

Abstract

An asymmetric Mannich reaction has been developed to generate chiral β-amino esters in good yields with excellent enantiomeric excesses (ee, up to 99%) using a chiral bifunctional thiourea catalyst derived from (R,R)-cyclohexyldiamine. This is the first report on the addition of 3-indolinone-2-carboxylates to N-Boc-benzaldimines generated in situ from α-amidosulfones, which proceeds under mild conditions.
Introduction
Chiral ß-amino esters are useful building blocks for the synthesis of ß-lactams and unnatural peptides.1 They found widespread applications in drug discovery.2 On the other hand, a chiral 2,2-disubstituted indolin-3-one skeleton3 is often present in several biologically active natural products such as (+)-isatisine A, trigonoliimine C, mersicarpine, etc.4,5 They are known to exhibit potent antiviral properties.6 Consequently, a few methods have been reported for the enantioselective conversion of 3-indolinone-2-carboxylates into chiral compounds.7 However, the construction of a chiral quaternary stereocenter is a challenging task for a synthetic chemist.8 Furthermore, an asymmetric Mannich reaction is a powerful strategy to produce chiral ß-amino ketones and ß-amino esters.9 Inspired by their fascinating structural features and potent biological activities, we were interested in producing chiral 2,2-disubstituted indolin-3-one derivatives. Indeed, there are no reports on the direct asymmetric Mannich-type addition of 3-indolinone-2-carboxylates to N-Boc-benzaldimines generated in situ from α-amidosulfones Figure 1.
Figure 1.

Examples of 3-indolinone natural products.
Following our interest in asymmetric synthesis,10 we herein report an enantioselective Mannich reaction of 2-substituted indolin-3-ones for the synthesis of chiral β-amino esters, using a thiourea catalyst derived from trans-(R,R)-1,2-diaminocyclohexane. Our investigation began with the reaction of 3-indolinone-2-carboxylate (1) with N-Boc-benzaldimine (2) using quinidine 4a as a catalyst in the presence of Na2CO3 in toluene (Table 1, entry a) at room temperature. Interestingly, the desired product 3a was obtained in 60% yield with 10% enantiomeric excesses (ee) and 92:8 diastereoselectivity. Next, we attempted the same reaction with dihydroquinidine 4b as a catalyst, and no significant improvement in yield and ee of product 3a was observed. Furthermore, the reaction was performed using a bifuntional Takemoto’s catalyst 4c, 5 mol %, and Na2CO3 in toluene at room temperature. Interestingly, the desired product 3a was obtained in 80% yield with 55% ee and 93:7 diastereoselectivity (Table 1, entry c). To enhance the enantio- and diastereoselectivity, the reaction was further performed with 5 mol % catalyst 4d under similar conditions (Table 1, entry d). A slight improvement was observed in enantio- and diastereoselectivity. Therefore, the reaction was further performed using a 5 mol % catalyst 4e under identical conditions. To our delight, the yield and ee were improved significantly (Table 1, entry e). To evaluate other thiourea catalysts (Scheme 1), the reaction was further carried out using a 5 mol % bis-thiourea catalyst 4f derived from trans-(R,R)-1,2-diaminocyclohexane. However, the product 3a was obtained with low enantio- and diastereoselectivity. To improve the enantio- and diastereoselectivity, the reaction was repeated using 5 mol % thiourea catalysts 4g and 4h derived from trans-1,2-diaminoindane and trans-1-amino-2-indanol, respectively. The desired product 3a was obtained in poor yield and with low selectivity (Table S1, entries g and h). Therefore, the next reaction was carried out using a thiourea catalyst 4i derived from 1,1′-binaphthyl-2,2′-diamine (Table 1, entry i). However, the catalyst 4i was found to be inferior than other catalysts. To know the effect of base, the reaction was conducted in toluene using different bases like Na2CO3, K2CO3, Cs2CO3, NaOH, and CsOH (Table 1, entries i–m). Among them, Na2CO3 in toluene was found to be the best for this transformation (Table 1, entry m). To our surprise, only 30% conversion was observed using 10% Na2CO3 solution and 60% conversion was observed with 50% Na2CO3 solution. Interestingly, 98% conversion was obtained with a sat. Na2CO3 solution, which was prepared using 32 g of Na2CO3 in 100 mL of water at 27 °C. Furthermore, we examined the effect of different solvents such as o-xylene, methyl tert-butyl ether, benzene, and dichloroethane on the conversion under similar reaction conditions (Table 1, entries n–r). None of these solvents produced better results than o-xylene (Table 1, entry n). Finally, we tested the effect of temperature, ranging from 25 to −78 °C, on the conversion. The best results were obtained using 5 mol % catalyst 4e and Na2CO3 in xylene at 0 °C. Due to the low freezing point of xylene (−25 °C), further reactions were conducted in toluene. However, there was no reaction in toluene at −78 °C under similar conditions (Table 1, entry u).
Table 1. Optimization of Reaction Conditions in the Formation of 3aa.

| entry | catalyst | base (aq.) | solvent T (°C) | time (days) | yield (%)b | dr (3a:3aa)c | ee (%)c | entry | catalyst | base (aq.) | solvent | T (°C) | time (days) | yield (%)b | dr (3a:3aa)c | ee (%)d,e |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a | 4a | Na2CO3 | toluene 25 | 3 | 45 | 92:8 | 10 | l | 4e | NaOH | toluene | 25 | 1 | 20 | 90:10 | 5 |
| b | 4b | Na2CO3 | toluene 25 | 2 | 60 | 93:7 | 5 | m | 4e | CsOH | toluene | 25 | 1 | 25 | 95:5 | 10 |
| c | 4c | Na2CO3 | toluene 25 | 4 | 80 | 93:7 | 55 | n | 4e | Na2CO3 | xylene | 0 | 3 | 98 | 99:1 | 99 |
| d | 4d | Na2CO3 | toluene 25 | 3 | 85 | 98:2 | 60 | o | 4e | Na2CO3 | toluene | 0 | 3 | 60 | 99:1 | 65 |
| e | 4e | Na2CO3 | toluene 25 | 2 | 90 | 99:1 | 85 | p | 4e | Na2CO3 | MTBE | 0 | 3 | 50 | 99:1 | 30 |
| f | 4f | Na2CO3 | toluene 25 | 4 | 40 | 95:5 | 50 | q | 4e | Na2CO3 | benzene | 0 | 3 | 55 | 99:1 | 40 |
| g | 4g | Na2CO3 | toluene 25 | 5 | 45 | 89:11 | 30 | r | 4e | Na2CO3 | DCE | 0 | 3 | 35 | 99:1 | 20 |
| h | 4h | Na2CO3 | toluene 25 | 4 | 50 | 88:22 | 25 | s | 4e | Na2CO3 | xylene | –20 | 4 | 35 | 99:1 | 40 |
| i | 4i | Na2CO3 | toluene 25 | 5 | 40 | 90:10 | 30 | t | 4e | Na2CO3 | toluene | –40 | 5 | 30 | 99:1 | 30 |
| j | 4e | K2CO3 | toluene 25 | 2 | 80 | 99:1 | 70 | u | 4e | Na2CO3 | toluene | –78 | 2 | |||
| k | 4e | Cs2CO3 | toluene 25 | 2 | 65 | 95:5 | 20 |
All reactions were performed at 0.21 mmol of 1, 0.25 mmol of 2, 5 mol % 4e and 0.2 mL of aqueous base in 5 mL of solvent.
Isolated yields after column chromatography.
Diastereomeric ratio was determined by 1H NMR.
Enantiomeric excess was determined by chiral high-performance liquid chromatography (HPLC).
Enantiomeric ratio of the major diastereomer.
Scheme 1. Catalyst Screening in the Asymmetric Mannich Reaction.

After having optimized conditions in hand, the scope of this method was examined with different substrates, and the results are summarized in Table 2. The substituent present on the aromatic ring of aldimine had shown some effect on the conversion. Ortho- and meta-substituted benzaldimines afforded the corresponding product in excellent yield with excellent enantioselectivity (Table 2, entries 3b, 3c, 3i, and 3k). The substrate bearing electron-withdrawing substituents such as nitro- and cyano- on the aromatic ring of aldimine gave the desired product in high yield with excellent enantiomeric excess (Table 2, entries 3h, 3i, 3j, and 3k). Conversely, the substrate having electron-donating groups like methyl- and methoxy on the aromatic ring gave the product with low ee (Table 2, entries 3f and 3g). Next, we examined the effect of substituents that are present at the 5th position of methyl 3-oxoindoline-2-carboxylate. The desired products were obtained in good yields but with low ee (Table 2, entries 3o, 3p, and 3q). The scope of this process was further extended to polyaromatic and heterocyclic systems. Interestingly, the aldimines derived from heteroaromatic aldehydes gave the products in excellent yield with excellent enantiomeric excess (Table 2, entries 3m and 3r). Furthermore, a sterically hindered naphthyl derivative also gave the desired product in good yield with excellent selectivity (Table 2, entry 3s). In addition, we have screened the N-substituted 3-oxo-indoline-2-carboxylate; in this, free NH and N-phenyl-substituted substrates failed to give the product, whereas the N-benzoyl 3-oxo-indoline-2-carboxylate participated smoothly in the present reaction and afforded the corresponding product (Table 2, entry 3n) in good yield and with excellent enantio- and diastereoselectivity. However, a slight decrease in the enantioselectivity was observed in the case of N-benzoyl 3-oxo-indoline-2-carboxylate (Table 2, entry 3n) compared to that of N-acetyl 3-oxo-indoline-2-carboxylate (Table 2, entry 3c).
Table 2. Substrate Scopea.


All reactions were performed at 0.21 mmol of 1, 0.25 mmol of 2, 5 mol % 4e, and 0.2 mL of aqueous base in 5 mL of solvent.
Isolated yields after column chromatography.
Diastereomeric ratio was determined by 1H NMR.
Enantiomeric excess was determined by chiral HPLC.
Enantiomeric ratio of the major diastereomer.
According to X-ray crystallography (CCDC 1842015), the structure and relative configuration of 3h were determined (Figure 2).11
Figure 2.

Oak Ridge thermal-ellipsoid plot program (ORTEP) diagram of 3h.
The absolute stereochemistry was established by single-crystal X-ray crystallography of 3t, which is having heavy atoms in its structure (Figure 3).11 As depicted in the ORTEP diagram, the absolute stereochemistry of 3t was assigned as R,S.
Figure 3.
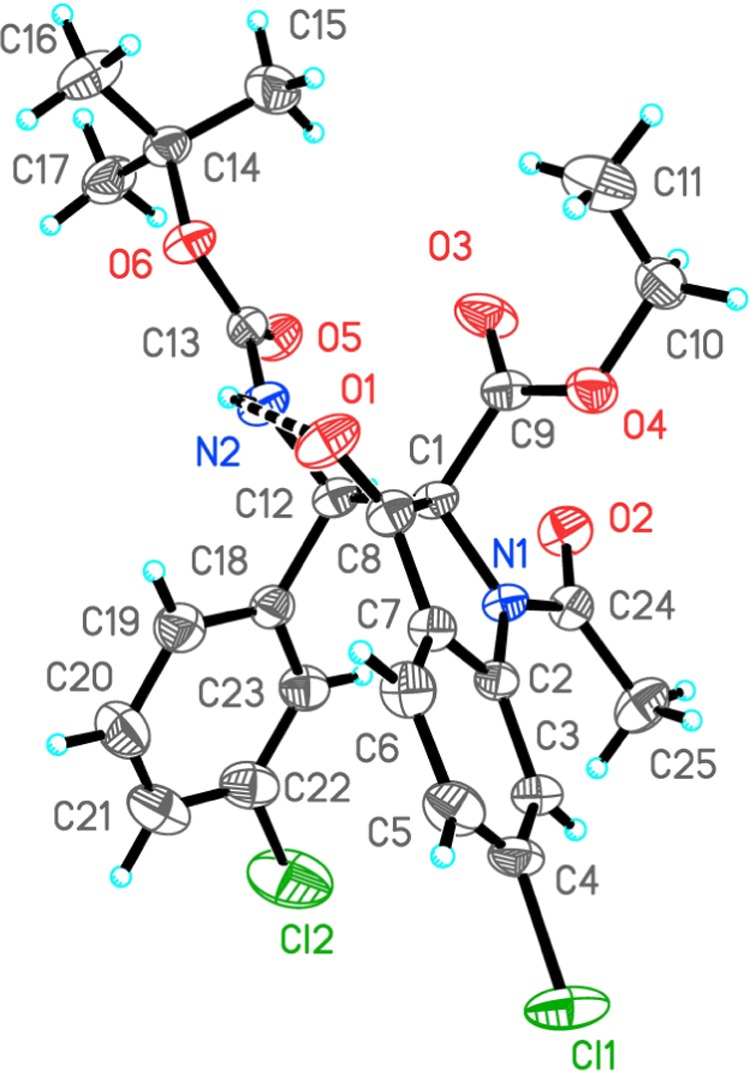
ORTEP diagram of 3t.
Finally, we tried to convert the product 3a into a spirolactam to demonstrate its synthetic application. To our surprise, the compound 3a failed to undergo cyclization under basic conditions (Scheme 2).
Scheme 2. Spirolactam Formation.

Mechanistically, the reaction proceeds through the formation of an enolate ion from 3-indolinone-2-carboxylate 3a by a tertiary amine moiety of the ligand 4e through deprotonation. A subsequent activation of N-Boc-benzaldimine by a thiourea moiety of the ligand 4e through hydrogen bonding generates a ternary complex, which is shown in Figure S3. A preferential Re-face attack of the enolate formed from 3-indolinone-2-carboxylate onto N-Boc-benzaldimine would give the desired product 3a, Figure 4.
Figure 4.

Plausible transition state.
Conclusions
In summary, we have successfully developed an organocatalytic asymmetric Mannich reaction for the synthesis of chiral β-amino esters, which are key intermediates for the synthesis of biologically active molecules. The reaction proceeds under mild conditions and is compatible with diverse range of substituents that are present on the aromatic ring of aldimines. This method works with a wide range of substrates including aryl, naphthyl, and heteroaryl cyclic ß-ketoesters.
General Methods
All solvents were dried according to standard literature procedures. The reactions were conducted under a nitrogen atmosphere. Melting points (mp) were obtained on Buchi B-540. 1H and 13C NMR (proton-decoupled) spectra were recorded in the CDCl3 solvent at 300, 400, or 500 MHz on an NMR spectrometer. Chemical shifts (δ) were reported in parts per million (ppm) with respect to tetramethylsilane as an internal standard. Coupling constants (J) are quoted in hertz (Hz). Mass spectra and high-resolution mass spectrometry (HRMS) were recorded on a mass spectrometer by the electrospray ionization (ESI) or atmospheric pressure chemical ionization technique. Optical rotations were recorded on an Anton Paar MCP-200 polarimeter. Enantiomeric excesses (ee’s) were determined by HPLC analysis using DAICEL Chiralpak OD-H, AS-H, IC, IA columns. The precursors were prepared according to the procedure reported in the literature.12
General Procedure for the Asymmetric Mannich Reaction (3a)
To a suspension of 2-substituted 3-indolinones 11 (50 mg, 1.0 equiv, 0.21 mmol %) and α-amidosulfones 22 (1.2 equiv, 0.25 mmol %) in xylene (5 mL) at 0 °C were added catalyst 4c3 (5 mol %) and saturated Na2CO3 (0.2 mL) successively. The resulting mixture was stirred for 3 days at 0 °C and then with water and extracted with ethyl acetate. The organic layer was dried over Na2SO4 and concentrated under vacuum to give the crude product, which was purified by column chromatography using a gradient mixture of ethyl acetate/hexane (2:8) as the eluent to afford the product 3a.
All other reactions were carried out according to the general procedure for the synthesis of 3a to 3s. The racemic samples were prepared by the following general procedure, using quinine and quinidine (1:1) as catalysts instead of thiourea 4c. In the absence of quinine and quinidine, the racemic reaction was very sluggish and takes more than 5 days for the completion with low yield.
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(phenyl)methyl)-3-oxoindoline-2-carboxylate (3a)
(0.084 g, 92% yield) white solid. Mp 130–132 °C. IR (neat) ν 3409.7, 3007.4, 2978.2, 1763.6, 1706.2, 1476.0, 1339.9, 1291.4, 1164.4, 1048.1, 770.8, 693.3 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.2 Hz, 1H), 7.50–7.41 (m, 1H), 7.10 (t, J = 7.5 Hz, 1H), 6.98 (s, 6H), 6.70 (s, 1H), 5.97 (s, 1H), 3.76 (s, 3H), 2.38 (s, 3H), 1.45 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 194.5, 167.8, 165.2, 155.2, 137.8, 127.8, 127.6, 126.9, 126.6, 124.8, 124.0, 114.5, 79.5, 74.4, 55.9, 53.4, 28.4, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C24H26N2O6Na [M + Na]+: 461.1683. Found: 461.1696. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (7.23 min), minor (8.50 min), ee: 97.25; [α]D20 −40.2 (c = 0.15, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-(2-bromophenyl)((tert-butoxycarbonyl)amino)methyl)-3-oxoindoline-2-carboxylate (3b)
(0.106 g, 96% yield) white solid. Mp 130–132 °C. IR (neat) ν 3415.7, 3007.5, 2978.3, 1765.0, 1706.8, 1607.1, 1499.7, 1470.3, 1333.3, 1266.2, 1166.0, 971.1, 770.9, 609.2 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 5.6 Hz, 1H), 7.47 (t, J = 7.4 Hz, 1H), 7.32 (s, 1H), 7.2–6.9 (m, 3H), 6.83 (s, 2H), 6.63 (s, 1H), 6.30 (s, 1H), 3.76 (s, 3H), 2.47 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.9, 168.2, 165.4, 155.0, 152.7, 137.6, 132.6, 129.3, 129.1, 126.5, 124.9, 124.2, 114.9, 79.7, 74.3, 55.3, 53.2, 28.4, 25.7. HRMS (Orbitrap ESI): exact mass calcd for C24H25N2O6BrNa [M + Na]+: 539.0788. Found: 539.0810. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 85:15, 1 mL/min, minor (11.55 min), ee: 96.83; [α]D20 −88 (c = 0.15, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-3-oxoindoline-2-carboxylate (3c)
(0.099 g, 98% yield) white solid. Mp 130–132 °C. IR (neat) ν 3411.1, 3011.5, 2978.1, 1761.6, 1716.1, 1606.7, 1500.1, 1434.3, 1374.2, 1334.2, 1265.9, 1165.6, 761.5, 614.5 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.5 Hz, 1H), 7.53–7.48 (m, 1H), 7.14 (t, J = 7.5 Hz, 1H), 7.02–6.84 (m, 5H), 6.69 (s, 1H), 5.94 (s, 1H), 3.76 (s, 3H), 2.41 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz,) δ 194.1, 167.8, 165.0, 155.2, 152.0, 139.5, 138.0, 133.6, 128.9, 128.0, 127.3, 125.4, 125.0, 124.2, 114.5, 79.8, 74.0, 55.4, 53.4, 28.4, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C24H25N2O6ClNa [M + Na]+: 495.1293. Found: 495.1312. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (5.58 min), minor (8.8 min), ee: 98.90; [α]D20 −61 (c = 0.5, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(4-fluorophenyl)methyl)-3-oxoindoline-2-carboxylate (3d)
(0.086 g, 88% yield) white solid. Mp 135–137 °C. IR (neat) ν 3410.5, 3010.5, 2979.8, 1764.5, 1707.2, 1606.5, 1471.9, 1371.4, 1324.3, 1238.3, 1164.1, 1065.5, 752.8, 619.1 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.66 (d, J = 5.6 Hz, 1H), 7.47 (t, J = 7.4 Hz, 1H), 7.32 (s, 1H), 7.08–6.91 (m, 3H), 6.83 (s, 2H), 6.63 (s, 1H), 6.30 (s, 1H), 3.76 (s, 3H), 2.47 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.5, 167.8, 165.3, 155.2, 140.6, 140.4, 137.8, 132.9, 130.3, 128.7, 127.4, 127.3, 126.8, 126.3, 124.8, 124.0, 114.7, 79.6, 55.7, 53.4, 28.4, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C24H25N2O6FNa [M + Na]+: 479.1588. Found: 479.1605. HPLC analysis (DAICEL Chiralpak IC), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm major (6.24 min), minor (9.40 min), 96.97; [α]D20 −74 (c = 0.1, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(4-chloro-2-fluorophenyl)methyl)-3-oxoindoline-2-carboxylate (3e)
(0.096 g, 92% yield) white solid. Mp 170–172 °C. IR (neat) ν 3410.7, 3032.4, 2956.5, 1760.4, 1728.9, 1684.5, 1606.2, 1463.5, 1376.2, 1339.6, 1297.6, 1155.0, 1093.9, 759.9, 647.7 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.73 (d, J = 7.4 Hz, 1H), 7.54–7.46 (m, 1H), 7.14 (t, J = 7.5 Hz, 1H), 7.00–6.84 (m, 4H), 6.69 (s, 1H), 5.95 (s, 1H), 3.76 (s, 3H), 2.41 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.5, 191.9, 167.9, 165.3, 155.2, 140.6, 140.4, 137.8, 128.7, 127.4, 126.8, 126.3, 124.8, 124.0, 114.7, 79.6, 74.4, 55.7, 53.4, 28.4, 25.7. HRMS (Orbitrap ESI): exact mass calcd for C24H24ClFN2O6Na [M + Na]+: 513.1198. Found: 513.1225. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (5.02 min), minor (6.83 min), ee: 88.65; [α]D20 −34.2 (c = 0.15, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(p-tolyl)methyl)-3-oxoindoline-2-carboxylate (3f)
(0.077 g, 80% yield) white solid. Mp 147–149 °C. IR (neat) ν 3399.8, 3021.3, 2969.8, 1735.6, 1710.6, 1680.3, 1585.6, 1465.8, 1369.7, 1322.5, 1165.6, 1029.5, 765.6, 660.7 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 6.6 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.11 (t, J = 7.4 Hz, 1H), 6.81–6.74 (m, 5H), 6.66 (s, 1H), 5.93 (s, 1H), 3.76 (s, 3H), 2.38 (s, 3H), 2.12 (s, 3H), 1.45 (s, 9H). 13C NMR (101 MHz) δ 194.6, 167.8, 165.3, 155.1, 152.1, 137.7, 137.4, 134.2, 128.3, 126.8, 124.8, 123.9, 114.5, 79.4, 74.5, 55.6, 53.3, 28.4, 25.6, 20.9. HRMS (Orbitrap ESI): exact mass calcd for C25H28N2O6Na [M + Na]+: 475.1839. Found: 475.1850. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (4.88 min), minor (6.09 min), ee: 66.97; [α]D20 −49 (c = 0.15, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(3,4-dimethoxyphenyl)methyl)-3-oxoindoline-2-carboxylate (3g)
(0.102 g, 96% yield) white solid. Mp 130–132 °C. IR (neat) ν 3411.5, 2956.9, 2924.1, 1785.7, 1727.3, 1689.1, 1589.4, 1464.7, 1379.9, 1345.6, 1224.7, 1155.1, 770.9 cm–1. 1H NMR (500 MHz, CDCl3) 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 7.3 Hz, 1H), 7.51 (ddd, J = 8.6, 7.3, 1.4 Hz, 1H), 7.14 (t, J = 7.5 Hz, 2H), 6.67 (d, J = 5.3 Hz, 1H), 6.14–6.09 (m, 3H), 5.92 (s, 1H), 3.76 (s, 3H), 3.55 (s, 6H), 2.39 (s, 3H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 13C NMR (101 MHz, 3>) δ 194.6, 191.9, 167.7, 165.2, 160.1, 155.2, 152.1, 139.6, 137.9, 124.7, 124.0, 114.7, 107.1, 105.0, 100.4, 79.6, 74.2, 56.10, 55.0, 53.4, 28.4, 28.2, 25.7. HRMS (Orbitrap ESI): exact mass calcd for C26H30N2O8Na [M + Na]+: 521.1894. Found: 521.1910. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (6.67 min), minor (11.68 min), ee: 82.73; [α]D20 −17 (c = 0.1, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(4-nitrophenyl)methyl)-3-oxoindoline-2-carboxylate (3h)
(0.091 g, 88% yield) yellow solid. Mp 158–160 °C. IR (neat) ν 3409.5, 3079.5, 2979.0, 1763.8, 1708.0, 1606.1, 1523.3, 1498.6, 1471.6, 1344.8, 1237.7, 1163.4, 1027.7, 752.3, 611.6 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.88 (d, J = 8.7 Hz, 2H), 7.74 (d, J = 7.6 Hz, 1H), 7.54–7.49 (m, 1H), 7.18 (m, 4H), 6.81 (s, 1H), 6.08 (d, J = 4.9 Hz, 1H), 3.77 (s, 3H), 2.42 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 193.7, 168.0, 164.7, 155.2, 151.8, 147.4, 145.1, 138.4, 128.0, 125.2, 124.6, 124.3, 122.9, 114.7, 80.1, 73.7, 55.7, 53.6, 28.3, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C24H25N3O8Na [M + Na]+: 506.1533. Found: 506.1553. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm major (12.60 min), minor (19.34 min), ee: 95.63; [α]D20 −57.5 (c = 0.5, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(2-nitrophenyl)methyl)-3-oxoindoline-2-carboxylate (3i)
(0.093 g, 90% yield) yellow solid. Mp 160–162 °C. IR (neat) ν 3409.9, 3076.2, 2978.1, 1764.6, 1706.8, 1606.5, 1525.1, 1499.5, 1470.1, 1343.2, 1235.2, 1165.2, 1022.6, 759.3, 615.2 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.60–7.49 (m, 3H), 7.41 (s, 1H), 7.21–7.10 (m, 4H), 6.65 (s, 2H), 3.73 (s, 3H), 2.43 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.0, 168.45, 165.2, 155.1, 152.2, 149.5, 137.6, 131.7, 131.4, 130.0, 128.6, 124.8, 124.5, 124.1, 116.1, 80.1, 7.6, 53.4, 52.0, 28.3, 24.7. HRMS (Orbitrap ESI): exact mass calcd for C24H25N3O8Na [M + Na]+: 506.1533. Found: 506.1551. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (10.19 min), minor (12.78 min), ee: 91.21; [α]D20 −52.3 (c = 0.15, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(4-cyanophenyl)methyl)-3-oxoindoline-2-carboxylate (3j)
(0.087 g, 90% yield) yellow solid. Mp 156–158 °C. IR (neat) ν 3401.6, 3009.8, 2978.9, 2230.1, 1763.7, 1707.8, 1676.5, 1606.3, 1499.8, 1417.3, 1339.5, 1239.2, 1164.2, 1025.6, 754.7, 615.6 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.5 Hz, 1H), 7.54 (ddd, J = 8.6, 7.4, 1.4 Hz, 1H), 7.31 (d, J = 8.2 Hz, 2H), 7.20–7.09 (m, 4H), 6.77 (s, 1H), 6.01 (s, 1H), 3.76 (s, 3H), 2.40 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 193.8, 168.0, 164.8, 155.2, 151.9, 143.0, 138.4, 131.5, 127.8, 125.1, 124.5, 118.3, 114.7, 111.7, 80.0, 73.7, 55.8, 53.6, 28.3, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C25H25N3O6Na [M + Na]+: 486.1635. Found: 486.1645. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (12.27 min), minor (16.12 min), ee: 97.63; [α]D20 −68.5 (c = 0.5, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(2-cyanophenyl)methyl)-3-oxoindoline-2-carboxylate (3k)
(0.091 g, 90% yield) yellow solid. Mp 162–164 °C. IR (neat) ν 3404.0, 3008.5, 2978.5, 2230.8, 1763.7, 1708.6, 1677.2, 1606.6, 1472.2, 1340.1, 1241.1, 1165.4, 1025.9, 758.2, 616.5 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.6 Hz, 1H), 7.54 (ddd, J = 8.6, 7.3, 1.4 Hz, 1H), 7.31 (d, J = 8.3 Hz, 2H), 7.20–7.08 (m, 4H), 6.77 (s, 1H), 6.01 (s, 1H), 3.76 (s, 3H), 2.40 (s, 3H), 1.44 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 193.7, 168.0, 164.7, 155.2, 151.9, 143.1, 138.4, 131.5, 127.8, 125.1, 124.5, 118.3, 114.7, 111.7, 80.0, 73.7, 55.8, 53.6, 28.3, 25.6. HRMS (Orbitrap ESI): exact mass calcd for C25H25N3O6Na [M + Na]+: 486.1635. Found: 486.1640. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (9.99 min), minor (10.98 min), ee: 97.76; [α]D20 −79.8 (c = 0.5, CHCl3).
Methyl (R)-2-((S)-[1,1′-Biphenyl]-4-yl((tert-butoxycarbonyl)amino)methyl)-1-acetyl-3-oxoindoline-2-carboxylate (3l)
(0.095 g, 85% yield) yellow solid. Mp 118–120 °C. IR (neat) ν 3403.5, 3009.2, 2924.3, 1762.3, 1729.9, 1682.6, 1606.6, 1463.2, 1375.3, 1337.6, 1297.4, 1259.5, 1155.0, 1126.5, 750.4, 698.6 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.72 (d, J = 6.4 Hz, 1H), 7.46–7.41 (m, 1H), 7.38–7.34 (m, 4H), 7.30 (dd, J = 6.1, 2.4 Hz, 1H), 7.22 (d, J = 8.0 Hz, 2H), 7.13–7.01 (m, 4H), 6.73 (s, 1H), 6.03 (s, 1H), 3.77 (s, 3H), 2.40 (s, 3H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.5, 191.9, 167.8, 165.3, 155.2, 140.6, 140.4, 137.8, 130.3, 128.7, 127.4, 127.3, 126.8, 126.3, 124.9, 124.8, 124.0, 114.7, 101.5, 79.6, 55.7, 53.46, 28.4, 25.7. HRMS (Orbitrap ESI): exact mass calcd for C30H30N2O6Na [M + Na]+: 537.1996. Found: 537.2018. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (9.46 min), minor (14.22 min), ee: 83.25; [α]D20 −48.6 (c = 0.2, CHCl3).
Methyl (R)-1-Acetyl-2-((R)-(4-bromothiophen-2-yl)((tert-butoxycarbonyl)amino)methyl)-3-oxoindoline-2-carboxylate (3m)
(0.106 g, 95% yield) yellow solid. Mp 118–120 °C. IR (neat) ν 3441.2, 3015.3, 2976.5, 1771.5, 1730.6, 1690.5, 1602.9, 1499.8, 1383.8, 1334.6, 1270.4, 1167.4, 1098.6, 761.9, 650.6 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.73 (d, J = 7.5 Hz, 1H), 7.62 (t, J = 7.5 Hz, 1H), 7.39 (s, 1H), 7.18 (t, J = 7.5 Hz, 1H), 6.84 (s, 1H), 6.66 (d, J = 9.9 Hz, 2H), 6.22 (d, J = 5.5 Hz, 1H), 3.75 (s, 3H), 2.49 (s, 3H), 1.48 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 193.8, 167.9, 164.7, 155.0, 152.7, 142.2, 138.2, 128.6, 125.5, 125.2, 124.4, 122.1, 114.9, 108.6, 80.1, 74.0, 53.5, 51.9, 28.3, 25.7. HRMS (Orbitrap ESI): exact mass calcd for C22H23BrN2O6SNa [M + Na]+: 545.0340. Found: 545.0368. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (8.66 min), minor (10.14 min), ee: 98.71; [α]D20 −80.5 (c = 0.15, CHCl3).
Methyl (R)-1-Benzoyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-3-oxoindoline-2-carboxylate (3n)
(0.086 g, 96% yield) yellow solid. Mp 172–174 °C. IR (neat) ν 3417.7, 3009.8, 2978.1, 1762.2, 1710.0, 1607.3, 1500.8, 1471.1, 1338.6, 1235.4, 1166.2, 1048.1, 752.5, 611.6 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.70 (d, J = 7.5 Hz, 1H), 7.55 (t, J = 7.5 Hz, 1H), 7.42 (t, J = 7.9 Hz, 2H), 7.24–6.79 (m, 9H), 6.12 (s, 1H), 5.80 (d, J = 8.4 Hz, 1H), 3.82 (s, 3H), 1.47 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.2, 167.8, 164.9, 155.2, 153.0, 139.9, 137.1, 134.6, 133.9, 131.8, 129.3, 129.1, 128.1, 127.8, 127.4, 125.3, 124.6, 124.3, 124.1, 115.6, 79.7, 74.2, 55.7, 53.6, 28.4. HRMS (Orbitrap ESI): exact mass calcd for C29H27ClN2O6Na [M + Na]+: 557.1449. Found: 557.1428. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (11.54 min), minor (15.97 min), ee: 80.79; [α]D20 −45 (c = 0.7, CHCl3).
Methyl (R)-1-Acetyl-5-bromo-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-3-oxoindoline-2-carboxylate (3o)
(0.070 g, 80% yield) yellow solid. Mp 168–170 °C. IR (neat) ν 3412.0, 3073.3, 2922.5, 1759.5, 1730.5, 1695.3, 1574.7, 1429.8, 1340.1, 1260.5, 1142.6, 1074.1, 771.9, 667.9 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.82 (s, 1H), 7.58 (dd, J = 8.9, 2.1 Hz, 1H), 6.99 (dt, J = 11.1, 8.1 Hz, 4H), 6.86 (d, J = 7.6 Hz, 1H), 6.59 (s, 1H), 5.93 (s, 1H), 3.76 (s, 3H), 2.39 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 192.9, 167.7, 164.5, 155.1, 150.9, 140.5, 139.2, 134.4, 133.8, 130.4, 128.2, 127.4, 125.0, 117.3, 116.2, 80.0, 74.5, 55.5, 53.6, 28.3, 25.5. HRMS (Orbitrap ESI): exact mass calcd for C24H24BrClN2O6Na [M + Na]+: 573.0398. Found: 573.0423. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm major (6.57 min), minor (7.35 min), ee: 40; [α]D20 −148 (c = 1.6, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-5-methyl-3-oxoindoline-2-carboxylate (3p)
(0.090 g, 92% yield) yellow solid. Mp 147–149 °C. IR (neat) ν 3402.6, 3073.3, 2925.5, 1753.6, 1721.5, 1681.1, 1586.5, 1488.5, 1378.9, 1344.7, 1230.6, 1123.6, 1069.3, 770.4, 640.7 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.51 (s, 1H), 7.31 (d, J = 8.1 Hz, 1H), 7.08–6.83 (m, 5H), 6.72 (s, 1H), 5.94 (s, 1H), 3.75 (s, 3H), 2.38 (s, 3H), 2.33 (s, 3H), 1.46 (s, 9H). 13C NMR (126 MHz, CDCl3) δ 194.1, 167.7, 164.8, 155.2, 150.2, 139.5, 139.2, 134.4, 133.5, 128.9, 127.9, 127.3, 125.1, 124.6, 114.3, 79.7, 74.1, 55.42, 53.4, 28.4, 25.5, 20.4. HRMS (Orbitrap ESI): exact mass calcd for C25H27ClN2O6Na [M + Na]+: 509.1449. Found: 509.1471. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm major (8.03 min), minor (11.46 min), ee: 90.47; [α]D20 −103 (c = 0.55, CHCl3).
Methyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-5-chloro-3-oxoindoline-2-carboxylate (3q)
(0.080 g, 85% yield) yellow solid. Mp 166–168 °C. IR (neat) ν 3410.5, 3007.8, 2978.3, 1765.6, 1710.2, 1680.3, 1501.1, 1470.8, 1371.1, 1337.1, 1290.1, 1166.3, 1049.2, 765.0, 686.9 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.66 (s, 1H), 7.44 (dd, J = 8.9, 2.3 Hz, 1H), 7.13–6.91 (m, 4H), 6.86 (d, J = 7.6 Hz, 1H), 6.59 (s, 1H), 5.92 (s, 1H), 3.76 (s, 3H), 2.40 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 193.0, 167.7, 164.8, 155.3, 150.6, 139.1, 137.0, 133.1, 130.7, 129.8, 128.3, 127.8, 125.6, 124.9, 124.2, 115.5, 79.7, 74.5, 55.0, 53.1, 28.8, 25.4. HRMS (Orbitrap ESI): exact mass calcd for C24H24Cl2N2O6Na [M + Na]+: 529.0898. Found: 529.0930. HPLC analysis (DAICEL Chiralpak OD-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (6.08 min), minor (7.19 min), ee: 57.6; [α]D20 −110 (c = 0.15, CHCl3).
Methyl (R)-4-Acetyl-5-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-6-oxo-5,6-dihydro-4H-thieno[3,2-b]pyrrole-5-carboxylate (3r)
(0.092 g, 92% yield) yellow solid. Mp 178–180 °C. IR (neat) ν 3394.7, 3009.9, 2978.5, 1761.5, 1685.5, 1512.2, 1454.9, 1371.6, 1323.3, 1235.9, 1129.7, 989.5, 752.6, 582.4 cm–1. 1H NMR (400 MHz, CDCl3) δ 7.89 (d, J = 5.1 Hz, 1H), 7.09–6.96 (m, 4H), 6.84 (d, J = 6.5 Hz, 1H), 6.67 (d, J = 5.0 Hz, 1H), 5.95 (d, J = 6.4 Hz, 1H), 3.79 (s, 3H), 2.29 (s, 3H), 1.46 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 184.2, 165.8, 164.4, 163.6, 155.2, 145.5, 139.5, 133.7, 128.9, 128.1, 127.3, 125.2, 122.8, 114.9, 79.7, 79.5, 55.3, 53.5, 28.4, 24.0. HRMS (Orbitrap ESI): exact mass calcd for C22H23ClN2O6SNa [M + Na]+: 501.0881. Found: 501.0880. HPLC analysis (DAICEL Chiralpak AS-H), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (9.71 min), minor (11.68 min), ee: 96.97; [α]D20 −205 (c = 0.55, CHCl3).
Methyl (R)-1-Benzoyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-3-oxo-2,3-dihydro-1H-benzo[f]indole-2-carboxylate (3s)
(0.097 g, 94% yield) yellow solid. Mp 148–150 °C. IR (neat) ν 3411.9, 3011.5, 2979.6, 1776.7, 1740.5, 1690.8, 1610.5, 1501.6, 1465.7, 1465.7, 1395.6, 1335.7, 1260.5, 1225.6, 1166.5, 1096.4, 776.1, 649.6 cm–1. 1H NMR (500 MHz, CDCl3) δ 8.30 (s, 1H), 7.85 (d, J = 8.1 Hz, 1H), 7.60 (t, J = 7.5 Hz, 1H), 7.53–7.28 (m, 6H), 7.20–7.15 (m, 2H), 7.04–6.96 (m, 4H), 6.18 (s, 1H), 6.04 (s, 1H), 3.81 (s, 3H), 1.49 (s, 9H). 13C NMR (101 MHz, CDCl3) δ 194.7, 168.4, 165.2, 155.2, 145.6, 140.1, 137.8, 134.8, 134.0, 131.7, 130.3, 130.3, 129.4, 129.0, 128.1, 128.0, 127.9, 127.5, 126.5, 126.1, 125.2, 124.0, 112.3, 79.7, 74.5, 55.6, 53.6, 28.4. HRMS (Orbitrap ESI): exact mass calcd for C33H29ClN2O6Na [M + Na]+: 607.1606. Found: 607.1631. HPLC analysis (DAICEL Chiralpak IA), n-hexane/2-PrOH = 80:20, 1 mL/min, 254 nm, major (6.80 min), minor (7.43 min), ee: 90.27; [α]D20 −150 (c = 0.15, CHCl3).
Ethyl (R)-1-Acetyl-2-((S)-((tert-butoxycarbonyl)amino)(3-chlorophenyl)methyl)-6-chloro-3-oxoindoline-2-carboxylate (3t)
(0.070 g, 76% yield) pale yellow solid. Mp 135–137 °C. IR (neat) ν 3309.8, 3006.6, 2968.2, 1765.3, 1711.5, 1688.2, 1502.1, 1471.3, 1372.0, 1337.2, 1291.1, 1164.2, 1049.1, 765.2, 682.8 cm–1. 1H NMR (500 MHz, CDCl3) δ 7.65 (d, J = 8.1 Hz, 1H), 7.11 (dd, J = 8.3, 1.4 Hz, 1H), 7.05 (d, J = 7.7 Hz, 1H), 7.00–6.95 (m, 2H), 6.88 (d, J = 7.6 Hz, 1H), 6.67 (s, 1H), 5.91 (s, 1H), 4.40–4.12 (m, 2H), 2.40 (s, 3H), 1.45 (s, 9H), 1.24 (t, J = 7.1 Hz, 3H). 13C NMR (101 MHz, CDCl3) δ 193.0, 167.7, 164.2, 155.1, 144.7, 133.8, 129.1, 128.2, 127.0, 125.6, 125.0, 114.8, 79.9, 74.7, 62.9, 55.5, 28.4, 25.5, 13.9. HRMS (Orbitrap ESI): exact mass calcd for C25H26Cl2N2O6Na [M + Na]+: 543.1055. Found: 543.1049. HPLC analysis (DAICEL Chiralpak IA), n-hexane/2-PrOH = 85:15, 1 mL/min, 254 nm, major (24.55 min), minor (21.23 min), ee: 90.00; [α]D20 −38 (c = 0.3, CHCl3).
Acknowledgments
G.R.K. thanks UGC, New Delhi, for the award of a fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02132.
The authors declare no competing financial interest.
Supplementary Material
References
- a Weiner B.; Szymański W.; Janssen D. B.; Minnaard A. J.; Feringa B. L. Recent advances in the catalytic asymmetric synthesis of β-amino acids. Chem. Soc. Rev. 2010, 39, 1656. 10.1039/b919599h. [DOI] [PubMed] [Google Scholar]; b Ashfaq M.; Tabassum R.; Ahmad M. M.; Hassan N. A.; Oku H.; Rivera G. Enantioselective synthesis of β-amino acids: A review. Med. Chem. 2015, 5, 7. 10.4172/2161-0444.1000278. [DOI] [Google Scholar]; c Li L.; Song B.-A.; Bhadury P. S.; Zhang Y.-P.; Hu D.-Y.; Yang S. Enantioselective synthesis of β-amino esters bearing a benzothiazole moiety via a Mannich-type reaction catalyzed by a Cinchona alkaloid derivative. Eur. J. Org. Chem. 2011, 4743. 10.1002/ejoc.201100351. [DOI] [Google Scholar]; d Josephson K.; Hartman M. C. T.; Szostak J. W. Ribosomal synthesis of unnatural peptides. J. Am. Chem. Soc. 2005, 127, 11727. 10.1021/ja0515809. [DOI] [PubMed] [Google Scholar]; e Cabrele C.; Martinek T. A.; Reiserg O.; Berlicki L. Peptides containing β-amino acid patterns: Challenges and successes in medicinal chemistry. J. Med. Chem. 2014, 57, 9718. 10.1021/jm5010896. [DOI] [PubMed] [Google Scholar]
- a Scott W. L.; Martynow J. G.; Huffman J. C.; O’Donnell M. J. Solid-phase synthesis of multiple classes of peptidomimetics from versatile resin-bound aldehyde intermediates. J. Am. Chem. Soc. 2007, 129, 7077. 10.1021/ja069188y. [DOI] [PubMed] [Google Scholar]; b Elashal H. E.; Sim Y. E.; Raj M. Serine promoted synthesis of peptide thioester-precursor on solid support for native chemical ligation. Chem. Sci. 2017, 8, 117. 10.1039/C6SC02162J. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kamath A.; Ojima I. Advances in the chemistry of β-lactam and its medicinal applications. Tetrahedron 2012, 68, 10640. 10.1016/j.tet.2012.07.090. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jamieson A. G.; Boutard N.; Sabatino D.; Lubell W. D. Peptide scanning for studying structure-activity relationships in drug discovery. Chem. Biol. Drug Des. 2013, 81, 148. 10.1111/cbdd.12042. [DOI] [PubMed] [Google Scholar]
- a The Chemistry of Indoles; Sundberg R. J., Ed.; Academic press: New York, 1996. [Google Scholar]; b Yap W.-S.; Gan C.-Y.; Low Y.-Y.; Choo Y.-M.; Etoh T.; Hayashi M.; Komiyama K.; Kam T.-S. Grandilodines A-C, biologically active indole alkaloids from Kopsiagrandifolia. J. Nat. Prod. 2011, 74, 1309. 10.1021/np200008g. [DOI] [PubMed] [Google Scholar]; c Umehara A.; Ueda H.; Tokuyama H. Total syntheses of Leuconoxine, Leuconodine B, and Melodinine E by oxidative cyclic aminal formation and diastereoselective ring-closing metathesis. Org. Lett. 2014, 16, 2526. 10.1021/ol500903e. [DOI] [PubMed] [Google Scholar]; d Bass P. D.; Gubler D. A.; Judd T. C.; Williams R. M. Mitomycinoid alkaloids: Mechanism of action, biosynthesis, total syntheses, and synthetic approaches. Chem. Rev. 2013, 113, 6816. 10.1021/cr3001059. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Lim K.-H.; Kam T.-S. New indole alkaloids from Kopsia. Alkaloid variation in Kopsiasingapurensis. Helv. Chim. Acta 2007, 90, 31. 10.1002/hlca.200790018. [DOI] [Google Scholar]; f Xu Z.; Wang Q.; Zhu J. Total syntheses of (−)-Mersicarpine, (−)-Scholarisine G, (+)-Melodinine E, (−)-Leuconoxine, (−)-Leuconolam, (−)-Leuconodine A, (+)-Leuconodine F, and (−)-Leuconodine C: Self-induced diastereomeric anisochronism (SIDA) phenomenon for Scholarisine G and Leuconodines A and C. J. Am. Chem. Soc. 2015, 137, 6712. 10.1021/jacs.5b03619. [DOI] [PubMed] [Google Scholar]; g Steven A.; Overman L. E. Total synthesis of complex cyclotryptamine alkaloids: Stereocontrolled construction of quaternary carbon stereocenters. Angew. Chem., Int. Ed. 2007, 46, 5488. 10.1002/anie.200700612. [DOI] [PubMed] [Google Scholar]; h Pearson W. H.; Mi Y.; Lee I. Y.; Stoy P. Total synthesis of the Kopsialapidilecta alkaloid (±)-Lapidilectine B. J. Am. Chem. Soc. 2001, 123, 6724. 10.1021/ja016007d. [DOI] [PubMed] [Google Scholar]; i Guengerich F. P.; Sorrells J. L.; Schmitt S.; Krauser J. A.; Aryal P.; Meijer L. Generation of new protein kinase inhibitors utilizing cytochrome p450 mutant enzymes for indigoid synthesis. J. Med. Chem. 2004, 47, 3236. 10.1021/jm030561b. [DOI] [PubMed] [Google Scholar]; j Petkovic M.; Nasufovic V.; Djukanovic D.; Vujosevic Z. T.; Jadranin M.; Matovic R.; Savic V. Cyclative cascades of allenamides derived from amino acids: Synthesis of annulated indoxyl derivatives. Eur. J. Org. Chem. 2016, 1279. 10.1002/ejoc.201600067. [DOI] [Google Scholar]; k Patel P.; Reddy B. N.; Ramana C. V. The synthesis of the central tricyclic core of the isatisine A: Harmonious orchestration of [metal]-catalyzed reactions in a sequence. Tetrahedron 2014, 70, 510. 10.1016/j.tet.2013.11.026. [DOI] [Google Scholar]
- a Tan C.-J.; Di Y.-T.; Wang Y.-H.; Zhang Y.; Si Y.-K.; Zhang Q.; Gao S.; Hu X.-J.; Fang X.; Li S.-F.; Hao X.-J. Three new indole alkaloids from Trigonostemon lii. Org. Lett. 2010, 12, 2370. 10.1021/ol100715x. [DOI] [PubMed] [Google Scholar]; b Han S.; Movassaghi M. Concise total synthesis and stereochemical revision of all (−)-trigonoliimines. J. Am. Chem. Soc. 2011, 133, 10768. 10.1021/ja204597k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Qi X.; Bao H.; Tambar U. K. Total synthesis of (±)-Trigonoliimine C via oxidative rearrangement of an unsymmetrical bis-tryptamine. J. Am. Chem. Soc. 2011, 133, 10050. 10.1021/ja203960b. [DOI] [PubMed] [Google Scholar]; d Reddy B. N.; Ramana C. V. A modular total synthesis of (±)-trigonoliimine C. Chem. Commun. 2013, 49, 9767. 10.1039/c3cc45512b. [DOI] [PubMed] [Google Scholar]; e Han S.; Morrison K. C.; Hergenrother P. J.; Movassaghi M. Total synthesis, stereochemical assignment, and biological activity of all known (−)-trigonoliimines. J. Org. Chem. 2014, 79, 473. 10.1021/jo4020358. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Liu S.; Hao X.-J. A biomimetic synthesis of the skeleton of trigonoliimine C. Tetrahedron Lett. 2011, 52, 5640. 10.1016/j.tetlet.2011.08.087. [DOI] [Google Scholar]
- a Wu P.-L.; Hsu Y.-L.; Jao C.-W. Indole alkaloids from Cephalanceropsisgracilis. J. Nat. Prod. 2006, 69, 1467. 10.1021/np060395l. [DOI] [PubMed] [Google Scholar]; b Dend Z.; Peng X.; Huang P.; Jiang L.; Ye D.; Liu L. A multi-functionalized strategy of indoles to C2-quaternary indolin-3-ones via a TEMPO/Pd-catalyzed cascade process. Org. Biomol. Chem. 2017, 15, 442. 10.1039/C6OB02285E. [DOI] [PubMed] [Google Scholar]; c Blunt J. W.; Copp B. R.; Keyzers R. A.; Munro M. H. G.; Prinsep M. R. Marine natural products. Nat. Prod. Rep. 2014, 31, 160. 10.1039/c3np70117d. [DOI] [PubMed] [Google Scholar]; d Baran P. S.; Corey E. J. A short synthetic route to (+)-austamide, (+)-deoxyisoaustamide, and (+)-hydratoaustamide from a common precursor by a novel palladium-mediated indole → dihydroindoloazocine Cyclization. J. Am. Chem. Soc. 2002, 124, 7904. 10.1021/ja026663t. [DOI] [PubMed] [Google Scholar]
- a Liu J.-F.; Jiang Z.-Y.; Wang R.-R.; Chen Y.-T.; Zheng J.-J.; Zhang X.-M.; Ma Y.-B. Isatisine A, a novel alkaloid with an unprecedented skeleton from leaves of Isatisindigotica. Org. Lett. 2007, 9, 4127. 10.1021/ol701540y. [DOI] [PubMed] [Google Scholar]; b Karadeolian A.; Kerr M. A. Total synthesis of (+)-isatisine A. Angew. Chem., Int. Ed. 2010, 49, 1133. 10.1002/anie.200906632. [DOI] [PubMed] [Google Scholar]; c Lee J.; Panek J. S.Total synthesis of the Hsp90 inhibitor Geldanamycin. Org. Lett. 2011, 13, 502. 10.1021/ol102848w. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Kumar C. V. S.; Puranik V. G.; Ramana C. V. InCl3-mediated addition of indole to isatogens: An expeditious synthesis of 13-deoxy-isatisine A. Chem. - Eur. J. 2012, 18, 9601. 10.1002/chem.201103604. [DOI] [PubMed] [Google Scholar]; e Gu W.; Zhang Y.; Hao X.-J.; Yang F. M.; Sun Q.-Y.; Morris-Natschke S. L.; Lee K.-H.; Wang Y.-H.; Long C.-L. Indole alkaloid glycosides from the aerial parts of Strobilanthescusia. J. Nat. Prod. 2014, 77, 2590. 10.1021/np5003274. [DOI] [PubMed] [Google Scholar]
- a Chen S.; Wang Y.; Zhou Z. Organo-catalyzed asymmetric Michael addition of 1-acetylindolin-3-ones to β,γ-unsaturated α-ketoesters: An access to chiral indolin-3-ones with two adjacent tertiary stereogenic centers. J. Org. Chem. 2016, 81, 11432. 10.1021/acs.joc.6b02070. [DOI] [PubMed] [Google Scholar]; b Chen T.-G.; Fang P.; Hou X.-L.; Dai L.-X. Palladium-Catalyzed asymmetric allylic alkylation reaction of 2-monosubstituted indolin-3-ones. Synthesis 2015, 47, 134. 10.1055/s-0034-1379043. [DOI] [Google Scholar]; c Yin Q.; You S.-L. Chiral phosphoric acid-catalysed Friedel–Crafts alkylation reaction of indoles with racemic spiro-indolin-3-ones. Chem. Sci. 2011, 2, 1344. 10.1039/c1sc00190f. [DOI] [Google Scholar]; d Guo J.; Lin Z.-H.; Chen K.-B.; Xie Y.; Chen A. S. C.; Weng J.; Lui G. Asymmetric amination of 2-substituted indolin-3-ones catalyzed by natural cinchona alkaloids. Org. Chem. Front. 2017, 4, 1400. 10.1039/C7QO00129K. [DOI] [Google Scholar]; e Liu Y.-Z.; Zhang J.; Xu P.-F.; Luo Y.-C. Organocatalytic asymmetric Michael addition of 1-acetylindolin-3-ones to α,β-unsaturated aldehydes: Synthesis of 2-substituted indolin-3-ones. J. Org. Chem. 2011, 76, 7551. 10.1021/jo201123p. [DOI] [PubMed] [Google Scholar]; f Jin C.-Y.; Wang Y.; Liu Y.-Z.; Shen C.; Xu P.-F. Organocatalytic asymmetric Michael addition of oxindoles to nitroolefins for the synthesis of 2,2-disubstituted oxindoles bearing adjacent quaternary and tertiary stereocenters. J. Org. Chem. 2012, 77, 11307. 10.1021/jo301886j. [DOI] [PubMed] [Google Scholar]; g Zhao Y.-L.; Wang Y.; Cao J.; Liang Y.-M.; Xu P.-F. Organocatalytic asymmetric Michael-Michael cascade for the construction of highly functionalized N-fused piperidino-indoline derivatives. Org. Lett. 2014, 16, 2438. 10.1021/ol5008185. [DOI] [PubMed] [Google Scholar]; h Mahajan S.; Chauhan P.; Loh C. C. J.; Uzungelis S.; Raabe G.; Enders D. Organocatalytic asymmetric domino Michael/Henry reaction of indolin-3-ones with O-formyl-β-nitrostyrenes. Synthesis 2015, 47, 1024. 10.1055/s-0034-1379943. [DOI] [PMC free article] [PubMed] [Google Scholar]; i Sun W.; Hong L.; Wang R. Chem. - Eur. J. 2011, 17, 6030. 10.1002/chem.201100144. [DOI] [PubMed] [Google Scholar]; j Ni Q.; Raabe G.; Enders D.; Song X. N-Heterocyclic carbene-catalyzed enantioselective annulation of indolin-3-ones with bromoenals. Chem. - Asian J. 2014, 9, 1535. 10.1002/asia.201402052. [DOI] [PubMed] [Google Scholar]; k Rueping M.; Raja S.; Nunez A. Asymmetric Brønsted acid-catalyzed Friedel–Crafts reactions of indoles with cyclic imines: Efficient generation of nitrogen substituted quaternary carbon centers. Adv. Synth. Catal. 2011, 353, 563. 10.1002/adsc.201000952. [DOI] [Google Scholar]; l Nakamura S.; Matsuda N.; Ohara M. Organocatalytic enantioselective aza-Friedel-Crafts reaction of cyclic ketimines with pyrroles using imidazoline-phosphoric acid catalysts. Chem. - Eur. J. 2016, 22, 1. 10.1002/chem.201601573. [DOI] [PubMed] [Google Scholar]; m Parra A.; Alfaro R.; Marzo L.; Moreno-Carrasco A.; Ruano J. L. G.; Alema J. Enantioselective aza-Henry reactions of cyclic α-carbonyl ketimines under bifunctional catalysis. Chem. Commun. 2012, 48, 9759. 10.1039/c2cc34053d. [DOI] [PubMed] [Google Scholar]; n Yin Q.; You S.-L. Chiral phosphoric acid-catalysed Friedel–Crafts alkylation reaction of indoles with racemic spiro-indolin-3-ones. Chem. Sci. 2011, 2, 1344. 10.1039/c1sc00190f. [DOI] [Google Scholar]; o Schendera E.; Lerch S.; Drathen T. V.; Unkel L.-N.; Brasholz M. Phosphoric acid catalyzed 1,2-rearrangements of 3-hydroxyindolenines to indoxyls and 2-oxindoles: Reagent-controlled regioselectivity enabled by dual activation. Eur. J. Org. Chem. 2017, 22, 3134. 10.1002/ejoc.201700085. [DOI] [Google Scholar]; p Kumar C. V. S.; Puranik V. G.; Ramana C. V. InCl3-mediated addition of indole to isatogens: An expeditious synthesis of 13-deoxy-isatisine A. Chem. - Eur. J. 2012, 18, 9601. 10.1002/chem.201103604. [DOI] [PubMed] [Google Scholar]
- a Liu R.-R.; Ye S.-C.; Lu C.-J.; Gao G.-L.; Zhuang J.-R.; Jia Y.-X. Dual catalysis for the redox annulation of nitroalkynes with indoles: Enantioselective construction of indolin-3-ones bearing quaternary stereocenters. Angew. Chem., Int. Ed. 2015, 54, 11205. 10.1002/anie.201504697. [DOI] [PubMed] [Google Scholar]; b Gajulapalli V. P. R.; Jafari E.; Kundu D. S.; Mahajan S.; Peuronen A.; Rissanen K.; Enders D. Organocatalytic asymmetric synthesis of 2,3′-connected bis-indolinones by Mannich reactions of N-acetylindolin-3-ones with isatin N-Boc-ketimines. Synthesis 2017, 49, 4986. 10.1055/s-0036-1590823. [DOI] [Google Scholar]; c Rueping M.; Rasappan R.; Raja S. Asymmetric proline-catalyzed addition of aldehydes to 3H-indol-3-ones: Enantioselective synthesis of 2,3-dihydro-1H-indol-3-ones with quaternary stereogenic centers. Helv. Chim. Acta 2012, 95, 2296. 10.1002/hlca.201200498. [DOI] [Google Scholar]; d Yan W.; Wang D.; Feng J.; Li P.; Zhao D.; Wang R. Synthesis of N-alkoxycarbonyl ketimines derived from isatins and their application in enantioselective synthesis of 3-aminooxindoles. Org. Lett. 2012, 14, 2512. 10.1021/ol3007953. [DOI] [PubMed] [Google Scholar]; e Zhao K.; Shu T.; Jia J.; Raabe G.; Enders D. An organocatalytic Mannich/denitration reaction for the asymmetric synthesis of 3-ethylacetate-substitued 3-amino-2-oxindoles: Formal synthesis of AG-041R. Chem. - Eur. J. 2015, 21, 3933. 10.1002/chem.201406422. [DOI] [PubMed] [Google Scholar]; f Hara N.; Nakamura S.; Sano M.; Amura R.; Funahashi Y.; Shibata N. Enantioselective synthesis of AG-041R by using N-heteroarenesulfonyl cinchona alkaloid amides as organocatalysts. Chem. - Eur. J. 2012, 18, 9276. 10.1002/chem.201200367. [DOI] [PubMed] [Google Scholar]; g Liu Y.-L.; Zhou J. Organocatalytic asymmetric cyanation of isatin derived N-Boc-ketoimines. Chem. Commun. 2013, 49, 4421. 10.1039/C2CC36665G. [DOI] [PubMed] [Google Scholar]
- a Cai X.-H.; Xie B. Recent advances on organo-catalysed asymmetric Mannich reactions. ARKIVOC 2013, 264. 10.3998/ark.5550190.p007.839. [DOI] [Google Scholar]; b Ishitani H.; Ueno M.; Kobayashi S. Enantioselective Mannich-type reactions using a novel chiral zirconium catalyst for the synthesis of optically active β-amino acid derivatives. J. Am. Chem. Soc. 2000, 122, 8180. 10.1021/ja001642p. [DOI] [Google Scholar]; c Zhuang W.; Saaby S.; Jørgensen K. A. Direct organocatalytic enantioselective Mannich reactions of ketimines: An approach to optically active quaternary α-amino acid derivatives. Angew. Chem., Int. Ed. 2004, 43, 4476. 10.1002/anie.200460158. [DOI] [PubMed] [Google Scholar]; d Verkade J. M. M.; Hemert L. J. C. V.; Quaedflieg P. J. L. M.; Rutjes F. P. J. T. Organo-catalysed asymmetric Mannich reactions. Chem. Soc. Rev. 2008, 37, 29–41. 10.1039/B713885G. [DOI] [PubMed] [Google Scholar]; e Córdova A. The direct catalytic asymmetric Mannich reaction. Acc. Chem. Res. 2004, 37, 102. 10.1021/ar030231l. [DOI] [PubMed] [Google Scholar]; f Trost B. M.; Saget T.; Hung C.-I. Direct catalytic asymmetric Mannich reactions for the construction of quaternary carbon stereocenters. J. Am. Chem. Soc. 2016, 138, 3659. 10.1021/jacs.6b01187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yarlagadda S.; Reddy C. R.; Ramesh B.; Kumar G. R.; Sridhar B.; Reddy B. V. S. Organocatalytic enantioselective Michael addition of 3-indolinone-2-carboxylates to maleimides. Eur. J. Org. Chem. 2018, 1364. 10.1002/ejoc.201701670. [DOI] [Google Scholar]; b Yarlagadda S.; Sridhar B.; Reddy B. V. S. Oxidative asymmetric aza-Friedel–Crafts alkylation of indoles with 3-indolinone-2-carboxylates catalyzed by a BINOL phosphoric acid and promoted by DDQ. Chem. - Asian J. 2018, 1327. 10.1002/asia.201800300. [DOI] [PubMed] [Google Scholar]; c Yarlagadda S.; Sankaram G. S.; Sridhar B.; Reddy B. V. S. Asymmetric Robinson annulation of 3-indolinone-2-carboxylates with cyclohexenone: Access to chiral bridged tricyclic hydrocarbazole. Org. Lett. 2018, 20, 4195. 10.1021/acs.orglett.8b01575. [DOI] [PubMed] [Google Scholar]
- CCDC 1842015 and 1887364 contains supplementary Crystallographic data for the structures 3h and 3t. These data can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
- a Yarlagadda S.; Ramesh B.; Reddy C. R.; Srinivas L.; Sridhar B.; Reddy B. V. S. Organocatalytic enantioselective amination of 2-substituted indolin-3-ones: A strategy for the synthesis of chiral α-hydrazino esters. Org. Lett. 2017, 19, 170. 10.1021/acs.orglett.6b03473. [DOI] [PubMed] [Google Scholar]; b Wang Q.; Leutzsch M.; Gemmeren M. V.; List B. Disulfonimide-catalyzed asymmetric synthesis of β-amino esters directly from N-Boc-amino sulfones. J. Am. Chem. Soc. 2013, 135, 15334. 10.1021/ja408747m. [DOI] [PubMed] [Google Scholar]; c Blom J.; Vidal-Albalat A.; Jørgensen J.; Barløse C. L.; Jessen K. S.; Iversen M. V.; Jørgensen K. A. Directing the activation of Donor–Acceptor cyclopropanes towards stereoselective 1,3-dipolar cycloaddition reactions by Brønsted base catalysis. Angew. Chem., Int. Ed. 2017, 56, 11831. 10.1002/anie.201706150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.