

Abstract
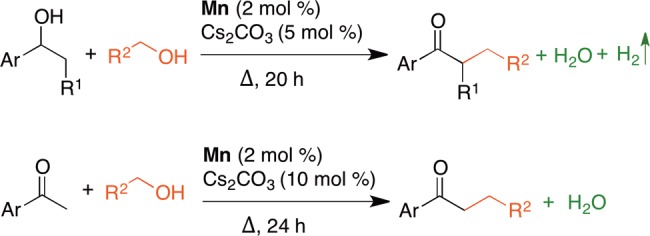
Catalytic cross-coupling of ketones and secondary alcohols with primary alcohols is reported. An abundant manganese-based pincer catalyst catalyzes the reactions. Low loading of catalyst (2 mol %) and catalytic use of a mild base (5–10 mol %) are sufficient for efficient cross-coupling. Various aryl and heteroaryl ketones are catalytically cross-coupled with primary alcohols to provide the selective α-alkylated products. Challenging α-ethylation of ketones is also attained using ethanol as an alkylating reagent. Further, direct use of secondary alcohols in the reaction results in in situ oxidation to provide the ketone intermediates, which undergo selective α-alkylation. The reaction proceeds via the borrowing hydrogen pathway. The catalyst oxidizes the primary alcohols to aldehydes, which undergo subsequent aldol condensation with ketones, promoted by catalytic amount of Cs2CO3, to provide the α,β-unsaturated ketone intermediates. The hydrogen liberated from oxidation of alcohols is used for hydrogenation of α,β-unsaturated ketone intermediates. Notably either water or water and dihydrogen are the only byproducts in these environmentally benign catalytic processes. Mechanistic studies allowed inferring all of the intermediates involved. Dearomatization–aromatization metal–ligand cooperation in the catalyst facilitates the facile O–H bond activation of both primary and secondary alcohols, and the resultant manganese alkoxide complexes produce corresponding carbonyl compounds, perhaps via β-hydride elimination. The manganese(I) hydride intermediate plays dual role as it hydrogenates α,β-unsaturated ketones and liberates molecular hydrogen to regenerate the catalytically active dearomatized intermediate. Metal–ligand cooperation allows all of the manganese intermediates to exist in same oxidation state (+1) and plays an important role in these catalytic cross-coupling reactions.
1. Introduction
Efficient and catalytic construction of C–C bond is an important transformation in organic synthesis. Among C–C bond formation methods, α-alkylation of ketones is one of the pivotal routes for the synthesis of several biologically active, heterocyclic compounds and natural product building blocks.1 Conventional methods involved use of reactive alkyl halides as alkylating reagents in the presence of stoichiometric amount of base that resulted in generation of toxic waste, which limited their sustainable applications.2 Advancement in catalysis, in particular introduction of “borrowing hydrogen” or “hydrogen autotransfer” methods, allowed to overcome these drawbacks. In recent years, borrowing hydrogen strategy for C–C bond formation has received significant attention for the construction of functionalized organic molecules.3 Typical borrowing hydrogen method involves catalytic dehydrogenation of alcohol to aldehyde and ketones, followed by base-mediated aldol reaction, which generates the corresponding α,β-unsaturated ketone intermediate with elimination of water. Further, catalytic and selective transfer hydrogenation of the enone intermediates using the hydrogen obtained from the oxidation of alcohols affords α-alkylated ketones with high atom efficiency. Liberated molecular hydrogen and water are the only byproducts from this transformation (Scheme 1).4
Scheme 1. Selective Catalytic Dehydrogenative Cross-Coupling of Ketones and Secondary Alcohols with Primary Alcohols Enabled by Borrowing Hydrogen Concept.

In general, this catalytic alkylation of ketones using alcohols is implemented using noble metals such as ruthenium,5 palladium,6 osmium,7 iridium,8 etc. Accordingly nonprecious first-row transition metals9 have drawn much attention. Recently, Beller and co-workers have reported an aliphatic electron-rich PNP tridentate manganese(I) catalyst for alkylation of ketones.10 More recently, α-alkylated ketones have been synthesized by direct dehydrogenative coupling of readily available and cheap secondary alcohols with primary alcohols.10b,10c Dehydrogenative cross-coupling of secondary alcohols and primary alcohols using Rh, Ru, and Ir catalysts has been reported.5,8b,11 Thus, earth-abundant, inexpensive, more eco-friendly base metals like Mn-, Fe-, and Co-catalyzed cross-coupling of primary and secondary alcohols to α-alkylated ketones is desirable. In this direction, recently, Lang and co-workers have reported ligand-controlled copper(I)-catalyzed cross-coupling of secondary and primary alcohols.11b
Eminent research groups have designed and developed various manganese pincer catalysts, which catalyzed an assortment of organic transformations.12−19 Manganese pincer-based catalysis has become more attractive because of new selectivity patterns in known reactions; different products are obtained just by altering the base.20 Recently, Yu and co-workers21 have reported manganese-catalyzed cross-coupling of secondary alcohols with primary alcohols, which provided the Guerbet β-alkylated secondary alcohol products in good yields. Kempe and co-workers14c reported the multicomponent synthesis of pyrimidines from alcohols and amidines that proceeded via β-alkylation of alcohols. Using ruthenium pincer catalyst, we have reported the cross-coupling of secondary alcohols.22a Selective α-alkylation and α-olefination of nitrile compounds with primary alcohols and secondary alcohols, respectively, were also reported using ruthenium catalyst.22b,22c Very recently, we have also reported manganese-catalyzed selective α-alkenylation of ketones using primary alcohols.22d In continuation of our quest to devise and develop atom-economical and sustainable catalytic transformations, herein, we report the selective cross-coupling of ketones and secondary alcohols with primary alcohols, which provided the α-alkylated ketone products in good to excellent yields. These dehydrogenative and dehydrative cross-coupling reactions are catalyzed by the manganese(I) pincer-based Kempe catalyst [(PN5P)Mn(CO)2Br] (1) together with catalytic amount of a mild base.
2. Results and Discussion
During the development of selective α-alkenylation of ketones using primary alcohols, we have found that catalyst 1 with phenyl substituent in the catalyst backbone favorably produced the α-alkylated ketone products, while similar catalyst with 4-methyl substitution promoted the selective α-olefination reaction.22d Thus, catalytic α-alkylation of ketones by primary alcohols using manganese pincer catalyst 1 was investigated. At the outset of our studies, acetophenone and benzyl alcohol were chosen as benchmark substrates to find the optimal reaction conditions for the PN5P triazine-based manganese catalyst 1, and the results are presented in Table 1. Reaction of acetophenone and benzyl alcohol (each 0.5 mmol) was performed in the presence of 2 mol % catalyst 1 and 5 mol % Cs2CO3 as base in tert-amyl alcohol solvent, which delivered the desired alkylated product in 63% yield (entry 1, Table 1). Upon increase of base load to 10 mol %, alkylated product 2a was isolated in 75% yield (entry 2, Table 1). When the amount of alcohol was increased from 1 to 1.2 equiv, the outcome was optimal, where the desired 2a was isolated in 86% yield with more than 99% conversion of acetophenone (entry 3, Table 1). Further, lowering the catalyst load to 1 mol % resulted in diminished yield of the product (entry 4, Table 1). Use of different solvents like 1,4-dioxane and toluene provided the alkylated product 2a in 84 and 51% yield, respectively (entries 5 and 6, Table 1). Reactions were tested with different bases. Upon use of sodium carbonate, product 2a was isolated in 11% yield (entry 7, Table 1). Although use of potassium carbonate as a base provided 97% conversion of acetophenone, the desired product 2a was isolated in 47% yield (entry 8, Table 1). Further, use of strong base sodium tert-butoxide provided the moderate conversion (80%) and yield (71%, entry 9, Table 1). When the reaction was carried out in the absence of catalyst 1 and in the presence of base alone, the product was obtained in 20% yield along with other undesired side products (entry 10, Table 1). When alkylation reactions were carried out with catalyst 1 in the absence of base and in the absence of both catalyst and base (entries 11 and 12, Table 1), no product was observed, implying that catalyst and base are essential for the alkylation of ketones using alcohols.
Table 1. Optimization of the Reaction Conditions for the α-Alkylation of Ketonesa.

| entry | 1 (mol %) | base (mol %) | alcohol (equiv) | conv. (%) | yield (%) |
|---|---|---|---|---|---|
| 1 | 2 | Cs2CO3 (5) | 1 | >99 | 63 |
| 2 | 2 | Cs2CO3 (10) | 1 | >99 | 75 |
| 3 | 2 | Cs2CO3 (10) | 1.2 | >99 | 86 |
| 4 | 1 | Cs2CO3 (10) | 1.2 | >99 | 71 |
| 5b | 2 | Cs2CO3 (10) | 1.2 | >99 | 84 |
| 6c | 2 | Cs2CO3 (10) | 1.2 | 98 | 51 |
| 7 | 2 | Na2CO3 (10) | 1.2 | 33 | 11 |
| 8 | 2 | K2CO3 (10) | 1.2 | 97 | 47 |
| 9 | 2 | NaOtBu (10) | 1.2 | 80 | 71 |
| 10 | Cs2CO3 (10) | 1.2 | 75 | 20 | |
| 11 | 2 | 1.2 | |||
| 12 | 1.2 |
Reaction conditions: acetophenone (0.5 mmol), benzyl alcohol, catalyst 1 (0.015 mmol), and the base in tert-amyl alcohol were heated at 140 °C for 24 h.
Dioxane was used as solvent.
Toluene was used as solvent.
Following the optimized experimental conditions, a wide range of alcohols was subjected to manganese-catalyzed α-alkylation reaction of acetophenone, which offered moderate to excellent yields of alkylated ketones (Table 2). In general, complete conversion of acetophenone was observed in all reactions. Upon using 4-methylbenzyl alcohol, the corresponding alkylated product 2b was isolated in 91% yield. While 4-fluorobenzyl alcohol provided the corresponding alkylated product 2c in 83% yield, 4-(trifluoromethyl)benzyl alcohol gave 2d only in 56% yield. A heteroaryl alcohol, 2-furanylmethanol, provided 84% of the alkylated product 2e. Further, a series of aliphatic primary alcohols such as 1-butanol, 1-octanol, 3,5,5-trimethylhexanol, and 2-cyclohexylethanol were investigated as alkylating partners, and in all of these experiments, excellent yields (91–97%) were obtained (2f, 2g, 2i, 2j). However, 2-ethyl-butanol provided 75% of the alkylated product 2h, perhaps due to steric hindrance in proximity to alcohol functionality.
Table 2. Manganese-Catalyzed α-Alkylation of Acetophenone Using Primary Alcoholsa.

Reaction conditions: ketone (0.5 mmol), alcohol (0.6 mmol), tert-amyl alcohol (2 mL), catalyst 1 (2 mol %), and Cs2CO3 (10 mol %) were heated at 140 °C under open condition with an argon flow. Yields were calculated for isolated products after column chromatography. Conversion of ketones was determined by gas chromatography (GC) analysis using toluene as an internal standard and given within parentheses.
Then, we explored the scope of different aryl ketones in the catalytic α-alkylation reactions using various primary alcohols (Table 3). Initially, different acetophenone derivatives bearing electron-withdrawing and electron-donating substituents were subjected to alkylation with benzyl alcohol. 4-Methylacetophenone provided the desired alkylated product 3a in 94% isolated yield. In general, aryl ketones having poor electron-aryl motifs provided the corresponding alkylated products in lower yields (3b–e) than electron-rich aryl ketones. Gratifyingly, heteroaryl ketone, 2-acetylthiophene, was transformed to the desired alkylated product. Upon reaction with benzyl alcohol, 65% of the alkylated product 3e was isolated. Despite 3-(pyridin-2-yl)propan-1-ol provided good conversion, its catalytic α-alkylation with 4-methylacetophenone resulted in 45% of 3f. With piperonyl alcohol as alkylating partner, 4-methoxyacetophenone yielded 62% of the corresponding alkylated product 3g. 1-Tetralone and 4-methyl-1-tetralone were converted quantitatively to the α-alkylated products 3h and 3i in 78 and 88% yields, respectively. Remarkably, this manganese-catalyzed α-alkylation reaction is highly chemoselective. Even in the presence of amine functionality, alkylation occurred exclusively on a carbon α to carbonyl group. Upon catalytic α-alkylation of 2′-aminoacetophenone with 1-hexanol and 1-heptanol, the corresponding alkylated products were obtained in 53 and 46% yields (3j and 3k). Notably, the N-alkylated product was not observed in this condition, which indicates the chemoselective C-alkylation over N-alkylation.
Table 3. Manganese-Catalyzed α-Alkylation of Ketones Using Alcohola.

To further explore the scope of manganese-catalyzed α-alkylation of ketones with primary alcohols, alkylation using ethanol is demonstrated (Table 4). Acetophenone and ethanol under the optimized catalytic conditions yielded 58% of the ethylated product 4a. 4′-Methylacetophenone and 4′-chloroacetophenone provided 65 and 63% yields of the corresponding ethylated product 4b and 4c. β-Napthyl methyl ketone provided the ethylated product 4d in 66% yield. However, when 3-acetyl quinoline was examined under standard experimental conditions, only 23% of the ethylated product 4e was isolated. Similar catalytic results were obtained by previously reported manganese-, iron-, and cobalt-catalyzed α-alkylation of ketones using primary alcohols.9,10a
Table 4. Manganese-Catalyzed α-Alkylation of Ketones Using Ethanola.

Reaction conditions: ketone (0.5 mmol), ethanol (1 mL), tert-amyl alcohol (1 mL), catalyst 1 (2 mol %), and Cs2CO3 (10 mol %) were heated at 140 °C in a sealed tube. Yields were calculated for isolated products after column chromatography. Conversion of ketones was determined by GC analysis using toluene as an internal standard and given within parentheses.
Exclusive chemoselectivity of primary alcohols over secondary alcohols on alkylation of ketone was investigated. Acetophenone (1 equiv) with cyclohexanol and 1-hexanol (each 1.2 equiv) was quantitatively converted to the α-alkylated product in the presence of catalyst 1 (2 mol %) and Cs2CO3 (10 mol %) to form 1-phenyloctan-1-one (2k, 86% isolated yield, Scheme 2a). A similar experiment using 3-pentanol also provided almost identical result (2k, 88% isolated yield Scheme 2b). Reaction of diols such as 1,5-hexanediol with acetophenone in the presence of catalyst 1 provided a mixture of products (Scheme 2c). Remarkably, when acetophenone (1 equiv) was reacted with 1-hexanol (1.2 equiv) and aniline (1 equiv) in the presence of catalyst 1 (2 mol %) and Cs2CO3 (10 mol %), quantitative conversion of ketone was observed to provide 1-phenyloctan-1-one (2k, 82% isolated yield, Scheme 2d).
Scheme 2. Manganese-Catalyzed Chemoselective α-Alkylation of Acetophenone.

In situ monitoring of the reaction progress by GC on the catalytic α-alkylation reaction of acetophenone using 4-methylbenzyl alcohol catalyzed by 1 allowed to infer the intermediates involved. As reaction proceeded, decrease in concentration of acetophenone (black line) can be corroborated with increasing concentration of product 2b (blue line). Notably, upon oxidation of 4-methylbenzyl alcohol, the in situ formed intermediate 4-methylbenzyl aldehyde (green line) was only short-lived as it undergoes rapid condensation with acetophenone to provide intermediate A (α,β-unsaturated ketones, red line). Reaction kinetics also indicated that hydrogenation of α,β-unsaturated ketone intermediate A to the corresponding alkylated product was rapid at the outset of the reaction and became slower over time (Figure 1).
Figure 1.
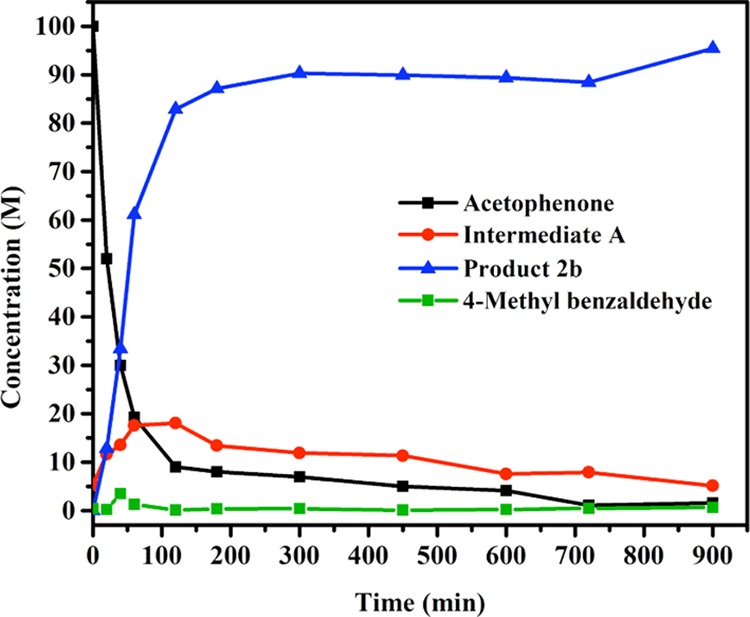
Monitoring the manganese-catalyzed α-alkylation using GC. Concentrations of acetophenone (black line), product 2b (blue line), intermediates α,β-unsaturated ketones (red line), and 4-methylbenzyl aldehyde (green line) in the catalytic α-alkylation of acetophenone.

In an attempt to further develop these benign catalytic alkylation reactions, cross-coupling of secondary alcohols with primary alcohols, which can result in selective β-alkylation of secondary alcohols leading to the exclusive formation of α-alkylated ketones, was envisaged. Thus, using catalyst 1, the cross-coupling reaction between 1-phenyl-1-ethanol and benzyl alcohol was subjected to optimization studies, and the results are presented in Table 5. Initial experiments with different loads of catalyst 1 (3–1 mol %) and base (Cs2CO3, 10 mol %) provided moderate to complete conversions (entries 1–3, Table 5). Spectral analyses of the isolated product clearly indicated the formation of α-alkylated ketone product, 1,3-diphenylpropan-1-one (2a). Upon performing the experiment using 2 mol % catalyst 1 and 5 mol % Cs2CO3, quantitative conversion of 1-phenyl-1-ethanol was observed and the alkylated product 2a was isolated in 90% yield (entry 4, Table 5). Further decrease of the catalyst and base loads turned out to be detrimental to the progress of the reaction as incomplete conversions were observed after 20 h (entries 5–6, Table 5). Control experiments using only catalyst 1, base (Cs2CO3) alone, and an experiment without catalyst and base (entries 7–9, Table 5) proved that this alkylation requires a catalyst and a base. Further, under similar reaction conditions, the effect of different bases was also explored. K2CO3 and KOH provided moderate yields of product (83 and 40%, respectively). However, use of bases such as NaH, NaOH, Na2CO3, and KOtBu resulted in poor conversion of secondary alcohols. Surprisingly, the in situ generation of ketones from the oxidation of secondary alcohols provided facile α-alkylation reactions by primary alcohols. Using only 5 mol % base, the reaction was completed in 20 h compared to the requirement of higher load of base, higher temperature, and longer reaction time required for the direct ketone alkylation by primary alcohols (see Table 1).
Table 5. Optimization of the Reaction Conditions for the β-Alkylation of Secondary Alcoholsa.

| entry | cat. 1 (mol %) | base (mol %) | conv. (%)b | yield (%)c |
|---|---|---|---|---|
| 1 | 3 | Cs2CO3 (10) | >99 | 80 |
| 2 | 2 | Cs2CO3 (10) | >99 | 88 |
| 3d | 1 | Cs2CO3 (10) | 87 | 54 |
| 4 | 2 | Cs2CO3 (5) | >99 | 90 |
| 5d | 1 | Cs2CO3 (5) | 37 | 23 |
| 6d | 2 | Cs2CO3 (2.5) | 51 | 42 |
| 7 | 2 | 5 | 0 | |
| 8 | Cs2CO3 (5) | 15 | 0 | |
| 9 | 0 | 0 |
Reaction conditions: 1-phenyl-1-ethanol (0.5 mmol), benzyl alcohol (0.6 mmol), tert-amyl alcohol (2 mL), catalyst 1, and base were heated at 135 °C with open condition under an argon flow.
Conversion of 1-phenyl-1-ethanol was determined by GC using toluene as an internal standard.
Isolated yields after column chromatography.
A minor amount of (E)-chalcone formation also observed.
As shown in Table 6, a wide range of primary alcohols was subjected to manganese-catalyzed β-alkylation of 1-phenyl-1-ethanol, which provided α-alkylated ketones in moderate to excellent yields. Reaction of different benzyl alcohols with 2 mol % catalyst 1 and 5 mol % Cs2CO3 resulted in good conversion of 1-phenyl-1-ethanol, and the β-alkylated products (2a–c and 2l–n) were isolated in 73–90% yields. Further, a series of primary aliphatic alcohols (nonbenzylic) such as 4-phenyl-1-butanol, 3-(pyridin-2-yl)propan-1-ol, 1-hexanol, 1-octanol, 3,5,5-trimethylhexanol, and 2-cyclohexylethanol were investigated as alkylating partners and, in general, excellent conversions as well as yields were obtained (2o, 2p, 2g, 2i, and 2j; Table 6). Manganese-catalyzed cross-coupling of 1-phenyl-1-ethanol with benzyl alcohols was reported using strong base KOtBu.10b
Table 6. Manganese-Catalyzed Cross-Coupling of 1-Phenyl-1-ethanol with Primary Alcoholsa.

Reaction conditions: 1-phenyl-1-ethanol (0.5 mmol), primary alcohol (0.6 mmol), tert-amyl alcohol (2 mL), catalyst 1 (0.01 mmol), and Cs2CO3 (0.025 mmol) were heated at 135 °C in open condition under an argon flow. Yield of the products corresponds to isolated pure compounds after column chromatography. Conversion of 1-phenyl-1-ethanol was determined by GC using toluene as an internal standard and given within parentheses.
Next, substrate scope of structurally diverse secondary alcohols was investigated in the catalytic cross-coupling reactions with various primary alcohols. Initially, different secondary alcohol derivatives bearing electron-donating and electron-withdrawing substituents were subjected to coupling with benzylic and aliphatic alcohol (Table 7). In general, nonactivated primary aliphatic alcohols provided the corresponding alkylated products 3m–p in higher yields than aryl alcohols. Notably, electron-withdrawing halogen-containing secondary alcohols underwent facile β-alkylation reaction and provided the corresponding alkylated products 3q–s and 3c in 66–95% yields. Remarkably, as observed in ketone alkylation reactions, the amine functionality is tolerated in the catalytic alkylation reactions. When (4-bromophenyl)-1-ethanol was reacted with (4-aminophenyl)methanol, the desired alkylated product 3t was obtained in 61% yield. Notably, the N-alkylated product was not observed in this condition, which indicates the chemoselective C-alkylation over N-alkylation. Heteroaryl secondary alcohols were also successfully alkylated using this manganese catalysis and alkylated heteroaryl ketones 3u, 3v, and 3e were isolated in moderate to good yields (Table 7). Nickel-catalyzed cross-coupling of secondary alcohols with primary alcohols is reported, which required the use of higher load of base (KOH, 50 mol %).10c
Table 7. Manganese-Catalyzed Dehydrogenative Cross-Coupling of Secondary Alcohols with Primary Alcoholsa.

Further, the substrate scope was extended to cyclic secondary alcohols (Table 8). Thus, the reactions of 1-indanol with 1-hexanol and 1-octanol resulted in the corresponding alkylated products 5a and 5b in 98 and 97% isolated yields. Gratifyingly, 1,2,3,4-tetrahydronaphthalen-1-ol and its derivatives efficiently reacted with 1-hexanol and 4-phenyl-1-butanol and afforded the alkylated products 5c, 3h, and 3i in 85–95% isolated yields, respectively (Table 8).
Table 8. Manganese-Catalyzed Cross-Coupling of Cyclic Secondary Alcohol with Primary Alcoholsa.

The reaction progress on β-alkylation reaction of 1-phenyl-1-ethanol with 3-phenyl-1-propanol catalyzed by 1 was monitored using GC, which indicated zero-order kinetics for the conversion of secondary alcohols as well for the formation of β-alkylated ketone product 2o (Figure 2). Notably, upon oxidation of 3-phenyl-1-propanol, the in situ formed intermediate hydrocinnamaldehyde was observed in a trace amount. Further, the rapid condensation of aldehyde with in situ generated acetophenone from 1-phenyl-1-ethanol provides α,β-unsaturated ketone intermediate. However, ketone and α,β-unsaturated ketone intermediates were not observed in the reaction; perhaps they undergo concomitant condensation and hydrogenation reactions, respectively.
Figure 2.
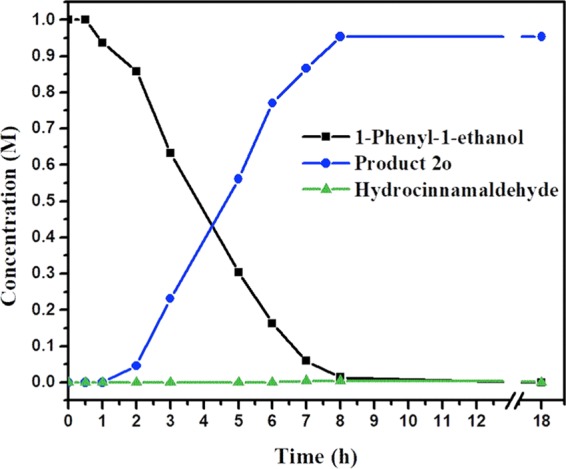
Monitoring the manganese-catalyzed β-alkylation of secondary alcohols by primary alcohols using GC; concentrations of 1-phenyl-1-ethanol (black line), product 2o (blue line), and hydrocinnamaldehyde (green line).

In a further attempt to identify the intermediates involved in this transformation, the catalytic reaction of 1-phenyl-1-ethanol with benzyl alcohol was intervened and the reaction mixture was subjected to 1H NMR analysis, which resonated a characteristic singlet at δ 10.05 ppm and a distinct doublet at δ 7.85 ppm with a coupling constant J = 16 Hz that corresponds to aldehyde proton of benzaldehyde and olefin protons of (E)-chalcone, respectively.23
Moreover, a mercury poisoning experiment was performed to examine the involvement of any metal nanoparticles formed from complex 1. When the dehydrogenative and dehydrative coupling of 1-phenyl-1-ethanol with benzyl alcohol was carried out in the presence of mercury (2 equiv, relative to substrate), nearly 93% conversion of 1-phenyl-1-ethanol was observed and the alkylated ketone 2a was obtained in 87% (see Scheme 3), indicating the involvement of molecular intermediates. Further, when 1-(4-methoxyphenyl)ethanol independently reacted with catalyst 1 under optimized experimental conditions, exclusive formation of 1-(4-methoxyphenyl)ethanone was observed in 74% yield, confirming the catalytic oxidation of secondary alcohols to ketones.
Scheme 3. Mercury Poisoning Test and Observation of Ketone Intermediate.

On the basis of these observations and the experimental studies involving catalyst 1 in recent reports,14b,22d the plausible catalytic cycle is proposed in Scheme 4. In the presence of catalytic amount of base, the dearomatized coordinatively unsaturated intermediate I is generated from catalyst 1 upon amine deprotonation and debromination from the metal center. O–H activation of the primary and secondary alcohols by intermediate I via proton transfer to the dearomatized imine nitrogen results in the formation of alkoxo intermediate II, which undergoes β-hydride elimination to release the aldehyde and ketone, and generates a saturated monohydrido manganese complex III. Notably, Liu24a and Milstein24b have previously isolated similar manganese alkoxo monohydrido complexes. Deprotonation of the α-acidic protons of carbonyl compound by base possibly generates the carbon nucleophile. Nucleophilic attack by the deprotonated methyl or methylene group of the ketone on the in situ formed aldehyde intermediate leads to an aldol condensation, resulting in the formation of α,β-unsaturated ketone intermediate (observed experimentally) and water. Interestingly, deprotonation of the methyl or methylene groups requires only catalytic amount of a mild base. The manganese hydride complex III concomitantly hydrogenates α,β-unsaturated ketone intermediates to afford the redox neutral alkylated product along with the regeneration of catalytically active intermediate I. The dearomatization–aromatization metal–ligand cooperation operative in manganese intermediates maintains the same oxidation state (+1) at all intermediate complexes and plays an important role in this selective catalytic coupling of ketones and secondary alcohols with primary alcohols.
Scheme 4. Proposed Reaction Mechanism for Cross-Coupling of Secondary and Primary Alcohols.

3. Conclusions
In summary, manganese pincer 1 catalyzed cross-coupling of ketones and secondary alcohols with primary alcohols leading to the selective α-alkylation of ketones. Primary alcohols serve as alkylating reagents. Remarkably, using a minimal catalyst load (2 mol %) and base (5–10 mol %), various ketones and secondary alcohols can be efficiently alkylated with an assortment of linear primary alcohols in excellent yields. Ethylation of ketones using ethanol is also demonstrated. Chemoselective α-alkylation of ketones is observed with primary alcohol in the presence of nonbenzylic secondary alcohol. Moreover, chemoselective catalytic C-alkylations of ketone and secondary alcohols over N-alkylation of aryl amine are achieved. Mechanistic studies confirm the formation of aldehyde, ketone, and α,β-unsaturated ketone intermediates. Accordingly, a catalytic cycle incorporating the formation of these intermediates eventually leading to the α-alkylated ketones is proposed. One equivalent of hydrogen liberated from oxidation of primary and secondary alcohols is utilized by the catalyst for the hydrogenation of α,β-unsaturated ketones to deliver the β-alkylated ketones. Overall, the catalytic coupling of ketones with alcohol releases 1 equiv of water, while the cross-coupling of secondary and primary alcohols produces 1 equiv of water and molecular hydrogen from the reactions as the only byproducts.
4. Experimental Section
4.1. General Information
All stoichiometric reactions were performed in nitrogen atmosphere. All catalytic reactions were performed under nitrogen atmosphere using standard Schlenk techniques. Dry solvents were prepared according to standard procedures. 1H, 13C{1H} NMR chemical shifts were reported in ppm downfield from tetramethyl silane. Multiplicity is abbreviated as: s, singlet; d, doublet; dd, doublet of doublets; dt, doublet of triplets; t, triplet; q, quartet; dq, doublet of quartets; td, triplet of doublets; m, multiplet; br, broad. IR spectra were recorded in an Fourier transform infrared spectrometer by using KBr pellets. Mass spectra were recorded on a micrOTOF-Q II Spectrometer.
4.2. Experimental Procedure
4.2.1. Synthesis of Ligand (4-Ph)Tr(NHP(iPr)2)214d
Inside a glovebox, a 15 mL sealed tube was charged with a stir bar, 2,4-diamino-6-phenyl-1,3,5-triazine (2 mmol, 0.374 g), and dry tetrahydrofuran (THF) (8 mL) under nitrogen atmosphere. The solution was cooled to 0 °C and then chlorodiisopropylphosphine (4.2 mmol, 0.665 mL) was added dropwise into the reaction mixture. Triethylamine (8 mmol, 1.112 mL) was added to the reaction mixture. The reaction mixture was allowed to warm to room temperature and stirred vigorously for 16 h at 60 °C. The suspension was filtered over a glass filter frit with a pad of Celite (3 cm) and washed with 10 mL of THF. The combined organic phase was concentrated in vacuo yielding (4-Ph)Tr(NHP(iPr)2)2 as a colorless solid (712 mg, 85%). 1H NMR (400 MHz, CDCl3): δ 8.38 (m, 2H), 7.40–7.45 (m, 3H), 5.17 (s, 2H), 1.81–1.87 (m, 4H), 1.07–1.11 (m, 24H) ppm. 13C NMR (176 MHz, CDCl3): δ 171.6, 169.8 (d, J = 9.0 Hz), 136.8, 131.5, 128.6, 128.3, 26.2 (d, J = 14.1 Hz), 18.8 (d, J = 19.4 Hz), 17.7 (br) ppm.
4.2.2. Synthesis of [(4-Ph)Tr(NHP(iPr)2)2Mn(CO)2Br] (1)14d
To the orange-yellow suspension of [MnBr(CO)5] (1 mmol, 275 mg) in toluene (15 mL), a solution of (4-Ph)Tr(NHP(iPr)2)2 (1 mmol, 420 mg) in toluene was added dropwise. The suspension turned into a clear yellow solution within 10 min. The reaction mixture was heated to 100 °C and further stirred for 16 h under nitrogen leading to the formation of a yellow precipitate. The reaction mixture was cooled to room temperature, and the supernatant solution was filtered off and the precipitate was dried in vacuo at 100 °C to afford [(4-Ph)Tr(NHP(iPr2))2Mn(CO)2Br] (1) as a bright yellow powder (501 mg, 82%). 1H NMR (400 MHz, CDCl3): δ = 8.17–8.18 (m, 2H), 7.37–7.43 (m, 3H), 6.05 (s br, 2H), 3.52 (s br, 2H), 2.66 (s br, 2H), 1.36 (s br, 24H) ppm. 31P NMR (162 MHz, CDCl3): δ 135.5, 134.2. IR (KBr, pellet, cm–1): 1937, 1867 (ν2CO symmetric + ν2CO antisymmetric).
4.3. General Procedure for α-Alkylation of Acetophenone Using Alcohols
Inside a nitrogen atmosphere glovebox, a 25 mL Schlenk tube was charged with a stir bar, catalyst 1 (0.01 mmol), base (0.05 mmol), acetophenone (0.5 mmol), solvent (2 mL), and alcohol (0.6 mmol) in the same order. The reaction flask was taken out of the glovebox and then refluxed (oil bath temperature, 140 °C) with stirring in an open system under argon flow for 24 h. The reaction mixture was then cooled down to room temperature. The solvent was evaporated under reduced pressure, and 0.5 mmol internal standard (toluene) was added into the reaction mixture and then diluted with 0.8 mL of acetone. An aliquot of the solution was passed through a small Celite plug and analyzed by GC. The solvent was evaporated under reduced pressure and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of acetophenone was calculated using GC analysis, and yields of pure products were determined after column chromatography.
4.4. General Procedure for Dehydrogenative Cross-Coupling of Alcohols
Inside a nitrogen atmosphere glovebox, a 25 mL Schlenk tube was charged with a stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.025 mmol), secondary alcohol (0.5 mmol), tert-amyl alcohol (2 mL), and primary alcohol (0.6 mmol) in the same order. The reaction flask was taken out of the glovebox and refluxed (oil bath temperature 135 °C) with stirring in an open system under argon flow for 24 h. The reaction mixture was then cooled down to room temperature. The solvent was evaporated under reduced pressure and 0.5 mmol internal standard (toluene) was added into the reaction mixture and then diluted with 0.8 mL of acetone. An aliquot of the solution was passed through a small Celite plug and analyzed by GC. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of secondary alcohol was calculated using GC analysis, and yields of pure products were determined after column chromatography.
4.5. Spectral Data of α-Alkylated Ketone Products
4.5.1. 1,3-Diphenylpropan-1-one8c (2a)
White solid. Yield for Table 1 (90 mg, 86%). Yield for Table 6 (95 mg, 90%). IR (dichloromethane (DCM)): 3061, 3025, 2950, 2922, 2864, 1682, 1602, 1595, 1580, 1495, 1448, 1410, 1364, 1209, 1075, 744, 701, 689 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 7.2 Hz, 2H), 7.48 (t, J = 7.2 Hz, 1H), 7.38 (t, J = 7.6 Hz, 2H), 7.11–7.25 (m, 5H), 3.23 (t, J = 8.0 Hz, 2H), 3.00 (t, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.3, 141.4, 137.0, 133.2, 128.7, 128.6, 128.5, 128.1, 126.2, 40.5, 30.2.
4.5.2. 1-Phenyl-3-(p-tolyl)propan-1-one8c (2b)
White solid. Yield for Table 2 (102 mg, 91%). Yield for Table 6 (90 mg, 80%). IR (DCM): 3053, 3022, 2921, 2861, 1686, 1597, 1580, 1514, 1203, 973, 811, 742 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.0 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.11–7.17 (m, 4H), 3.29 (t, J = 7.6 Hz, 2H), 3.04 (t, J = 7.6 Hz, 2H), 2.33 (s, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.5, 138.3, 137.0, 135.8, 133.2, 129.3, 128.7, 128.4, 128.2, 40.8, 29.9, 21.1.
4.5.3. 3-(4-Fluorophenyl)-1-phenylpropan-1-one25 (2c)
White solid. Yield for Table 2 (95 mg, 83%). Yield for Table 6 (91 mg, 79%). IR (DCM): 3062, 2930, 1685, 1598, 1509, 1448, 1221, 1206, 828, 743, 689 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.46 (t, J = 7.2 Hz, 2H), 7.19–7.23 (m, 2H), 6.98 (t, J = 8.4 Hz, 2H), 3.28 (t, J = 7.6 Hz, 2H), 3.05 (t, J = 7.2 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.2, 161.5, 137.0, 136.9, 133.3, 130.0 (d, 3JC–F = 8.0 Hz), 128.8, 128.1, 115.4 (d, 2JC–F = 20.0 Hz), 40.5, 29.4.
4.5.4. 1-Phenyl-3-(4-(trifluoromethyl)phenyl)propan-1-one10a (2d)
White solid. Yield (77 mg, 56%). IR (DCM): 3061, 2930, 1686, 1618, 1597, 1449, 1325, 1163, 1121, 1067, 1018, 828, 743 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 7.2 Hz, 2H), 7.54–7.59 (m, 3H), 7.37 (d, J = 8.0 Hz, 2H), 3.33 (t, J = 7.6 Hz, 2H), 3.14 (t, J = 7.2 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 198.7, 145.6, 136.8, 133.4, 128.9, 128.8, 128.1, 125.6 (q, 1JC–F = 4.0 Hz), 40.0, 29.9.
4.5.5. 3-(Furan-2-yl)-1-phenylpropan-1-one8c (2e)
Brown solid. Yield (84 mg, 84%). IR (DCM): 3060, 2920, 2359, 2341, 1716, 1683, 1597, 1448, 1211, 1018, 1001, 745 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.9 (d, J = 7.2 Hz, 2H), 7.49 (tt, 1J = 7.6 Hz, 2J = 1.2 Hz, 1H), 7.38 (t, J = 7.2 Hz, 2H), 7.23 (d, J = 1.2 Hz, 1H), 6.20–6.21 (m, 1H), 5.97–5.98 (m, 1H), 3.26 (t, J = 7.6 Hz, 2H), 3.01 (t, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 198.8, 154.9, 141.2, 136.8, 133.3, 128.7, 128.1, 110.4, 105.4, 37.0, 22.6.
4.5.6. 1-Phenylhexan-1-one10a (2f)
Colorless oil. Yield (85 mg, 97%). IR (DCM): 3057, 2929, 2861, 1681, 1597, 1465, 1448, 1405, 1370, 745, 725 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.95–7.97 (m, 2H), 7.55 (t, J = 7.2 Hz, 1H), 7.46 (t, J = 7.2 Hz, 2H), 2.96 (t, J = 7.6 Hz, 2H), 1.73 (q, J = 4 Hz, 2H), 1.34–1.39 (m, 4H), 0.89–0.93 (m, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.8, 137.2, 133.0, 128.7, 128.2, 38.7, 31.7, 24.2, 22.7, 14.1.
4.5.7. 1-Phenyldecan-1-one8c (2g)
Colorless oil. Yield for Table 2 (108 mg, 93%). Yield for Table 6 (104 mg, 90%). IR (DCM): 3084, 3056, 3026, 2948, 2916, 2849, 1686, 1474, 1462, 1447, 1406, 1376, 1072, 750, 734 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.87–7.89 (m, 2H), 7.47 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 9.2 Hz, 2H), 2.88 (t, J = 7.6 Hz, 2H), 1.66 (pentet, J = 7.6 Hz, 2H), 1.19–1.28 (m, 12H), 0.80 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.7, 137.2, 133.0, 128.7, 128.2, 38.8, 32.0, 29.6, 29.5, 29.4, 24.5, 22.8, 14.2.
4.5.8. 4-Ethyl-1-phenylhexan-1-one (2h)
Colorless oil. Yield (74 mg, 75%). IR (DCM): 2961, 2930, 2873, 1686, 1620, 1597, 1459, 1448, 1379, 1205, 735 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.95–7.97 (m, 2H), 7.53–7.58 (m, 1H), 7.46 (t, J = 7.6 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 1.67–1.72 (m, 2H), 1.25–1.38 (m, 5H) 0.87 (t, J = 7.6 Hz, 6H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 201.1, 137.2, 133.0, 128.7, 128.2, 40.3, 36.2, 27.3, 25.4, 11.0. High-resolution mass spectrometry (HRMS) (electrospray ionization (ESI)) m/z calcd for C14H21O (M + H)+: 205.1587, found: 205.1567.
4.5.9. 5,7,7-Trimethyl-1-phenyloctan-1-one (2i)
Pale yellow oil. Yield for Table 2 (112 mg, 91%). Yield for Table 6 (119 mg, 96%). IR (DCM): 2953, 2902, 2866, 1688, 1597, 1580, 1448, 1363, 1232, 1206, 1001, 752, 730 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 7.2 Hz, 2H), 7.48 (t, J = 7.6 Hz, 1H), 7.38 (t, J = 7.2 Hz, 2H), 2.87 (t, J = 7.2 Hz, 2H), 1.61–1.71 (m, 2H), 1.40–1.45 (m, 1H), 1.23–1.32 (m, 1H), 1.14–1.19 (m, 2H), 0.97 (dd, 1J = 14.0 Hz, 2J = 6.4 Hz, 1H), 0.86 (d, J = 6.4 Hz, 3H), 0.81 (s, 9H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.7, 137.3, 133.0, 128.7, 128.2, 51.3, 39.3, 39.0, 31.2, 30.2, 29.4, 22.7, 22.2. HRMS (ESI) m/z calcd for C17H27O (M + H)+: 247.2056, found: 247.2051.
4.5.10. 4-Cyclohexyl-1-phenylbutan-1-one26 (2j)
Colorless liquid. Yield for Table 2 (112 mg, 97%). Yield for Table 6 (87 mg, 76%). IR (DCM): 2953, 2902, 2866, 1687, 1597, 1465, 1448, 1363, 1232, 1206, 731 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 7.3 Hz, 2H), 7.52–7.56 (m, 1H), 7.45 (t, J = 7.2 Hz, 2H), 2.93 (t, J = 7.2 Hz, 2H), 1.65–1.74 (m, 7H), 1.15–1.27 (m, 6H), 0.87–0.92 (m, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.8, 137.3, 133.0, 128.7, 128.2, 39.1, 37.7, 37.3, 33.4, 26.8, 26.5, 21.9.
4.5.11. 1-Phenyloctan-1-one28 (2k)
Colorless oil. Yield (97 mg, 95%). IR (DCM): 2954, 2927, 2855, 1687, 1597, 1448, 1220, 750, 690 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 7.2 Hz, 2H), 7.53–7.59 (m, 1H), 7.44–7.47 (m, 2H), 2.96 (t, J = 7.2 Hz, 2H), 1.73 (quintet, J = 7.2 Hz, 2H), 1.29–1.35 (m, 12H, 6 × CH2), 0.88 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100 MHz, CDCl3): δ 200.8, 137.3, 133.0, 128.7, 128.2, 38.8, 31.9, 29.5, 29.3, 24.5, 22.8, 14.2.
4.5.12. 1-Phenyl-3-(o-tolyl)propan-1-one8a (2l)
White solid. Yield (87 mg, 79%). IR (DCM): 3061, 3023, 2927, 1686, 1597, 1580, 1492, 1448, 1315, 1203, 1026, 974, 746, 690 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.97–7.99 (m, 2H), 7.55–7.59 (m, 1H), 7.45–7.49 (m, 2H), 7.14–7.19 (m, 4H), 3.26 (t, J = 7.0 Hz, 2H), 3.07 (t, J = 8.0 Hz, 2H), 2.36 (s, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.5, 139.5, 137.0, 136.1, 133.2, 130.5, 128.9, 128.8, 128.2, 126.5, 126.3, 39.2, 27.6, 19.5.
4.5.13. 3-(4-Methoxyphenyl)-1-phenylpropan-1-one10a (2m)
White solid. Yield (88 mg, 73%). IR (DCM): 2957, 2933, 2835, 1681, 1609, 1512, 1205, 1033, 825, 743, 689 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 7.6 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 7.18 (d, J = 8.0 Hz, 2H), 6.85 (d, J = 8.0 Hz, 2H), 3.79 (s, 3H), 3.28 (t, J = 7.2 Hz, 2H), 3.02 (t, J = 8.0 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.5, 158.1, 137.0, 133.4, 133.1, 129.5, 128.7, 128.1, 114.0, 55.4, 40.8, 29.4.
4.5.14. 3-(Benzo[d][1,3]dioxol-5-yl)-1-phenylpropan-1-one11b (2n)
White solid. Yield (114 mg, 90%). IR (DCM): 2894, 1681, 1502, 1444, 1244, 1203, 1037, 927, 742 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 7.2 Hz, 2H), 7.56 (t, J = 7.6 Hz, 1H), 7.46 (t, J = 7.6 Hz, 2H), 6.69–6.75 (m, 3H), 5.91 (s, 2H), 3.26 (t, J = 7.6 Hz, 2H), 2.99 (t, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.3, 147.7, 145.9, 136.9, 135.2, 133.2, 128.7, 128.1, 121.3, 109.0, 108.4, 100.9, 40.8, 29.9.
4.5.15. 1,5-Diphenylpentan-1-one27 (2o)
Colorless liquid. Yield (99 mg, 83%). IR (DCM): 3060, 3025, 2933, 1685, 1597, 1448, 1220, 749, 698 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.91 (d, J = 7.2 Hz, 2H), 7.51 (t, J = 7.2 Hz, 1H), 7.42 (t, J = 7.6 Hz, 2H), 7.22–7.24 (m, 2H), 7.14–7.16 (m, 3H), 2.95 (t, J = 7.2 Hz, 2H), 2.64 (t, J = 7.6 Hz, 2H), 1.65–1.79 (m, 4H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.4, 142.4, 137.2, 133.0, 128.7, 128.5, 128.4, 125.9, 38.5, 35.9, 31.2, 24.1.
4.5.16. 1-Phenyl-5-(pyridin-2-yl)pentan-1-one (2p)
Colorless oil. Yield (88 mg, 74%). IR (DCM): 3061, 2935, 2860, 1684, 1595, 1474, 1448, 1434, 1219, 751, 691 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.50 (d, J = 4.8 Hz, 1H), 7.92–7.94 (m, 2H), 7.51–7.59 (m, 2H), 7.43 (t, J = 8.0 Hz, 2H), 7.15 (d, J = 8.0 Hz, 1H), 7.07–7.10 (m, 1H), 3.00 (t, J = 7.2 Hz, 2H), 2.84 (t, J = 7.2 Hz, 2H), 1.80–1.83 (m, 4H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.3, 161.9, 149.3, 137.1, 136.5, 133.0, 128.7, 128.1, 122.9, 121.1, 38.5, 38.2, 29.5, 24.0. HRMS (ESI) m/z calcd for C16H19NO (M + 2H)+: 241.1461, found: 241.1468.
4.5.17. 3-Phenyl-1-(p-tolyl)propan-1-one8c (3a)
White solid. Yield (105 mg, 94%). IR (DCM): 3061, 3026, 2920, 1679, 1606, 1494, 1451, 1408, 1179, 825, 774, 743, 699 cm–1. 1H NMR (700 MHz, CDCl3): δ 7.78 (d, J = 7.7 Hz, 2H), 7.21–7.23 (m, 2H), 7.16–7.18 (m, 4H), 7.11–7.13 (m, 1H), 3.19 (t, J = 7.7 Hz, 2H), 2.98 (t, J = 7.7 Hz, 2H), 2.32 (s, 3H). 13C{1H} NMR (176 MHz, CDCl3): δ199.0, 144.0, 141.5, 134.5, 129.4, 128.64, 128.55, 128.4, 126.2, 40.5, 30.3, 21.8.
4.5.18. 1-(4-Chlorophenyl)-3-phenylpropan-1-one9a (3b)
White solid. Yield (107 mg, 88%). IR (DCM): 3085, 3059, 3026, 2950, 2925, 2861, 1681, 1587, 1491, 1405, 1281, 1095, 1075, 850, 832, 779, 745 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 8.4 Hz, 2H), 7.31 (d, J = 8.4 Hz, 2H), 7.19–7.23 (m, 2H), 7.10–7.16 (m, 3H), 3.17 (t, J = 8.0 Hz, 2H), 2.96 (t, J = 8.0 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ198.0, 141.1, 139.6, 135.2, 129.5, 129.0, 128.7, 128.5, 126.3, 40.5, 30.1.
4.5.19. 1-(4-Bromophenyl)-3-phenylpropan-1-one9a (3c)
White solid. Yield for Table 3 (120 mg, 84%). Yield for Table 7 (95 mg, 66%). IR (DCM): 3083, 3057, 3024, 2929, 1682, 1583, 1493, 1451, 843, 829, 778 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.84 (d, J = 8.8 Hz, 2H), 7.62 (d, J = 8.8 Hz, 2H), 7.31–7.35 (m, 2H), 7.22–7.26 (m, 3H), 3.29 (t, J = 7.2 Hz, 2H), 3.06 (t, J = 7.2 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 198.3, 141.1, 135.6, 132.0, 129.7, 128.7, 128.5, 128.36, 126.35, 40.5, 30.1.
4.5.20. 1-(Naphthalen-2-yl)-3-phenylpropan-1-one9a (3d)
White solid. Yield (97 mg, 75%). IR (DCM): 3059, 3025, 2948, 2928, 1681, 1494, 1365, 1289, 1173 1123, 865, 819, 749 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.37 (s, 1H), 7.95 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 1H), 7.77–7.85 (m, 3H), 7.43–7.53 (m, 2H), 7.20–7.26 (m, 4H), 7.14–7.16 (m, 1H), 3.35 (t, J = 7.6 Hz, 2H), 3.04 (t, J = 7.2 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.3, 141.5, 135.7, 134.2, 132.6, 129.8, 129.7, 128.7, 128.59, 128.58, 128.57, 127.9, 126.9, 126.3, 123.9, 40.7, 30.4.
4.5.21. 3-Phenyl-1-(thiophen-2-yl)propan-1-one9a (3e)
Pale yellow oil. Yield (70 mg, 65%). IR (DCM): 3085, 3061, 3026, 2925, 1660, 1517, 1495, 1415, 1238, 1209, 1079, 1062, 852, 723, 699 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.60 (dd, J1 = 4.0 Hz, J2 = 1.2 Hz, 1H), 7.52 (dd, J1 = 4.8 Hz, J2 = 1.2 Hz, 1H), 7.20–7.23 (m, 2H), 7.10–7.17 (m, 3H), 7.02 (dd, J1 = 5.2 Hz, J2 = 4.0 Hz, 1H), 3.15 (t, J = 7.6 Hz, 2H), 2.98 (t, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 192.3, 144.2, 141.1, 133.7, 131.9, 128.6, 128.5, 128.2, 126.3, 41.2, 30.4.
4.5.22. 5-(Pyridin-2-yl)-1-(p-tolyl)pentan-1-one (3f)
Yellow oil. Yield (57 mg, 45%). IR (DCM): 2927, 2860, 1680, 1606, 1569, 1474, 1434, 1180, 819, 767 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.50 (d, J = 4.4 Hz, 1H), 7.83 (d, J = 8.0 Hz, 2H), 7.57 (td, J1 = 7.6 Hz, J2 = 1.2 Hz, 1H), 7.23 (d, J = 8.0 Hz, 2H), 7.14 (d, J = 7.6 Hz, 1H), 7.08 (m, 1H), 2.97 (t, J = 7.2 Hz, 2H), 2.83 (t, J = 7.2 Hz, 2H), 2.38 (s, 3H), 1.81–1.83 (m, 4H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.0, 162.0, 149.3, 143.7, 136.4, 134.6, 129.3, 128.3, 122.9, 121.1, 38.4, 38.2, 29.5, 24.2, 21.7. HRMS (ESI) m/z calcd for C17H20NO (M + H)+: 254.1545, found: 254.1572.
4.5.23. 3-(Benzo[d][1,3]dioxol-5-yl)-1-(4-methoxyphenyl)propan-1-one29 (3g)
Colorless oil. Yield for Table 3 (88 mg, 62%). Yield for Table 7 (132 mg, 93%). IR (DCM): 2925, 2840, 1474, 1600, 1575, 1503, 1488, 1443, 1245, 1209, 1170, 1037, 979, 837, 809 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.8 Hz, 2H), 6.91 (d, J = 8.8 Hz, 2H), 6.68–6.74 (m, 3H), 5.91 (s, 2H), 3.86 (s, 3H), 3.19 (t, J = 7.6 Hz, 2H), 2.97 (d, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 197.9, 163.6, 147.7, 145.9, 135.3, 130.4, 130.1, 121.2, 113.8, 109.0, 108.3, 100.9, 55.5, 40.4, 30.2.
4.5.24. 2-Hexyl-3,4-dihydronaphthalen-1(2H)-one (3h)
Yellow oil. Yield for Table 3 (90 mg, 78%). Yield for Table 8 (109 mg, 95%). IR (DCM): 2925, 2856, 1683, 1601, 1445, 1289, 1223, 915, 774, 741 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 7.6 Hz, 1H), 7.37 (t, J = 7.2 Hz, 1H), 7.21 (t, J = 7.6 Hz, 1H), 7.15 (d, J = 7.6 Hz, 1H), 2.88–2.93 (m, 2H), 2.35–2.42 (m, 1H), 2.12–2.18 (dq, J1 = 13.2 Hz, J2 = 4.8 Hz, 1H), 1.81–1.87 (m, 2H), 1.22–1.43 (m, 9H), 0.81 (t, J = 6.8 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.6, 144.1, 133.2, 132.7, 128.6, 127.6, 126.6, 47.6, 31.9, 29.6, 29.5, 28.4, 28.3, 27.1, 22.8, 14.2. HRMS (ESI) m/z calcd for C16H23NO (M + H)+: 231.1743, found: 231.1753.
4.5.25. 4-Methyl-2-(4-phenylbutyl)-3,4-dihydronaphthalen-1(2H)-one (3i)
Mixture of two diastereoisomers, ca. 1:1. Colorless oil. Yield (129 mg, 88%). IR (DCM): 3061, 3025, 2929, 2856, 1682, 1600, 1495, 1479, 1454, 1228, 752, 772, 699 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.92–7.97 (m, 2H), 7.38–7.45 (m, 2H), 7.30–7.32 (m, 1H), 7.16–7.24 (m, 7H), 7.07–7.12 (m, 6H), 3.06–3.10 (m, 1H), 2.54–2.62 (m, 5H), 2.37–2.42 (m, 1H), 1.95–2.09 (m, 5H), 1.55–1.63 (m, 5H), 1.38–1.46 (m, 6H), 1.33 (d, 3H). 1H NMR (400 MHz, CDCl3): δ 7.92–7.97 (m, 2H), 7.38–7.45 (m, 2H), 7.30–7.32 (m, 1H), 7.16–7.24 (m, 7H), 7.07–7.12 (m, 6H), 3.06–3.10 (m, 1H), 2.54–2.62 (m, 5H), 2.37–2.42 (m, 1H), 1.95–2.09 (m, 5H), 1.55–1.63 (m, 5H), 1.38–1.46 (m, 6H), 1.30 (d, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.6, 200.5, 148.7, 148.1, 142.8, 142.82, 142.75, 133.5, 133.4, 132.7, 131.7, 128.53, 128.52, 128.4, 128.1, 127.6, 127.5, 126.6, 126.5, 126.4, 125.7, 48.0, 43.0, 38.1, 36.0, 35.9, 35.2, 33.2, 31.8, 31.6, 31.2, 29.7, 29.5, 26.8, 26.7, 21.6, 20.4. HRMS (ESI) m/z calcd for C21H25O (M + H)+: 293.1905, found: 293.1933.
4.5.26. 1-(2-Aminophenyl)octan-1-one (3j)
Yellow oil. Yield (58 mg, 53%). IR (DCM): 3465, 3342, 2920, 2926, 2854, 1646, 1614, 1581, 1549, 1451, 1218, 1160, 747, 668 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 7.7 Hz, 1H), 7.27 (m, 1H), 6.66 (d, J = 7.0 Hz, 2H), 6.29 (s, 2H), 2.95 (t, J = 7.7 Hz, 2H), 1.74 (quintate, J = 7.7 Hz, 2H), 1.30–1.40 (m, 8H), 0.91 (t, J = 7.7 Hz, 3H). 13C{1H} NMR (176.0 MHz, CDCl3): δ 203.4, 150.5, 134.2, 131.4, 118.2, 117.5, 115.9, 39.5, 31.9, 29.6, 29.6, 29.3, 25.1, 22.8, 14.2. HRMS (ESI) m/z calcd for C14H22NO (M + H)+: 220.1696, found: 220.1689.
4.5.27. 1-(2-Aminophenyl)nonan-1-one (3k)
Yellow oil. Yield (53 mg, 46%). IR (DCM): 3467, 3343, 2925, 2854, 1650, 1614, 1581, 1484, 1451, 1214, 1160, 747 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.67 (d, J = 8.4 Hz, 1H), 7.16–7.19 (m, 1H), 6.55–6.58 (m, 2H), 6.19 (s, 2H), 2.85 (t, J = 8.4 Hz, 2H), 1.64 (quintet, J = 7.6 Hz, 2H), 1.20–1.27 (m, 10H), 0.81 (t, J = 6.8 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 203.4, 150.5, 134.2, 134.4, 118.2, 117.5, 115.8, 39.5, 32.0, 29.6, 29.3, 25.1, 22.8, 14.2. HRMS (ESI) m/z calcd for C15H24NO (M + H)+: 234.1852, found: 234.1834.
4.5.28. 1-(5-Methylfuran-2-yl)octan-1-one (3l)
Brown oil. Yield for Table 3 (88 mg, 85%). Yield for Table 7 (90 mg, 86%). IR (DCM): 2927, 2856, 1714, 1515, 1466, 1209, 1168, 930 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.06 (d, J = 3.2 Hz, 1H), 6.12 (d, J = 2.4 Hz, 1H), 2.73 (t, J = 7.6 Hz, 2H), 2.37 (s, 3H), 1.66–1.73 (m, 2H), 1.27–1.32 (m, 8H), 0.87 (t, J = 4.8 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 189.3, 157.7, 151.7, 119.0, 108.9, 38.3, 31.6, 29.4, 29.2, 24.9, 22.7, 14.2, 14.1. HRMS (ESI) m/z calcd for C13H21O2 (M + H)+: 209.1542, found: 209.1555.
4.5.29. 1-(p-Tolyl)heptan-1-one30 (3m)
White solid. Yield (82 mg, 81%). IR (DCM): 2924, 2857, 1677, 1607, 1466, 1406, 1181, 827, 791, 728 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.78 (d, J = 8.0 Hz, 2H), 7.17 (d, J = 8.0 Hz, 2H), 2.85 (t, J = 7.6 Hz, 2H), 2.32 (s, 3H), 1.60–1.68 (m, 2H), 1.21–1.29 (m, 6H), 0.81 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.4, 143.7, 134.7, 129.3, 128.3, 38.6, 31.8, 29.2, 24.6, 22.7, 21.7, 14.2.
4.5.30. 5-Phenyl-1-(p-tolyl)pentan-1-one27 (3n)
Colorless oil. Yield (105 mg, 83%). IR (DCM): 3023, 2931, 2858, 1681, 1460, 1450, 1407, 827, 781, 741, 700 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 8.4 Hz, 2H), 7.15–7.21 (m, 4H), 7.07–7.11 (m, 3H), 2.87 (t, J = 7.2 Hz, 2H), 2.58 (t, J = 7.6 Hz, 2H), 2.32 (s, 3H), 1.61–1.73 (m, 4H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.1, 143.8, 142.4, 134.7, 129.4, 128.5, 128.4, 128.3, 125.8, 38.4, 35.9, 31.3, 24.2, 21.7.
4.5.31. 1-(4-Methoxyphenyl)heptan-1-one31 (3o)
White solid. Yield (102 mg, 93%). IR (DCM): 2931, 2858, 1672, 1603, 1509, 1468, 1449, 1258, 1176, 1034, 837, 821, 798, 731 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H), 2.89 (t, J = 7.6 Hz, 2H), 1.71 (quintate, J = 7.2 Hz, 2H), 1.30–1.35 (m, 6H), 0.88 (t, J = 6.8 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.3, 163.4, 130.4, 130.3, 113.8, 55.5, 38.4, 31.8, 29.2, 24.7, 22.7, 14.2.
4.5.32. 1-(4-Methoxyphenyl)octan-1-one32 (3p)
White solid. Yield (107 mg, 92%). IR (DCM): 2953, 2932, 2852, 1670, 1603, 1509, 1468, 1255, 1199, 1177, 1034, 847, 828, 738 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.93 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 8.8 Hz, 2H), 3.85 (s, 3H), 2.89 (t, J = 7.6 Hz, 2H), 1.71 (quintate, J = 7.6 Hz, 2H), 1.28–1.34 (m, 8H), 0.87 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.4, 163.4, 130.4, 130.3, 113.8, 55.5, 38.4, 31.8, 29.5, 29.3, 24.8, 22.7, 14.2.
4.5.33. 1-(4-Fluorophenyl)-3-phenylpropan-1-one8c (3q)
Colorless oil. Yield (88 mg, 77%). IR (DCM): 3062, 3027, 2927, 1635, 1598, 1505, 1452, 1408, 1231, 1270, 1156, 1098, 840, 699, 603 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.90–7.94 (m, 2H), 7.13–7.24 (m, 5H), 7.03–7.07 (m, 2H), 3.21 (t, J = 8.0 Hz, 2H), 3.00 (t, J = 8.0 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 197.7, 165.8 (d, 1JC–F = 253.0 Hz), 141.3, 133.4, 130.7 (d, 3JC–F = 9.2 Hz), 128.7, 128.5, 126.3, 115.8 (d, 2JC–F = 21.7 Hz), 40.5, 30.2.
4.5.34. 1-(4-Chlorophenyl)hexan-1-one32 (3r)
White solid. Yield (100 mg, 95%). IR (DCM): 2954, 2932, 2861, 1682, 1589, 1487, 1402, 1265, 1091, 1029, 776, 698 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.88 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.8 Hz, 2H), 2.91 (t, J = 7.2 Hz, 2H), 1.68–1.73 (m, 2H), 1.33–1.36 (m, 4H), 0.90 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.4, 139.4, 135.5, 129.6, 129.0, 38.7, 31.6, 24.1, 22.6, 14.1.
4.5.35. 1-(4-Chlorophenyl)-3-phenylpropan-1-one9a (3s)
White solid. Yield (93 mg, 76%). IR (DCM): 3061, 3085, 3027, 2927, 1687, 1588, 1452, 1203, 1091, 1029, 776, 698 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.83 (d, J = 8.8 Hz, 2H), 7.36 (d, J = 8.8 Hz, 2H), 7.13–7.24 (m, 5H), 3.21 (t, J = 8.4 Hz, 2H), 3.00 (t, J = 8.0 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 198.1, 141.2, 139.6, 135.3, 129.6, 129.0, 128.7, 128.5, 126.3, 40.5, 30.2.
4.5.36. 3-(4-Aminophenyl)-1-(4-bromophenyl)propan-1-one (3t)
Orange solid. Yield (90 mg, 61%). IR (DCM): 3446, 3365, 3029, 2923, 1682, 1624, 1584, 1516, 1396, 1201, 1070, 978, 815 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 8.8 Hz, 2H), 7.58 (d, J = 8.4 Hz, 2H), 7.02 (d, J = 8.4 Hz, 2H), 6.63 (d, J = 8.0 Hz, 2H), 3.52 (br s, 2H), 3.20 (t, J = 8.0 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 198.7, 144.7, 135.8, 132.0, 131.1, 129.7, 129.3, 128.2, 115.5, 40.9, 29.4. HRMS (ESI) m/z calcd for C15H15BrNO (M + H)+: 304.0332, found: 304.0339.
4.5.37. 1-(Thiophen-2-yl)octan-1-one33 (3u)
Yellowish oil. Yield (90 mg, 86%). IR (DCM): 2954, 2926, 2855, 1663, 1416, 1234, 720, 857 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.69–7.70 (m, 1H), 7.59–7.61 (m, 1H), 7.10–7.12 (m, 1H), 2.85–2.90 (m, 2H), 1.69–1.75 (m, 2H), 1.23–1.33 (m, 8H), 0.85–0.89 (m, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 193.7, 144.7, 133.4, 131.8, 128.1, 39.6, 31.8, 29.4, 29.2, 24.9, 22.7, 14.2.
4.5.38. 1-(5-Methylfuran-2-yl)octan-1-one (3v)
Yellow oil. Yield (81 mg, 78%). IR (DCM): 2955, 2926, 2855, 1672, 1516, 1206, 1026, 795 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.08 (d, J = 3.2 Hz, 1H), 6.13 (d, J = 3.2 Hz, 1H), 2.72–2.75 (m, 2H), 2.38 (s, 3H), 1.63–1.71 (m, 2H), 1.27–1.32 (m, 8H), 0.87 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 189.4, 157.7, 151.7, 119.0, 108.9, 38.4, 31.8, 29.5, 29.2, 24.9, 22.7, 14.20, 14.19. HRMS (ESI) m/z calcd for C13H21O2 (M + H)+: 209.1542, found: 209.1555.
4.6. General Procedure for α-Ethylation of Ketones Using Ethanol
Inside a nitrogen atmosphere glovebox, a 38 mL sealed tube was charged with a stir bar, catalyst 1 (0.01 mmol, 2 mol %), Cs2CO3 (0.05 mmol, 10 mol %), ketone (0.5 mmol), tert-amyl alcohol (1 mL), and ethanol (1 mL) in the same order. The reaction mixture was refluxed (oil bath temperature, 140 °C) with stirring for 24 h under closed condition. After cooling, the solvent was evaporated under reduced pressure and the crude mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent.
4.6.1. 1-Phenylbutan-1-one10a (4a)
Colorless oil. Yield (43 mg, 58%). IR (DCM): 2962, 2932, 2873, 1686, 1597, 1448, 1273, 1213, 1002, 753, 735, 690 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.96 (d, J = 8.4 Hz, 2H), 7.53–7.57 (m, 1H), 7.44–7.47 (m, 2H), 2.94 (t, J = 7.6 Hz, 2H), 1.73–1.82 (m, 2H), 1.01 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.6, 137.3, 133.0, 128.7, 128.2, 40.7, 17.9, 14.0.
4.6.2. 1-(p-Tolyl)butan-1-one34 (4b)
Colorless oil. Yield (53 mg, 65%). IR (DCM): 2961, 2931, 2873, 1683, 1607, 1456, 1408, 1222, 1207, 1180, 809 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.89 (d, J = 8.0 Hz, 2H), 7.28 (d, J = 8.0 Hz, 2H), 2.94 (t, J = 7.6 Hz, 2H), 2.43 (s, 3H), 1.74–1.84 (m, 2H), 1.03 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.2, 143.7, 134.8, 129.3, 128.3, 40.5, 21.7, 18.0, 14.0.
4.6.3. 1-(4-Chlorophenyl)butan-1-one35 (4c)
Colorless oil. Yield (57 mg, 63%). IR (DCM): 2963, 2933, 2874, 1686, 1588, 1487, 1401, 1366, 1210, 1092, 1013, 998, 988, 817 cm–1. 1H NMR (700 MHz, CDCl3): δ 7.89 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.4 Hz, 2H), 2.91 (t, J = 7.7 Hz, 2H), 1.77 (m, 2H), 1.00 (t, J = 7.7 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.3, 139.4, 135.5, 129.6, 129.0, 40.6, 17.8, 14.0.
4.6.4. 1-(Naphthalen-2-yl)butan-1-one (4d)
Colorless oil. Yield (65 mg, 66%). IR (DCM): 3058, 2961, 2931, 2873, 1681, 1627, 1467, 1371, 1304, 1277, 1210, 1185, 1125, 855, 822, 753 cm–1. 1H NMR (400 MHz, CDCl3): δ 8.4 (s, 1H), 8.04 (dd, J1 = 8.8, 1.6 Hz, 1H), 7.96 (d, J = 8.0 Hz, 1H), 7.86–7.90 (m, 2H), 7.53–7.61 (m, 2H), 3.08 (t, J = 7.2 Hz, 2H), 1.79–1.89 (m, 2H), 1.05 (t, J = 7.6 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 200.5, 135.6, 134.6, 132.7, 129.7, 129.6, 129.5, 128.4, 127.9, 126.8, 124.1, 40.7, 18.1, 14.1. HRMS (ESI) m/z calcd for C14H15O (M + H)+: 199.1123, found: 199.1132.
4.6.5. 1-(Quinolin-3-yl)butan-1-one (4e)
Yellow solid. Yield (23 mg, 23%). IR (DCM): 2959, 2929, 2871, 1683, 1618, 1595, 1463, 1402, 1381, 1178, 963, 787, 751 cm–1. 1H NMR (400 MHz, CDCl3): δ 9.42 (d, J = 2.0 Hz, 1H), 8.70 (d, J = 2.0 Hz, 1H), 8.15 (d, J = 8.4 Hz, 1H), 7.93 (d, J = 8.0 Hz, 1H), 7.80–7.84 (m, 1H), 7.62 (t, J = 7.2 Hz, 1H), 3.07 (t, J = 7.2 Hz, 2H), 1.79–1.88 (m, 2H), 1.05 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.2, 149.8, 149.3, 137.1, 132.0, 129.5, 129.45, 129.4, 127.6, 127.0, 41.0, 17.7, 14.0. HRMS (ESI) m/z calcd for C13H14O (M + H)+: 200.1075, found: 200.1083.
4.7. Chemoselective α-C-Alkylation of Acetophenone by Primary Alcohols over Secondary Alcohols
Inside a glovebox, a 25 mL Schlenk tube was charged with a magnetic stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.05 mmol), acetophenone (0.5 mmol), tert-amyl alcohol (2 mL), 1-hexanol (0.6 mmol), and cyclohexanol (0.6 mmol) sequentially under nitrogen atmosphere. The reaction flask was taken out of the glovebox and then refluxed (oil bath temperature, 140 °C) with stirring in an open system under argon flow for 24 h. The reaction mixture was then cooled to room temperature. The solvent was evaporated under reduced pressure, and 0.5 mmol internal standard (toluene) was added into the reaction mixture and then diluted with 0.8 mL of acetone. Afterward, an aliquot of this reaction mixture was quickly filtered through a small plug of Celite and analyzed by GC. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of product was calculated using GC analysis, and the yield of pure product was determined after column chromatography.
4.8. Chemoselective α-C-Alkylation of Acetophenone by Primary Alcohols over Secondary Alcohols
Inside a glovebox, a 25 mL Schlenk tube was charged with a magnetic stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.05 mmol), acetophenone (0.5 mmol), tert-amyl alcohol (2 mL), 1-hexanol (0.6 mmol), and 3-pentanol (0.6 mmol) sequentially under nitrogen atmosphere. The reaction flask was taken out of the glovebox and then refluxed (oil bath temperature, 140 °C) with stirring in an open system under argon flow for 24 h. The reaction mixture was then cooled to room temperature. The solvent was evaporated under reduced pressure, and 0.5 mmol internal standard (toluene) was added into the reaction mixture and then diluted with 0.8 mL of acetone. Afterward, an aliquot of this reaction mixture was quickly filtered through a small plug of Celite and analyzed by GC. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of product was calculated using GC analysis, and the yield of pure product was determined after column chromatography.
4.9. Chemoselective α-C-Alkylation of Acetophenone by Primary Alcohols over N-Alkylation of Aniline
Inside a nitrogen atmosphere glovebox, a 25 mL Schlenk tube was charged with a stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.05 mmol), acetophenone (0.5 mmol), tert-amyl alcohol (2 mL), 1-hexanol (0.6 mmol), and aniline (0.5 mmol) in the same order. The reaction flask was taken out of the glovebox, equipped with a condenser, and the reaction mixture was refluxed (oil bath temperature, 140 °C) with stirring in an open system under the flow of argon for 24 h. The reaction mixture was then cooled down to room temperature. The solvent was evaporated under reduced pressure and 0.5 mmol internal standard (toluene) was added into the reaction mixture and the solution was quickly filtered through a small plug of Celite. Afterward, an aliquot of this reaction mixture was dissolved in 0.8 mL of acetone and analyzed by GC analysis. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of product was calculated using GC analysis, and the yield of pure product was determined after column chromatography.
4.9.1. 2-Hexyl-2,3-dihydro-1H-inden-1-one36 (5a)
Yellow oil. Yield (106 mg, 98%). IR (DCM): 2924, 2853, 1713, 1609, 1464, 1294, 1276, 750, 721 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.74 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 7.2 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.35 (t, J = 7.2 Hz, 1H), 3.32 (dd, J1 = 17.2 Hz, J2 = 8.0 Hz, 1H), 2.81 (dd, J1 = 17.2 Hz, J2 = 3.6 Hz, 1H), 2.62–2.68 (m, 1H), 1.92–1.98 (m, 1H), 1.28–1.46 (m, 9H), 0.87 (t, J = 6.4 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 209.4, 154.0, 137.0, 134.7, 127.4, 126.7, 124.0, 47.6, 33.0, 31.8, 31.6, 29.4, 27.5, 22.7, 14.2.
4.9.2. 2-Octyl-2,3-dihydro-1H-inden-1-one37 (5b)
Yellow oil. Yield (120 mg, 97%). IR (DCM): 2924, 2853, 1713, 1609, 1464, 1294, 1276, 750, 721 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.75 (d, J = 7.6 Hz, 1H), 7.55–7.59 (m, 1H), 7.45 (d, J = 7.6 Hz, 1H), 7.35 (t, J = 7.6 Hz, 1H), 3.32 (dd, J1 = 17.2 Hz, J2 = 7.6 Hz, 1H), 2.81 (dd, J1 = 16.8 Hz, J2 = 4 Hz, 1H), 2.62–2.68 (m, 1H), 1.92–1.95 (m, 1H), 1.26–1.43 (m, 13H), 0.87 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 209.3, 153.9, 137.0, 134.7, 127.4, 126.7, 124.0, 47.6, 33.0, 32.0, 31.6, 29.8, 29.6, 29.4, 27.6, 22.8, 14.2.
4.9.3. 6-Methoxy-2-octyl-3,4-dihydronaphthalen-1(2H)-one (5c)
Colorless oil. Yield (120 mg, 85%). IR (DCM): 2924, 2854, 1675, 1600, 1572, 1464, 1354, 1251, 1227, 1133, 1028, 846, 830 cm–1. 1H NMR (400 MHz, CDCl3): δ 7.99 (d, J = 8.8 Hz, 1H), 6.80 (dd, J1 = 8.8 Hz, J2 = 2 Hz, 1H), 6.67 (d, J = 2 Hz, 1H), 3.84 (s, 3H), 2.91–2.95 (m, 2H), 2.39–2.43 (m, 1H), 2.18–2.22 (m, 1H), 1.87–1.92 (m, 2H), 1.26–1.47 (m, 13H), 0.87 (t, J = 7.2 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 199.5, 163.4, 146.5, 130.0, 126.3, 113.1, 112.5, 55.5, 47.3, 32.0, 29.9, 29.7, 29.6, 29.4, 28.7, 28.3, 27.2, 22.8, 14.2. HRMS (ESI) m/z calcd for C19H29O2 (M + H)+: 289.2162, found: 289.2148.
4.9.4. 1-(4-Methoxyphenyl)ethanone37 (6)
Pale yellow solid. Yield (56 mg, 74%). 1H NMR (400 MHz, CDCl3): δ 7.93 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 2H), 6.92 (dd, J1 = 8.8 Hz, J2 = 1.6 Hz, 2H), 3.86 (d, J1 = 2.0 Hz, 3H), 2.55 (d, J1 = 2.0 Hz, 3H). 13C{1H} NMR (100.6 MHz, CDCl3): δ 197.0, 163.6, 130.7, 130.4, 113.8, 55.6, 26.5.
4.10. GC Monitoring of the Reaction Mixture 2o
Inside a nitrogen atmosphere glovebox containing a 25 mL Schlenk tube equipped with a stir bar were added catalyst 1 (0.02 mmol), Cs2CO3 (0.05 mmol), 1-phenyl-1-ethanol (1 mmol), 3-phenyl-1-propanol (1.2 mmol), mesitylene (1 mmol, as an internal standard), and tert-amyl alcohol (4 mL) sequentially. The reaction flask was taken out of the glovebox and equipped with a condenser, and the solution was refluxed (oil bath temperature, 135 °C) with stirring in open system under the flow of argon. Aliquots were collected from reaction mixture periodically, further diluted with acetone (0.5 mL), and analyzed by GC. The conversion of 1-phenyl-1-ethanol and yield of 2h were calculated using GC analysis.
4.11. Mercury Poisoning Test
Inside a nitrogen atmosphere glovebox, a 25 mL Schlenk tube was charged with a stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.025 mmol), 1-phenyl-1-ethanol (0.5 mmol), solvent (2 mL), benzyl alcohol (0.6 mmol), and mercury (2 equiv 200 mg) in the same order. The reaction flask was taken out of the glovebox and then refluxed (oil bath temperature 135 °C) with stirring in an open system under argon flow for 24 h. The reaction mixture was then cooled down to room temperature. The solvent was evaporated under reduced pressure and 0.5 mmol of an internal standard (toluene) was added into the reaction mixture and then diluted with 0.8 mL of acetone. An aliquot of the solution was passed through a small Celite plug and analyzed by GC. The solvent was evaporated under reduced pressure, and the crude reaction mixture was purified by column chromatography over silica gel (60–120 mesh) using ethyl acetate/pet ether mixture as an eluent. The conversion of 1-phenyl-1-ethanol was calculated using GC analysis, and the yield of pure product was determined after column chromatography.
4.12. Observation of Ketone Intermediate
In a glovebox, a 25 mL Schlenk tube was charged with a stir bar, catalyst 1 (0.01 mmol), Cs2CO3 (0.025 mmol), 1-(4-methoxyphenyl)ethanol (0.5 mmol), and solvent (2 mL) under nitrogen atmosphere. The flask was taken out of the glovebox, equipped with a condenser, and the solution was refluxed (oil bath temperature 135 °C) with stirring in an open system under a flow of argon for 20 h. After cooling to room temperature, 0.5 mmol of an internal standard (dodecane) was added and the reaction mixture was subjected GC analysis. The solvent was evaporated, and the crude reaction mixture was purified by column chromatography over silica gel (100–200 mesh) using ethyl acetate/pet ether as an eluent. The conversion of 1-(4-methoxyphenyl)ethanol was calculated using GC analysis, and yield of pure products after column chromatography.
Acknowledgments
The authors acknowledge SERB New Delhi (EMR/2016/002517), DAE, and NISER for the financial support. S.S.G. acknowledges CSIR, and S.P. and B.K.P. acknowledge NISER for research fellowships.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01246.
1H and 13C NMR spectra of the products (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- For selected reviews, see:; a Huang F.; Liu Z.; Yu Z. C-Alkylation of Ketones and Related Compounds by Alcohols: Transition-Metal-Catalyzed Dehydrogenation. Angew. Chem., Int. Ed. 2016, 55, 862–875. 10.1002/anie.201507521. [DOI] [PubMed] [Google Scholar]; b Obora Y. C-Alkylation by hydrogen autotransfer reactions. Top. Curr. Chem. 2016, 374, 11. 10.1007/s41061-016-0012-8. [DOI] [PubMed] [Google Scholar]; c Obora Y. Recent Advances in α-Alkylation Reactions using Alcohols with Hydrogen Borrowing Methodologies. ACS Catal. 2014, 4, 3972–3981. 10.1021/cs501269d. [DOI] [Google Scholar]; d Dobereiner G. E.; Crabtree R. H. Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition-Metal Catalysis. Chem. Rev. 2010, 110, 681–703. 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]
- Representative reviews on α-alkylation of ketones with alkylhalides:; a Reetz M. T. Lewis Acid Induced α-Alkylation of Carbonyl Compounds. Angew. Chem., Int. Ed. 1982, 21, 96–108. 10.1002/anie.198200961. [DOI] [Google Scholar]; b Caine D. In Comprehensive Organic Chemistry; Trost B. M., Fleming I., Pattenden G., Eds.; Pergamon: Oxford, 1991; Vol. 3, pp 1–63. [Google Scholar]; c Otera J., Ed. Modern Carbonyl Chemistry; Wiley-VCH: Weinheim, 2000. [Google Scholar]
- For reviews, see:; a Corma A.; Navas J.; Sabater M. J. Advances in One-Pot Synthesis Through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. 10.1021/acs.chemrev.7b00340. [DOI] [PubMed] [Google Scholar]; b Faisca Phillips A. M.; Pombeiro A. J. L.; Kopylovich M. N. Recent Advances in Cascade Reactions Initiated by Alcohol Oxidation. ChemCatChem 2017, 9, 217–246. 10.1002/cctc.201601176. [DOI] [Google Scholar]; c Hamid M. H. S. A.; Slatford P. A.; Williams J. M. J. Borrowing Hydrogen in the Activation of Alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575. 10.1002/adsc.200600638. [DOI] [Google Scholar]
- For reviews, see:; a Corma A.; Navas J.; Sabater M. J. Advances in One-Pot Synthesis Through Borrowing Hydrogen Catalysis. Chem. Rev. 2018, 118, 1410–1459. 10.1021/acs.chemrev.7b00340. [DOI] [PubMed] [Google Scholar]; b Crabtree R. H. Homogeneous Transition Metal Catalysis of Acceptorless Dehydrogenative Alcohol Oxidation: Applications in Hydrogen Storage and to Heterocycle Synthesis. Chem. Rev. 2017, 117, 9228–9246. 10.1021/acs.chemrev.6b00556. [DOI] [PubMed] [Google Scholar]; c Khusnutdinova J. R.; Milstein D. Metal-Ligand Cooperation. Angew. Chem., Int. Ed. 2015, 54, 12236–12273. 10.1002/anie.201503873. [DOI] [PubMed] [Google Scholar]; d Gunanathan C.; Milstein D. Bond Activation and Catalysis by Ruthenium Pincer Complexes. Chem. Rev. 2014, 114, 12024–12087. 10.1021/cr5002782. [DOI] [PubMed] [Google Scholar]; e Gunanathan C.; Milstein D. Applications of Acceptorless Dehydrogenation and Related Transformations in Chemical Synthesis. Science 2013, 341, 1229712 10.1126/science.1229712. [DOI] [PubMed] [Google Scholar]; f Watson A. J. A.; Williams J. M. J. The Give and Take of Alcohol Activation. Science 2010, 329, 635–636. 10.1126/science.1191843. [DOI] [PubMed] [Google Scholar]; g Bähn S.; Imm S.; Neubert L.; Zhang M.; Neumann H.; Beller M. The Catalytic Amination of Alcohols. ChemCatChem 2011, 3, 1853–1864. 10.1002/cctc.201100255. [DOI] [Google Scholar]; h Guillena G.; Ramón D. J.; Yus M. Hydrogen Autotransfer in the N-Alkylation of Amines and Related Compounds Using Alcohols and Amines as Electrophiles. Chem. Rev. 2010, 110, 1611–1641. 10.1021/cr9002159. [DOI] [PubMed] [Google Scholar]; i Hamid M. H. S. A.; Slatford P. A.; Williams J. M. J. Borrowing Hydrogen in the Activation of Alcohols. Adv. Synth. Catal. 2007, 349, 1555–1575. 10.1002/adsc.200600638. [DOI] [Google Scholar]
- Cao X.-N.; Wan X.-M.; Yang F.-L.; Li K.; Hao X.-Q.; Shao T.; Zhu X.; Song M.-P. NNN Pincer Ru(II)-Complex-Catalyzed α-Akylation of Ketones with Alcohol. J. Org. Chem. 2018, 83, 3657–3668. 10.1021/acs.joc.8b00013. [DOI] [PubMed] [Google Scholar]
- Mamidala R.; Samser S.; Sharma N.; Lourderaj U.; Venkatasubbaiah K. Isolation and Characterization of Regioisomers of Pyrazole-Based Palladacycles and Their Use in α-Alkylation of Ketones Using Alcohols. Organometallics 2017, 36, 3343–3351. 10.1021/acs.organomet.7b00478. [DOI] [Google Scholar]
- Buil M. L.; Esteruelas M. A.; Herrero J.; Izquierdo S.; Pastor I. M.; Yus M. Osmium Catalyst for the Borrowing Hydrogen Methodology: Alkylation of Arylacetonitriles and Methyl Ketones. ACS Catal. 2013, 3, 2072–2075. 10.1021/cs4005375. [DOI] [Google Scholar]
- a Wang R.; Fan H.; Zhao W.; Li F. Acceptorless Dehydrogenative Cyclization of o-Aminobenzyl Alcohols with Ketones to Quinolines in Water Catalyzed by Water-Soluble Metal-Ligand Bifunctional Catalyst [Cp*(6,6′-(OH)2bpy)(H2O)][OTf]2. Org. Lett. 2016, 18, 3558–3561. 10.1021/acs.orglett.6b01518. [DOI] [PubMed] [Google Scholar]; b Liu P.; Liang R.; Lu L.; Yu Z.; Li F. Use of a Cyclometalated Iridium(III) Complex Containing a NCN Coordinating Terdentate Ligand as a Catalyst for the N-Alkylation of Ketones and N-Alkylation of Amines with Alcohols. J. Org. Chem. 2017, 82, 1943–1950. 10.1021/acs.joc.6b02758. [DOI] [PubMed] [Google Scholar]; c Genç S.; Günnaz S.; Çetinkaya B.; Gülcemal S.; Gülcemal D. Iridium(I)-Catalyzed Alkylation Reactions To Form α-Alkylated Ketones. J. Org. Chem. 2018, 83, 2875–2881. 10.1021/acs.joc.8b00043. [DOI] [PubMed] [Google Scholar]
- a Elangovan S.; Sortais J.-B.; Beller M.; Darcel C. Iron-Catalyzed α-Alkylation of Ketones with Alcohols. Angew. Chem., Int. Ed. 2015, 54, 14483–14486. 10.1002/anie.201506698. [DOI] [PubMed] [Google Scholar]; b Deibl N.; Kempe R. General and Mild Cobalt-Catalyzed C-Alkylation of Unactivated Amides and Esters with Alcohols. J. Am. Chem. Soc. 2016, 138, 10786–10789. 10.1021/jacs.6b06448. [DOI] [PubMed] [Google Scholar]; c Zhang G.; Wu J.; Zeng H.; Zhang S.; Yin Z.; Zheng S. Cobalt-Catalyzed α-Alkylation of Ketones with Primary Alcohol. Org. Lett. 2017, 19, 1080–1083. 10.1021/acs.orglett.7b00106. [DOI] [PubMed] [Google Scholar]
- a Peña-López M.; Piehl P.; Elangovan S.; Neumann H.; Beller M. Manganese-Catalyzed Hydrogen-Autotransfer C-C Bond Formation: α-Alkylation of Ketones with Primary Alcohols. Angew. Chem., Int. Ed. 2016, 55, 14967–14971. 10.1002/anie.201607072. [DOI] [PubMed] [Google Scholar]; b Chakraborty S.; Daw P.; Ben-David Y.; Milstein D. Manganese-Catalyzed α-Alkylation of Ketones, Esters, and Amides Using Alcohols. ACS Catal. 2018, 8, 10300–10305. 10.1021/acscatal.8b03720. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zhang M.-J.; Li H.-X.; Young D. J.; Li H.-Y.; Lang J.-P. Reaction Condition Controlled Nickel(II)-Catalyzed C–C Cross-Coupling of Alcohols. Org. Biomol. Chem. 2019, 17, 3567–3574. 10.1039/C9OB00418A. [DOI] [PubMed] [Google Scholar]
- a Yu X.; Wang Q. Y.; Wu Q. J.; Wang D. W. Rhodium-Catalyzed Alkylation of Ketones and Alcohols with Alcohols. Russ. J. Gen. Chem. 2016, 86, 178–183. 10.1134/S107036321601028X. [DOI] [Google Scholar]; b Tan D.-W.; Li H.-X.; Zhu D.-L.; Li H.-Y.; Young D. J.; Yao J.-L.; Lang J.-P. Ligand-Controlled Copper(I)-Catalyzed Cross-Coupling of Secondary and Primary Alcohols to α-Alkylated Ketones, Pyridines, and Quinolines. Org. Lett. 2018, 20, 608–611. 10.1021/acs.orglett.7b03726. [DOI] [PubMed] [Google Scholar]
- a Mukherjee A.; Nerush A.; Leitus G.; Shimon L. J. W.; Ben David Y.; Jalapa N. A. E.; Milstein D. Manganese-Catalyzed Environmentally Benign Dehydrogenative Coupling of Alcohols and Amines to Form Aldimines and H2: A Catalytic and Mechanistic Study. J. Am. Chem. Soc. 2016, 138, 4298–4301. 10.1021/jacs.5b13519. [DOI] [PubMed] [Google Scholar]; b Chakraborty S.; Daw P.; Ben-David Y.; Milstein D. Manganese-Catalyzed α-Alkylation of Ketones, Esters, and Amides Using Alcohols. ACS Catal. 2018, 8, 10300–10305. 10.1021/acscatal.8b03720. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zell T.; Langer R. From Ruthenium to Iron and Manganese-A Mechanistic View on Challenges and Design Principles of Base-Metal Hydrogenation Catalysts. ChemCatChem 2018, 10, 1930–1940. 10.1002/cctc.201701722. [DOI] [Google Scholar]; d Mukherjee A.; Milstein D. Homogeneous Catalysis by Cobalt and Manganese Pincer Complexes. ACS Catal. 2018, 8, 11435–11469. 10.1021/acscatal.8b02869. [DOI] [Google Scholar]
- a Elangovan S.; Topf C.; Fischer S.; Jiao H.; Spannenberg A.; Baumann W.; Ludwig R.; Junge K.; Beller M. Selective Catalytic Hydrogenations of Nitriles, Ketones, and Aldehydes by Well-Defined Manganese Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 8809–8814. 10.1021/jacs.6b03709. [DOI] [PubMed] [Google Scholar]; b Peña-López M.; Piehl P.; Elangovan S.; Neumann H.; Beller M. Manganese-Catalyzed Hydrogen-Autotransfer C-C Bond Formation: α-Alkylation of Ketones with Primary Alcohols. Angew. Chem., Int. Ed. 2016, 55, 14967–14971. 10.1002/anie.201607072. [DOI] [PubMed] [Google Scholar]
- a Kallmeier F.; Dudziec B.; Irrgang T.; Kempe R. Manganese-Catalyzed Sustainable Synthesis of Pyrroles from Alcohols and Amino Alcohols. Angew. Chem., Int. Ed. 2017, 56, 7261–7265. 10.1002/anie.201702543. [DOI] [PubMed] [Google Scholar]; b Zhang G.; Irrgang T.; Dietel T.; Kallmeier F.; Kempe R. Manganese-Catalyzed Dehydrogenative Alkylation or α-Olefination of Alkyl-Substituted N-Heteroarenes with Alcohols. Angew. Chem., Int. Ed. 2018, 57, 9131–9135. 10.1002/anie.201801573. [DOI] [PubMed] [Google Scholar]; c Deibl N.; Kempe R. Manganese-Catalyzed Multicomponent Synthesis of Pyrimidines from Alcohols and Amidines. Angew. Chem., Int. Ed. 2017, 56, 1663–1666. 10.1002/anie.201611318. [DOI] [PubMed] [Google Scholar]; d Kallmeier F.; Irrgang T.; Dietel T.; Kempe R. Highly Active and Selective Manganese C=O Bond Hydrogenation Catalysts: The Importance of the Multidentate Ligand, the Ancillary Ligands, and the Oxidation State. Angew. Chem., Int. Ed. 2016, 55, 11806–11809. 10.1002/anie.201606218. [DOI] [PubMed] [Google Scholar]; e Kallmeier F.; Kempe R. Manganese Complexes for (De)Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem., Int. Ed. 2018, 57, 46–60. 10.1002/anie.201709010. [DOI] [PubMed] [Google Scholar]
- Jang Y. K.; Krückel T.; Rueping M.; El-Sepelgy O. Sustainable Alkylation of Unactivated Esters and Amides with Alcohols Enabled by Manganese Catalysis. Org. Lett. 2018, 20, 7779–7783. 10.1021/acs.orglett.8b03184. [DOI] [PubMed] [Google Scholar]
- a Mastalir M.; Glatz M.; Pittenauer E.; Allmaier G.; Kirchner K. Sustainable Synthesis of Quinolines and Pyrimidines Catalyzed by Manganese PNP Pincer Complexes. J. Am. Chem. Soc. 2016, 138, 15543–15546. 10.1021/jacs.6b10433. [DOI] [PubMed] [Google Scholar]; b Mastalir M.; Pittenauer E.; Allmaier G.; Kirchner K. Manganese-Catalyzed Aminomethylation of Aromatic Compounds with Methanol as a Sustainable C1 Building Block. J. Am. Chem. Soc. 2017, 139, 8812–8815. 10.1021/jacs.7b05253. [DOI] [PubMed] [Google Scholar]; c Gorgas N.; Kirchner K. Isoelectronic Manganese and Iron Hydrogenation-Dehydrogenation Catalysts: Similarities and Divergences. Acc. Chem. Res. 2018, 51, 1558–1569. 10.1021/acs.accounts.8b00149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bruneau-Voisine A.; Wang D.; Dorcet V.; Roisnel T.; Darcel C.; Sortais J.-B. Transfer Hydrogenation of Carbonyl Derivatives Catalyzed by an Inexpensive Phosphine-Free Manganese Precatalyst. Org. Lett. 2017, 19, 3656–3659. 10.1021/acs.orglett.7b01657. [DOI] [PubMed] [Google Scholar]; b Filonenko G. A.; van Putten R.; Hensen E. J. M.; Pidko E. A. Catalytic (de)hydrogenation promoted by non-precious metals - Co, Fe and Mn: recent advances in an emerging field. Chem. Soc. Rev. 2018, 47, 1459–1483. 10.1039/C7CS00334J. [DOI] [PubMed] [Google Scholar]; c Nguyen D. H.; Trivelli X.; Capet F.; Paul J-F.; Dumeignil F.; Gauvin R. M. Manganese Pincer Complexes for the Base-Free, Acceptorless Dehydrogenative Coupling of Alcohols to Esters: Development, Scope, and Understanding. ACS Catal. 2017, 7, 2022–2032. 10.1021/acscatal.6b03554. [DOI] [Google Scholar]; d Maji B.; Barman M. K. Recent Developments of Manganese Complexes for Catalytic Hydrogenation and Dehydrogenation Reactions. Synthesis 2017, 49, 3377–3393. 10.1055/s-0036-1590818. [DOI] [Google Scholar]
- Tondreau A. M.; Boncella J. M. 1,2-Addition of Formic or Oxalic Acid to −N{CH2CH2(PiPr2)}2-Supported Mn(I) Dicarbonyl Complexes and the Manganese-Mediated Decomposition of Formic Acid. Organometallics 2016, 35, 2049–2052. 10.1021/acs.organomet.6b00274. [DOI] [Google Scholar]
- a Garbe M.; Junge K.; Beller M. Homogeneous Catalysis by Manganese-Based Pincer Complexes. Eur. J. Org. Chem. 2017, 4344–4362. 10.1002/ejoc.201700376. [DOI] [Google Scholar]; b Trovitch R. J. The Emergence of Manganese-Based Carbonyl Hydrosilylation Catalysts. Acc. Chem. Res. 2017, 50, 2842–2852. 10.1021/acs.accounts.7b00422. [DOI] [PubMed] [Google Scholar]; c Kallmeier F.; Kempe R. Manganese Complexes for (De)Hydrogenation Catalysis: A Comparison to Cobalt and Iron Catalysts. Angew. Chem., Int. Ed. 2018, 57, 46–60. 10.1002/anie.201709010. [DOI] [PubMed] [Google Scholar]
- Fertig R.; Irrgang T.; Freitag F.; Zander J.; Kempe R. Manganese-Catalyzed and Base-Switchable Synthesis of Amines or Imines via Borrowing Hydrogen or Dehydrogenative Condensation. ACS Catal. 2018, 8, 8525–8530. 10.1021/acscatal.8b02530. [DOI] [Google Scholar]
- Liu T.; Wang L.; Wu K.; Yu Z. Manganese-Catalyzed β-Alkylation of Secondary Alcohols with Primary Alcohols under Phosphine-Free Conditions. ACS Catal. 2018, 8, 7201–7207. 10.1021/acscatal.8b01960. [DOI] [Google Scholar]
- a Thiyagarajan S.; Gunanathan C. Catalytic Cross-Coupling of Secondary Alcohols. J. Am. Chem. Soc. 2019, 141, 3822–3827. 10.1021/jacs.9b00025. [DOI] [PubMed] [Google Scholar]; b Thiyagarajan S.; Gunanathan C. Facile Ruthenium(II)-Catalyzed α-Alkylation of Arylmethyl Nitriles Using Alcohols Enabled by Metal–Ligand Cooperation. ACS Catal. 2017, 7, 5483–5490. 10.1021/acscatal.7b01427. [DOI] [Google Scholar]; c Thiyagarajan S.; Gunanathan C. Ruthenium-Catalyzed α-Olefination of Nitriles Using Secondary Alcohols. ACS Catal. 2018, 8, 2473–2478. 10.1021/acscatal.7b04013. [DOI] [Google Scholar]; d Gawali S. S.; Pandia B. K.; Gunanathan C. Manganese(I)-Catalyzed α-Alkenylation of Ketones Using Primary Alcohols. Org. Lett. 2019, 21, 3842–3847. 10.1021/acs.orglett.9b01327. [DOI] [PubMed] [Google Scholar]
- a Schmink J. R.; Holcomb J. L.; Leadbeater N. E. Testing the Validity of Microwave-Interfaced, in Situ Raman Spectroscopy as a Tool for Kinetic Studies. Org. Lett. 2009, 11, 365–368. 10.1021/ol802594s. [DOI] [PubMed] [Google Scholar]; b Jiang N.; Ragauskas A. J. Copper(II)-Catalyzed Aerobic Oxidation of Primary Alcohols to Aldehydes in Ionic Liquid [bmpy]PF6. Org. Lett. 2005, 7, 3689–3692. 10.1021/ol051293+. [DOI] [PubMed] [Google Scholar]
- a Fu S.; Shao Z.; Wang Y.; Liu Q. Manganese-Catalyzed Upgrading of Ethanol into 1-Butanol. J. Am. Chem. Soc. 2017, 139, 11941–11948. 10.1021/jacs.7b05939. [DOI] [PubMed] [Google Scholar]; b Das U. K.; Ben-David Y.; Diskin-Posner Y.; Milstein D. N-Substituted Hydrazones by Manganese-Catalyzed Coupling of Alcohols with Hydrazine: Borrowing Hydrogen and Acceptorless Dehydrogenation in One System. Angew. Chem., Int. Ed. 2018, 57, 2179–2182. 10.1002/anie.201712593. [DOI] [PubMed] [Google Scholar]
- Sahoo A. R.; Lalitha G.; Murugesh V.; Bruneau C.; Sharma G. V. M.; Suresh S.; Achard M. Ruthenium Phosphine–Pyridone Catalyzed Cross-Coupling of Alcohols To form α-Alkylated Ketones. J. Org. Chem. 2017, 82, 10727–10731. 10.1021/acs.joc.7b02042. [DOI] [PubMed] [Google Scholar]
- Jensen A. E.; Knochel P. Nickel-Catalyzed Cross-Coupling between Functionalized Primary or Secondary Alkylzinc Halides and Primary Alkyl Halides. J. Org. Chem. 2002, 67, 79–85. 10.1021/jo0105787. [DOI] [PubMed] [Google Scholar]
- Crawley M. L.; Phipps K. M.; Goljer I.; Mehlmann J. F.; Lundquist J. T.; Ullrich J. W.; Yang C.; Mahaney P. E. Efficient, Regioselective Palladium-Catalyzed Tandem Heck-Isomerization Reaction of Aryl Bromides and Non-Allylic Benzyl Alcohols. Org. Lett. 2009, 11, 1183–1185. 10.1021/ol900036y. [DOI] [PubMed] [Google Scholar]
- Ackermann L.; Kaspar L. T. TiCl4-Catalyzed Indirect Anti-Markovnikov Hydration of Alkynes and Its Application to the Synthesis of Benzo[b]furans. J. Org. Chem. 2007, 72, 6149–6153. 10.1021/jo070887i. [DOI] [PubMed] [Google Scholar]
- Seck C.; Mbaye M. D.; Coufourier S.; Lator A.; Lohier J.-F.; Poater A.; Ward T. R.; Gaillard S.; Renaud J.-L. Alkylation of Ketones Catalyzed by Bifunctional Iron Complexes: From Mechanistic Understanding to Application. ChemCatChem 2017, 9, 4410–4416. 10.1002/cctc.201701241. [DOI] [Google Scholar]
- Tseng M.-C.; Kan H.-C.; Chu Y.-H. Reactivity of trihexyl(tetradecyl)-phosphonium chloride, a room-temperature phosphonium ionic liquid. Tetrahedron Lett. 2007, 48, 9085–9089. 10.1016/j.tetlet.2007.10.131. [DOI] [Google Scholar]
- Scheiper B.; Bonnekessel M.; Krause H.; Fürstner A. Selective Iron-Catalyzed Cross-Coupling Reactions of Grignard Reagents with Enol Triflates, Acid Chlorides, and Dichloroarenes. J. Org. Chem. 2004, 69, 3943–3949. 10.1021/jo0498866. [DOI] [PubMed] [Google Scholar]
- Ruan J.; Saidi O.; Iggo J. A.; Xiao J. Direct Acylation of Aryl Bromides with Aldehydes by Palladium Catalysis. J. Am. Chem. Soc. 2008, 130, 10510–10511. 10.1021/ja804351z. [DOI] [PubMed] [Google Scholar]
- Adak L.; Bhadra S.; Ranu B. C. Palladium(0) nanoparticle-catalyzed sp2 C–H activation: a convenient route to alkyl–aryl ketones by direct acylation of aryl bromides and iodides with aldehydes. Tetrahedron Lett. 2010, 51, 3811–3814. 10.1016/j.tetlet.2010.05.067. [DOI] [Google Scholar]
- Koppolu S. R.; Naveen N.; Balamurugan R. Triflic Acid Promoted Direct α-Alkylation of Unactivated Ketones Using Benzylic Alcohols via in Situ Formed Acetals. J. Org. Chem. 2014, 79, 6069–6078. 10.1021/jo500759a. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yao B.; Deng C.-L.; Tang R.-Y.; Zhang X.-G.; Li J.-H. Palladium-Catalyzed Oxidative Coupling of Trialkylamines with Aryl Iodides Leading to Alkyl Aryl Ketones. Org. Lett. 2011, 13, 2184–2187. 10.1021/ol200404z. [DOI] [PubMed] [Google Scholar]
- Yoon I. C.; Kim T. G.; Cho C. S. α-Alkylation of Ketones by Trialkylamines under Heterogeneous Pd/C Catalysis. Organometallics 2014, 33, 1890–1892. 10.1021/om500228b. [DOI] [Google Scholar]
- Buchwald S. L.; Watson B. T.; Lum R. T.; Nugent W. A. A General Method for the Preparation of Zirconocene Complexes of Substituted Benzynes: In Situ Generation, Coupling Reactions, and Use in the Synthesis of Polyfunctionalized Aromatic Compounds. J. Am. Chem. Soc. 1987, 109, 7137–7141. 10.1021/ja00257a038. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.