

Abstract
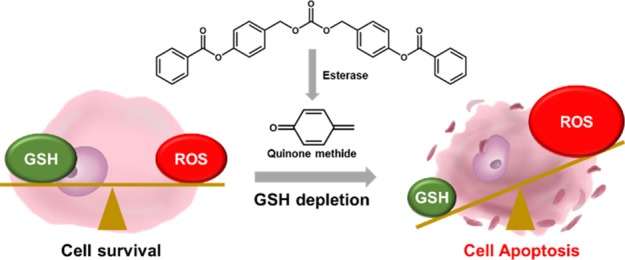
A main challenge in the development of anticancer drugs that eradicate cancer cells specifically with minimal toxicity to normal cells is to identify the cancer-specific properties. Cancer cells sustain a higher level of reactive oxygen species, owing to metabolic and signaling aberrations and unrestrained growth. Cancer cells are also furnished with a powerful reducing environment, owing to the overproduction of antioxidants such as glutathione (GSH). Therefore, the altered redox balance is probably the most prevailing property of cancer cells distinct from normal cells, which could serve as a plausible therapeutic target. In this work, we developed a GSH-depleting pro-oxidant, benzoyloxy dibenzyl carbonate, termed B2C, which is capable of rapidly declining GSH and elevating oxidative stress to a threshold level above which cancer cells cannot survive. B2C was designed to release quinone methide (QM) that rapidly depletes GSH through esterase-mediated hydrolysis. B2C was able to rapidly deplete GSH and induce an overwhelming level of oxidative stress in cancer cells, leading to mitochondrial disruption, activation of procaspase-3 and PARP-1, and cleavage of Bcl-2. In the study of tumor xenograft models, intravenously injected B2C caused apoptotic cell death in tumors and significantly suppressed tumor growth. These findings provide a new insight into the design of more effective anticancer drugs, which exploit altered redox balance in cancer cells.
Introduction
Reactive oxygen species (ROS) act crucial roles in biological processes as a secondary messenger in cellular signaling to activate proliferation and survival pathways.1,2 Despite their fundamental roles, excessive ROS induces oxidative stress, which provokes DNA damages, mutagenesis, and even cell death.3,4 Cells therefore should sustain redox homeostasis to maintain normal cellular functions and ensure cell survival.1 Contrary to normal cells, cancer cells sustain an elevated level of ROS resulting from metabolic and signaling aberrations and unrestrained growth.1,5,6 The high level of ROS drives cancer cell proliferation and enhances the aggressive phenotypes of cancer cells such as promotion of mutations, invasion, and metastasis.7,8 Therefore, cancer cells must upregulate multiple antioxidant defense systems to overwhelm the high level of ROS and overcome detrimental effects of oxidative stress.6 The upregulation of antioxidants in adjustment to inherent oxidative stress is also known to confer resistance to chemotherapeutics and radiation.3 A recent study has reported that cancer cells extremely depend on their antioxidant systems to sustain redox balance and are hypersensitive to exogenous agents that damage antioxidant capability.5
Although the roles of ROS in cancer cells still remain controversial, it is reasonable to employ the biochemical differences in redox balance between normal and cancer cells as an effective basis for selective toxicity toward cancer cells.5,9 A simple and promising strategy to target cancer cells by ROS-mediated mechanism is the suppression of antioxidant systems in cancer cells. Among various endogenous antioxidants, GSH, a tripeptide of Glu-Cys-Gly, is a major ROS-scavenging system and plays fundamental roles in sustaining redox homeostasis.6 GSH is also known to be the largest source of nonprotein thiol groups in cells and is considered to be the most abundant and important antioxidant.10,11 In addition, the GSH antioxidant pathway is desired for tumor initiation and development.1 Therefore, depletion of antioxidant GSH alters cell’s ability to modulate ROS and causes accumulation of ROS to a level or the threshold above which cells cannot survive.5 In contrast, normal cells are less susceptible to GSH depleting agents, owing to a low basal level of ROS and well-maintained redox balance.2
There is cumulative evidence demonstrating that GSH inhibition induces severe ROS accumulation and raises oxidative stress, causing cancer cell death.3,5,12 β-Phenethyl isocyanate (PEITC) is a naturally occurring isothiocyanate abundant in cruciferous vegetables such as watercress and is also known to accumulate ROS in cancer cells.5,13 It was reported that PEITC conjugates with GSH for export from cancer cells and elevate the level of ROS, consequently resulting in cell death.14,15 QM, an α,β-unsaturated ketone, is an intermediate generated from hydrolysis of phenolic ester compounds and is also well known to readily alkylate GSH.2,16 Hulsman et al. reported that an unsubstituted QM generated immediately after esterase-catalyzed carboxylic ester hydrolysis reacts selectively with cellular GSH, triggering cell death.17 This GSH scavenger exhibited potent anticancer activity, although one QM intermediate could deplete only one GSH. We therefore hypothesized that a GSH scavenger that is able to deplete two GSH molecules could elicit higher anticancer activity and a low dose is required to achieve sufficient anticancer therapeutic effects.
In this context, we developed a pro-oxidant, benzoyloxy dibenzyl carbonate (B2C), which is able to generate two QM intermediates and rapidly deplete two GSH molecules through alkylation of thiol. B2C was designed to undergo esterase-catalyzed hydrolysis of phenolic ester to produce two QM intermediates and enhance ROS accumulation, resulting in cancer cell death (Figure 1). We have undertaken extensive studies to determine the anticancer therapeutic efficacy and the mode of action of pro-oxidant B2C using cell cultures and xenograft mouse models. Herein, we report that multiple GSH-depleting B2C holds remarkable translational potential as a targeted anticancer therapeutic agent.
Figure 1.

Synthetic route and chemical structure of B2C.
Results
Design and Synthesis of B2C
B2C was designed to undergo esterase-catalyzed hydrolysis to release two QM intermediates, which then deplete two GSH molecules. The synthetic route and chemical structure are shown in Figure 1. Hydroxybenzyl alcohol was reacted with benzoyl chloride to generate 4-(hydroxymethyl)phenyl benzoate (1). 1 was then reacted with 1,1-dicarbonyldiimidazole to yield 4-(benzoyloxy)benzyl 1H-imidazole-1-carboxylate (2). From the reaction of 1 and 2, B2C, 4,4′-(carbonylbis(oxy)bis(methylene))bis(4,1-phenylene) dibenzoate, was obtained in the form of a white powder (60% yield). The chemical structure of B2C was verified by NMR spectroscopy (Figure S1). B2C was highly stable, as demonstrated by no change in NMR spectra after 3 days of incubation under aqueous conditions (Figure S2). For comparison purposes, we also synthesized B1C, which is able to generate one QM intermediate. In addition, DBC was synthesized, which does not generate a QM intermediate (Figure S3).
Esterase-Triggered QM Generation from B2C
B2C could be easily attacked by esterase to undergo carboxylic ester hydrolysis. As shown in Figure 2a, esterase-triggered hydrolysis of carboxylic ester of B2C generates two QM intermediates through energetically favorable 1,6-elimination of phenolates. We first examined the ability of B2C to deplete GSH in the presence or absence of esterase. Figure 2b shows the level of GSH determined 24 h after the addition of B2C. B2C significantly declined the level of GSH in the presence of esterase in a concentration-dependent manner. However, in the absence of esterase, slight GSH depletion was induced by a high concentration of B2C (1 mM). These results indicate that carboxylic ester of B2C is readily attacked by esterase to release GSH-depleting QM intermediates. We also compared B2C with B1C and DBC (Figure 2c). As expected, B1C could also deplete GSH in the presence of esterase but to a substantially less extent than B2C, and DBC was unable to deplete GSH.
Figure 2.

Esterase-triggered GSH depletion by B2C. (a) Mechanism of esterase-triggered GSH depletion through the formation of QM intermediates. (b) Depletion of GSH by B2C in the presence or absence of esterase. The concentration of GSH is 500 μM. (c) Comparison of the GSH-depleting ability of B2C with B1C and DBC. The concentrations of B2C, B1C, and DBC are 1 mM. Values are mean ± SD (n = 4). ***p < 0.001 relative to a group of esterase only. †††p < 0.001 relative to B1C. Level of GSH in (d) SW620 cells, (e) DU145 cells, and (f) A549 cells after B2C treatment. Values are mean ± SD (n = 4). ***p < 0.001 relative to a group of 0 μM.
We next studied the ability of B2C to scavenge intracellular GSH in esterase-triggered manners using various cancer cells (SW620, DU145, and A549 cell lines). B2C dramatically reduced the level of intracellular GSH in a concentration-dependent manner (Figure 2d–f). B2C at a concentration of 50 μM depleted a majority of GSH. In particular, SW620 cells exhibited the most reduction in the level of GSH after B2C treatment.
B2C-Induced Cell Death
MTT assay was performed to evaluate the cytotoxicity of B2C toward various cancer cells. After 24 h of B2C treatment, the cell viability markedly decreased with increasing concentration of B2C (Figure 3). B2C exhibited the strongest toxicity against SW620 cells, which underwent the most reduction in the level of GSH (Figure 2d). In addition, B2C displayed significantly higher toxicity than B1C and DBC. Normal cells (RAW 264.7) were also killed by B2C but to less extent than cancer cells (Figure S4) probably because of their well-maintained redox balance. We next compared the anticancer activity of B2C with PEITC that is known to kill cancer cells by exporting GSH from cells. As shown in Figure S5, B2C exerted stronger anticancer activity than PEITC at the same concentration.
Figure 3.

Cytotoxicity of B2C, B1C, and DBC against various cancer cells. (a) SW620 cells, (b) DU145 cells, and (c) A549 cells. Values are mean ± SD (n = 4). **p < 0.01, ***p < 0.001 relative to a group of 0 μM. ††p < 0.01, †††p < 0.001 relative to the same group of DBC.
B2C-Mediated Elevation of Oxidative Stress
To confirm whether B2C depletes GSH to elevate oxidative stress, the level of intracellular ROS was evaluated by flow cytometry. After incubation with various concentrations of B2C, cells were stained with 2′,7′-dichlorodihydrofluorescein-diacetate (DCFH-DA) as a probe of ROS. B2C treatment increased markedly the level of intracellular ROS, evidenced by the rightward shift of fluorescence (Figure 4a–c and Figure S6). However, the antioxidant N-acetylcysteine (NAC) suppressed B2C-induced accumulation of ROS (Figure S7). These results indicate that B2C depletes GSH to allow cancer cells to accumulate more ROS.
Figure 4.

B2C-induced cell death through the elevation of oxidative stress. Flow cytometric analysis of intracellular ROS in cancer cells treated with B2C. (a) SW620 cells, (b) DU145 cells, and (c) A549 cells. Cytotoxicity of B2C against (d) SW620 cells, (e) DU145 cells, and (f) A549 cells in the absence or presence of NAC. Values are mean ± SD (n = 4). ***p < 0.001 relative to a group of 0 μM; †††p < 0.001 relative to the same concentration of B2C + NAC (1 mM).
To further verify whether B2C provokes cancer cell death through the elevation of oxidative stress, the cell viability was measured in the presence of NAC. Although NAC (1 mM) could not completely prevent B2C-induced cell death, the cytotoxicity of B2C was significantly diminished by NAC (Figure 4d–f). These observations demonstrate that B2C kills cancer cells through the elevation of oxidative stress resulting from the depletion of antioxidant GSH.
B2C-Mediated Apoptotic Cell Death
Cell cycle assay was performed to examine whether B2C affects cell cycle distribution of cancer cells (Figure 5a and Figure S8). B2C caused no significant changes in the percentage in G2/M phase but significantly increased the portion in the sub-G0 phase, which is indicative of possible induction of cellular apoptosis.18,19 To substantiate B2C-induced cell death, flow cytometry was employed to analyze cancer cells with FITC-conjugated Annexin V as an apoptosis marker and propidium iodide as a viability marker. As shown in Figure 5b and Figure S9, the populations of the upper right quadrant increased with B2C concentrations, demonstrating that B2C induces apoptosis in a concentration-dependent manner. Cells were also analyzed using JC-1 because the mitochondrial disruption is one of features of early apoptosis.2 B2C induced a substantial loss of mitochondrial membrane in concentration-dependent manner (Figure 5c).
Figure 5.

B2C-induced apoptotic cell death on SW620 cells. (a) Cell cycle assay of SW620 cells after B2C treatment. (b) Flow cytometric analysis of cells stained with Annexin V-FITC and propidium iodide. (c) Flow cytometric analysis for mitochondrial membrane potential using JC-1. Data are representative of three independent experiments.
To gain mechanistic insight into how B2C triggers apoptosis, we carried out Western blotting for Bcl-2, caspase 3, and PARP-1. As shown in Figure 6 and Figure S10, B2C effectively suppressed the expression of Bcl-2 and induced the cleavage of pro-apoptotic proteins caspase 3 and PARP-1. These observations demonstrate that B2C induces significant mitochondrial disruption, leading to apoptotic cascades.
Figure 6.

Effects of B2C on the expression of apoptosis-related proteins in SW620 cells.
Antitumor Activity of B2C
Therapeutic activity of B2C was evaluated using a mouse xenograft model. B2C was intravenously injected through a tail vein every other day, and the tumor volume and body weight were monitored for 25 days (Figure 7). B2C at doses less than 5 mg/kg exerted insignificant inhibitory effects on tumor growth. However, B2C remarkably suppressed the tumor growth without changes in body weight at doses higher than 10 mg/kg. Histological examination was carried out further to verify anticancer activity of B2C. Untreated groups showed a large number of tumor cells that sustain their normal morphology with apparent membrane and nuclear structure. However, a number of dead cells without nuclei were observed in tumors of the B2C-treated group (Figure 8a). A large number of terminal deoxynucleotidyl transferase dUTP nick end (TUNEL)-positive cells were observed in B2C-treated groups in a dose-dependent manner (Figure 8b and Figure S11). The results suggest that B2C causes apoptotic cancer cell death in tumors.
Figure 7.

Therapeutic anticancer activity of B2C. (a) Photographs of tumor-bearing mice treated with various doses of B2C and representative tumor images excised after 25 days. Red dotted lines indicate inoculation sites of SW620 cells. (b) Changes in tumor volumes. Arrows indicate the date of B2C injection. (c) Body weight changes of tumor-bearing mice during the B2C treatment. Values are mean ± SD (n = 4).
Figure 8.

Histological evaluation of tumors after B2C treatment. (a) Micrographs of tumor sections stained with H&E. (b) Micrographs of tumor sections stained with TUNEL.
Finally, to study the potential cumulative toxicity of B2C, we administrated B2C (20 mg/kg) for 2 weeks. B2C administration caused no significant changes in the level of alanine aminotransferase (ALT) (Figure S12a). In addition, no substantial histological evidence of accumulated toxicity in organs after B2C administration was observed (Figure S12b–d). The results demonstrate that B2C has excellent safety profiles at therapeutic doses.
Discussion
In the development of anticancer drugs that eradicate cancer cells specifically with minimal toxicity to normal cells, a main challenge is to identify the cancer-specific properties.20 Although there have been enormous efforts to exploit the differences between normal and cancer cells as a plausible therapeutic target, many of these strategies have failed to demonstrate sufficient therapeutic activities.21 Among the various unique features of cancer cells distinct from normal cells, the most prevailing property is probably the redox state.20 Compared to normal cells, cancer cells have not only a higher level of ROS but also a strong reducing environment due mainly to the overproduction of GSH.3,22,23 It has been well accepted that normal cells are less likely to be damaged by elevated oxidative stress, owing to their high level of antioxidant systems, which provides a logical basis of cancer cell-selective toxicity.2,11,24 Notably, most conventional chemotherapeutic drugs are known to induce oxidative insults to eradicate cancer cells.20 On the basis of the redox potential gradient between cancer cells and normal cells, oxidative therapy has recently been emerging as an anticancer treatment regimen.18,22,23,25
GSH is considered the major mechanism used to control the cellular redox balance.2 The functions of GSH include reduction of ROS such as H2O2 generated in mitochondria and detoxification of increased production of lipid peroxidase.20 Cancer cells greatly need a high level of GSH for their proliferation and survival.18 We therefore developed B2C that could deplete two GSH molecules to elevate oxidative stress and preferentially kill cancer cells based on the hypothesis that GSH depletion effectively provokes selective toxicity against a wide variety of cancer cells. To deliver precise proof of this hypothesis, we also synthesized model compounds, one GSH-depleting B1C and one GSH-nondepleting DBC.
B2C was synthesized by coupling two benzoyloxy-substituted benzyl alcohols (QM-generating moiety) using a carbonate bond that functions as a leaving group (Figure 1). Therefore, the design of B2C fulfills the requirements for esterase-triggered QM generation that a phenolic ester (QM-generating moiety) is substituted with a p-methylene group attached to a good leaving group.2,17 In the presence of esterase, B2C depleted GSH significantly more than B1C and DBC (Figure 2b,c) at the same concentrations. In good accordance with these results, B2C dramatically reduced the intracellular level of GSH in a concentration-dependent manner. B2C showed more potent GSH-depleting activity and higher toxicity against cancer cells compared to B1C and DBC. Interestingly, 50 μM B1C exerted almost the same level of cytotoxicity as 25 μM B2C (Figure S13). These observations obviously support our hypotheses that esterase-catalyzed ester hydrolysis of B2C affords QM intermediates to deplete GSH and the toxicity of QM-generating compounds is dictated by the number of QM intermediates generated from esterase-triggered hydrolysis of phenolic ester bonds.
To further demonstrate the advantages of B2C over B1C and the significance of GSH depleting capability, direct comparison of therapeutic efficacy was made between B2C and B1C. As expected, B2C showed significantly higher anticancer activity than B1C at the same dose of 20 mg/kg (Figure S14). The higher therapeutic efficacy of B2C can be explained by their different biodistribution and GSH-depleting capability. As aforementioned, B2C was designed to release QM twice as much as B1C, and the molecular weight of B1C is higher than half of B2C. Therefore, assuming that they have the same cancer cell-targeting ability and are administrated at the same dose, B1C needs to be accumulated in tumors more than twice as much as B2C to exert the same therapeutic efficacy as B2C. However, B1C has no ability to target cancer cells more than twice as much as B2C. We therefore reason that the superior therapeutic efficacy of B2C over B1C is attributed mainly to its more potent GSH-depleting capability and coupling of two GSH-depleting moieties is a promising chemical strategy for developing anticancer therapeutics.
As shown in Figure S15, the level of intracellular GSH varied with cells. We found that cancer cells (SW620, DU145, and A549) showed a higher level of GSH than noncancerous cells (RAW264.7) when cultured under the same condition. Among cancer cells tested, A549 cells had the highest level of GSH. The extent of B2C-mediated GSH depletion varied with cells because of their different levels of intrinsic GSH. B2C at a concentration of 50 μM depleted a majority of GSH. In particular, SW620 cells possessing a relatively lower GSH level showed the most reduction in the level of GSH after B2C treatment. As expected from these findings, the SW620 cell line was the most susceptible to GSH-depleting B2C, evidenced by the lowest cell viability (Figures 3a and 4d).
Our data illustrate that esterase-catalyzed activation of B2C generates two QM intermediates. We observed that B2C induces an overwhelming level of oxidative stress in cancer cells to trigger mitochondrial disruption, activation of procaspase-3 and PARP-1, and cleavage of Bcl-2, resulting in apoptotic cell death. Inhibition of B2C-induced cell death by antioxidant NAC strongly suggests that elevation of oxidative stress contributes predominantly to B2C-induced apoptotic cell death. Proof-of-concept animal studies further demonstrate that B2C as a GSH-scavenging pro-oxidant elicits substantial apoptotic cell death in tumors and inhibits tumor growth without conspicuous side effects to the liver and heart. However, further studies including pharmacokinetics, pharmacodynamics, and toxicology are greatly needed to fully determine the translational potential of B2C as an anticancer drug. In particular, the effects of long-term use of B2C should be investigated because deregulation of GSH production is tightly related to several diseases such as diabetes.
Conclusions
We developed the GSH-depleting pro-oxidant B2C as a new family of oxidative anticancer therapeutics that can destroy preferentially cancer cells through oxidative stress elevation. B2C was synthesized by coupling benzoyloxy-substituted benzyl alcohols (QM-generating moiety) through a carbonate bond. In the presence of esterase, B2C rapidly generated QM that depletes antioxidant GSH to significantly increase the level of ROS. B2C generated an overwhelming level of oxidative stress in cancer cells to trigger mitochondrial disruption, activation of procaspase-3 and PARP-1, and cleavage of Bcl-2, leading to apoptotic cell death. In tumor-bearing mouse models, intravenously injected B2C significantly suppressed the tumor growth without noticeable side effects. We believe that GSH-depleting B2C provides a promising strategy in the design of selective anticancer drugs and holds great potential as anticancer therapeutics.
Materials and Methods
Synthesis of B2C
4-Hydroxybenzyl alcohol (80.0 mmol) and triethylamine (80.0 mmol) were dissolved in 250 mL of dichloromethane (DCM). The solution was stirred at 0 °C for 30 min, to which benzoyl chloride (80.0 mmol) in 50 mL of DCM was added dropwise. The resulting solution was stirred at room temperature for 5 h and mixed with saturated aqueous solution of sodium carbonate (100 mL). After phase separation, the organic phase was obtained and dried over magnesium sulfate, followed by filtering. The solvent was taken away under diminished pressure using a rotary evaporator. 4-(Hydroxymethyl)phenyl benzoate (1) was obtained as a white powder (60% yield) from silica gel chromatography and crystallization. 4-(Benzoyloxy)benzyl 1H-imidazole-1-carboxylate (2) was synthesized from the reaction of 1 and 1,1′-carbonyldiimidazole. 1 (4.38 mmol) was dissolved in 15 mL of DCM, to which 1,1′-carbonyldiimidazole (8.76 mmol) was added. The reaction was allowed at room temperature for 1 h. 2 was obtained as a white powder (60% yield) from silica gel chromatography. 4,4′-(Carbonylbis(oxy)bis(methylene))bis(4,1-phenylene) dibenzoate (B2C; 3) was synthesized from the reaction of 1 and 2. 1 (3.10 mmol), 2 (3.10 mmol), and 4-(dimethylamino)pyridine (3.10 mmol) were dissolved in 15 mL of dry tetrahydrofuran. The solution was stirred at 40 °C for 10 h. B2C was obtained as a white powder (25% yield) from flash silica gel chromatography and recrystallization. The chemical structure of 3 was verified by NMR (JNM-AL400, JEOL Ltd., Tokyo, Japan) and liquid chromatography–tandem mass spectrometry (6410, Agilent, Santa Clara, CA). 1H NMR: δ 8.19 (d, J = 7.2 Hz, 4H), 7.63 (t, J = 7.2 Hz, 2H), 7.51 (t, J = 7.2 Hz, 4H), 7.46 (d, J = 8.4 Hz, 4H), 7.22 (d, J = 8.4 Hz, 4H), 5.19 (s, 4H); 13C NMR: δ 165.15, 155.06, 151.18, 133.79, 132.90, 130.30, 129.86, 129.46, 128.70, 122.06, 69.24; ESI-MS (m/z): [M + H]+ calcd. for C29H22O7, 482.14; found, 483.16; elemental analysis (calcd., found for C29H22O7): C (72.19, 72.16), H (4.60, 4.60).
Synthesis of B1C
Benzyl alcohol (9.24 mmol) and 1,1′-carbonyldiimidazole (13.86 mmol) were dissolved in 15 mL of DCM and stirred at room temperature for 1 h. Benzyl 1H-imidazole-1-carboxylate (4) was obtained as a white powder (80% yield) by silica gel chromatography. 4 (2.47 mmol), 1 (2.47 mmol), and N,N-diisopropylethylamine (2.47 mmol) were dissolved in 15 mL of dry DCM. The solution was stirred at 40 °C for 10 h. B1C ((benzyloxycarbonyloxy)methyl)phenyl benzoate; 5) was obtained as a white powder (40% yield) by flash column chromatography.
Synthesis of DBC
Sodium hydride (22.2 mmol) was added to 20 mL of tetrahydrofuran including benzyl alcohol (8.5 mmol) at 0 °C, and the solution was stirred at room temperature for 1 h. 1,1′-Carbonyldiimidazole (9.75 mmol) in 10 mL of tetrahydrofuran was added to the solution dropwise at 0 °C, and the resulting mixture was stirred at 0 °C for 5 h. The reaction mixture was extracted with ethyl acetate and aqueous solution of NaH4Cl. The organic phase was dried over magnesium sulfate and filtered, and the solvent was removed under diminished pressure. Dibenzyl carbonate (DBC) (6) was obtained as a yellowish oil (40% yield) by silica gel chromatography and crystallization.
Cytotoxicity of B2C
SW620, DU145, A549, and RAW 264.7 cells were incubated in a 24-well plate for 24 h. Cells with 90% confluency were incubated with B2C (10, 25, or 50 μM) for 12 h. Cells were then added to 100 μL of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) solution and incubated for 4 h. Formazan crystals were dissolved in 1 mL of dimethyl sulfoxide (DMSO). The absorbance at 570 nm was measured after 10 min of incubation to determine the cell viability using a microplate reader (Biotek Instruments, Winooski, VT).
Measurement of the Level of GSH
One milliliter of GSH solutions (500 μM) was mixed with B2C (10, 25, or 50 μM) for 1 h with or without esterase (100 μg/mL). For another set of experiments, B2C was added to the cells. One hour later, cells were harvested and lysed on ice with 100 μL of lysis buffer. Cell lysates were centrifuged (9800g), and the supernatant (10 μL) was mixed with 50 μL of Ellman’s reagent (0.5 mM, 5,5′-dithiobis-(2-nitrobenzoic acid)). The microplate reader was employed to measure the level of remaining GSH solution and cellular GSH by measuring the optical density at 405 nm.
Flow Cytometry
SW620, DU145, and A549 cells were incubated with B2C (10, 25, or 50 μM). For the detection of intracellular ROS, cells were incubated with 2 μM DCFH-DA (Sigma-Aldrich) for 15 min at 37 °C in the dark. For the analysis of apoptosis, the cells were treated with Annexin V-FITC and propidium iodide purchased from BD Biosciences Pharmigen (San Diego, CA). To demonstrate the effect of B2C on the cell cycle, B2C-treated cells were stained with propidium iodide (10 μg/mL) in the presence of RNase A. JC-1 assay kit (Molecular Probes, Eugene, OR) was used to study the mitochondrial membrane potential. The stained cells were transferred to 5 mL round-bottom tubes and examined using a flow cytometer (FACS Caliber, Becton Dickinson, San Jose, CA).
Immunoblotting
Cells were treated with various concentrations of B2C. Proteins were extracted from cells using a lysis buffer. Electrophoresis was carried out using proteins (30 μg) on a 12% polyacrylamide gel, and proteins were transferred to poly(vinylidene difluoride) membranes (Bio-Rad, Hercules, CA). After being blocked with 5% solution of nonfat milk for 1 h, the blots were incubated with primary antibodies (procaspase 3, PARP-1, and Bcl-2) purchased from Santa Cruz Biotechnology (Dallas, TX) and HRP-conjugated anti-goat secondary antibody (BD Biosciences, Mississauga, Canada). The immunoblot signals were developed using SuperSignal Ultra chemiluminescent reagent (Pierce, Rockford, IL).
Tumor-Bearing Mouse Model
SW620 cells were inoculated subcutaneously into the dorsa of nude BALB/c mice (6 weeks old, Orient Bio, Korea). When the inoculated tumor reached ∼50 mm3 in size, B2C dissolved in aqueous DMSO solution (20 mg/mL) was intravenously injected seven times into mice at a dose of 5, 10, and 20 mg/kg 2 days apart for 25 days. The body weight and tumor volume of mice were measured 2 days apart. The tumor volume was determined using the following formula: (width2 × length)/2. At the end of the experimentation, mice were sacrificed, and solid tumors and organs were excised for histological examination. Tissues were sliced and stained with hematoxylin and eosin (H&E) and TUNEL. Animal experiments were approved by the Institution Animal Ethical Committee of Chonbuk National University (CBU2014-00024) and carried out in compliance with the national guidelines.
Statistical Analysis
Values were expressed as mean ± SD (standard deviation). One-way ANOVA (analysis of variation) using GraphPad Prism 5.0 was conducted to make comparison between groups. A difference of p < 0.05 was considered significant statistically.
Acknowledgments
This work was supported by a grant of Korean Health Technology R&D Project (HI15C1619), Ministry of Health & Welfare, and Mid-Career Research Program through National Research Foundation (2016R1A2B4008489), Ministry of Science, ICT and Future Planning, Republic of Korea.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00140.
Synthesis and characterization of B2C, quantification of the GSH level and apoptotic cell death, and biocompatibility of B2C (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Harris I. S.; Treloar A. E.; Inoue S.; Sasaki M.; Gorrini C.; Lee K. C.; Yung K. Y.; Brenner D.; Knobbe-Thomsen C. B.; et al. Glutathione and Thioredoxin Antioxidant Pathways Synergize to Drive Cancer Initiation and Progression. Cancer Cell 2015, 27, 211–222. 10.1016/j.ccell.2014.11.019. [DOI] [PubMed] [Google Scholar]
- Han E.; Kwon B.; Yoo D.; Kang C.; Khang G.; Lee D. Dual Stimuli-Activatable Oxidative Stress Amplifying Agent as a Hybrid Anticancer Prodrug. Bioconjugate Chem. 2017, 28, 968–978. 10.1021/acs.bioconjchem.6b00683. [DOI] [PubMed] [Google Scholar]
- Trachootham D.; Alexandre J.; Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach?. Nat. Rev. Drug Discovery 2009, 8, 579–591. 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- Watson J. Oxidants, antioxidants and the current incurability of metastatic cancers. Open Biol. 2013, 3, 120144. 10.1098/rsob.120144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachootham D.; Zhou Y.; Zhang H.; Demizu Y.; Chen Z.; Pelicano H.; Chiao P. J.; Achanta G.; Arlinghaus R. B.; et al. Selective killing of oncogenically transformed cells through a ROS-mediated mechanism by β-phenylethyl isothiocyanate. Cancer Cell 2006, 10, 241–252. 10.1016/j.ccr.2006.08.009. [DOI] [PubMed] [Google Scholar]
- Gorrini C.; Harris I. S.; Mak T. W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discovery 2013, 12, 931–947. 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- Kumar B.; Koul S.; Khandrika L.; Meacham R. B.; Koul H. K. Oxidative stress is inherent in prostate cancer cells and mediates aggressive phenotype. J. Urology 2008, 179, 192–193. 10.1016/S0022-5347(08)60559-5. [DOI] [PubMed] [Google Scholar]
- Trachootham D.; Lu W.; Ogasawara M. A.; Valle N. R.-D.; Huang P. Redox regulation of cell survival. Antioxid. Redox Signaling 2008, 10, 1343–1374. 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Raedt T.; Walton Z.; Yecies J. L.; Li D.; Chen Y.; Malone C. F.; Maertens O.; Jeong S. M.; Bronson R. T.; et al. Exploiting Cancer Cell Vulnerabilities to Develop a Combination Therapy for Ras-Driven Tumors. Cancer Cell 2011, 20, 400–413. 10.1016/j.ccr.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A. Selective Modification of Glutathione Metabolism. Science 1983, 220, 472–477. 10.1126/science.6836290. [DOI] [PubMed] [Google Scholar]
- Davison K.; Côté S.; Mader S.; Miller W. H. Jr. Glutathione depletion overcomes resistance to arsenic trioxide in arsenic-resistant cell lines. Leukemia 2003, 17, 931–940. 10.1038/sj.leu.2402876. [DOI] [PubMed] [Google Scholar]
- Maeda H.; Hori S.; Ohizumi H.; Segawa T.; Kakehi Y.; Ogawa O.; Kakizuka A. Effective treatment of advanced solid tumors by the combination of arsenic trioxide and L-buthionine-sulfoximine. Cell Death Differ. 2004, 11, 737–746. 10.1038/sj.cdd.4401389. [DOI] [PubMed] [Google Scholar]
- Khor T. O.; Keum Y.-S.; Lin W.; Kim J.-H.; Hu R.; Shen G.; Xu C.; Gopalakrishnan A.; Reddy B.; et al. Combined inhibitory effects of curcumin and phenethyl isothiocyanate on the growth of human PC-3 prostate xenografts in immunodeficient mice. Cancer Res. 2006, 66, 613–621. 10.1158/0008-5472.CAN-05-2708. [DOI] [PubMed] [Google Scholar]
- Xu K.; Thornalley P. J. Involvement of glutathione metabolism in the cytotoxicity of the phenethyl isothiocyanate and its cysteine conjugate to human leukaemia cells in vitro. Biochem. Pharmacol. 2001, 61, 165–177. 10.1016/S0006-2952(00)00526-8. [DOI] [PubMed] [Google Scholar]
- Kumari V.; Dyba M. A.; Holland R. J.; Liang Y.-H.; Singh S. V.; Ji X. Irreversible Inhibition of Glutathione S-Transferase by Phenethyl Isothiocyanate (PEITC), a Dietary Cancer Chemopreventive Phytochemical. PLoS One 2016, 11, e0163821 10.1371/journal.pone.0163821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dufrasne F.; Gelbcke M.; Neve J.; Kiss R.; Kraus J. L. Quinone Methides and their Prodrugs: A Subtle Equilibrium Between Cancer Promotion, Prevention, and Cure. Curr. Med. Chem. 2011, 18, 3995–4011. 10.2174/092986711796957301. [DOI] [PubMed] [Google Scholar]
- Hulsman N.; Medema J. P.; Bos C.; Jongejan A.; Leurs R.; Smit M. J.; de Esch I. J. P.; Richel D.; Wijtmans M. Chemical insights in the concept of hybrid drugs: The antitumor effect of nitric oxide-donating aspirin involves a quinone methide but not nitric oxide nor aspirin. J. Med. Chem. 2007, 50, 2424–2431. 10.1021/jm061371e. [DOI] [PubMed] [Google Scholar]
- Chang H.-H.; Guo M.-K.; Kasten F. H.; Chang M.-C.; Huang G.-F.; Wang Y.-L.; Wang R.-S.; Jeng J.-H. Stimulation of glutathione depletion, ROS production and cell cycle arrest of dental pulp cells and gingival epithelial cells by HEMA. Biomaterials 2005, 26, 745–753. 10.1016/j.biomaterials.2004.03.021. [DOI] [PubMed] [Google Scholar]
- Chew E.-H.; Nagle A. A.; Zhang Y.; Scarmagnani S.; Palaniappan P.; Bradshaw T. D.; Holmgren A.; Westwell A. D. Cinnamaldehydes inhibit thioredoxin reductase and induce Nrf2: potential candidates for cancer therapy and chemoprevention. Free Radical Biol. Med. 2010, 48, 98–111. 10.1016/j.freeradbiomed.2009.10.028. [DOI] [PubMed] [Google Scholar]
- Pei S.; Minhajuddin M.; Callahan K. P.; Balys M.; Ashton J. M.; Neering S. J.; Lagadinou E. D.; Corbett C.; Ye H.; et al. Targeting Aberrant Glutathione Metabolism to Eradicate Human Acute Myelogenous Leukemia Cells. J. Biol. Chem. 2013, 288, 33542–33558. 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzin J. Cancer drugs - Smart weapons prove tough to design. Science 2002, 298, 522–525. 10.1126/science.298.5593.522. [DOI] [PubMed] [Google Scholar]
- Luo C.; Sun J.; Liu D.; Sun B.; Miao L.; Musetti S.; Li J.; Han X.; Du Y.; et al. Self-Assembled Redox Dual-Responsive Prodrug-Nanosystem Formed by Single Thioether-Bridged Paclitaxel-Fatty Acid Conjugate for Cancer Chemotherapy. Nano Lett. 2016, 16, 5401–5408. 10.1021/acs.nanolett.6b01632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda H. Controlling oxidative stress: therapeutic and delivery strategies Preface. Adv. Drug Delivery Rev. 2009, 61, 285–286. 10.1016/j.addr.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Noh J.; Kwon B.; Han E.; Park M.; Yang W.; Cho W.; Yoo W.; Khang G.; Lee D. Amplification of oxidative stress by a dual stimuli-responsive hybrid drug enhances cancer cell death. Nat. Commun. 2015, 6, 6907. 10.1038/ncomms7907. [DOI] [PubMed] [Google Scholar]
- Yin W.; Li J.; Ke W.; Zha Z.; Ge Z. Integrated Nanoparticles To Synergistically Elevate Tumor Oxidative Stress and Suppress Antioxidative Capability for Amplified Oxidation Therapy. ACS Appl. Mater. Interfaces 2017, 9, 29538–29546. 10.1021/acsami.7b08347. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.