Scholar]
- Van Geem K. M.; Reyniers M. F.; Marin G. B.; Song J.; Green W. H.; Matheu D. M. Automatic Reaction Network Generation Using RMG for Steam Cracking of N-Hexane. AIChE J. 2006, 52, 718–730. 10.1002/aic.10655. [DOI] [Google Scholar]
- Goldsmith C. F.; West R. H. Automatic Generation of Microkinetic Mechanisms for Heterogeneous Catalysis. J. Phys. Chem. C 2017, 121, 9970–9981. 10.1021/acs.jpcc.7b02133. [DOI] [Google Scholar]
- Prickett S. E.; Mavrovouniotis M. L. Construction of Complex Reaction Systems - III. An Example: Alkylation of Olefins. Comput. Chem. Eng. 1997, 21, 1325–1337. 10.1016/S0098-1354(97)00004-5. [DOI] [Google Scholar]
- Prickett S. E.; Mavrovouniotis M. L. Construction of Complex Reaction Systems - II. Molecule Manipulation and Reaction Application Algorithms. Comput. Chem. Eng. 1997, 21, 1237–1254. 10.1016/S0098-1354(97)00003-3. [DOI] [Google Scholar]
- Prickett S. E.; Mavrovouniotis M. L. Construction of Complex Reaction Systems - I. Reaction Description Language. Comput. Chem. Eng. 1997, 21, 1219–1235. 10.1016/S0098-1354(97)00002-1. [DOI] [Google Scholar]
- Aghalayam P.; Park Y. K.; Vlachos D. G. Construction and Optimization of Complex Surface-Reaction Mechanisms. AIChE J. 2000, 46, 2017–2029. 10.1002/aic.690461013. [DOI] [Google Scholar]
- Ulissi Z. W.; Medford A. J.; Bligaard T.; Nørskov J. K. To Address Surface Reaction Network Complexity Using Scaling Relations Machine Learning and DFT Calculations. Nat. Commun. 2017, 8, 14621. 10.1038/ncomms14621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reymond J. L.; Awale M. Exploring Chemical Space for Drug Discovery Using the Chemical Universe Database. ACS Chem. Neurosci. 2012, 3, 649–657. 10.1021/cn3000422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruddigkeit L.; Van Deursen R.; Blum L. C.; Reymond J. L. Enumeration of 166 Billion Organic Small Molecules in the Chemical Universe Database GDB-17. J. Chem. Inf. Model. 2012, 52, 2864–2875. 10.1021/ci300415d. [DOI] [PubMed] [Google Scholar]
- Ramakrishnan R.; Dral P. O.; Rupp M.; von Lilienfeld O. A. Quantum Chemistry Structures and Properties of 134 Kilo Molecules. Sci. Data 2014, 1, 1–7. 10.1038/sdata.2014.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo H.; Rupp M.. Unified Representation of Molecules and Crystals for Machine Learning. 2017, arXiv:1704.06439 [Google Scholar]
- Husch T.; Reiher M. Comprehensive Analysis of the Neglect of Diatomic Differential Overlap Approximation. J. Chem. Theory Comput. 2018, 14, 5169–5179. 10.1021/acs.jctc.8b00601. [DOI] [PubMed] [Google Scholar]
- Landrum G.RDKit: Open-Source Cheminformatics. https://www.rdkit.org/.
- Frenking G.; Hermann M. Critical Comments on “One Molecule, Two Atoms, Three Views, Four Bonds?”. Angew. Chem., Int. Ed. 2013, 52, 5922–5925. 10.1002/anie.201301485. [DOI] [PubMed] [Google Scholar]
- Grunenberg J. Quadruply Bonded Carbon. Nat. Chem. 2012, 4, 154–155. 10.1038/nchem.1274. [DOI] [PubMed] [Google Scholar]
- Grzybowski B. A.; Bishop K. J. M.; Kowalczyk B.; Wilmer C. E. The “Wired” Universe of Organic Chemistry. Nat. Chem. 2009, 1, 31–36. 10.1038/nchem.136. [DOI] [PubMed] [Google Scholar]
- Fialkowski M.; Bishop K. J. M.; Chubukov V. A.; Campbell C. J.; Grzybowski B. A. Architecture and Evolution of Organic Chemistry. Angew. Chem., Int. Ed. 2005, 44, 7263–7269. 10.1002/anie.200502272. [DOI] [PubMed] [Google Scholar]
- Bishop K. J. M.; Klajn R.; Grzybowski B. A. The Core and Most Useful Molecules in Organic Chemistry. Angew. Chem., Int. Ed. 2006, 45, 5348–5354. 10.1002/anie.200600881. [DOI] [PubMed] [Google Scholar]
- Eyring H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. 10.1063/1.1749604. [DOI] [Google Scholar]
- Weininger D. SMILES, a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules. J. Chem. Inf. Model. 1988, 28, 31–36. 10.1021/ci00057a005. [DOI] [Google Scholar]
- Margraf J. T.; Ranasinghe D. S.; Bartlett R. J. Automatic Generation of Reaction Energy Databases from Highly Accurate Atomization Energy Benchmark Sets. Phys. Chem. Chem. Phys. 2017, 19 (15), 9798–9805. 10.1039/C7CP00757D. [DOI] [PubMed] [Google Scholar]
- Stamatakis M.; Vlachos D. G. Unraveling the Complexity of Catalytic Reactions via Kinetic Monte Carlo Simulation: Current Status and Frontiers. ACS Catal. 2012, 2, 2648–2663. 10.1021/cs3005709. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.