

Abstract

Anthracenenitrile oxide undergoes 1,3-dipolar cycloaddition reactions with 1-substituted-4-(prop-2-yn-1-yloxy)benzene affording the expected isoxazoles in good yields and as single regioisomers. N–O Bond cleavage and boron complexation afforded the corresponding boron complexes, derivatized with either a triple bond for click-chemistry applications or an oxime group for nitrile oxide 1,3-dipolar cycloaddition. The optical properties of these compounds show potential for employment as fluorescent tags in imaging techniques. The activity-based protein profiling assays showed good reactivity for the synthesized probes even with short peptide chains (epoxomicin). Scope and limitations are discussed in the light of the obtained results.
Introduction
1,3-Dipolar cycloadditions can be undoubtedly considered a pillar of the synthesis of isoxazoles. The versatility of this strategy was recently applied to synthesize structures designed for imaging. Albeit the number of works devoted to this topic is increasing, the applications of the probes constructed via this pathway remain quite low so far.1 In a recent conceptual work, we successfully employed 1,3-dipolar cycloaddition of stable aromatic nitrile oxides to afford novel fluorescent compounds. The reported strategy is the cornerstone to furnish boron-substituted complexes suitable for biochemical applications; the nitrile oxide-based protocol is cleaner and selective with dipolarophiles and can be a useful alternative to the use of azides. Indeed, once properly derivatized, it can find proper use in the activity-based protein profiling (ABPP).2
ABPP is one of the most powerful tools to gain insight into complex biological systems, e.g., the activity of enzymes in complex proteasomes.3 The aim of ABPP resides in the visualization of the active forms of the enzymes using chemical probes directed to the active site of a target protein, resulting in the selective labeling of the sole catalytically active form of the enzyme.4
Structurally, chemical probes consist of three different parts: recognition tag, variable length linker, and warhead (ligation handles) containing the functional groups to link the probe with the target substrate with highly specific interactions that make the probe selective for a well-defined biological structure (Scheme 1). The ligation strategy we wish to exploit is based on 1,3-dipolar cycloaddition for the two-step activity-based labeling of endogenously expressed enzymes in complex biological samples.5
Scheme 1. From Nitrile Oxides 1 to 5-Substituted Isoxazoles 3, N–O Bond Cleavage and Boron Complexation: Synthetic Route to Fluorescent Probes of Type 5 and 6.

The investigated probes aimed to be applied in a labeling procedure that enabled us to label active proteasome β-subunits selectively in cellular extracts and in living cells.
In our previous work, we detailed the synthetic strategy and the fluorescence study of compound 5 that structurally follows the aforementioned paradigms. The compound is synthesized starting from a 1,3-dipolar cycloaddition reaction between 1-iodo-4-(prop-2-yn-1-yloxy)benzene (2), affording the 5-substituted isoxazole 3 in very good yields as a single regioisomer. The outcome of the transformation nicely follows the predictions based on the frontier orbital theory.2 Cleavage of the N–O bond and BF3 complexation furnish the corresponding fluorescent boron complex of type 5 (Scheme 1). The molecule bears an anthryl substituent (Ar = 9-anthryl) on the tag and a triple C≡C bond on the warhead end terminus (R = −C≡CH) of the probe, suitable for click-chemistry applications and late-stage functionalization.
Here, we present a strategy to prepare chemical probes that maintain the same tag structure, while bearing variable ligation handles prone to orthogonal functionalization with a proteasome inhibitor peptide. In compound 5, the triple bond requires the use of a substrate bearing a dipole (typically an azido-derivative) to connect the two sides of the chemical reporter (Scheme 2). The newly designed compound 6 bears an oxime moiety on the warhead part that is suitable to be oxidized to nitrile oxide. Hence, the probe is another 1,3-dipole that can be attached to a double (or triple) bond located on a target substrate. One of the great advantages of the use of nitrile oxides is the fact that the cycloaddition reactions are cleaner than those in the presence of azides; these latter ones require metals (copper ions, typically) that are detrimental for the cellular environment.
Scheme 2. Probe Structures: Dipolarophile and Dipole Precursor Ligation Handle Structures.

The two chemical probes 5 and 6 will be compared from the photophysical point of view and tested by coupling them with differently functionalized epoxomicin derivatives. Ultimately, competitive ABPP assays will be performed to verify the maintenance of proteasome inhibitor properties and possible differences in terms of selectivity.
Results and Discussion
The commercially available 4-hydroxybenzaldehyde 7 was derivatized with freshly distilled propargyl chloride in the presence of a base under reflux in acetonitrile (Scheme 3). The ethynyl derivative 8 is a known compound6 and was transformed into the corresponding oxime by treatment with hydroxylamine in a hydro-alcoholic solution, to afford compound 10.7 The oxime dipolarophile was allowed to react with an equimolecular amount of anthracenenitrile oxide 9 in anhydrous dichloromethane (DCM) at room temperature for 48 h. After purification, the 5-substituted isoxazole 11 was obtained in 66% yield.8 In the 1H NMR spectrum (dimethyl sulfoxide, DMSO), the diagnostic signal of the H4 proton of the isoxazole ring was detected at δ 7.03 ppm. The inclusion of the dipolarophile moiety into the cycloadduct was also confirmed by the presence of the methyleneoxy group at δ 5.53 ppm (singlet) and by the highly deshielded oxime OH signal at δ 11.06 ppm.
Scheme 3. Probe Synthesis: From 4-Hydroxybenzaldehyde (7) to 4-((4-(Anthracen-9-yl)-2,2-difluoro-2,3-dihydro-1λ3,3,2λ4-oxazaborinin-6-yl)methoxy)benzaldehyde Oxime (6).

The selective N–O bond cleavage9 in the isoxazole moieties was performed using Mo(CO)6 to give the 4-[(4-amino-4-(anthracen-9-yl)-2-oxobut-3-en-1-yl)oxy] benzaldehyde oxime 12 as a reddish solid in 68% yield. As previously reported,2 the signals detected in the 1H NMR spectrum denote the existence of an intramolecular hydrogen bond between the amino group and the carbonyl functionality obtained from the N–O bond cleavage. As a consequence, one of the NH2 protons is found at δ 8.47 ppm whereas the other is strongly deshielded at δ 10.25 ppm. The latter is the direct evidence of the 6-membered cyclic array kept together by the hydrogen bond.
The enaminoketone 12 was used as ligand for boron complexation,10 achieved by adding BF3·Et2O in an anhydrous DCM solution, in the presence of Et3N (Scheme 3). Boron complex 6 was obtained as a yellow solid in 40% yield. In the 1H NMR spectrum, a single NH proton signal is detected at δ 7.62 ppm, as a consequence of the newly formed bond between the nitrogen and the boron.
To verify the reactivity of the fluorescent probe 6 as 1,3-dipole, we oxidized the oxime group with ([bis(trifluoroacetoxy)iodo]benzene) (PIFA) in the presence of an excess of norbornene (Scheme 4).11 Norbornene, an excellent trap for nitrile oxides,12 is able to attach the in situ formed nitrile oxide from compound 6, affording the corresponding 1,3-dipolar cycloadduct 13 in 81% yield, and the reaction does not require any metal promoter such as copper that is normally used with azides. Hence, the reaction is cleaner, and after simple chromatographic purification, compound 13 was fully characterized; the presence in the 1H NMR spectrum of the signals corresponding to the isoxazoline protons at δ 3.64 and 4.55 ppm clearly indicates the successful coupling between the nitrile oxide moiety and the C=C double bond of the norbornene.
Scheme 4. 1,3-Dipolar Cycloaddition Reaction of Probe 6 with Norbornene.

To evaluate the potential use of the new boron complex 6 as a chemical probe in biological studies, we performed some UV–vis and fluorescence studies in solvents of different polarities, comparing the results with those previously obtained for compound 5.2
Figure 1 shows the UV–vis spectra of compounds 5 and 6 in DCM as solvent. The oxime derivative 6 showed an absorption band at 384 nm (ε = 5.00 × 103 M–1 cm–1) with a shoulder at 238 nm and vibrational peaks. Analogously, the BF2 complex 5 shows an absorption band at 387 nm (ε = 8.20 × 103 M–1 cm–1) along with more structured vibrational peaks compared with compound 6. The change in functional groups on the phenyl ring does not drastically shift the absorption peak in the near-UV region. It however results in a broadening of the absorption band around 250 nm that merges with the blue-shifted shoulder.
Figure 1.

UV–vis spectra of compounds 5 (A) and 6 (B) in DCM (2 × 10–5 M solutions).
The fluorescence emission spectra of 5 and 6 are shown in Figure 2. The fluorescence maxima for both the compounds appear at 490 nm, indicating a Stokes’ shift of ca. 5630 cm–1 for the two complexes. The emission spectra are structureless for both compounds and present a single intense peak. The complete loss of the vibrational features, along with the considerable Stokes’ shift, could be indicative of a structural rearrangement occurring at the excited state. This feature is less pronounced in more rigid fluorescent boron derivatives.13 Indeed, the first excited-state optimized structure of compound 6 at the SMD(DCM)-TD-CAM-B3LYP/6-31G(d,p)//TD-B3LYP/6-31G(d,p) level of theory differs from the Franck–Condon one, due to the torsion along the bond connecting the boracycle to the anthryl moiety.14 The associated dihedral angle varies from ca. 66 to 41°. Such a structural change is an indication of the nature of the electronic transition associated to fluorescence. The excited state populates an antibonding orbital that spans from the anthryl moiety to the boracycle with an electron coming from the anthracene (see Figure 3). Consequently, the two connected cycles involved in the transition bend to increase the conjugation in the molecule.
Figure 2.
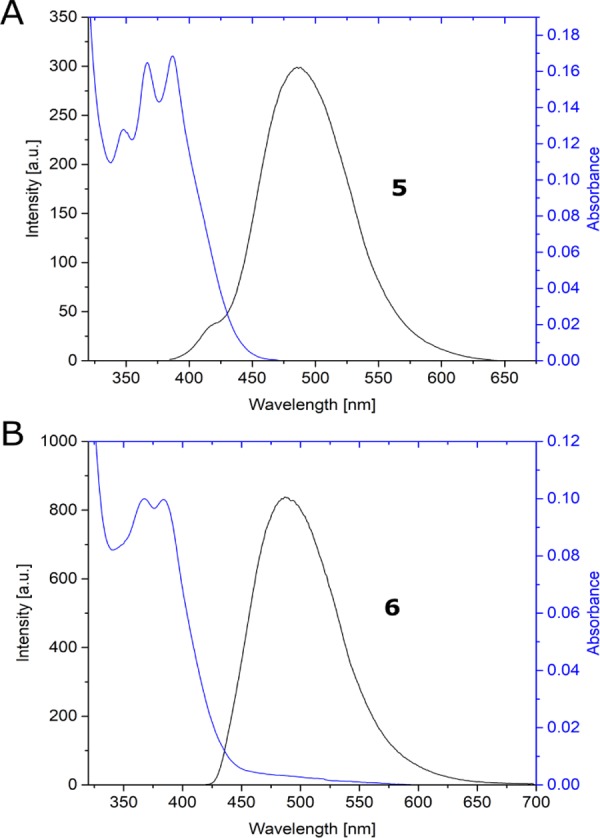
Fluorescence spectra of compounds 5 (A) and 6 (B) in DCM (blue line, absorption; black line, emission).
Figure 3.

Optimized structure of the ground state of compound 6 (A) and its first excited state (B). Orbitals involved in the first excite state of 6 without (C, D) and with (E, F) protic interactions taken into account. All calculations are at the SMD(DCM)-(TD)-CAM-B3LYP/6-31G(d,p)//(TD)-B3LYP/6-31G(d,p) level, and the structures correspond to energy minima.
In DCM, compound 6 is characterized by fluorescence quantum yield ΦF = 0.07 whereas compound 5 exhibits ΦF = 0.602 (both values were determined relative to the fluorescent intensity of a boron-substituted reference compound; for further details refer to the Supporting Information SI).10 The low quantum yields of compound 6 even in DCM could be the indication of intermolecular quenching of fluorescence. It is known that a protic environment can influence the excited-state nature of fluorophores.15 The source of this interaction could be the hydroxylamino derivative in compound 6, absent in compound 5. To test our hypothesis, we optimized compound 6 at the SMD(DCM)-TD-CAM-B3LYP/6-31G(d,p)//TD-B3LYP/6-31G(d,p) level, in the presence of a water molecule, as a prototypical protic compound. In this case, the excited-state changes in nature and a charge-transfer electron transfer from the phenoxy moiety to the anthryl one quenches the fluorescence. Hence, we can conclude that the interaction of 6 with a protic solvent or with its hydroxylamino moiety can be the culprit for the low quantum yields of 6 (see Figure 3).
The solvent effect on the absorption and fluorescence properties of the new boron complex 6 was examined (spectra reported in the SI), and Table 1 reports the ΦF values obtained. The absorption (λmax) values of 6 are not affected by solvent polarity, suggesting that the dipole moments of the molecule in the ground and excited states are almost the same.16 On the other hand, the maximum of fluorescence emission (λem) values showed a blue shift of about 40 nm in polar solvents with respect to the value in DCM. This determined a Stokes’ shift reduction at just about ca. 3820 cm–1. These results show the same trend as that observed for compound 5.2 Significantly, the ΦF values dramatically diminish upon polarity changes as well as viscosity; remarkable drops in quantum yields are observed in MeOH (0.01) and in ethylene glycol (0.02). The same happens in the 1:1 DMSO/H2O mixture with ΦF value = 0.01. This could suggest some drawbacks when the compound is used in a biological environment.17
Table 1. Absorption λmax (nm) and Fluorescence Quantum Yield (ΦF) Values for Complex 6 in the Listed Solventsa.
| solvent | dielectric constant | viscosity18 (cP at 20 °C) | λmax (nm) | λem (nm) | ΦF |
|---|---|---|---|---|---|
| DCM | 8.9 | 0.44 | 384 | 490 | 0.07 |
| MeOH | 33 | 0.55 | 380 | 488 | 0.01 |
| ethylene glycol | 39 | 23.5 | 384 | 455 | 0.02 |
| DMSO/H2O | 55 | 3.30 | 384 | 450 | 0.01 |
Concentration 2 × 10–5 M in all solvents.
On the basis of novel and previous observations and with the aim to employ compounds 5 and 6 in ABPP assays, we performed a series of coupling experiments with suitable epoxomicin derivatives. Azido-substituted epoxomicin 14, a proteasome inhibitor peptide, was coupled with the ethynyl-probe 5 under standard click-reaction conditions (Scheme 5).3,4 The expected product 15 was isolated and purified by column chromatography. Retaining of water in the final compound required a prolonged drying step. After the drying process, the product was obtained in 55% yield and the mass spectrum (MS) reported a mass value of 1023.30 m/z corresponding to the molecular weight of 15 (1006.94 m/z) plus OH (MW + OH). This discrepancy in the MS spectra values can be tentatively attributed to the harsh purification protocol followed (even with standard methods) that presumably left on the polar peptide chain residues of the basic treatment. We cannot exclude the epoxide ring opening to give a diol or even the stabilization of the hydrate forms of the carbonyl group adjacent to the oxirane ring.
Scheme 5. Coupling of Ethynyl-Probe 5 with Azido-Substituted Epoxomicin 14; Inset Shows the Possible Structure for the Oxirane Fragment Located at the End of Compound 15.

On the other hand, norbornene-substituted epoxomicin 16 was allowed to react with oxime 6 under oxidative conditions (using phenyliodine diacetate (PIDA) as an oxidant in dimethylformamide (DMF) at room temperature). The in situ-generated nitrile oxide of 6 was added exo-selectively19 to the C=C bond of the norbornene moiety to afford the corresponding 1,3-dipolar cycloadduct 17 (Scheme 6), presumably as a mixture of regioisomers that would not affect the biological behavior. Due to the known reactivity of nitrile oxides with nucleophilic centers and the nature of the dipolarophile, the purification step was quite problematic and several steps of chromatographic purification were needed. Nevertheless, the final cycloadduct was obtained in 45% yield and the MS spectrum confirmed the structure as drawn in Scheme 6 with MW = 1199.30 (theor. 1199.15) m/z.
Scheme 6. Coupling of Oxime-Probe 6 with Norbornene-Substituted Epoxomicin 16.

The coupling products 15 and 17 were then tested in competitive ABPP assays (sodium dodecyl sulfate polyacrylamide gel electrophoresis, SDS-PAGE) to check the maintenance of the proteasome inhibitor properties of epoxomicin20 and the display of fluorescence properties. The competitive SDS-PAGE assay was performed with some other marked proteasome inhibitors (details in the SI).
The analysis was done on Raji cell lysate (1 μL), containing both immuno and constitutive proteasome ([proteasome] = 20 μg μL–1). Different samples of lysate were incubated for half an hour at 37 °C along with 9 μL of 26S assay buffer and 1 μL of a DMSO solution of compounds 15 or 17 at different concentrations (0, 0.1, 1, 10, and 100 μM). Then, 1 μL of the 10 × ABP cocktail was added and the samples were incubated for other 30 min. Finally, 5 μL of the 4× sample buffer to denature and reduce proteins was added and the samples were boiled at 100 °C for 5 min.
The samples were fixed in the different lanes of a SDS-PAGE stacking gel and then were made to run by gel electrophoresis on the running gel. Figure 4 shows the SDS-PAGE gel results where in the lane 1, it is possible to see the different β subunits of the proteasome, highlighted with specific fluorescent markers that bind them selectively: in blue, Cy5-NC-001 linked to the β1 subunits, in green, BODIPY(FL)-LU-112 was selective to the β2 ones, and in red, BODIPY(TMR)-NC-005-VS linked to the β5 ones.
Figure 4.
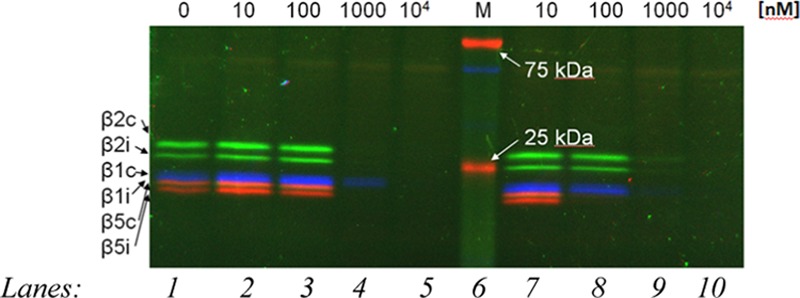
SDS-PAGE gel of compounds 15 and 17 (lanes 2, 3, 4, and 5, for increasing concentrations of probe 15; lanes 7, 8, 9, and 10 for increasing concentrations of probe 17; lane 1, specific fluorescent markers for β subunits; and lane 6, a molecular weight marker for reference).
In lane 2, 3, 4, and 5, increasing concentrations of probe 15 (from 10 to 104 nM) are reported, whereas in lane 7, 8, 9, and 10, the same increasing concentration of probe 17 (from 10 to 104 nM) can be found. For a concentration of 1000 nM, both probes show a very good competition in binding the proteasome; in fact, in lanes 4 and 5 and 9 and 10 the colored lines relative to the other marked inhibitors are not visible. In lane 8, the red stripes relative to the β5 subunits disappeared, suggesting a higher activity of the probe 17 for these subunits.
To detect the fluorescence of probes 15 and 17 while they bind the proteasome, several different light sources were tried to excite the fluorophores but unluckily they did not match the excitation wavelength of our probes and no fluorescence were registered. Some possible explanations can be considered. We cannot state if these probes maintain their fluorescence during the interaction with the proteasome; it is also possible that the fluorophores maintain their optical properties but the excitation was not efficient. Alternatively, the probes 15 and 17 were not strong enough from the fluorescence point of view to be detected with the instruments employed. We cannot exclude potential quenching or degradation (instability of the boron complex) during the interaction with the proteasome.
Conclusions
In conclusion, this work has shown the possible and reliable application of nitrile oxide chemistry in the biomedical field, especially as starting materials for the synthesis of some fluorescent probes. The chemistry of isoxazoles,21 dating to many decades ago, in the field of imaging probes seems to be a promising and valuable alternative to azides.22 Azides, in fact, do suffer from the need of copper to perform correctly the cycloaddition. The use of copper and metals in general is detrimental in a biological environment, and the chemistry we offer by replacing azides with nitrile oxides is cleaner and safer; the construction of the boron complexes is simple, reliable, and robust. The methodology can be applied by taking advantage of the variety of structures having the aldehydes as starting compounds, to prepare corresponding nitrile oxides. In light of this, we have indeed demonstrated the possibility to expand the application of 1,3-dipolar cycloaddition reactions to obtain a group of easily derivatizable fluorescent probes. We were able to obtain two novel boron fluorescent probes 15 and 17 that enhance the range of reactions suitable for the coupling with proteasome inhibitors. For this purpose, the optical properties of these compounds were investigated and suggest the potential for employment as fluorescent tags in imaging techniques. The drop of fluorescence quantum yield values for compound 6 with respect to 5 remains a problem whose solution is not far from the synthetic route we have followed, while maintaining the validity of the chemical strategy. The moderate quantum yield values maintained in the DMSO/H2O mixture confirm the suitability of these compounds in the cellular environment. The ABPP assays performed did not give us the ultimate answer about the behavior of compounds 15 and 17 in SDS-PAGE analyses. The coupling tests performed showed good reactivity for the probes 15 and 17, even with short peptide chains such as epoxomicin. Improvement of the fluorescence properties of enaminoketone-based boron complexes is needed and actively pursued. At present, the tuning of the optical properties of the probes is a crucial point for making these compounds competitive with commercial ABPP probes. The stability of the boron complex within the lisate environment is also a key point for its application in imaging techniques.
Design, synthesis, and application: these aspects must be synchronized and to do that, brand new isoxazole derivatives are currently under preparation by functionalizing the anthryl moiety with several aromatic rings bearing donor and acceptor substituents by means of the Suzuki coupling; moreover, selected heterocycles have been inserted too (Scheme 7).23
Scheme 7. Synthetic Strategy toward Newly Designed Fluorescent Probes.

The Spacer can be also deleted, leaving a fully conjugated system.
This revised and enlarged library of isoxazoles was already verified to show unexpected intense fluorescent properties, even stronger than the corresponding boron complexes. Fluorescent studies are actively pursued to compare the photochemical behavior of the uncomplexed and complexed probe couples (see Scheme 7) to determine the real need of a boron complex to display optical properties in this type of compound. These findings will be communicated in a full paper that is under preparation.
Experimental Section
General Methods and Materials
Melting points (mp) are uncorrected. Elemental analyses were performed on a FlashSmart elemental analyzer. 1H NMR and 13C NMR were registered on Bruker AVANCE 300 spectrometers in deuterated solvent solutions. The chemical shifts are expressed in ppm, using tetramethylsilane as the internal reference. IR spectra were registered using a FT-IR PerkinElmer RX-1 spectrometer dissolving the analyzed products in DCM (film).
Chromatographic columns were obtained using Kieselgel 60, 70–230 (Merck), with a BIOTAGE MPLC or BIOTAGE Isolera One with KP-SIL columns; the elution solvents were from cyclohexane/ethyl acetate, 9:1, to pure ethyl acetate. UV–vis spectra were registered with a Jasco V-550 UV–vis spectrophotometer; the fluorescence spectra were registered with a PerkinElmer LS 55 luminescence spectrometer.
Anthracenenitrile oxide 9 was prepared from the corresponding commercially available oxime according to the known procedure.24,3 Solvents and other reagents were purchased and used without any further purification.
Synthesis of 4-(Prop-2-yn-1-yloxy)benzaldehyde oxime (10)
A solution of 3.20 g (46.0 mmol) of hydroxylamine hydrochloride in 20 mL of water was added to 4.83 g (30.1 mmol) of 4-(prop-2-ynyloxy) benzaldehyde (8) dissolved in 125 mL of ethanol. NaOH solution was added to adjust the pH at 5, and the reaction was heated at 40 °C for 3 h. The mixture was then refluxed for 20 min and kept under stirring overnight. Ethanol was partially evaporated until the formation of a white solid, and water was added afterward. The mixture was extracted with diethyl ether, and the combined organic phases were dried over MgSO4 and evaporated after filtration. The product was recrystallized from cyclohexane/ethyl acetate, obtaining a white solid of 10 in 87% yield (4.60 g). Mp: 68–70 °C from cyclohexane/ethyl acetate. FT-IR: νC=N 1603 cm–1; νOH 3262 cm–1. 1H NMR (δ, DMSO): 3.59 (s, 1H, C≡C–H); 4.83 (s, 2H, CH2); 7.01 (AA′BB′, 2H, arom.); 7.54 (AA′BB′, 2H, arom.); 8.08 (s, 1H, N=C–H); 11.02 (s, 1H, OH). 13C NMR (δ, DMSO): 55.4; 78.4; 79.0; 115.1; 126.3; 127.7; 147.5; 158.0. Elemental analysis: calculated for C10H9NO2 (MW = 175.06): C, 68.56; H, 5.18; N, 8.08; found: C, 68.55; H, 5.15; N, 8.09.
Synthesis of 4-((3-(Anthracen-9-yl)isoxazol-5-yl)methoxy)benzaldehyde Oxime (11)
In a 250 mL round-bottom flask, 2.27 g (10.4 mmol) of anthracenenitrile oxide (9) was added dropwise to a solution of 1.89 g (10.8 mmol) of 4-(prop-2-ynyloxy) benzaldehyde oxime (10) in 125 mL of anhydrous DCM. The reaction was kept in the dark and under stirring at room temperature for 48 h. Then, the mixture was diluted with DCM (100 mL) and the solution was washed with brine (3 × 50 mL) and dried over anhydrous Na2SO4.
The solvent was evaporated and the crude residue was purified by column chromatography, giving a yellowish solid of 11 in 66% yield (2.69 g). Mp: 128–131 °C from cyclohexane/ethyl acetate. FT-IR: νC=N 1598 cm–1; νOH 3265 cm–1. 1H NMR (δ, DMSO): 5.54 (s, 2H, CH2); 7.04 (s, 1H, C=C–H); 7.19 (AA′BB′, 2H, arom.); 7.58 (m, 6H, anthr.); 7.72 (m, 2H, anthr.) 8.14 (s, 1H, N=C–H); 8.20 (AA′BB′, 2H, arom.); 8.83 (s, 1H, anthr.); 11.07 (s, 1H, OH).
13C NMR (δ, DMSO): 18.5; 60.7; 107.7; 115.3; 122.5; 124.9; 125.7; 126.7; 127.0; 127.9; 128.6; 129.0; 129.9; 130.7; 147.5; 158.4; 160.3; 168.4. Elemental analysis: calculated for C25H18N2O3 (MW = 394.13): C, 76.13; H, 4.60; N, 7.10; found: C, 76.15; H, 4.63; N, 7.09.
Synthesis of 4-((4-Amino-4-(anthracen-9-yl)-2-oxobut-3-en-1-yl)oxy)benzaldehyde Oxime (12)
Compound 11 (1.80 mmol, 720 mg) was dissolved in 120 mL of a 9:1 acetonitrile/water mixture in a three-necked round-bottom flask. The solution was stirred under nitrogen for 15 min, and then 605 mg of molybdenum hexacarbonyl (2.30 mmol) was added. The reaction was stirred at 70 °C for 4 h.
DCM was then added, and the mixture was washed with brine (3 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and evaporated. The crude product was purified on column chromatography, giving a beige-reddish solid of 12 in 68% yield (491 mg). Mp: 183–189 °C from cyclohexane/ethyl acetate. FT-IR: νC=N 1579 cm–1; νOH/NH2 3265 cm–1. 1H NMR (δ, DMSO): 4.66 (s, 2H, CH2); 5.40 (s, 1H, C=C–H); 6.93 (AA′BB′, 2H, arom.); [7.11] (AA′BB′, 2H, arom.); 7.53 (AA′BB′, 2H, arom.); 7.53 (m, 4H, anthr.); [7.85] (AA′BB′, 2H, arom.); 7.97 (m, 2H, anthr.); 8.07 (s, 1H, N=C–H); 8.13 (m, 2H, anthr.); 8.47 (s, 1H, NH2); 8.71 (s, 1H, ant.); 9.88 (s, 1H, OH); 10.25 (s, 1H, NH2); [10.95] (s, 1H, OH). 13C NMR (δ, DMSO): 70.8; 93.4; 93.5; 114.5; 114.8; 124.8; 125.2; 125.5; 126.2; 127.4; 127.5; 128.0; 129.5; 130.2; 130.7; 130.7; 131.3; 147.2; 158.5; 161.5; 161.7; 190.9; 191.2; 192.0. Elemental analysis: calculated for C25H20N2O3 (MW = 396.15): C, 75.74; H, 5.08; N, 7.07; found: C, 75.75; H, 5.03; N, 7.08.
Synthesis of 4-((4-(Anthracen-9-yl)-2,2-difluoro-2,3-dihydro-1λ3,3,2λ4-oxazaborinin-6-yl)methoxy)benzaldehyde Oxime (6)
Compound 12 (2.30 mmol, 916 mg) was dissolved in 100 mL of anhydrous DCM along with 10 mL of distilled triethylamine in a 250 mL three-necked bottom flask. The solution was stirred under a nitrogen atmosphere for 10 min, and then 4 mL of boron trifluoride etherate (32.4 mmol) was added. The reaction was stirred under a nitrogen atmosphere overnight. The mixture was then diluted with DCM (20 mL) and washed with brine (3 × 50 mL). The organic phase was dried over anhydrous Na2SO4 and evaporated, obtaining a dark oil. The crude product was then purified on column chromatography, giving a yellow solid of 6 in 40% yield (409 mg). Mp: 179–184 °C from from cyclohexane/ethyl acetate. FT-IR: νNH 3362 cm–1; νOH 3272 cm–1; νC=N 1614 cm–1. 1H NMR (δ, DMSO): 5.05 (s, 2H, CH2); 6.06 (s, 1H, C=C–H); 7.03 (AA′BB′, 2H, arom.); 7.51 (AA′BB′, 2H, arom.); 7.62 (m, 4H, anthr.); 7.62 (bs, 1H, NH); 7.71 (m, 2H, anthr.); 8.07 (s, 1H, N=C–H); 8.23 (m, 2H, anthr.); 8.86 (s, 1H, anthr.); 11.03 (s, 1H, OH); [11.16] (s, 1H, OH). 13C NMR (δ, DMSO): 30.7; 67.0; 97.6; 115.0; 123.8; 125.9; 126.5; 127.2; 127.7; 127.8; 128.8; 129.5; 130.4; 147.4; 158.1; 172.0; 175.2. Elemental analysis: calculated for C25H19BF2N2O3 (MW = 444.15): C, 67.59; H, 4.31; N, 6.31; found: C, 67.55; H, 4.33; N, 6.30.
Synthesis of 3-(4-((4-(Anthracen-9-yl)-2,2-difluoro-2,3-dihydro-1λ3,3,2λ4-oxazaborinin-6-yl)methoxy)phenyl)-3a,4,5,6,7,7a-hexahydro-4,7-methanobenzo[d]isoxazole (13)
PIFA (20 mg) was dissolved in 50 mL of DMSO along with boron complex 6 (123 mg). After 5 min, 150 mg of 2-norbornene was added and the mixture was stirred for 4 h. The mixture was then extracted with DCM, and the combined organic phases was dried over Na2SO4. The solvent was evaporated and the product was purified by a chromatographic column, giving a yellow solid (13) in 81% yield (122 mg). Mp: >210 °C from ethyl acetate. FT-IR: νC=N 1598 cm–1; νNH 3361 cm–1. 1H NMR (δ, DMSO): 1.15 (d, 1H, norb.); 1.26 (d, 1H, norb.); 1.48 (m, 4H, norb.); 3.64 (d, J = 8, 1H, norb.); 4.55 (d, J = 8, 1H, norb.); 5.08 (s, 2H, CH2); 6.06 (s, 1H, C=C-H); 7.07 (AA′BB′, 2H, arom.); 7.63 (AA′BB′, 2H, arom.); 7.63 (m, 4H, anthr.); 7.73 (m, 2H, anthr.); 8.85 (s, 1H, anthr.); 11.18 (s, 1H, NH). 13C NMR (δ, DMSO): 22.1; 26.6; 31.8; 56.2; 67.0; 86.7; 97.6; 115.1; 122.3; 123.8; 125.8; 125.9; 127.2; 127.6; 128.2; 128.8; 129.5; 130.4; 130.7; 137.1; 156.0; 158.4; 172.0; 175.1. Elemental analysis: calculated for C32H27BF2N2O3 (MW = 536.21): C, 71.66; H, 5.07; N, 5.22; found: C, 71.65; H, 5.03; N, 5.20.
Synthesis of (2S,3R)-2-(2-(4-(4-((4-(Anthracen-9-yl)-2,2-difluoro-2,3-dihydro-1λ3,3,2λ4-oxazaborinin-6-yl)methoxy)phenyl)-1H-1,2,3-triazol-1-yl)acetamido)-N-((2S,3R)-1-(((2S,3S)-3-hydroxy-1-(((S)-4-methyl-1-((R)-2-methyloxiran-2-yl)-1-oxopentan-2-yl)amino)-1-oxobutan-2-yl)amino)-3-methyl-1-oxopentan-2-yl)-3-methylpentanamide (15)
Alkynyl fluorescent probe 5 (2.71 mg, 6.37 μmol) was dissolved in 1 mL of argon-degassed DMF along with 3.64 mg (6.26 μmol) of azido-epoxomicin 14. An aqueous solution of CuSO4 (1 mg, 4 μmol) and sodium ascorbate (1 mg, 5 μmol) was added to the mixture, and the solution was then stirred overnight at room temperature under an argon atmosphere.
The crude product was purified by silica gel column chromatography (DCM/MeOH 9:1) to give 8.59 mg of cycloadduct 15 (6.37 μmol).
The product weight was initially higher than expected probably due to some water residue that was very difficult to remove. After a series of careful drying processes, the product, obtained in 55% yield, was submitted for relative characterization by mass spectrometry.
The mass spectrum (MS) reporting a mass value of 1023.30 m/z corresponds to the molecular weight of 15 (1006.94 m/z) plus OH (MW + OH).
Synthesis of (3aS,4R,6S,7S,7aS)-3-(4-((4-(Anthracen-9-yl)-2,2-difluoro-2,3-dihydro-1λ3,3,2λ4-oxazaborinin-6-yl)methoxy)phenyl)-N-((1-((4S,7S,10S,13S)-4,7-di((R)-sec-butyl)-10-((S)-1-hydroxyethyl)-15-methyl-13-((R)-2-methyloxirane-2-carbonyl)-2,5,8,11-tetraoxo-3,6,9,12-tetraazahexadecyl)-1H-1,2,3-triazol-4-yl)methyl)-3a,4,5,6,7,7a-hexahydro-4,7-methanobenzo[d]isoxazole-6-carboxamide (17)
To a solution of 1.59 mg (3.58 μmol) of oxime fluorescent probe 6 in argon-degassed DMF was added 1.74 mg (5.40 μmol) of PIDA. The solution was stirred for 10 min under an argon atmosphere, and then 1.98 mg (2.62 μmol) of norbornene-epoxomicin 16 was added to the solution. The mixture was stirred overnight at room temperature.
The crude product was purified by silica gel column chromatography (DCM/MeOH 9:1) to give 1.87 mg (1.56 μmol) of cycloadduct 17 (45% yield).
Due the known reactivity of nitrile oxides and the nature of the dipolarophile, the purification step was quite problematic and several steps of chromatographic purification were needed. The final cycloadduct 15 was obtained, and the MS spectrum confirmed its structure with MW = 1199.30 (theor. 1199.15) m/z.
Density Functional Theory (DFT) and Time-Dependent DFT Calculations
Calculations were performed using Gaussian 09, Revision E.01. All optimizations were performed using the SMD(DCM)-(TD)-CAM-B3LYP/6-31G(d,p)//(TD)-B3LYP/6-31G(d,p) level of theory (over the first 10 singlet states). The identity of the stationary points was confirmed by frequency analysis due to the absence imaginary frequencies.
Acknowledgments
Financial support by the University of Pavia and COST Action (CM1004) “Synthetic Probes for Chemical Proteomics and Elucidation of Biosynthetic Pathways” is gratefully acknowledged. We also thank “VIPCAT, Value Added Innovative Protocols for Catalytic Transformations” project (CUP: E46D17000110009) for valuable financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00672.
Synthesis of 4-(prop-2-yn-1-yloxy)benzaldehyde (8); competitive ABPP assays (SDS-PAGE); UV–vis and fluorescent spectra of compounds 6, 5, 15, and 17 in DCM, MeOH, ethylene glycol, and DMSO/H2O 1:1, as well as the corresponding fluorescent spectra and the quantum yield data; 1H and 13C spectra of all new synthesized compounds; MS spectra of the final adducts 15 and 17 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Frath D.; Massue J.; Ulrich G.; Ziessel R. Luminescent Materials: Locking π-Conjugated and Heterocyclic Ligands with Boron(III). Angew. Chem., Int. Ed. 2014, 53, 2290–2310. 10.1002/anie.201305554. [DOI] [PubMed] [Google Scholar]
- Minuti L. F.; Crespi S.; Memeo M. G.; Quadrelli P. Fluorescent Probes from Stable Aromatic Nitrile Oxides. Eur. J. Org. Chem. 2016, 821–829. 10.1002/ejoc.201501478. [DOI] [Google Scholar]
- Li N.; Overkleeft H. S.; Florea B. I. Activity-Based Protein Profiling: an Enabling Technology in Chemical Biology Research. Curr. Opin. Chem. Biol. 2012, 16, 227–233. 10.1016/j.cbpa.2012.01.008. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Overkleeft H. S.; van Kasteren S. I. Current Developments in Activity-Based Protein Profiling. Bioconjugate Chem. 2014, 25, 1181–1191. 10.1021/bc500208y. [DOI] [PubMed] [Google Scholar]
- Willems L. I.; Verdoes M.; Bogdan F. I.; van der Marel G. A.; Overkleeft H. S. Two-Step Labeling of Endogenous Enzymatic Activities by Diels–Alder Ligation. ChemBioChem 2010, 11, 1769–1781. 10.1002/cbic.201000280. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Zhao J.; Wu S.; Wang Z.; Wu W.; Ma J.; Guo S.; Huang L. Intramolecular RET Enhanced Visible Light-Absorbing Bodipy Organic Triplet Photosensitizers and Application in Photooxidation and Triplet-Triplet Annihilation Upconversion. J. Am. Chem. Soc. 2013, 135, 10566–10578. 10.1021/ja405170j. [DOI] [PubMed] [Google Scholar]
- Dey T.; Praveena K. S. S.; Pal S.; Mukherjee A. K. Three Oxime Ether Derivatives: Synthesis, Crystallographic Study, Electronic Structure and Molecular Electrostatic Potential Calculation. J. Mol. Struct. 2017, 1137, 615–625. 10.1016/j.molstruc.2017.02.089. [DOI] [Google Scholar]
- Houk K. N.; Sims J.; Watt C. R.; Luskus L. J. Origin of Reactivity, Regioselectivity, and Periselectivity in 1,3-Dipolar Cycloadditions. J. Am. Chem. Soc. 1973, 95, 7301–7315. 10.1021/ja00803a018. [DOI] [Google Scholar]
- Koyama Y.; Matsumura T.; Yui T.; Ishitani O.; Takata T. Fluorescence Control of Boron Enaminoketonate Using a Rotaxane Shuttle. Org. Lett. 2013, 15, 4686–4689. 10.1021/ol401984j. [DOI] [PubMed] [Google Scholar]
- Kubota Y.; Tanaka S.; Funabiki K.; Matsui M. Synthesis and Fluorescence Properties of Thiazole-Boron Complexes Bearing a β-Ketoiminate Ligand. Org. Lett. 2012, 14, 4682–4685. 10.1021/ol302179r. [DOI] [PubMed] [Google Scholar]
- Turner C. D.; Ciufolini M. A. Oxidation of oximes with hypervalent iodine reagents: opportunities, development, and applications. ARKIVOC 2011, 410–428. 10.3998/ark.5550190.0012.108. [DOI] [Google Scholar]
- Truong V. X.; Zhou K.; Simon G. P.; Forsythe J. S. Nitrile Oxide-Norbornene Cycloaddition as a Bioorthogonal Crosslinking Reaction for the Preparation of Hydrogels. Macromol. Rapid Commun. 2015, 36, 1729–1734. 10.1002/marc.201500314. [DOI] [PubMed] [Google Scholar]
- Jędrzejewskaa B.; Grabarzb A.; Bartkowiakb W.; Ośmiałowski B. Spectral and Physicochemical Properties of Difluoroboranyls Containing N,N-Dimethylamino Group Studied by Solvatochromic Methods. Spectrochim. Acta, Part A 2018, 199, 86–95. 10.1016/j.saa.2018.03.048. [DOI] [PubMed] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; Nakatsuji H.; Caricato M.; Li X.; Hratchian H. P.; Izmaylov A. F.; Bloino J.; Zheng G.; Sonnenberg J. L.; Hada M.; Ehara M.; Toyota K.; Fukuda R.; Hasegawa J.; Ishida M.; Nakajima T.; Honda Y.; Kitao O.; Nakai H.; Vreven T.; Montgomery J. A. Jr.; Peralta J. E.; Ogliaro F.; Bearpark M.; Heyd J. J.; Brothers E.; Kudin K. N.; Staroverov V. N.; Kobayashi R.; Normand J.; Raghavachari K.; Rendell A.; Burant J. C.; Iyengar S. S.; Tomasi J.; Cossi M.; Rega N.; Millam J. M.; Klene M.; Knox J. E.; Cross J. B.; Bakken V.; Adamo C.; Jaramillo J.; Gomperts R.; Stratmann R. E.; Yazyev O.; Austin A. J.; Cammi R.; Pomelli C.; Ochterski J. W.; Martin R. L.; Morokuma K.; Zakrzewski V. G.; Voth G. A.; Salvador P.; Dannenberg J. J.; Dapprich S.; Daniels A. D.; Farkas Ö.; Foresman J. B.; Ortiz J. V.; Cioslowski J.; Fox D. J.. Gaussian 09, revision E.01; Gaussian, Inc.: Wallingford CT, 2009.
- Zhao G.-J.; Han K.-L. Ultrafast Hydrogen Bond Strengthening of the Photoexcited Fluorenone in Alcohols for Facilitating the Fluorescence Quenching. J. Phys. Chem. A 2007, 111, 9218–9223. 10.1021/jp0719659. [DOI] [PubMed] [Google Scholar]
- Vendrell M.; Zhai D.; Er J. C.; Chang Y. T. Combinatorial Strategies in Fluorescent Probe Development. Chem. Rev. 2012, 112, 4391–4420. 10.1021/cr200355j. [DOI] [PubMed] [Google Scholar]
- Ośmiałowski B.; Zakrzewska A.; Jedrzejewska B.; Grabarz A. M.; Zalesny R.; Bartkowiak W.; Kolehmainen E. The Influence of Substituent and Benzoannulation on Photophysical Properties of 1-Benzoylmethyleneisoquinoline Difluoroborates. J. Org. Chem. 2015, 80, 2072–2080. 10.1021/jo502244j. [DOI] [PubMed] [Google Scholar]
- Cowie J. M. G.; Toporowski P. M. Association in the Binary Liquid System Dimethylsulphoxide-Water. Can. J. Chem. 1961, 39, 2240–2243. 10.1139/v61-296. [DOI] [Google Scholar]
- Gucma M.; Gołębiewski W. M.; Krawczyk M. NMR Studies on 1,3-Dipolar Cycloaddition of Nitrile Oxides to Norbornenes. J. Braz. Chem. Soc. 2013, 24, 805–813. 10.5935/0103-5053.20130106. [DOI] [Google Scholar]
- Epoxomicin: proteasome inhibitor presenting an α′,β′-epoxyketone warhead able to interact with the N-terminal threonine of the proteotically active β units of the proteasome. The interaction leads to the formation of a 1,4-oxazepane ring, inhibiting the proteolytic activity of the protein complex. See:Goldberg A. L. Development of Proteasome Inhibitors as Research Tools and Cancer Drugs. J. Cell. Biol. 2012, 199, 583–588. 10.1083/jcb.201210077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Search: topic, “isoxazole”; refine, “fluorescent probes”; Dec. 2018: 29 references found, including ref (2). - From SciFinder.
- Brandhofer T.; Özdemir A.; Gini A.; García Mancheco O. Double Cu-Catalyzed Direct Csp3-H Azidation/ CuAAC Reaction: A Direct Approach towards Demanding Triazole Conjugates. Chem. - Eur. J. 2019, 25, 4077–4086. 10.1002/chem.201806288. [DOI] [PubMed] [Google Scholar]
- Moiola M.; Bova A.; Crespi S.; Memeo M. G.; Mella M.; Overkleeft H. S.; Florea B. I.; Quadrelli P.. Fluorescent Probes from Aromatic Polycyclic Nitrile Oxides: Isoxazoles vs. Dihydro-1λ3,3,2λ4-oxazaborinines. Manuscript submitted. [DOI] [PMC free article] [PubMed]
- Grundmann C.; Dean J. M. Nitrile Oxides. V. Stable Aromatic Nitrile Oxides. J. Org. Chem. 1965, 30, 2809–2812. 10.1021/jo01019a074. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.