

Summary
A significant proportion of cancer patients do not respond to immune checkpoint blockade. To better understand the molecular mechanisms underlying these treatments, we explored the role of CD4+Foxp3− T cells expressing PD-1 (4PD1hi) and observed that 4PD1hi accumulate intratumorally as a function of tumor burden. Interestingly, CTLA-4 blockade promotes intratumoral and peripheral 4PD1hi increases in a dose-dependent manner, while combination with PD-1 blockade mitigates this effect and improves anti-tumor activity. We found that lack of effective 4PD1hi reduction after anti-PD-1 correlates with poor prognosis. Mechanistically, we provide evidence that mouse and human circulating and intra-tumor 4PD1hi inhibit T cell functions in a PD-1/PD-L1 dependent fashion and resemble follicular-helper-T-cell(TFH)-like cells. Accordingly, anti-CTLA-4 activity is improved in TFH deficient mice.
In Brief
Zappasodi et al. show that a subset of CD4+Foxp3− T cells with high PD-1 expression, designated 4PD1hi cells, inhibits T cell functions. CTLA-4 blockade increases intratumoral and systemic 4PD1hi cells, while combination with PD-1 blockade reduces the increase of 4PD1hi cells and improves anti-tumor activity.
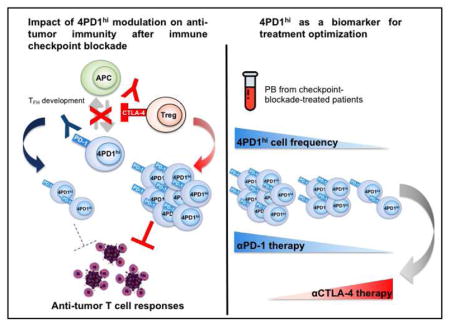
Introduction
CTLA-4 (cytotoxic T-lymphocyte-associated protein-4) and PD-1 (programmed cell death protein-1) are the best-characterized immune co-inhibitory receptors successfully targeted to promote and reinvigorate immune responses to cancer. Both molecules are induced on T cells upon T-cell receptor (TCR) signaling activation, but with different kinetics. CTLA-4 is up-regulated during the initial stage of T-cell activation and competes with CD28 for the same ligands (CD86 and CD80) expressed on antigen presenting cells (APCs), thus limiting excessive T-cell priming(Fife and Bluestone, 2008; Pentcheva-Hoang et al., 2004). PD-1 is induced later and controls previously activated T cells, typically at the effector sites of immune responses, and is considered the prototype marker of T-cell exhaustion(Fife and Bluestone, 2008; Keir et al., 2008). CTLA-4 is also constitutively up-regulated on regulatory T cells (Tregs) and constitutes one of their immunosuppressive mechanisms(Wing et al., 2008). CTLA-4 and PD-1 checkpoints are particularly deregulated in tumor-bearing hosts, where chronic ineffective immune responses usually predominate and result in T-cell exhaustion and Treg induction(Wing et al., 2008). These observations led to the development of strategies to block CTLA-4 and PD-1 for cancer immunotherapy(Dong et al., 2002; Iwai et al., 2002; Leach et al., 1996; Strome et al., 2003).
Antibodies blocking CTLA-4 and PD-1 (αCTLA-4 and αPD-1 Abs) have become a standard of care for metastatic melanoma, producing tumor regression in about 20–45% of patients as monotherapies, and in up to 60% of the cases in combination(Hodi et al., 2010; Larkin et al., 2015; Robert et al., 2015; Weber et al., 2015; Wolchok et al., 2017). PD-1 blockade has also achieved impressive clinical results in advanced non-small cell lung cancer (NSCLC) patients, where it is being investigated in combination with CTLA-4 blockade(J Clin Oncol 34, 2016 (suppl; abstr 3001))(Hellmann et al., 2016). Despite these successes, checkpoint blockade still does not benefit a significant proportion of patients with metastatic cancer, and poses a potentially high risk for developing severe immune-related toxicities, in particular when αCTLA-4 and αPD-1 are combined(Friedman et al., 2016). Except for tumor-associated PD-L1 expression, which can help enrich for patients more likely to respond to PD-1 pathway blockade(Herbst et al., 2014; Topalian et al., 2012), there are no validated biomarkers guiding selection of optimal checkpoint blockade combinations across different tumor types. This underscores the need to better characterize the biological activity of αCTLA-4 and αPD-1 for more precise utilization of these therapies.
CTLA-4 and PD-1 blockade have shown differing activity profiles, which can potentially complement each other(Larkin et al., 2015; Postow et al., 2015; Wolchok et al., 2017; Wolchok et al., 2013). Given the dominant immune evasion associated with PD-L1 overexpression in tumors, PD-1 pathway blockade yields superior therapeutic activity(Larkin et al., 2015; Postow et al., 2015; Robert et al., 2015; Wolchok et al., 2017). However, αPD-1 as a monotherapy or in combination with αCTLA-4 can be effective even against tumors with very low levels of PD-L1(Brahmer et al., 2015; Larkin et al., 2015; Wolchok et al., 2017), pointing to the existence of multiple non-redundant immune effects.
To better understand the mechanisms underlying CTLA-4 and PD-1 blockade, here we investigate modulation of CD4+Foxp3−PD-1hi T cells (4PD1hi) during these treatments. We previously reported that lack of intra-tumor 4PD1hi accumulation was associated with improved therapeutic activity of an alphavirus-based anti-melanoma vaccine (VRP-TRP2) in combination with immunomodulatory Abs in B16-bearing mice(Avogadri et al., 2014; Zappasodi and Merghoub, 2015). CTLA-4 blockade, which produced the greatest increases in intra-tumor 4PD1hi, was the least efficient modality to enhance VRP-TRP2 activity(Avogadri et al., 2014). We thus hypothesized that 4PD1hi could limit anti-tumor activity of immunotherapy. Here, we show that 4PD1hi contribute to tumor immune evasion, as they accumulate intratumorally as a function of tumor progression and limit effector T-cell (Teff) functions. We found that αCTLA-4 promotes increases in 4PD1hi while αPD-1 mitigates this effect and counteracts 4PD1hi inhibitory function, and persistence of elevated 4PD1hi frequencies after PD-1 blockade is a negative prognostic factor. Immunosuppressive 4PD1hi express a follicular helper T-cell (TFH)-like phenotype and therapeutic activity of CTLA-4 blockade is improved in TFH deficient mice.
Our study illustrates a mechanism underlying response/resistance to checkpoint blockade therapy and indicates that prospective assessment of circulating 4PD1hi during checkpoint blockade treatment can guide timely and personalized optimization of regimens and treatments.
Results
CD4+Foxp3− T cells expressing PD-1 (4PD1hi) accumulate at the tumor site in mice and humans
To begin investigating the role of 4PD1hi, we assessed 4PD1hi tissue distribution in untreated naive and B16 melanoma-bearing mice and observed that these cells are significantly enriched at the tumor site (Figure 1A) and accumulated in the tumor as a function of tumor size (Figure 1B and S1A). In addition, the ratios between CD4+Foxp3−PD-1− (hereafter abbreviated as 4PD1−) or CD8+ T cells and 4PD1hi inversely correlate with tumor burden (Figure 1B and S1A). When the same analyses were performed with Tregs, correlations were not statistically significant (Figure 1B). We substantiated the association between intra-tumor 4PD1hi accumulation and tumor progression in genetically engineered mice that develop melanoma spontaneously (Grm1-TG)(Pollock et al., 2003) and observed that peripheral 4PD1hi increases preceded their intra-tumor accumulation (Figure S1B). We also found that 4PD1hi proliferate more actively in tumor-draining lymph nodes (Figure S1C), display a similar effector memory phenotype independent of anatomic location (Figure S1D) and have a less diverse TCR repertoire compared to 4PD1−, especially at the tumor site (Figure S1E).
Figure 1. 4PD1hi accumulate intratumorally in mice and humans.

(A,B) Mice were injected with 0.25×105 (n=5, tumor onset in 4 of 5 injected mice), 0.5×105 (n=5), 1×105 (n=5, one mouse died before FACS analysis), 2×105 B16 cells (n=5, one mouse died before FACS analysis), and 2 weeks later 4PD1hi and Tregs (percentage of total CD4+) were analyzed in spleen (SP), tumor-draining lymph nodes (DLNs) and tumor (TM) in comparison with spleens from naive mice (SP naive) (mean ± SEM; unpaired t test) (A). Pearson correlation analyses of tumor burden and intra-tumor 4PD1hi %, Tregs % and the indicated intra-tumor T-cell ratios (B). (C) Percentage of 4PD1hi among CD4+ T cells in healthy donors’ PB (n=7), advanced melanoma patients’ PB (n=47) and tumors (TM, n=10); NSCLC patients’ PB (n=51) and tumors (TM, n=10) (mean ± SEM; unpaired t test), and representative plots of Foxp3 and PD-1 expression in CD4+CD45+ T cells, and CD25 expression in 4PD1hi, Tregs and 4PD1− from the indicated samples. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S1.
In immunotherapy-naive melanoma and NSCLC patients, we observed that 4PD1hi frequency is significantly higher in tumor compared to peripheral blood (PB) (Figure 1C), indicating that these cells accumulate intratumorally in humans, as they do in mice. Importantly, 4PD1hi lack both Foxp3 and CD25 expression, thus confirming their non-Treg phenotype (Figure 1C, right).
These results indicate that 4PD1hi are a pool of mature – likely antigen-experienced – cells that exist in naive and tumor-bearing hosts, and preferentially expand in the periphery and accumulate at the tumor site as a function of tumor burden. However, their function in this context remains elusive.
Mouse and human 4PD1hi limit T-cell effector functions
To determine whether 4PD1hi contribute to tumor immune escape, we tested these cells in both in vitro and in vivo suppression assays. 4PD1hi were isolated along with 4PD1− and Tregs from Foxp3-GFP transgenic mice, where the transcription factor Foxp3 can be tracked by GFP expression. We first tested 4PD1hi from spleens of naive Foxp3-GFP mice to clarify their function at the steady state (Figure 2A). Naive splenic 4PD1hi significantly reduced T-cell proliferation, activation, and production of IFN-γ, TNF-α and IL-2, although to a lesser extent than Tregs (Figure 2B,C and S2A,B). These results cannot be attributed to cell competition for proliferation, because 4PD1hi were not capable of sustained division in culture (Figure S2C), or to the acquisition of a Treg phenotype, because Foxp3 or CD25 were not up-regulated in 4PD1hi (Figure 2D). To verify these effects in vivo, we monitored proliferation and activation of Pmel-1/gp100-specific CD8+ T cells adoptively transferred with 4PD1hi or Tregs from tumor-bearing mice and stimulated in vivo by injection of irradiated B16 (Figure 2E schema). Co-transfer of 4PD1hi or Tregs similarly reduced proliferation and expression of activation markers in Pmel-1 CD8+ T cells (Figure 2E).
Figure 2. Mouse 4PD1hi limit T-cell effector functions.

(A) Schema of in vitro suppression assay of CD45.1+ T cells (target) with 4PD1hi, 4PD1− or PD-1−Tregs FACS-sorted from spleens of naive Foxp3-GFP mice (CD45.1−, effectors). (B) CTV dilution and frequency of CD44+CD25+ in total CD45.1+CD4+ target T cells in the indicated co-cultures at the indicated effector:target ratios after 48-hr incubation (mean ± SD; n=2; 2-way ANOVA, 4PD1hi and Tregs vs 4PD1−). (C) Quantification of IFN-γ, TNF-α and IL-2 by FACS-based bead immunoassay in culture supernatants of CD4+ cells alone or co-cultured with the indicated cells for 48 hr (ratio 1:1; mean ± SD; n=2; unpaired t test). (D) Foxp3, CD25 and PD-1 MFI in effector CD45.1−CD4+ T-cell subsets co-cultured with CD45.1+CD4+ target T cells (ratio 1:1) for 48 hr (mean ± SD; n=2; unpaired t test). (E) In vivo suppression assay with 4PD1hi or Tregs FACS-sorted from B16-bearing Foxp3-GFP mice and co-transferred with CFSE-labeled Pmel/gp100-TCR-specific CD8+ T cells (Pmels) (1:1 ratio) into irradiated CD45.1+ recipients and stimulated in vivo with irradiated B16 cells (schema). Proliferation (CFSE dilution) and activation (CD44 and CD25 expression) of CD45.1−Thy1.1+CD8+ Pmels in recipient spleens (mean ± SEM; n=4–5; unpaired t test). * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S2.
We then took advantage of differential CD25 expression between 4PD1hi and Tregs (Figure 1C) to separate these two cell subsets from human samples and compare them in functional assays. Circulating donor-derived 4PD1hi significantly reduced T-cell proliferation, activation, and production of pro-inflammatory cytokines in comparison with 4PD1− (Figure 3A and S3A) and did not acquire expression of Treg-associated markers (Figure S3B). Similarly, 4PD1hi from melanoma and NSCLC lesions consistently limited proliferation, activation and production of pro-inflammatory cytokines of either autologous tumor-infiltrating (TILs) or donor-derived peripheral T cells (Figure 3B,C and Figure S3C) and maintained a distinct phenotype in culture (Figure S3D). The inhibitory capacity of human 4PD1hi, as for mouse 4PD1hi, was consistent yet not always as potent as that of Tregs.
Figure 3. Human 4PD1hi limit T-cell effector function.

(A) FACS gating strategy to sort human 4PD1hi, 4PD1− and Tregs based on PD-1 and CD25 expression in CD4+ T cells and related Foxp3 expression (left). Proliferation (CTVlow) and activation (CD25 MFI) of autologous target CD4+ T cells co-cultured with donor-derived 4PD1hi, 4PD1− and Tregs at 1:1 ratio for 72 hr (middle) (mean ± SD; n=3; unpaired t test, 4PD1hi and Tregs vs 4PD1−). Heatmap with unsupervised hierarchical clustering of the indicated cytokines in co-culture supernatants as assessed by Luminex-based bead immunoassay (right). (B,C) Proliferation (CTVlow) of target autologous (auto) CD4+ TILs (B) or donor-derived allogeneic circulating CD8+ T cells (C) co-cultured with human tumor-infiltrating 4PD1hi, 4PD1− or Tregs (1:1 ratio) for 72 (B) or 96 hr (C), and cytokine production by Luminex-based bead immunoassay in the same cultures. Mean ± SD (B melanoma, n=2 with Tregs and 4PD1hi, n=6 with 4PD1−; B NSCLC, n=2 with 4PD1hi, n=3 with 4PD1− and Tregs; C, n=3); unpaired t test, 4PD1hi and Tregs vs 4PD1−. * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S3.
These results indicate that human and mouse 4PD1hi constitutively limit Teff functions. Given this functional role, we reasoned that their relevance in therapeutic settings should be investigated.
4PD1hi modulation during immune checkpoint blockade
To evaluate the relevance of 4PD1hi in anti-tumor immunity in vivo, we monitored this cell subset in cancer patients treated with immune checkpoint blockade. To detect human PD-1, we employed an Ab whose binding is not cross-blocked by the therapeutic αPD-1 Abs nivolumab or pembrolizumab (Figure S4A). In metastatic NSCLC patients, we found that nivolumab monotherapy reduced peripheral 4PD1hi (Figure 4A, nivo3, blue, n=10). Interestingly, this effect was abrogated by concurrent administration of the αCTLA-4 ipilimumab (Figure 4A, nivo3+ipi1, green, n=21). Moreover, addition of a relatively low (Figure 4A, nivo1+ipi1, black, n=11) or higher dose (Figure 4A, nivo1+ipi3, red, n=8) of ipilimumab to a lower dose of nivolumab (1 mg/kg) produced proportional increases in circulating 4PD1hi compared to the patients treated with nivolumab monotherapy (Figure 4A). We confirmed the capacity of αCTLA-4 monotherapy to increase 4PD1hi in a dose-dependent manner in B16-bearing mice (Figure 4B and S4B). The presence of tumor contributed to αCTLA-4-mediated induction of 4PD1hi, as 4PD1hi did not significantly increase upon treatment with the lower αCTLA-4 dose (100 μg) in non-tumor-bearing C57BL/6J or Balb/c mice (Figure S4C,D).
Figure 4. 4PD1hi modulation and efficacy of immune checkpoint blockade.

(A) Fold changes in circulating 4PD1hi (percentage of total CD4+) in advanced NSCLC patients during treatment with nivo3 (nivolumab 3 mg/kg, once every 2 weeks (q2wks), n=10), nivo3+ipi1 (nivolumab 3 mg/kg + ipilimumab 1 mg/kg, q3wks, q6wks+q2wks, or q12wks+q2wks, n=21), nivo1+ipi1 (nivolumab 1 mg/kg + ipilimumab 1mg/kg, q3wks, or q6wks, n=11) or nivo1+ipi3 (nivolumab 1mg/kg + ipilimumab 3mg/kg, q3wks, n=8) (average ± SEM; 2-way ANOVA with Bonferroni correction, nivo3 vs nivo1+ipi1 and nivo3 vs nivo1+ipi3). Representative FACS plots of Foxp3 and PD-1 expression in CD4+ T cells from NSCLC patients treated with nivo1+ipi3 (red) or nivo3 (blue) at the indicated time points. (B) Circulating 4PD1hi (percentage of CD4+) in B16-bearing mice treated with αCTLA-4 monotherapy (100 μg or 300 μg/cycle; average ± SEM, n=7–10) relative to naive mice (n=5) (2-way ANOVA with Bonferroni correction, treated vs naive mice). (C) Pairwise comparison of 4PD1hi (percentage of CD4+) at the indicated time points relative to baseline in advanced melanoma patients during ipilimumab (ipi, 3 mg/kg, q3wks; n=47) or pembrolizumab treatment (pembro, 2 or 10 mg/kg, q3wks; n=52) (average ± SEM) (left). Representative FACS plots of Foxp3 and PD-1 expression in CD4+ T cells from melanoma patients treated with ipi (red) or pembro (blue) at the indicated time points (middle). Pairwise comparison of circulating 4PD1hi/CD4+ % at baseline and 3 weeks after pembro (right). (D) Average ± SEM tumor diameter (left; n=10; 2-way ANOVA with Bonferroni correction) and Kaplan-Meier tumor-free survival curves (right; pooled data from 3 independent experiments, n=30; log-rank test; number of tumor-free mice approximately 100 days after tumor implantation is reported for each group) of B16-bearing mice treated with VRP-TRP2 and αCTLA-4 and/or αPD-1 as indicated. (E) Intra-tumor 4PD1hi and Tregs frequencies one day after treatment completion in B16-bearing mice treated as in D (average ± SEM; n=9–10; unpaired t test). * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S4.
We confirmed the results achieved in NSCLC patients in larger cohorts of metastatic melanoma patients treated with ipilimumab (Figure 4C, red, n=47) or the αPD-1 pembrolizumab (Figure 4C, blue, n=52, 50/52 upon relapse on ipilimumab). αCTLA-4 increased circulating 4PD1hi, while administration of αPD-1 reduced their frequency (Figure 4C). We further substantiated the capability of αPD-1 (pembrolizumab) to down-regulate 4PD1hi in an independent cohort of melanoma patients(Huang et al., 2017) (Figure S4E). These data indicate that αCTLA-4 and αPD-1 modulate 4PD1hi frequency in opposing directions in cancer patients and suggest that combining different dosages (as in Figure 4A) may differentially affect 4PD1hi, with αPD-1 being able to antagonize the effects of αCTLA-4 as long as αCTLA-4 dose is not in relative excess.
4PD1hi are a biomarker of activity of immune checkpoint blockade
To clarify the clinical implication of these observations, we tested whether the potency of αPD-1 to down-regulate 4PD1hi correlated with the clinical outcome. We assessed the prognostic importance of 4PD1hi frequency and modulation in advanced melanoma patients during pembrolizumab treatment (Figure 4C right) and found that elevated 4PD1hi frequencies and/or lack of significant 4PD1hi down-modulation after PD-1 blockade resulted in a significantly higher risk of death (Table 1, hazard ratio= 1.4 and 4.4 respectively).
Table 1. Post-therapy 4PD1hi and clinical benefit of pembrolizumab.
Hazard ratios (risk of death) for post-therapy 4PD1hi% and 4PD1hi fold reduction by the Cox regression model in advanced melanoma patients treated with pembrolizumab (Figure 4C).
| Variable | n | Hazard Ratio | 95% CI | Cox p value |
|---|---|---|---|---|
| 4PD1hi % (3 weeks post-therapy) | 52 (24 deaths) | 1.4 | (1.16, 1.70) | .0005 *** |
| FoldChange in 4PD1hi% | 52 (24 deaths) | 4.4 | (1.03, 19.14) | .046 * |
We confirmed the therapeutic impact of targeting 4PD1hi in mice by testing the effects of PD-1 blockade in the setting of αCTLA-4 in combination with the anti-melanoma vaccine VRP-TRP2, which we previously found to increase 4PD1hi in association with suboptimal therapeutic effects (Avogadri et al., 2014). The triple combination (VRP-TRP2+αCTLA-4+αPD-1) promoted B16 tumor shrinkage and durable tumor control compared to the individual Abs plus the vaccine (Figure 4D) and reduced intra-tumor 4PD1hi (Figure 4E), as assessed by the anti-PD-1 Ab RMP1-30 that is not cross-blocked by the therapeutic clone RMP1-14 (Figure S4F). VRP-TRP2 plus αPD-1 alone, while preventing an increase in 4PD1hi, promoted intra-tumor accumulation of Tregs (Figure 4E). Concomitant CTLA-4 and PD-1 inhibition in the triple combination counteracted reciprocal induction of 4PD1hi and Tregs by each checkpoint blockade therapy (Figure 4E), thus providing one possible explanation for its increased therapeutic effects (Figure 4D).
Selective PD-1 pathway blockade in 4PD1hi counteracts their inhibitory function
We next questioned whether PD-1 constituted a functional target of 4PD1hi and tested the effect of PD-1 pathway blockade on 4PD1hi inhibition of T-cell tumoricidal function in a 3D killing assay(Budhu et al., 2010). First, we verified the capacity of 4PD1hi to suppress anti-tumor CD8+ T-cell cytotoxicity in this assay (Figure S5A). We then reduced the number and ratios of 4PD1hi cells relative to CD8+ T cells to enable a parallel analysis of 4PD1hi in multiple conditions. Even in this suboptimal setting, 4PD1hi limited B16 killing (Figure 5A). Importantly, PD-1 or PD-L1 blockade restored CD8+ T-cell-mediated B16 killing in the presence of 4PD1hi but did not augment baseline CD8+ T-cell cytotoxicity (Figure 5A), pointing to a specific role of PD-1/PD-L1 inhibition on 4PD1hi for this effect. Given the high PD-1 and/or PD-L1 expression on CD8+ TILs and B16 cells (Figure S5B), we could not exclude a contribution from blocking the PD-1 pathway on those cells. We therefore selectively blocked PD-1 or PD-L1 on 4PD1hi, or 4PD1− as control, before adding these cells to CD8+ TIL-B16 co-cultures. We found that selective blockade of either PD-1 or PD-L1 on 4PD1hi was sufficient to abolish their inhibitory function (Figure 5B), and that 4PD1hi overexpressed PD-L1 in addition to PD-1, particularly at the tumor site (Figure 5C). This suggests that the PD-1 pathway mediates 4PD1hi inhibitory activity.
Figure 5. PD-1/PD-L1 blockade counteracts 4PD1hi inhibitory function.

(A,B) 3D killing assays with B16 cells and intra-tumor 4PD1hi or 4PD1− and CD8+ TILs (CD8:CD4=5×104:1×104, suboptimal conditions) FACS-sorted from untreated B16-bearing Foxp3-GFP mice. Mean ± SD percent of killed B16 in co-cultures treated with αPD-1 or αPD-L1 or matched isotype IgGs after 48-hr incubation (n=2–3) (A). Mean ± SD percent of killed B16 in culture with CD8+ TILs and αPD-1- or αPD-L1-pre-treated 4PD1hi or 4PD1− after 48-hr incubation (n=2–3) (B). (C) PD-L1 MFI in 4PD1hi in comparison with 4PD1− and CD8+ T cells from spleen, tumor-draining lymph nodes (DLNs) and tumor in B16-bearing mice (mean ± SEM; n=10). (D) Human NSCLC-derived 4PD1hi, Tregs and 4PD1− were pre-treated with αPD-1 or control isotype IgG and cultured with stimulated autologous (auto) CD8+ TILs at 1:1 ratio for 72 hr. Quantification by Luminex-based bead immunoassay of IFN-γ and IL-2 in CD8+ TIL cultures with αPD-1- or IgG-pre-treated CD4+ T-cell subsets or incubated with αPD-1 or the matched isotype IgG (mean ± SD; n=2 with Tregs and 4PD1hi, n=3 with CD8+ TILs alone; n=4 with IgG-treated 4PD1−; n=6 with αPD-1-treated 4PD1−). Unpaired t test: * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S5.
We next asked whether PD-1 blockade on 4PD1hi from human tumors affects their inhibitory function. In the absence of TILs and clonogenic tumor cell lines from the same patients to perform 3D killing assays, we adapted the suppression assay described above to measure activation of T cells co-cultured with PD-1-blocked or control 4PD1hi. Human NSCLC-derived 4PD1hi, Tregs and 4PD1− were pre-incubated with αPD-1 or control IgG and co-cultured with stimulated autologous target CD8+ TILs (Figure 5D top). We found increased IFN-γ and IL-2 production in culture of CD8+ TILs with PD-1-blocked 4PD1hi, but not from CD8+ TILs cultured alone with αPD-1 (Figure 5D bottom), suggesting that blocking PD-1 on 4PD1hi may favor the development of cytotoxic anti-tumor T-cell responses.
4PD1hi express a TFH-like phenotype
To investigate the identity of 4PD1hi, we compared RNAseq gene expression profiles of functionally validated mouse and human 4PD1hi, Tregs and 4PD1−. Principal component analysis of variably expressed genes showed that these three CD4+ T-cell subsets are transcriptionally distinct populations both in mice and humans (Figure S6A). Gene set enrichment analysis in 4PD1hi of gene signatures from known CD4+ T-cell subsets revealed extensive overlap with TFH and exhausted T cells (Figure S6B). However, the greatest number of genes shared with 4PD1hi were unique to the TFH phenotype (Figure S6B). Accordingly, 4PD1hi and conventional TFH transcriptomes(Miyauchi et al., 2016) showed overlapping profiles when a comprehensive set of genes previously found differentially expressed (up-regulated and down-regulated) in bona fide TFH(Choi et al., 2015; Kenefeck et al., 2015; Liu et al., 2012; Miyauchi et al., 2016) was analyzed (Figure S6C). Moreover, both mouse and human 4PD1hi were accurately distinguished from 4PD1− and Tregs by genes typically overexpressed in TFH(Kenefeck et al., 2015; Sahoo et al., 2015) (Figure 6A,B).
Figure 6. Mouse and human 4PD1hi are a distinct CD4+ T-cell subset with a TFH-like phenotype.

(A–B) Heatmaps with unsupervised hierarchical clustering and single-sample gene set enrichment analysis (ssGSEA) scores of TFH-associated genes in gene expression data sets from functionally validated mouse splenic (A) and donor-derived (B) 4PD1hi, 4PD1− and Tregs. *, Wilcoxon matched-pairs signed-rank test p=0.03125 (mean ± SEM; A, n=3; B, n=5). (C) 4PD1hi (percent of CD4+ T cells, left; proportion of total cells, right) in tumors from B16-bearing Batf KO or wild-type (WT) mice treated with αCTLA-4 or control isotype IgG (100 μg x4) as assessed by FACS (mean ± SEM; WT mice, n=10; Batf KO mice, n=6–7; unpaired t test) or immunofluorescence staining (IF; mean ± SEM; n=3; unpaired t test). (D) CD86 MFI on circulating B220+CD45+ B cells from B16-bearing mice treated with αCTLA-4 or control isotype IgG (100 μg x4) (left; mean ± SEM; n=9–10, unpaired t test), and on circulating CD19+CD45+ B cells before and during ipilimumab treatment (ipi) in metastatic melanoma patients (right; 3 mg/kg, q3wks; n=16; paired t test). (E,F) In vitro suppression assays with 4PD1hi, memory CD4+ T cells (CD44hiPD-1−Foxp3−CD4+ T cells, Tmem) and Tregs from tumors (E) and spleens (F) of αCTLA-4-treated (100 μg x4) B16-bearing Foxp3-GFP mice at the indicated effector:target ratios. 4PD1hi, Tmem and Tregs FACS gating strategy and baseline CD44 expression after sorting is depicted (E top). Data show mean ± SD proliferation (CTVlow) and activation (CD25+CD44+) of target CD8+ and CD4+ T cells from 48- and 72-hr co-cultures respectively (n=3; E, unpaired t test; F, 2-way ANOVA, 4PD1hi and Tregs vs Tmem). ** = p<0.01, **** = p<0.0001. See also Figure S6.
TFH are a specialized subset of CD4+ T cells – generally defined by CXCR5, Bcl6, ICOS and PD-1 expression – which mature to assist germinal center (GC) B cells to produce high-affinity Abs, mainly via IL-4 and IL-21 release(Akiba et al., 2005; Crotty, 2014; Sage et al., 2013; Sahoo et al., 2015). In both mice and humans, TFH can down-regulate Bcl6 and CXCR5, exit GCs and recirculate in the periphery as memory TFH(Hale and Ahmed, 2015; He et al., 2013; Rao et al., 2017; Sage et al., 2014), and circulating CD4+CXCR5+ T cells have been shown to mirror active TFH responses in secondary lymphoid organs(He et al., 2013). This highlights the plasticity of TFH phenotype according to anatomic location. TFH are also defined by the lack of IL-2Rα (CD25) expression, as IL-2 is a potent inhibitor of their differentiation(Ballesteros-Tato et al., 2012; Johnston et al., 2012). Our findings that 4PD1hi express an effector memory phenotype, lack CD25 and Foxp3 expression, and expand preferentially in secondary lymphoid organs were all in agreement with these TFH features. Consistently, TFH markers were generally expressed at higher levels in 4PD1hi than in Tregs and 4PD1− from mice (Figure S6C–F), healthy donors and cancer patients (Figure S6G–I). However, outside of secondary lymphoid organs, such as in PB and tumor, 4PD1hi less markedly overexpressed these TFH markers and did not always preferentially co-express them, with ICOS as an example being predominantly detected on Tregs in those anatomic locations (Figure S6D,E,G). This would point to a phenotype of GC-experienced TFH in peripheral 4PDhi, which is distinguished by reduced expression of Bcl6, CXCR5 and ICOS(He et al., 2013; Sage et al., 2014). Interestingly, in B16-bearing mice, CTLA-4 blockade up-regulated CXCR5 and Bcl6 in intra-tumor 4PD1hi (Figure S6F top). According to our initial observations, CD25 and Foxp3 were selectively overexpressed in Tregs in these analyses (Figure 6A,B; Figure S6C,G-I).
To corroborate these findings, we tested whether αCTLA-4 could still increase 4PD1hi in Batf knockout (KO) mice, which have profound defects in GC reactions, but a functional T-bet-IFN-γ axis and normal PD-1 expression(Murphy et al., 2013). Although TH17 differentiation is also defective in Batf KO mice(Murphy et al., 2013), the fact that 4PD1hi did not preferentially express the TH17-lineage-defining genes Rorc and Il17a (Figure S6J) quite confidently suggested that eventual differences in 4PD1hi modulation in Batf KO mice could not depend on TH17 deficiency. In accordance with our hypothesis, CTLA-4 blockade loses its capacity to induce intra-tumor 4PD1hi in B16-bearing Batf KO mice (Figure 6C and S6K).
We thus reasoned that 4PD1hi, when increased upon CTLA-4 blockade, could be mechanistically linked to disinhibition of the CTLA-4-mediated control of CD86 expression on APCs (in particular B cells), which is also responsible for Treg suppression of TFH expansion(Hou et al., 2015; Wing et al., 2014). In line with this hypothesis, αCTLA-4-treated B16-bearing mice and melanoma patients up-regulated CD86 on circulating B cells together with 4PD1hi (Figure 6D), suggesting that these effects may be interdependent in vivo. Furthermore, the αCTLA-4 Ab used in our in vivo experiments was able to counteract Treg-mediated inhibition of CD86 expression on B cells and T-cell proliferation in vitro (Figure S6L). However, acquisition of suppressive function was not a general feature of all antigen-experienced CD4+Foxp3− T cells induced upon CTLA-4 blockade. In fact, CD44+ antigen-experienced PD-1−CD4+Foxp3− T cells (Tmem) from the periphery or the tumor of αCTLA-4-treated mice enhanced T-cell proliferation and activation in contrast to 4PD1hi and Tregs (Figure 6E,F).
Dual opposing immune activity of 4PD1hi
If excessive T-cell priming upon CTLA-4 blockade is at the basis of enhanced production of inhibitory TFH-like 4PD1hi, we questioned whether conventional TFH responses could generate a similar T-cell population. We thus induced GC reactions by immunizing mice with sheep red blood cells (sRBC) and analyzed 4PD1hi modulation and function (Figure 7A schema). sRBC modestly promoted PD-1 expression in Foxp3−CD4+ T cells and induced TFH differentiation in splenic and intra-tumor 4PD1hi subset (Figure S7A). Comparison of 4PD1hi from sRBC-treated (sRBC-4PD1hi) and untreated (NT-4PD1hi) B16-bearing mice in functional assays showed that sRBC-4PD1hi inhibit T cells even more powerfully than NT-4PD1hi (Figure 7A and S7B,C). Of note, stronger T-cell inhibitory activity was coupled with higher PD-1 expression levels in sRBC-4PD1hi (Figure 7A).
Figure 7. TFH-like 4PD1hi limit anti-tumor immunity.

(A) In vitro suppression assays with 4PD1hi, Tregs and 4PD1− from spleens of sRBC-immunized or control B16-bearing Foxp3-GFP mice at the indicated ratios. Mean ± SD proliferation (CTVlow) and activation (CD25+CD44+) of target CD4+ T cells, and Foxp3 and PD-1 MFI in CD45.1− 4PD1hi, 4PD1− or Tregs from the same co-cultures after 48-hr incubation (n=2–3; 2-way ANOVA, NT-4PD1hi vs sRBC-4PD1hi). (B) In vitro suppression assays with CXCR5+ and CXCR5− 4PD1hi, 4PD1− and Tregs from spleens (SP) and tumors (TM) of naive and B16-bearing (TB) Foxp3-GFP mice immunized with sRBC. Mean ± SD proliferation (CTVlow) of target CD45.1+CD4+ T cells and quantification of IL-2 by Luminex-based bead immunoassay in culture supernatants after 72-hr incubation with effector cells (1:1 ratio; 4×104 cells from SP; 1×104 cells from TM; n=2–4; unpaired t test). (C) In vitro suppression assays with 4PD1hi, CD44hiPD-1−Foxp3−CD4+ Tmem and Foxp3+ Tregs from spleens of sRBC-immunized Foxp3-GFP mice at the indicated effector:target ratios. Mean ± SD proliferation (CTVlow) and activation (CD25+CD44+) of target CD8+ and CD4+ T cells from 48- and 72-hr co-cultures respectively (n=2–3; 2-way ANOVA, 4PD1hi and Tregs vs Tmem). (D) Average ± SEM tumor diameter of B16-bearing Sh2d1a (SAP) KO and WT C57BL/6J mice treated with αCTLA-4 or control isotype IgG (100 μg x4) starting on day 7 after tumor implantation (suboptimal treatment) (n=4–5; 2-way ANOVA with Bonferroni correction). * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001. See also Figure S7.
We next tested the effects of 4PD1hi on B-cell activation using a T-cell dependent B-cell activation assay, in which B cells mature as a function of the signals released by activated T cells over a short period of time (Figure S7D) (Wing et al., 2014). Both spleen- and tumor-derived 4PD1hi promoted B-cell activation, similar to 4PD1− and in contrast to Tregs, as revealed by B-cell upregulation of CD86 and MHC-II (Figure S7E). To understand whether B-cell stimulatory and T-cell inhibitory activities were retained by the same cells within the 4PD1hi pool independent of the degree of TFH differentiation, and/or were modulated by the presence of tumor, we compared functions of CXCR5+ (mature GC TFH) and CXCR5− 4PD1hi from B16-bearing and naive mice immunized with sRBC (Figure S7F). In all conditions, both 4PD1hi subsets consistently sustained B-cell activation (Figure S7G) and limited Teff functions (Figure 7B), pointing to dual opposing immune modulating activities of 4PD1hi independent of the degree of TFH differentiation. Once again, the suppressive function of 4PD1hi was not shared by PD-1− antigen-experienced memory T cells upon sRBC immunization (Figure 7C). Overall, these findings suggest that exacerbated priming or TFH responses (with αCTLA-4 or immunization with sRBC) can come at the expense of impaired T-cell function, which in tumor-bearing hosts may promote immune evasion. To formally prove this hypothesis, we tested CTLA-4 blockade in Sh2d1a (SAP) KO mice, which specifically lack TFH due to selective abrogation of B-T cell interactions and GC formation(Qi et al., 2008). We found that αCTLA-4 monotherapy, starting when B16 tumors are established (a regimen which is ineffective in wild-type animals, Figure 7D left), could still control tumor growth in Sh2d1a KO mice (Figure 7D right). The mechanism underlying this effect may be multifactorial, as indicated by the multiple immune inhibitory genes overexpressed by TFH-like 4PD1hi cells, including HAVCR2, TGFB and IL10 in addition to PDCD1 (Figure S7H,I). Dissecting the relative contribution of these immunosuppressive molecules and their interplay with the PD-1 pathway will thus be important to deepen the understanding of 4PD1hi biology.
Discussion
A significant proportion of patients still do not benefit from checkpoint blockade. Here, we investigate non-conventional inhibitory cells (4PD1hi) as a potential mechanism limiting activity of immunotherapy. We find that 4PD1hi are present at low frequency in the circulation of normal hosts, accumulate at the tumor site as a function of tumor burden, and constitutively inhibit Teff functions. CTLA-4 blockade promotes increases in 4PD1hi in a dose-dependent manner and counteracting this effect with PD-1 blockade achieves major tumor regression in mice and is associated with a better outcome in patients. PD-1 pathway blockade abolishes 4PD1hi suppressive activity, thus controlling 4PD1hi both quantitatively and functionally. 4PD1hi express TFH-associated genes and stimulate B cells in contrast to Tregs that prevent both Teff function and B-cell activation. This dual opposing activity of 4PD1hi is displayed by both CXCR5+ and CXCR5− 4PD1hi subtests, pointing to a major role of PD-1 expression in defining T-cell-inhibitory/B-cell-stimulatory Foxp3−CD4+ T cells. Accordingly, similar transcriptional programs and B-helper functions have been recently described in PD1hiCD4+ T cells irrespective to CXCR5 expression in patients with rheumatoid arthritis(Rao et al., 2017).
Intratumoral TFH infiltration was previously proposed as a positive prognostic factor in breast and colorectal cancer patients, based on correlation of TFH gene signatures with prolonged survival(Bindea et al., 2013; Gu-Trantien et al., 2013). These TFH gene signatures were selected or found to positively correlate with abundant intratumoral T-cell infiltration(Bindea et al., 2013; Gu-Trantien et al., 2013), which may per se favor prolonged survival(Ascierto et al., 2011). However, expression of TFH markers in the context of a specific type of immune infiltrate and localization within the tumor was also found associated with unfavorable outcome(Bindea et al., 2013), but T-cell inhibitory function of tumor-derived TFH-like cells was not tested. The immune-cell-attracting potential of tumor-associated TFH may be counterbalanced by the T-cell inhibitory function that we identify here in cells with a similar phenotype in melanoma and lung cancer patients, thus helping to explain these apparently discordant results. To add to this complexity, B-cell responses in tumor-bearing hosts can potentially deliver both inhibitory and stimulatory signals for immune-mediated tumor killing(Yuen et al., 2016). These studies, together with our findings, are delineating an intriguing role of TFH and TFH-like cells in anti-tumor immunity, which may vary depending on the tumor type and/or immunogenicity of tumor-associated B-cell epitopes. Here, we show that limiting priming of immunosuppressive TFH-like cells may be important to maximize the anti-tumor activity of immune checkpoint blockade.
CTLA-4 controls priming of CD4+ T cells, in particular TFH, by modulating expression and accessibility of CD86 and CD80 for CD28 co-stimulation(Wang et al., 2015; Wing et al., 2014). Here, we show that αCTLA-4 increases CD86 expression on B cells both in vivo and in vitro and promotes CD4+ T-cell proliferation in vitro; however, only 4PD1hi, and not all antigen-experienced CD44+Foxp3−CD4+T cells induced upon CTLA-4 blockade, acquire T-cell inhibitory function. Accordingly, selective abrogation of TFH differentiation in Sh2d1a KO mice improves anti-tumor activity of suboptimal αCTLA-4. Previous studies reported an increase in ICOS+ T cells upon ipilimumab treatment(Chen et al., 2009; Wei et al., 2017). As ICOS is a TFH marker, these cells could include 4PD1hi. However, elevation in ICOS+ T cells (both CD4+ and CD8+) was associated with a positive outcome of immune checkpoint blockade and was not diminished by αPD-1(J Clin Oncol 31, 2013 (suppl: abstr3003)), as opposed with what we observe for 4PD1hi. Moreover, a recent study has shown that ICOS+CD4+ T cells expanded by checkpoint blockade express a Th1-like effector phenotype(Wei et al., 2017). This suggests that ICOS does not uniquely and specifically distinguish the inhibitory TFH-like 4PD1hi cells described here, and points to ICOS up-regulation as a marker of T-cell activation upon checkpoint blockade.
As 4PD1hi increase and accumulate within the tumor microenvironment as a function of tumor burden, persistent tumor-antigen exposure may facilitate and sustain their generation. Chronic antigen stimulation is a prerequisite for both conventional TFH development(Baumjohann et al., 2013) and T-cell exhaustion(Wherry and Kurachi, 2015) and these two outcomes may result from common molecular pathways. In chronic infection models, partially exhausted CXCR5+CD8+ T cells with a TFH-like phenotype are the preferential target of PD-1-pathway-blockade-mediated reinvigoration, and respond by increasing proliferation and eventually differentiating into terminally exhausted CXCR5−CD8+ T cells(He et al., 2016; Im et al., 2016). The immunosuppressive TFH-like 4PD1hi described here are instead functionally and quantitatively counteracted by PD-1 blockade.
Suppressive CD4+Foxp3− T-cell subsets were reported in previous studies(Gagliani et al., 2013; Liu et al., 2014); however, these regulatory populations were not found to express TFH-like profiles and their distinctive markers are not co-expressed by 4PD1hi, pointing to 4PD1hi induction as a distinct T-cell inhibitory mechanism. The possibility that the TFH differentiation program is coupled with the acquisition of T-cell inhibitory function is not completely counterintuitive if we reason that, in secondary lymphoid organs, T cells and TFH should not divide despite receiving positive stimuli in an immunologically active microenvironment (GCs) that needs to preferentially sustain B-cell proliferation and affinity maturation. Although follicular Tregs (TFR) do control TFH development during GC reactions, the distinctive TFH expression of co-inhibitory molecules, such as PD-1, BTLA, SLAMF6 and TIGIT(Cubas et al., 2013; Kageyama et al., 2012; Sage et al., 2013; Seth et al., 2009) may also provide negative T-cell signals that prevent excessive T-cell proliferation and ensure TCR responsiveness in the follicles despite constant antigen exposure. Accordingly, TFH-like 4PD1hi not only inhibit activation of naive T cells in vitro but also display limited proliferation capacity when cultured alone. Further dissection of the mechanism(s) of suppression of 4PD1hi is important to more broadly understand their biology and the strategies that can counteract their functions.
As activity and tolerability of checkpoint blockade can vary depending on the tumor type, determining the optimal regimen in each individual case is a clinical priority(J Thorac Oncol 10, 2015 (suppl2; abstr786)(Hammers et al., 2017; Hellmann et al., 2016; Larkin et al., 2015; Postow et al., 2015; Wolchok et al., 2013). Given that 4PD1hi are modulated by checkpoint blockade in a dose dependent manner, such a biomarker may be valuable toward this aim. Of note, in advanced NSCLC patients, combination checkpoint blockade achieves a maximal clinical benefit:toxicity balance when exposure to αCTLA-4 is limited to one administration every 12 weeks(Hellmann et al., 2016). Here, we find that patients treated with pembrolizumab after progression on ipilimumab, who started with an increased frequency of 4PD1hi (data not shown), had an unfavorable outcome if 4PD1hi were not efficiently reduced. We were unfortunately unable to test the prognostic value of 4PD1hi in melanoma patients treated with ipilimumab monotherapy due limited sample availability and imbalanced groups of long-term responders/non-responders in this cohort. As checkpoint blockade therapy is becoming available for more cancer patients, monitoring this parameter in larger and more controlled series of patients with different types of malignancy will be important.
In B16-bearing mice vaccinated with VRP-TRP2, therapeutic improvements with αPD-1 in combination with αCTLA-4 was associated with reciprocal control of 4PD1hi and Tregs. Understanding the mechanism responsible for Treg induction by αPD-1 monotherapy and investigation of this effect in cancer patients warrants attention in future studies. PD-1 can control Treg homeostasis by restraining Treg peripheral conversion(Ellestad et al., 2014) as well as TFR development(Sage et al., 2013). PD-1 blockade may thus remove this control and promote the generation of tumor-associated Tregs. As the PD-1 blocking Abs used in this study do not deplete targeted cells, 4PD1hi reduction after αPD-1 may be the result of activation-induced cell death or Treg expansion, which may limit 4PD1hi priming. In support of αPD-1-mediated inhibition of TFH-like 4PD1hi, we found that anti-tumor humoral immunity is hampered in mice treated with PD-1 blockade (data not shown). Overall these observations point to the capability of αCTLA-4 and αPD-1 to perturb GC reactions in tumor-bearing hosts. Investigating these mechanisms in the context of B-cell malignancies would thus be a logical next step.
In summary, we demonstrate that 4PD1hi constitute an unconventional T-cell inhibitory subset with TFH-like features, which can affect the outcome of cancer immunotherapy. Importantly, PD-1/PD-L1 blocking Abs are already an option to control these cells. Monitoring circulating 4PD1hi in patients receiving immune checkpoint blockade will allow for corroborating the clinical value of this parameter in multiple settings and potentially lead to more precise and personalized design of combination immunotherapies.
STAR METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Purified anti-mouse CTLA-4 blocking antibody (clone 9D9) | BioXcell | Cat# BE0146 |
| Purified anti-mouse PD-1 blocking antibody (clone RMP1-14) | BioXcell | Cat# BE0164 |
| Purified anti-mouse PD-L1 blocking antibody (clone 10F.9G2) | BioXcell | Cat# BE0101 |
| Mouse IgG2b isotype control (clone MPC-11) | BioXcell | Cat# BE0086 |
| Rat IgG2a isotype control (clone 2A3) | BioXcell | Cat# BE0089 |
| Rat IgG2b isotype control (LTF-2) | BioXcell | Cat# BE0090 |
| Purified agonist anti-mouse CD3 (clone 145-2C11) | MSKCC antibody core facility | N/A |
| anti-mouse CD16/CD32 Ab (clone 2.4G2) | BD Biosciences | Cat# 553141 |
| APCCy7-labeled anti-mouse CD45 (clone 30-F11) | BD Biosciences | Cat# 557659 |
| AlexaFluor780-labeled anti-mouse CD45.1 (clone A20) | eBioscience | Cat# 47-0453-82 |
| PECy-labeled anti-mouse CD4 (clone RM4-5) | BD Biosciences | Cat# 552775 |
| BV650-labeled anti-mouse CD8a (clone 53-6.7) | Biolegend | Cat# 100741 |
| PE-Texas Red anti-mouse CD8a (clone 5H10) | Invitrogen | Cat# MCD0817 |
| PE-labeled anti-mouse Thy1.1 (clone OX-7) | BD Biosciences | Cat# 554898 |
| AlexaFluor700-labeled anti-mouse B220 (clone RA3-6B2) | BD Biosciences | Cat# 557957 |
| APC-labeled anti-mouse CD19 (clone 1D3) | BD Biosciences | Cat# 550992 |
| PerCPCy5.5-labeled anti-mouse CD19 (clone 1D3) | BD Biosciences | Cat# 551001 |
| ef450-labeled anti-mouse PD-1 (clone RMP1-30) | eBioscience | Cat# 48-9981-82 |
| PE-labeled anti-mouse PD-1 (clone RMP1-30) | eBioscience | Cat# 12-9981-83 |
| APC-labeled anti-mouse PD-1 (clone RMP1-30) | eBioscience | Cat# 17-9981-82 |
| AlexaFluor700-labeled anti-mouse CD44 (clone 1M7) | eBioscience | Cat# 56-0441-82 |
| PE-labeled anti-mouse CD62L (clone MEL-14) | BD Biosciences | Cat# 01265B |
| PerCPCy5.5-labeled anti-mouse CD25 (clone PC61.5) | BD Biosciences | Cat# 551071 |
| PE-labeled anti-mouse CD86 (clone GL-1) | BD Biosciences | Cat# 553692 |
| APC-labeled anti-mouse CD86 (clone GL-1) | BD Biosciences | Cat# 558703 |
| AlexaFluor700-labeled anti-mouse CD86 (clone GL-1) | Biolegend | Cat# 105024 |
| PerCPCy5.5.-labeled anti-mouse H-2Kb (clone AF6-88.5) | BD Bioscience | Cat# 562831 |
| ef450-labeled anti-mouse I-A/I-E (clone M5/114.15.2) | eBioscience | Cat# 48-5321-82 |
| PE-labeled anti-mouse PD-L1 (clone MIH5) | BD Biosciences | Cat# 558091 |
| FITC-labeled anti-mouse ICOS (clone C398.4A) | eBioscience | Cat# 11-9949-82 |
| Biotin-conjugated anti-moue CXCR5 (clone 2G8) | BD Biosciences | Cat# 551960 |
| PECF594-labeled anti-Bcl6 (clone K112-91) | BD Biosciences | Cat# 562401 |
| PECy7-labeled anti-Ki67 (clone B56) | BD Biosciences | Cat# 561283 |
| PECy7-labeled anti-Tbet (clone 4B10) | eBioscience | Cat# 25-5825-82 |
| FITC-labeled anti-mouse Foxp3 (clone FJK-16s) | eBioscience | Cat# 11-5773-82 |
| PerCPCy5.5-labeled anti-mouse Foxp3 (clone FJK-16s) | eBioscience | Cat# 45-5773-82 |
| APC-labeled anti-mouse IL-21 (clone FFA21) | eBioscience | Cat# 17-7211-82 |
| Purified anti-mouse CD4 | R&D Systems | Cat# AF554 |
| Purified anti-mouse Foxp3 (clone FJK-16s) | eBioscience | Cat# 14-5773-82 |
| Purified anti-mouse PD-1 | Sino Biological | Cat# 50124-RP02 |
| Human FcR Blocking Reagent | Miltenyi Biotec. | Cat# 130-059-901 |
| APC-labeled anti-human CD45 (clone HI30) | Tonbo | Cat# 20-0459 |
| FITC-labeled anti-human CD45RA (clone HI100) | BD Biosciences | Cat# 555488 |
| AlexaFluor700 anti-human CD3 (clone UCHT1) | Biolegend | Cat# 300424 |
| BV570-labeled anti-human CD3 (clone UCHT1) | Biolegend | Cat# 300436 |
| FITC-labeled anti-human CD4 (clone RPA-T4) | Tonbo | Cat# 35-0049 |
| AlexaFluor700-labeled anti-human CD4 (clone RPA-T4) | eBioscience | Cat# 56-0049-42 |
| Qdot655-conjugated anti-human CD4 (clone S3.5) | Life Technology | Cat# Q10007 |
| APCCy7-labeled anti-human CD4 APCCy7 (clone RPA-T4) | BD Biosciences | Cat# 557871 |
| PE-Texas Red-labeled CD8 (clone 3B5) | Invitrogen | Cat# MHCD0817 |
| FITC-labeled anti-human PD-1 (clone MIH4) | BD Biosciences | Cat# 561035 |
| PE-labeled anti-human PD-1 (clone MIH4) | BD Biosciences | Cat# 557946 |
| PerCPef710-labeled anti-human PD-1 (clone J105) | eBioscience | Cat# 46-2799-42 |
| APCCy7-labeled anti0human CD25 (clone MA251) | BD Biosciences | Cat# 557753 |
| PECy7-labeled anti-human ICOS (clone ISA-3) | BD Biosciences | Cat# 25-9948-41 |
| AlexaFluor647-labeled anti-human CXCR5 (clone RF8B2) | BD Biosciences | Cat# 558113 |
| PE-labeled anti-human CD19 (clone HIB19) | BD Biosciences | Cat# 555413 |
| PECF594-labeled anti-human CD86 (clone FUN-1) | BD Biosciences | Cat# 562390 |
| eFluor506 fixable viability dye | eBioscience | Cat# 65-0866-14 |
| eFuor450-labeled anti-human Foxp3 (clone PCH101) | eBioscience | Cat# 48-4776-42 |
| PECy7-labeled anti-human CTLA-4 (clone 14D3) | eBioscience | Cat# 25-1529-42 |
| APC-labeled anti-human CTLA-4 (clone BNI3) | BD Biosciences | Cat# 560938 |
| Purified blocking anti-human PD-1 | provided by Bristol-Myers Squibb | N/A |
| Bacterial and Virus Strains | ||
| VRP-TRP2 | AlphaVax | N/A |
| Biological Samples | ||
| Sheep red blood cells, packed 10% | Innovative Research | Cat# IC10-0210 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Fibrinogen | American Diagnostica | Cat# 436/1 |
| Collagen I | BD Bioscience | Cat# 354236 |
| Thrombin | Sigma | Cat# T6884 |
| Collagenase | Sigma | Cat# C9891 |
| Trypsin | Sigma | Cat# T8003 |
| Methylene Blue | Sigma | Cat# M9140 |
| DNAse I | Roche | Cat# 10104159001 |
| Liberase TL | Roche | Cat# 05401020001 |
| Percoll density gradient media | GE Healthcare | N/A |
| Mouse IFNγ, recombinant | Peprotech | Cat# 315-05 |
| Mouse IL-2, recombinant | Peprotech | Cat# 212-12 |
| Leucoagglutinin PHA-L | Sigma | Cat# L2769 |
| BD™ CBA Mouse Th1/Th2/Th17 Cytokine Kit | BD Biosciences | Cat# 560485 |
| MILLIPLEX MAP Mouse TH17 Magnetic Bead Panel | Millipore | Cat# MTH17MAG-47K |
| Th1/Th2/Th9/Th17/Th22/Treg Cytokine 18-Plex Human ProcartaPlex™ Panel | Thermo Fisher Scientific | Cat# EPX180-12165-901 |
| Critical Commercial Assays | ||
| CD4 Microbeads, mouse | Miltenyi Biotec | Cat# 130-117-043 |
| CD8 Microbeads, mouse | Miltenyi Biotec | Cat# 130-117-044 |
| CD19 Microbeads, mouse | Miltenyi Biotec | Cat# 130-052-201 |
| CD4 Microbeads, human | Miltenyi Biotec | Cat# 130-045-101 |
| CellTrace CFSE cell proliferation kit | Life Technologies | Cat# C34554 |
| CellTrace Violet cell proliferation kit | Life Technologies | Cat# C34557 |
| FoxP3/Transcription Factor Staining Buffer Set | eBioscience | Cat# 00-5523-00 |
| Dynabeads™ Human T-Expander CD3/CD28 | Thermo Fisher Scientific | Cat# 11141D |
| Pharm Lyse Buffer | BD Biosciences | Cat# 555899 |
| Deposited Data | ||
| Mouse 4PD1hi, 4PD1neg and Treg RNAseq data sets | This paper | GSE95756 |
| Human 4PD1hi, 4PD1neg, Treg RNAseq dat sets | This paper | GSE95754 |
| Mouse TFH gene expression data set | Gene expression omnibus | GSE85316 |
| Mouse Th1, Th2, Th17, iTreg and nTreg gene expression data sets | Gene expression Omnibus | GSE14308 |
| Memory, effector and exhaustion mouse CD4+ T cell data sets | Gene expression Omnibus | GSE30431 |
| Mouse Tr1 data set | Gene expression Omnibus | GSE92940 |
| Experimental Models: Cell Lines | ||
| B16F10 | Originally provided by I. Fidler (M. D. Anderson Cancer Center, Houston, TX) | N/A |
| TUBO | Provided by Dr G Forni (University of Turin, Italy) | N/A |
| Experimental Models: Organisms/Strains | ||
| Balb/c | Jackson Laboratory | Stock n: 000651 |
| C57BL/6J | Jackson Laboratory | Stock n: 000664 |
| CD45.1+ C57BL/6J | Jackson Laboratory | Stock n: 002014 |
| Batf Knockout (B6.129S-Batftm1.1Kmm/J) | Jackson Laboratory | Stock n: 013758 |
| Sh2d1a (SAP) Knockout (B6.129S6-Sh2d1atm1Pls/J) | Jackson Laboratory | Stock n: 025754 |
| Foxp3-GFP C57BL/6J | Gift from Alexander Rudensky (MSKCC, New York, NY) | N/A |
| Pmel-1/gp100-specific CD8 TCR transgenic C57BL/6J | Gift from Nicholas Restifo (NCI, Bethesda, MD) | N/A |
| Grm1-TG mice | Gift from S. Chen (Rutgers, The State University of New Jersey, Piscataway, NJ) | N/A |
| Oligonucleotides | ||
| Primers for spectratyping, see Table 2 | ||
| Pdcd1 TaqMan gene expression assay | Thermo Fisher Scientific | Cat# Mm00452054_m1 |
| Bcl6 TaqMan gene expression assay | Thermo Fisher Scientific | Cat# Mm00477633_m1 |
| Cxcr5 TaqMan gene expression assay | Thermo Fisher Scientific | Cat# Mm00432086_m1 |
| Icos TaqMan gene expression assay | Thermo Fisher Scientific | Cat# Mm00497600_m1 |
| Il21 TaqMan gene expression assay | Thermo Fisher Scientific | Cat# Mm00517640_m1 |
| Gapdh TaqMan gene expression assay | Thermo Fisher Scientific | Cat# 4352932E |
| Software and Algorithms | ||
| FlowJo 10.2 | Tree Star Inc. | https://www.flowjo.com/solutions/flowjo |
| Prism 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| R Studio | https://www.rstudio.com/ | |
| Pannoramic Viewer | 3DHistech | https://www.3dhistech.com/pannoramic_viewer |
| FIJI/ImageJ software | https://fiji.sc/ | |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Jedd D. Wolchok (wolchokj@mskcc.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mouse procedures were performed in accordance with institutional protocol guidelines at MSKCC. Wild-type Balb/c and wild-type, CD45.1+ congenic, Batf KO, and Sh2d1a (SAP) KO C57BL/6J mice were obtained from the Jackson Laboratory. Foxp3-GFP transgenic mice were generously provided by Dr. Alexander Rudensky and backcrossed to C57BL/6J at MSKCC. Pmel-1/gp100-specific CD8 TCR transgenic mice were a gift from Nicholas Restifo (NCI, Bethesda, MD). Grm1-TG mice, where ectopic expression of the metabotropic receptor Grm1 (glutamate receptor 1) in melanocytes spontaneously drives melanomagenesis(Pollock et al., 2003), were provided by S. Chen (Rutgers, The State University of New Jersey, Piscataway, NJ). Mice were maintained according to NIH Animal Care guidelines, under a protocol approved by the MSKCC Institutional Animal Care Committee. Littermates of same age (6–8-week old, unless otherwise specified) and same sex were randomly assigned to experimental groups.
Tumor cell lines
The B16F10 mouse melanoma cell line was originally obtained from I. Fidler (M. D. Anderson Cancer Center, Houston, TX) and cultured in RPMI 1640 medium supplemented with 10% inactivated FBS, 1× nonessential amino acids and 2 mM l-glutamine. The BALB-neu derived mammary carcinoma cell line TUBO was kindly provided by Dr G Forni (University of Turin, Italy) and cultured in DMEM supplemented with 20% inactivated FBS, 1× nonessential amino acids and 2 mM l-glutamine. We confirmed expression of melanoma differentiation antigens in B16F10 melanoma cells and expression of rat Her-2/neu in TUBO breast carcinoma cells. Cell lines were routinely screened to avoid mycoplasma contamination and maintained in a humidified chamber with 5% CO2 at 37°C for up to 1 week after thawing before injection in mice.
Patient material
All patients and healthy donors signed an approved informed consent before providing tissue samples. Patient samples were collected on a tissue-collection protocol approved by the MSKCC Institutional Review Board. Donors’ PBMCs were obtained from whole blood using a density gradient (Ficoll Paque PLUS, GE Healthcare) and then processed for CD4+ or CD8+ T-cell isolation as described below or cryopreserved in 10% DMSO FBS. Patients’ PBMCs were isolated from whole blood collected in CPT tubes containing sodium heparin (BD Vacutainer) according to the manufacturer’s instruction and cryopreserved in 10% DMSO FBS until use. Single cell suspensions from patients’ tumors were obtained by digesting tumor samples with type I collagenase (2 mg/mL), type V hyaluronidase (2 mg/mL) and type IV deoxyribonuclease I (200 U/mL) in serum-free RPMI 1640 using a GentleMACS Octo Dissociator (Miltenyi Biotec)(Holmgaard et al., 2015). Advanced NSCLC (n=50) and melanoma patients (treated with ipilimumab, n=47; treated with pembrolizumab, n=52) were treated with checkpoint blockade as part of NCT01454102, NCT00495066 and NCT01295827 clinical trials respectively.
METHOD DETAILS
In vivo tumor injection and treatments
B16F10 melanoma cells were implanted intradermally (105 cells, for tumor-growth and survival analyses) or subcutaneously in matrigel (Matrigel Matrix Growth Factor Reduced, Becton Dickinson) (2×105 cells, for immune-cell infiltrate analyses). Vaccination with VRP-TRP2 (AlphaVax Inc.) was performed by injection of 1×106 virus-like replicon particles (VRPs)(Zappasodi and Merghoub, 2015) expressing mouse TRP2 into the plantar surface of each footpad for 3 times 1 week apart, starting 3 days after tumor implantation(Avogadri et al., 2014). Treatment with αCTLA-4 (clone 9D9, BioXcell, 100 μg or 300 μg/injection), αPD-1 (clone RMP1-14, BioXcell, 250 μg/injection) or the matched isotype IgGs (BioXcell) was started 3–4 (optimal treatment) or 6–7 days (suboptimal treatment) after tumor implantation for respectively 5 or 4 intraperitoneal (i.p.) administrations 3 days apart. Immunization with sRBC was performed i.p. with 200 μl 10% volume/volume sRBC solution (Innovative Research). TUBO breast carcinoma cells were implanted subcutaneously in Balb/c mice (106 cells/mouse) and αCTLA-4 treatment was started 10 days after. Animals were monitored at least twice a week and were considered tumor-free until lesions were palpable.
FACS and cell sorting
Tumors were dissociated after 30 min incubation with Liberase TL and DNAse I (Roche) to obtain single-cell suspensions. When tumor mass exceeded 0.1 gr, immune-cell infiltrates were enriched by Percoll (GE Healthcare) gradient centrifugation. Cells from tumor-draining lymph nodes and spleens were prepared by mechanical dissociation on 40 μM filters and RBC lysis (ACK buffer, Lonza). Mouse PB was collected by retro-orbital puncture and red blood cells were lysed with Pharm Lyse Buffer (BD Bioscences). Surface staining of mouse cells was performed after 15 min pre-incubation with anti-mouse CD16/CD32 Ab (clone 2.4G2; BD Biosciences) to block Fcγ receptor binding, with panels of appropriately diluted fluorochrome-conjugated Abs (from BD Biosciences, eBioscience or Invitrogen) against the following mouse proteins in different combinations: CD45 (clone 30-F11), CD45.1 (clone A20), CD4 (clone RM4-5), CD8a (clone 5H10), Thy1.1 (clone OX-7), B220 (clone RA3-6B2), CD19 (clone 1D3), PD-1 (clone RMP1-30), CD44 (clone IM7), CD62L (clone MEL-14), CD25 (clone PC61.5), CD86 (clone GL-1), H-2Kb (clone AF6-88.5), I-A/I-E (clone M5/114.15.2), PD-L1 (clone MIH5), ICOS (clone C398.4A), CXCR5 (biotin-conjugated clone 2G8, followed by PE-/APC-labeled streptaividin staining), and an eFluor506 fixable viability dye. For intracellular staining, mouse cells were fixed and permeabilized (Foxp3 fixation/permeabilization buffer, eBioscience) and incubated with appropriately diluted PECF594-labeled anti-Bcl6 (clone K112-91, BD Biosciences), PECy7-labeled anti-Ki67 (clone B56, BD Biosciences) or Tbet (clone 4B10, eBioscience) and FITC-labeled anti-Foxp3 (clone FJK-16s, eBioscience) Abs. Surface staining of human cells was performed in the presence of the Fcγ receptor Blocking Reagent (Miltenyi Biotec) with proper dilutions of fluorochrome-conjugated Abs (from BD Biosciences, eBioscience or Tonbo) against the following human proteins in different combinations: CD45 (clone HI30), CD45RA (clone HI100), CD3 (clone UCHT1), CD4 (clone RPA-T4), PD-1 (clone MIH4 or J105 in αPD-1-treatment naive samples), CD25 (clone MA251), ICOS (clone ISA-3), CXCR5 (clone RF8B2), CD19 (clone HIB19), and CD86 (clone FUN-1), and an eFluor506 fixable viability dye. For intracellular staining, human cells were fixed and permeabilized (Foxp3 fixation/permeabilization buffer, eBioscience) and then incubated with appropriately diluted eFuor450-labeled anti-Foxp3 (clone PCH101, eBiosciences), PECF594-labeled anti-Bcl6 (clone K112-91), and APC-labeled anti-CTLA-4 (clone BNI3, BD Biosciences) Abs.
For intracellular cytokine staining, mouse immune cells were re-stimulated with 500 ng/ml PMA and 1 μg/ml ionomycin in complete RPMI 1640 supplemented with 1 mM sodium pyruvate and 50 μM β-mercaptoethanol at 37°C. After 1 hour, 1x GolgiStop and 1x GolgiPlug (BD Biosciences) were added to the cultures and incubated for additional 4–5 hr at 37°C. Surface staining was performed after Fcγ receptor blockade by incubation with eFluor450-labeled anti-PD-1, AlexaFluor(AF)700-labeled anti-CD4 and APCCy7-labeled anti-CD45 (BD Biosciences) Abs and an eFluor506-labeled fixable viability dye (eBioscience). After 30 min incubation, cells were washed, fixed and permeabilized with the Foxp3 fixation/permeabilization buffer (eBioscience) according to the manufacturer’s instructions and stained for 45 min with FITC-labeled anti-Foxp3 and APC-labeled anti-IL-21 (clone FFA21) Abs (eBioscience).
Mouse T-cell subsets were sorted from Foxp3-GFP mice by using CD4-pre-enriched splenocytes (CD4 Microbeads, Miltenyi Biotec) or tumor immune infiltrate enriched by Percoll gradient centrifugation. Briefly, following incubation with anti-mouse CD16/CD32 Ab, samples were stained with anti-CD4, anti-CD8, anti-CD44 and anti-PD-1 Abs in different combinations depending on the populations to isolate. DAPI was added to stained samples immediately before acquisition. To isolate CXCR5+ and CXCR5− 4PD1hi and conventional TFH, cell suspensions were first incubated with a biotin-conjugated anti-CXCR5 Ab, washed, and then stained with fluorochrome-conjugated surface Ab cocktail including PE-labeled streptavidin. Human 4PD1−, Tregs, 4PD1hi and CD8+ T cells were sorted upon incubation with the Fcγ receptor Blocking Reagent (Miltenyi Biotec), and staining with FITC-labeled anti-CD4, PE-Texas Red CD8 (clone 3B5, Invitrogen), PerCPC-eF710-labeled anti-PD-1, APC-labeled anti-CD45 and APCCy-labeled anti-CD25 Abs, and DAPI immediately before acquisition. FACS sorting was conducted on a FACSAria II cell sorter (BD Biosciences). After gating according to lymphocyte morphology, excluding doublets and dead cells, CD4+ T cells were sub-gated into Foxp3-GFP−PD-1− (mouse 4PD1−), Foxp3-GFP−PD-1−CD44+ (mouse Tmem), Foxp3-GFP+ (total mouse Tregs) or Foxp3-GFP+PD-1− (conventional mouse Tregs), and Foxp3-GFP−PD1hi (mouse 4PD1hi), or CD25−PD-1− (human 4PD1−), CD25+ (human Tregs) and CD25−PD1hi (human 4PD1hi) to sort the indicated populations from mouse and human tissues respectively. Conventional TFH were sorted as CD4+Foxp3-GFP−CXCR5+PD-1hi T cells from spleens of sRBC-treated Foxp3-GFP mice.
In vitro assays
A 3D collagen–fibrin gel culture system previously described(Budhu et al., 2010) was adapted to study the function of suppressive T cells. Briefly, 0.1×105 viable B16F10 target cells were co-embedded into collagen–fibrin gels with 1×105 or 0.5×105 effector CD8+ T cells alone or together with 0.25×105 or 0.1×105 (4:1 or 5:1 ratio) 4PD1−, Tregs or 4PD1hi FACS-sorted from B16F10 nodules. CD8+ T cells were from the tumor or in vitro cultures of gp100-primed splenocytes (5-day stimulation with gp100 peptide, AnaSpec) from Pmel-1/gp100-specific TCR transgenic mice. B16F10 target cells were pre-incubated with 100 ng/ml IFN-γ to allow MHC-I and MHC-II up-regulation. Gels were lysed after 48 hr, and tumor cells were diluted and plated in 6-well plates for colony formation. After 7 days, plates were fixed with 3.7% formaldehyde and stained with 2% methylene blue before counting colonies as described(Budhu et al., 2010). Where indicated, 4PD1hi, and 4PD1− as control, were pre-incubated with 10 μg/ml αPD-1 (clone RMP1-14, BioXcell) or αPD-L1 (clone 10F.9G2, BioXcell) or matched isotype IgGs (BioXcell) for 30 min on ice and after extensive washes embedded into the gels. Alternatively, PD-1/PD-L1 blocking Abs (10 μg/ml) were directly added to the gels.
Suppression assays with mouse cells were performed by incubating at the indicated ratios 4PD1−, Tmem, Tregs or 4PD1hi from Foxp3-GFP mice with CellTrace Violet (CTV, Invitrogen)-labeled target T cells immunomagnetically purified (CD4 and CD8 Microbeads, Miltenyi Biotec) from spleens of CD45.1+ C57BL/6J congenic mice. Cultures were stimulated for 48–72 hr with 0.5 μg/ml soluble αCD3 Ab and irradiated splenocytes before analyses of target T-cell CTV dilution (proliferation) and CD25 and CD44 up-regulation (activation).
B-cell activation/T-cell proliferation assays(Wing et al., 2014) with CTLA-4 blockade were performed in a similar way by using, in place of irradiated splenocytes, live CD19+ B cells immunomagnetically purified from spleens (CD19 Microbeads, Miltenyi Biotec) of CD45.1+ C57BL/6J congenic mice, and treating cultures with 50 μg/ml αCTLA-4 (clone 9D9, BioXcell) or the matched isotype IgG.
T-cell dependent B-cell activation assays were adapted from Wing et al.(Wing et al., 2014) and performed by stimulating CD45.1+CD19+ B cells with 5 μg/ml PHA (Sigma) and 20 U/ml recombinant mouse IL-2 alone or in the presence of CD45.1−CD4+ T-cell subsets at 2:1 ratio for 48 hr. B-cell activation was measured by FACS analysis of CD86 and MHC-II expression.
Suppression assays with human cells were performed by incubating 4PD1−, Tregs or 4PD1hi FACS-sorted from PB or tumor cell suspensions with an equal amount of CTV-labeled autologous or allogeneic donor-derived T cells. Cultures were suboptimally stimulated with αCD3/αCD28 microbeads (Dynabeads Human T-Expander CD3/CD28, ThermoFisher) until CTV dilution was detected in control cultures (72–96 hr). Target T-cell CTV dilution (proliferation) and CD25 up-regulation (activation) were then quantified in all samples. Where indicated, αPD-1 (generously provided by Bristol-Myers Squibb, 10 μg/ml), or matched isotype IgG as control, was added in culture or used to pre-block PD-1 on human CD4+ T-cell subsets by 30 min incubation on ice before co-culturing them with target T cells.
Cytokine concentrations in culture supernatants were quantified by using either BD CBA Cytokine Kits (BD Biosciences) or Luminex-based bead multiplex immunoassays according to the manufacturers’ instructions (eBioscience and Millipore). Heatmaps showing cytokine production were generated in the R statistical environment using log2-transformed cytokine concentrations.
In vivo suppression assay
4PD1hi and Tregs were FACS-sorted from B16-bearing Foxp3-GFP transgenic mice and co-transferred with CFSE-labeled Pmel-1/gp100-specific CD8+ T cells, purified from the spleen of Pmel-1/gp100 TCR transgenic Thy1.1+ mice, at 1:1 ratio via tail vein injection into irradiated CD45.1+ recipients (600 cGy total body irradiation). The day after transfer, recipient mice were immunized with intradermal administration of 2×105 irradiated B16 cells to stimulate transferred T cells in vivo. Seven days later, recipient mice were sacrificed, and spleens processed for FACS analysis of CFSE dilution and activation markers in Pmel-1/gp100-specific Thy1.1+CD8+ T cells.
Immunofluorescence staining and image processing
Multiplex immunofluorescence stainings were performed at the Molecular Cytology Core Facility of MSKCC using the Discovery XT processor (Ventana Medical Systems), as previously reported(Yarilin et al., 2015). Briefly, tissue sections were deparaffinized with EZPrep buffer (Ventana Medical Systems) and antigen retrieval was performed with CC1 buffer (Ventana Medical Systems). Sections were blocked for 30 min with Background Buster solution (Innovex) followed by avidin/biotin blocking for 8 min. Stainings were performed sequentially starting with an anti-CD4 Ab (polyclonal, R&D Systems, 2 μg/ml) followed by an anti-Foxp3 Ab (clone FJK-16s, eBioscience, 0.5 μg/ml), and finally an anti-PD-1 Ab (polyclonal, Sino Biological, 1 μg/ml). Sections were incubated with primary Abs for 5–6 hr followed by incubation with appropriate biotin-conjugated secondary Abs (Vector labs, 1:200) for 60 min. Detection was performed with Streptavidin-HRP D (part of DABMap kit, Ventana Medical Systems), followed by incubation with AF488-, or AF568-, or AF647-labeled Tyramide (Invitrogen) prepared according to manufacturer’s instructions with predetermined dilutions. Slides were counterstained with DAPI (Sigma Aldrich, 5 μg/ml) for 10 min. Stained slides were scanned using Pannoramic Flash (Perkin Elmer) using customized AF488, AF568, AF647, and DAPI filters to separate the channels. Relevant tissue regions were drawn using Pannoramic Viewer (3DHistech) and exported as TIFF images at full resolution (0.325 μm/pixel). Image analysis was performed using the FIJI/ImageJ software (NIH). DAPI channel was used to segment and count the number of cells in each region. Each nuclear signal was dilated appropriately to cover the entire cell. Regions of interest were drawn around each cell and matched to signals detected in other channels in order to count the number of positive cells for each individual staining as well as for double or triple stainings.
Real time quantitative PCR
Total RNA was extracted from FACS-purified CD4+ T-cell subsets by using TRIZOL reagent (Invitrogen) and reverse-transcribed into cDNA using the High Capacity cDNA Transcription kit (Applied Biosystems). Expression of the indicated transcripts was quantified with the Fluidigm Biomark™ system by using the appropriate FAM-MGB-conjugated TaqMan primer probes (Applied Biosystem) upon target gene pre-amplification according to the manufacturer’s protocol. Gene expression was normalized relative to glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Data were analyzed by applying the 2^(-dCt) calculation method.
Spectratyping
RNA from FACS-purified 4PD1−, Tregs and 4PD1hi was prepared and used for cDNA synthesis. The cDNA was used as a template to amplify the TCR BV repertoire with 24 BV-specific primers and a common BC-specific primer pairs (Table S1). BV-BC PCR products were subjected to a cycle of elongation (run-off) with an internal FAM- or HEX-labeled BC-primer. Each PCR product, representing a different TCR BV family, was size separated by electrophoresis using a 48-capillary 3730 DNA Analyzer (Life Technologies), and the product lengths were identified using the Peak Scanner software 2 (Applied Biosciences).
RNAseq
Whole transcriptome libraries were generated from RNA extracted from FACS-sorted CD4+ T cell subsets, amplified using the SMARTer Universal Low Input RNA Kit (Clontech), and sequenced on a Proton sequencing system using 200bp version 2 chemistry at the Integrated Genomics Operation Core Facility at MSKCC. Briefly, after ribogreen quantification and quality control by the Agilent BioAnalyzer (RIN>7), cDNA was synthetized using the SMARTer Universal Low Input RNA Kit according to the manufacturer’s guidelines, and then fragmentated with covaris E220. The fragmented sample quality and yield were evaluated with the Agilent BioAnalyzer. Subsequently, the fragmented material underwent whole transcriptome library preparation according to the Ion Total RNA-Seq Kit v2 protocol (Life Technologies), with 12–16 cycles of PCR. Samples were barcoded, template-positive Ion PI™ and Ion Sphere™ Particles (ISPs) were prepared using the ion one touch system II and Ion PI™ Template OT2 200kit v2 Kit (Life Technologies). Enriched particles were sequenced on a Proton sequencing system using 200bp version-2 chemistry. An average of 70×106 to 80×106 reads was generated per sample.
The raw output BAM files were converted to FASTQ using PICARD (version 1.119) Sam2Fastq. Reads were then trimmed using fastq_quality_trimmer (version 0.0.13) with default settings. For analyses conducted in mouse cells, the trimmed reads were first mapped to the mouse genome using rnaStar (version 2.3.0e). The genome used was MM9 with junctions from ENSEMBL (Mus_musculus.NCBIM37.67) and a read overhang of 49. Any unmapped reads were mapped to MM9 using BWA MEM (version 0.7.5a). For analyses conducted in human cells, the genome used was HG19 with junctions from ENSEMBL (GRCh37.69_ENSEMBL) and a read overhang of 49. Any unmapped reads were mapped to HG19 using BWA MEM (version 0.7.5a). The two mapped BAM files were then merged and sorted and gene level counts were computed using htseq-count (options -s y -m intersection-strict) and the same gene models (Mus_musculus.NCBIM37.67 or GRCh37.69_ENSEMBL).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNAseq analysis
Heatmaps of expressed genes were generated using log2-transformed counts. Unsupervised hierarchical clustering was performed using hclust with Euclidean distance and Ward linkage. Heatmap and unsupervised hierarchical clustering of 4PD1hi, Treg and previously reported conventional TFH(Miyauchi et al., 2016) transcriptomes with respect to a broad list of TFH differentially expressed genes(Choi et al., 2015; Kenefeck et al., 2015; Liu et al., 2012; Miyauchi et al., 2016) (Table S2) were generated with log2-transformed counts normalized relative to the naive T-cell data set in each study. PCA was performed on log2-transformed gene counts using the prcomp package (with parameters center = TRUE, scale = TRUE). ssGSEA was implemented using the GSVA(Hanzelmann et al., 2013) package in R to measure the level of enrichment of a TFH gene signature(Kenefeck et al., 2015) in the different CD4+ T-cell subsets. ssGSEA takes as input the genome-wide transcriptional profile of a sample and computes an overexpression measure for a gene list of interest relative to all other genes in the genome(Barbie et al., 2009). All analyses after gene count generation were conducted in the R statistical environment (R development Core Team, 2008; ISBN 3-900051-07-0) (version 3.1.3).
FACS analysis
Samples were acquired on an LSRII or Fortessa flow cytometer (BD Biosciences) using BD FACSDiva software (BD Biosciences) and data analyzed with FlowJo 10.2 software (Tree Star Inc.).
Comparison between groups
Two-sided Student’s t test and 2-way ANOVA (with Bonferroni’s multiple comparisons test) were used to detect statistically significant differences between groups. P values for tumor-free survival analyses were calculated with log-rank (Mantel-Cox) test. Pearson correlation test was used to analyze dependency between variables. The Cox regression model was used to calculate significant hazard ratios of continuous variables. Statistical analyses were performed on the Prism 7.0a software (GraphPad Software) version for Macintosh Pro personal computer. Detailed information of the statistical test and number of observations/replicates used in each experiment, and the definition of center and dispersion is appropriately reported in the legend of each figure. Significance was defined as follows: * = p<0.05, ** = p<0.01, *** = p<0.001, **** = p<0.0001.
DATA AVAILABILITY
The data sets generated in this study have been submitted to the GEO (Gene Expression Omnibus) repository and will be publicly available after December 1st 2018 (GSE95754, GSE95756). Other data sets used in the study are available in the GEO repository, GSE14308, GSE30431, GSE85316, GSE92940.
Supplementary Material
Significance.
We demonstrate the immunosuppressive function of TFH-like cells that are modulated by PD-1 and CTLA-4 blockade in opposing directions, thus broadening our understanding of the incremental activity of combination checkpoint blockade. Our results indicate that these cells may serve as a pharmacodynamic and prognostic biomarker in patients treated with checkpoint blockade. Since activity and tolerability of these therapies can vary depending on the tumor type and determining the optimal regimen in each individual case is a clinical priority, monitoring 4PD1hi changes during checkpoint blockade may be important to guide optimization of combination schedules and dosages.
Highlights.
CD4+Foxp3−PD-1hi T cells (4PD1hi) negatively regulate T-cell responses
CTLA-4 and PD-1 blockade modulate 4PD1hi frequency in opposing directions
4PD1hi are a pharmacodynamic and negative prognostic factor of checkpoint blockade
Checkpoint blockade regimens may be optimized based on circulating 4PD1hi levels
Acknowledgments
We thank the Immune Monitoring, Flow Cytometry, Integrated Genomics Operation and Molecular Cytology Core Facilities at MSKCC for technical assistance; Dr Virginia Pascual and Dr Hideki Ueno for sharing their expertise on TFH biology. This research was funded in part through the NIH/NCI Cancer Center Support Grant P30 CA008748, the Swim Across America, Ludwig Institute for Cancer Research, Parker Institute for Cancer Immunotherapy and Breast Cancer Research Foundation. R.Z. is the recipient of the Parker Institute for Cancer Immunotherapy scholar award.
Footnotes
Author contribution
R.Z., T.M. and J.D.W. developed the concept and discussed experiments; R.Z. designed, performed, and analyzed experiments, and wrote the manuscript; S.B. contributed to the 3D killing assay design and helped with the related experiments; M.H. provided NSCLC patients’ material; M.P. and A.C.H. provided melanoma patients’ material; Y.S. and S.M. contributed to the bioinformatic analyses; B.G. processed patients’ samples and provided technical assistance; H.Z. maintained mouse colonies and provided technical assistance; C.L. and Y.L. provided technical assistance; D.H.C. contributed to the adoptive transfer experiments; K.S.P. performed statistical analyses of survival in patients; E.J.W. contributed to the phenotypic characterization of 4PD1hi; T.M. and J.D.W. supervised the progress of the study and edited the manuscript.
Declaration of Interests
RZ, TM and JDW are inventors on a patent application related to this work, filed by MSKCC.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akiba H, Takeda K, Kojima Y, Usui Y, Harada N, Yamazaki T, Ma J, Tezuka K, Yagita H, Okumura K. The role of ICOS in the CXCR5+ follicular B helper T cell maintenance in vivo. J Immunol. 2005;175:2340–2348. doi: 10.4049/jimmunol.175.4.2340. [DOI] [PubMed] [Google Scholar]
- Ascierto ML, De Giorgi V, Liu Q, Bedognetti D, Spivey TL, Murtas D, Uccellini L, Ayotte BD, Stroncek DF, Chouchane L, et al. An immunologic portrait of cancer. J Transl Med. 2011;9:146. doi: 10.1186/1479-5876-9-146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avogadri F, Zappasodi R, Yang A, Budhu S, Malandro N, Hirschhorn-Cymerman D, Tiwari S, Maughan MF, Olmsted R, Wolchok JD, Merghoub T. Combination of alphavirus replicon particle-based vaccination with immunomodulatory antibodies: therapeutic activity in the B16 melanoma mouse model and immune correlates. Cancer immunology research. 2014;2:448–458. doi: 10.1158/2326-6066.CIR-13-0220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros-Tato A, Leon B, Graf BA, Moquin A, Adams PS, Lund FE, Randall TD. Interleukin-2 inhibits germinal center formation by limiting T follicular helper cell differentiation. Immunity. 2012;36:847–856. doi: 10.1016/j.immuni.2012.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbie DA, Tamayo P, Boehm JS, Kim SY, Moody SE, Dunn IF, Schinzel AC, Sandy P, Meylan E, Scholl C, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–112. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumjohann D, Preite S, Reboldi A, Ronchi F, Ansel KM, Lanzavecchia A, Sallusto F. Persistent antigen and germinal center B cells sustain T follicular helper cell responses and phenotype. Immunity. 2013;38:596–605. doi: 10.1016/j.immuni.2012.11.020. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity. 2013;39:782–795. doi: 10.1016/j.immuni.2013.10.003. [DOI] [PubMed] [Google Scholar]
- Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–135. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budhu S, Loike JD, Pandolfi A, Han S, Catalano G, Constantinescu A, Clynes R, Silverstein SC. CD8+ T cell concentration determines their efficiency in killing cognate antigen-expressing syngeneic mammalian cells in vitro and in mouse tissues. J Exp Med. 2010;207:223–235. doi: 10.1084/jem.20091279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liakou CI, Kamat A, Pettaway C, Ward JF, Tang DN, Sun J, Jungbluth AA, Troncoso P, Logothetis C, Sharma P. Anti-CTLA-4 therapy results in higher CD4+ICOShi T cell frequency and IFN-gamma levels in both nonmalignant and malignant prostate tissues. Proc Natl Acad Sci U S A. 2009;106:2729–2734. doi: 10.1073/pnas.0813175106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Gullicksrud JA, Xing S, Zeng Z, Shan Q, Li F, Love PE, Peng W, Xue HH, Crotty S. LEF-1 and TCF-1 orchestrate T(FH) differentiation by regulating differentiation circuits upstream of the transcriptional repressor Bcl6. Nature immunology. 2015;16:980–990. doi: 10.1038/ni.3226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubas RA, Mudd JC, Savoye AL, Perreau M, van Grevenynghe J, Metcalf T, Connick E, Meditz A, Freeman GJ, Abesada-Terk G, Jr, et al. Inadequate T follicular cell help impairs B cell immunity during HIV infection. Nat Med. 2013;19:494–499. doi: 10.1038/nm.3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Strome SE, Salomao DR, Tamura H, Hirano F, Flies DB, Roche PC, Lu J, Zhu G, Tamada K, et al. Tumor-associated B7-H1 promotes T-cell apoptosis: a potential mechanism of immune evasion. Nat Med. 2002;8:793–800. doi: 10.1038/nm730. [DOI] [PubMed] [Google Scholar]
- Ellestad KK, Thangavelu G, Ewen CL, Boon L, Anderson CC. PD-1 is not required for natural or peripherally induced regulatory T cells: Severe autoimmunity despite normal production of regulatory T cells. Eur J Immunol. 2014;44:3560–3572. doi: 10.1002/eji.201444688. [DOI] [PubMed] [Google Scholar]
- Fife BT, Bluestone JA. Control of peripheral T-cell tolerance and autoimmunity via the CTLA-4 and PD-1 pathways. Immunol Rev. 2008;224:166–182. doi: 10.1111/j.1600-065X.2008.00662.x. [DOI] [PubMed] [Google Scholar]
- Friedman CF, Proverbs-Singh TA, Postow MA. Treatment of the Immune-Related Adverse Effects of Immune Checkpoint Inhibitors: A Review. JAMA Oncol. 2016;2:1346–1353. doi: 10.1001/jamaoncol.2016.1051. [DOI] [PubMed] [Google Scholar]
- Gagliani N, Magnani CF, Huber S, Gianolini ME, Pala M, Licona-Limon P, Guo B, Herbert DR, Bulfone A, Trentini F, et al. Coexpression of CD49b and LAG-3 identifies human and mouse T regulatory type 1 cells. Nat Med. 2013;19:739–746. doi: 10.1038/nm.3179. [DOI] [PubMed] [Google Scholar]
- Gu-Trantien C, Loi S, Garaud S, Equeter C, Libin M, de Wind A, Ravoet M, Le Buanec H, Sibille C, Manfouo-Foutsop G, et al. CD4(+) follicular helper T cell infiltration predicts breast cancer survival. J Clin Invest. 2013;123:2873–2892. doi: 10.1172/JCI67428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale JS, Ahmed R. Memory T follicular helper CD4 T cells. Front Immunol. 2015;6:16. doi: 10.3389/fimmu.2015.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammers HJ, Plimack ER, Infante JR, Rini BI, McDermott DF, Lewis LD, Voss MH, Sharma P, Pal SK, Razak ARA, et al. Safety and Efficacy of Nivolumab in Combination With Ipilimumab in Metastatic Renal Cell Carcinoma: The CheckMate 016 Study. J Clin Oncol. 2017;35:3851–3858. doi: 10.1200/JCO.2016.72.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S, Castelo R, Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Tsai LM, Leong YA, Hu X, Ma CS, Chevalier N, Sun X, Vandenberg K, Rockman S, Ding Y, et al. Circulating precursor CCR7(lo)PD-1(hi) CXCR5(+) CD4(+) T cells indicate Tfh cell activity and promote antibody responses upon antigen reexposure. Immunity. 2013;39:770–781. doi: 10.1016/j.immuni.2013.09.007. [DOI] [PubMed] [Google Scholar]
- He R, Hou S, Liu C, Zhang A, Bai Q, Han M, Yang Y, Wei G, Shen T, Yang X, et al. Follicular CXCR5-expressing CD8+ T cells curtail chronic viral infection. Nature. 2016 doi: 10.1038/nature19317. [DOI] [PubMed] [Google Scholar]
- Hellmann MD, Rizvi NA, Goldman JW, Gettinger SN, Borghaei H, Brahmer JR, Ready NE, Gerber DE, Chow LQ, Juergens RA, et al. Nivolumab plus ipilimumab as first-line treatment for advanced non-small-cell lung cancer (CheckMate 012): results of an open-label, phase 1, multicohort study. Lancet Oncol. 2016 doi: 10.1016/S1470-2045(16)30624-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, Sosman JA, McDermott DF, Powderly JD, Gettinger SN, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563–567. doi: 10.1038/nature14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363:711–723. doi: 10.1056/NEJMoa1003466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgaard RB, Zamarin D, Li Y, Gasmi B, Munn DH, Allison JP, Merghoub T, Wolchok JD. Tumor-Expressed IDO Recruits and Activates MDSCs in a Treg-Dependent Manner. Cell Rep. 2015;13:412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou TZ, Qureshi OS, Wang CJ, Baker J, Young SP, Walker LS, Sansom DM. A transendocytosis model of CTLA-4 function predicts its suppressive behavior on regulatory T cells. J Immunol. 2015;194:2148–2159. doi: 10.4049/jimmunol.1401876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545:60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature. 2016 doi: 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwai Y, Ishida M, Tanaka Y, Okazaki T, Honjo T, Minato N. Involvement of PD-L1 on tumor cells in the escape from host immune system and tumor immunotherapy by PD-L1 blockade. Proc Natl Acad Sci U S A. 2002;99:12293–12297. doi: 10.1073/pnas.192461099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston RJ, Choi YS, Diamond JA, Yang JA, Crotty S. STAT5 is a potent negative regulator of TFH cell differentiation. J Exp Med. 2012;209:243–250. doi: 10.1084/jem.20111174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Cannons JL, Zhao F, Yusuf I, Lao C, Locci M, Schwartzberg PL, Crotty S. The receptor Ly108 functions as a SAP adaptor-dependent on-off switch for T cell help to B cells and NKT cell development. Immunity. 2012;36:986–1002. doi: 10.1016/j.immuni.2012.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenefeck R, Wang CJ, Kapadi T, Wardzinski L, Attridge K, Clough LE, Heuts F, Kogimtzis A, Patel S, Rosenthal M, et al. Follicular helper T cell signature in type 1 diabetes. J Clin Invest. 2015;125:292–303. doi: 10.1172/JCI76238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med. 2015;373:23–34. doi: 10.1056/NEJMoa1504030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734–1736. doi: 10.1126/science.271.5256.1734. [DOI] [PubMed] [Google Scholar]
- Liu X, Yan X, Zhong B, Nurieva RI, Wang A, Wang X, Martin-Orozco N, Wang Y, Chang SH, Esplugues E, et al. Bcl6 expression specifies the T follicular helper cell program in vivo. J Exp Med. 2012;209:1841–1852. S1841–1824. doi: 10.1084/jem.20120219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Carlsson R, Comabella M, Wang J, Kosicki M, Carrion B, Hasan M, Wu X, Montalban X, Dziegiel MH, et al. FoxA1 directs the lineage and immunosuppressive properties of a novel regulatory T cell population in EAE and MS. Nat Med. 2014;20:272–282. doi: 10.1038/nm.3485. [DOI] [PubMed] [Google Scholar]
- Miyauchi K, Sugimoto-Ishige A, Harada Y, Adachi Y, Usami Y, Kaji T, Inoue K, Hasegawa H, Watanabe T, Hijikata A, et al. Protective neutralizing influenza antibody response in the absence of T follicular helper cells. Nature immunology. 2016;17:1447–1458. doi: 10.1038/ni.3563. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol. 2013;13:499–509. doi: 10.1038/nri3470. [DOI] [PubMed] [Google Scholar]
- Pentcheva-Hoang T, Egen JG, Wojnoonski K, Allison JP. B7–1 and B7–2 selectively recruit CTLA-4 and CD28 to the immunological synapse. Immunity. 2004;21:401–413. doi: 10.1016/j.immuni.2004.06.017. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Cohen-Solal K, Sood R, Namkoong J, Martino JJ, Koganti A, Zhu H, Robbins C, Makalowska I, Shin SS, et al. Melanoma mouse model implicates metabotropic glutamate signaling in melanocytic neoplasia. Nat Genet. 2003;34:108–112. doi: 10.1038/ng1148. [DOI] [PubMed] [Google Scholar]
- Postow MA, Chesney J, Pavlick AC, Robert C, Grossmann K, McDermott D, Linette GP, Meyer N, Giguere JK, Agarwala SS, et al. Nivolumab and ipilimumab versus ipilimumab in untreated melanoma. N Engl J Med. 2015;372:2006–2017. doi: 10.1056/NEJMoa1414428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi H, Cannons JL, Klauschen F, Schwartzberg PL, Germain RN. SAP-controlled T-B cell interactions underlie germinal centre formation. Nature. 2008;455:764–769. doi: 10.1038/nature07345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao DA, Gurish MF, Marshall JL, Slowikowski K, Fonseka CY, Liu Y, Donlin LT, Henderson LA, Wei K, Mizoguchi F, et al. Pathologically expanded peripheral T helper cell subset drives B cells in rheumatoid arthritis. Nature. 2017;542:110–114. doi: 10.1038/nature20810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med. 2015;372:2521–2532. doi: 10.1056/NEJMoa1503093. [DOI] [PubMed] [Google Scholar]
- Sage PT, Alvarez D, Godec J, von Andrian UH, Sharpe AH. Circulating T follicular regulatory and helper cells have memory-like properties. J Clin Invest. 2014;124:5191–5204. doi: 10.1172/JCI76861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage PT, Francisco LM, Carman CV, Sharpe AH. The receptor PD-1 controls follicular regulatory T cells in the lymph nodes and blood. Nature immunology. 2013;14:152–161. doi: 10.1038/ni.2496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo A, Alekseev A, Tanaka K, Obertas L, Lerman B, Haymaker C, Clise-Dwyer K, McMurray JS, Nurieva R. Batf is important for IL-4 expression in T follicular helper cells. Nat Commun. 2015;6:7997. doi: 10.1038/ncomms8997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth S, Ravens I, Kremmer E, Maier MK, Hadis U, Hardtke S, Forster R, Bernhardt G. Abundance of follicular helper T cells in Peyer’s patches is modulated by CD155. Eur J Immunol. 2009;39:3160–3170. doi: 10.1002/eji.200939470. [DOI] [PubMed] [Google Scholar]
- Strome SE, Dong H, Tamura H, Voss SG, Flies DB, Tamada K, Salomao D, Cheville J, Hirano F, Lin W, et al. B7-H1 blockade augments adoptive T-cell immunotherapy for squamous cell carcinoma. Cancer Res. 2003;63:6501–6505. [PubMed] [Google Scholar]
- Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, Powderly JD, Carvajal RD, Sosman JA, Atkins MB, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366:2443–2454. doi: 10.1056/NEJMoa1200690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CJ, Heuts F, Ovcinnikovs V, Wardzinski L, Bowers C, Schmidt EM, Kogimtzis A, Kenefeck R, Sansom DM, Walker LS. CTLA-4 controls follicular helper T-cell differentiation by regulating the strength of CD28 engagement. Proc Natl Acad Sci U S A. 2015;112:524–529. doi: 10.1073/pnas.1414576112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber JS, D’Angelo SP, Minor D, Hodi FS, Gutzmer R, Neyns B, Hoeller C, Khushalani NI, Miller WH, Jr, Lao CD, et al. Nivolumab versus chemotherapy in patients with advanced melanoma who progressed after anti-CTLA-4 treatment (CheckMate 037): a randomised, controlled, open-label, phase 3 trial. Lancet Oncol. 2015;16:375–384. doi: 10.1016/S1470-2045(15)70076-8. [DOI] [PubMed] [Google Scholar]
- Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang NAS, Andrews MC, Sharma P, Wang J, Wargo JA, Pe’er D, Allison JP. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade. Cell. 2017;170:1120–1133 e1117. doi: 10.1016/j.cell.2017.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wing JB, Ise W, Kurosaki T, Sakaguchi S. Regulatory T cells control antigen-specific expansion of Tfh cell number and humoral immune responses via the coreceptor CTLA-4. Immunity. 2014;41:1013–1025. doi: 10.1016/j.immuni.2014.12.006. [DOI] [PubMed] [Google Scholar]
- Wing K, Onishi Y, Prieto-Martin P, Yamaguchi T, Miyara M, Fehervari Z, Nomura T, Sakaguchi S. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322:271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, Lao CD, Wagstaff J, Schadendorf D, Ferrucci PF, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma. N Engl J Med. 2017;377:1345–1356. doi: 10.1056/NEJMoa1709684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, Segal NH, Ariyan CE, Gordon RA, Reed K, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369:122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarilin D, Xu K, Turkekul M, Fan N, Romin Y, Fijisawa S, Barlas A, Manova-Todorova K. Machine-based method for multiplex in situ molecular characterization of tissues by immunofluorescence detection. Sci Rep. 2015;5:9534. doi: 10.1038/srep09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen GJ, Demissie E, Pillai S. B lymphocytes and cancer: a love-hate relationship. Trends in cancer. 2016;2:747–757. doi: 10.1016/j.trecan.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappasodi R, Merghoub T. Alphavirus-based vaccines in melanoma: rationale and potential improvements in immunotherapeutic combinations. Immunotherapy. 2015;7:981–997. doi: 10.2217/imt.15.64. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data sets generated in this study have been submitted to the GEO (Gene Expression Omnibus) repository and will be publicly available after December 1st 2018 (GSE95754, GSE95756). Other data sets used in the study are available in the GEO repository, GSE14308, GSE30431, GSE85316, GSE92940.