

Summary
Control of protein activity in living cells can reveal the role of spatio-temporal dynamics in signaling circuits. Protein analogs with engineered allosteric responses can be particularly effective, as they can replace endogenous proteins with minimum perturbation of native interactions. However, it has been a challenge to identify allosteric sites in target proteins where insertion of responsive domains produces activation changes comparable to native proteins. Here we describe a detailed protocol to generate genetically-encoded analogs of proteins that can be allosterically controlled by either rapamycin or blue light, using computational methods to identify effective insertion sites. Rapamycin added to the medium activates the protein essentially irreversibly, while light-controlled allosteric switches provide reversible inactivation with higher spatio-temporal resolution. We discuss the computational framework to identify the insertion sites, and experimental procedures to produce and test the engineered proteins in vitro and in mammalian cell lines. This method has been successfully applied to catalytic domains of protein kinases, Rho family GTPase and guanine exchange factors, as well as binding domain of a guanine exchange factor Vav2. Computational tasks can be completed within a few hours, followed by 1–2 weeks of experimental validation. We provide protocols for computational design, cloning, and experimental testing of the engineered proteins, using Src tyrosine kinase, guanine exchange factor Vav2, and Rho GTPase Rac1 as examples.
Keywords: engineered allostery, optogenetics, chemogenetics, allosteric regulation, protein engineering, computational design, photo-activation, photo-inhibition, kinase, guanine nucleotide exchange factor, Rho GTPase
EDITORIAL SUMMARY
This protocol describes how to design proteins that can be controlled either irreversibly by rapamycin or reversibly by light. The procedures detail how to identify sites for insertion of engineered regulatory domains and how to test the analogs biochemically and in living cells.
TWEET
A protocol to generate protein allosteric switches controlled by rapamycin or light
INTRODUCTION
Cells precisely coordinate signal transduction in space and time using complex and dynamic protein networks. Understanding and controlling such tight regulation, especially for rapid behaviors such as cell motility, necessitates approaches that allow manipulation of signaling with high temporal and/or spatial resolution in living cells. Because many proteins function in more than one subcellular region, it is often valuable to control the activity of an intact protein, rather than targeting a protein fragment with specific activity to a particular subcellular location1, 2, 3. Engineering allostery offered a way to control protein activity without disrupting native interactions of the target protein, and it provided kinetic control. By using rapamycin activation, we could examine the role of different Src family isoforms even though their sequences and structures were closely related3. In contrast to genetic manipulation, which would have allowed compensation and upregulation of other family members, we could study cells immediately after altering the function of individual family members. Furthermore, by activating the kinase at precise times during trafficking, we could dissect out different roles of target proteins1, 2, 3. As in other structure-based computational design approaches, our protocol requires knowledge of the target protein’s structure, or at least the structure of homologous proteins, to insert elements that respond to artificial stimuli. Combinatorial approaches, not addressed here, can identify sites that are missed due to the limits of computational methods4, 5, 6, but design and implementation of the required assays can be challenging.
For the method described here, the choice of an inducer – ligand or light – has to be considered carefully, as ligand-based control is currently irreversible and takes place over seconds as rapamycin diffuses into the cell. In contrast, light provides reversibility, higher temporal and spatial resolution, and adjustable kinetics, but requires more complex microscope approaches and cannot readily reach deep into tissues. We illustrate the critical steps required to alter a target protein so that it responds allosterically to light or rapamycin analogs, including non-immunosuppressive analogues that do not interact with the mTOR pathway7, 8 (Fig. 1). We describe reversible protein photoinhibition (PI), and weakly reversible bimolecular (RapR) or unimolecular (uniRapR) rapamycin-induced activation (Fig. 2). Reversible photoactivation has also been accomplished through light control of autoinhibitory domains1.
Figure 1.
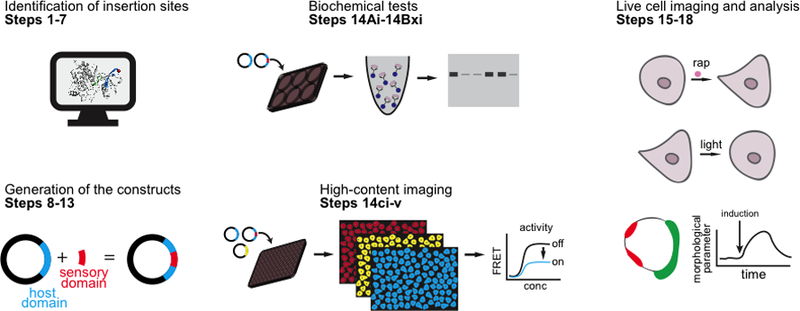
Outline of the procedure.
Figure 2. Strategies for engineered control of protein activity.
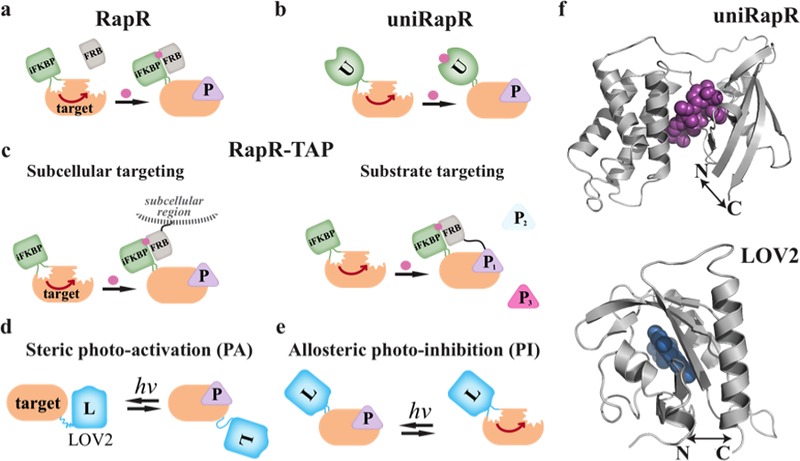
a) In rapamycin-regulated control (RapR), an insertable FKBP domain (iFKBP) is inserted at a surface loop where it allosterically inhibits the activity of the protein. Heterodimerization of iFKBP with co-expressed FRB is induced by addition of rapamycin (pink circle) to the medium, leading to activation of the target protein and productive interaction with downstream targets (P). Typically, constitutively active proteins are used so that activity is solely controlled by the user. b) The single chain/unimolecular version of this approach, called uniRapR, does not require co-expression of FRB, as the inserted domain (U) is a fusion of iFKBP and FRB. c) RapR-TAP enables allosteric activation of the target protein in a specified subcellular region, or when it is interacting specifically with one downstream target. Here iFKBP is inserted in the target, and the FRB required for activation is fused to the specific target protein, or to a subcellular targeting sequence. d) In protein photo-activation (PA), the LOV2 domain (L) is fused to the target protein, where it sterically blocks the active site. Blue light induces a LOV2 conformational change that reversibly uncovers the active site. e) In protein photo-inhibition (PI), LOV2 is inserted at a site where it allosterically perturbs the active site only in the light. f) The close proximity of the N and C termini of the iFKBP, uniRapR and LOV2 domains enables successful insertion into surface loops and subsequent allosteric regulation.
Strategies to make genetically-encoded, allosterically-controlled proteins
Protein activation by rapamycin: RapR and uniRapR.
In the presence of the small molecule rapamycin, the 12-kDa FK506-binding protein (FKBP12) interacts with the 11-kDa protein FKBP-rapamycin-binding (FRB). This interaction has been exploited to control the localization of proteins by fusing the target protein to one member of the interacting pair, and a cellular targeting sequence to the other2, 7, 9, 10, 11. Addition of rapamycin, which is cell-permeable, resulted in localization of the target protein to a specific cellular compartment. In the approach addressed here, insertable truncated FKBP12 (iFKBP) is inserted into the target proteins to control their conformation2, 10, 11, 12. iFKBP was created by removing the first 20 amino acids from the N-terminus of FKBP12, bringing the N- and C- termini close together (approximately 7 Å apart) and thereby enabling iFKBP insertion into target proteins without greatly changing the spacing across insertion sites (Fig. 2)10. Because the iFKBP domain is predominantly unstructured without rapamycin, as shown by molecular dynamics simulations10, insertion of iFKBP decreases the stability of the target protein structure and thereby results in inactivation of the target protein. Once iFKBP binds rapamycin and FRB, it becomes predominantly structured, resulting in activation of the target protein10. This approach requires the co-expression of FRB, so heterogeneity in expression levels can result in differences of activation level from cell to cell. To reduce this complexity, a unimolecular version of this system was developed by fusing a circularly permutated FRBinto the iFKBP structure13 (an engineered form of FRB with different amino acid connectivity but with a similar 3D structure). This fusion protein was named the uniRapR domain, and enabled ready generation of single-chain rapamycin-sensitive proteins1, 13. Rapamycin may have side effects in cells, especially during long term application, as it is an mTOR complex inhibitor. However, inert analogs that interact predominantly with a mutant form of exogenous FKBP12, iFKBP and uniRapR enable specific activation of the target proteins7, 8, 10, 13, 14, 15.
Activation results from a specific protein-protein interaction, or occurs at a selected subcellular site: RapR-TAP.
Using RapR-TAP it is possible to cause an activated protein to interact only with a specific downstream ligand (Fig. 1). In RapR-TAP, iFKBP is inserted into the protein to be activated, and FRB is attached to the downstream target. Activation can only occur when the target protein and the specific downstream partner are brought together. RapR-TAP can also be used to activate a protein at selected subcellular sites if FRB is tagged with a localization sequence, while iFKBP is inserted into the protein targeted for allosteric control. Using this strategy, it was shown that the effects of the tyrosine kinase Src are substantially different in the cytosol versus the plasma membrane2, and that specific events downstream of Src can be attributed to Src interaction with FAK versus p130Cas2.
Photo-activation (PA) and photo-inhibition (PI) with LOV2.
The light-oxygen-voltage-sensing domain (LOV2, 12 kDa) is used by plants, microalgae, fungi and bacteria to sense environmental cues. Its cofactor flavin, seated in the Per-Arnt-Sim (PAS) subdomain, absorbs light efficiently between 300–500 nm, with maximal effect at 450 nm. Upon light absorption, the flavin forms a covalent bond with a cysteine residue (C450) in the PAS domain16, 17. This leads to a conformational change that propagates from the PAS domain to the C-terminal Jα helix and to a short N-terminal helix, resulting in distortion and the unfolding of the Jα helix18. This light-induced allosteric regulation has been used for various modes of protein control. For example, a photo-activatable analogue of the small GTPase Rac1 (PA-Rac1) was generated by fusing the C-terminus of LOV2 to the N-terminus of Rac119. The crystal structure of PA-Rac1 revealed that in dark conditions LOV2 sterically blocked the Rac1 active site, but the active site was exposed when the Jα helix was unwound upon illumination18. PA-Rac1 has been used to study protein function in a wide variety of systems and animals: cellular protrusions in fibroblasts19, collective cell migration in Drosophila20, learning and memory in mice21, 22, 23. The LOV2 domain has been used in a variety of designs to activate proteins including Caspase-724, stromal interaction molecule 1 (Stim1)25, endonuclease PvuII26, ornithine decarboxylase-like degradation sequence27, formin mDia28, 29, and DNA binding proteins30, 31.
Because the termini of LOV2 are relatively close to each other (~15 Å) in the dark state, LOV2 can be inserted into the surface of target proteins through replacement of only a few amino acids in exposed loops or sequences. We have shown that surface insertion of LOV2 can provide light-induced inhibition1. We computationally identified surface loops that were “tight” (controlling the proximity of two internal structured units, e.g. the loop connecting interacting antiparallel helices or strands of a β pleated sheet), not evolutionarily conserved, and were allosterically coupled to the active site. By inhibiting auto-inhibitory domains, we could also achieve activation. We applied this approach to several domains including the catalytic domain of a kinase, the catalytic Dbl homology (DH) domain of Rho guanine exchange factors (GEFs), the autoinhibitory domain of GEF Vav2, and the catalytic domain of Rho GTPases1.
Comparison with other methods.
There are multiple genetically-encoded strategies to acutely control protein activity. Because many proteins function at specific subcellular regions, it has been possible to control protein function by controlling subcellular localization, for example using ligand-induced dimerizers including FKBP12 + FRB + Rapamycin7, 9, pyrabactin resistance (PYR)/PYR1-like (PYL)/regulatory component + abscisic acid(ABA) + protein phosphatase type 2Cs (PP2C)32, and gibberellin insensitive (GAI) + gibberellin (GA3) + gibberellin insensitive dwarf1 (GID1)33. It has also been possible to control protein localization using light-induced dimerizers such as (LOV2) + GIGANTAE (GI)34, cryptochrome2 (CRY2) + cryptochrome interacting basic helix-loop-helix (CIB)35, or phytochrome B (PhyB) + phytochrome interacting factor (PIF)36. These tools can be applied by attachment of the active form of the target protein to one part of the dimerizer and a localizing sequence to the other part. Alternately, the protein can be trapped in an irrelevant subcellular region, to be reversibly released by light. Examples of this strategy include LOVTRAP, implemented using a 6-kDa engineered Z domain (Zdk) that interacts only with the dark state of LOV237, as well as LARIAT38 and CRY2olig39, engineered to sequester target proteins as nonfunctional oligomers. Methods that rely on sterically blocking the active site include the PA-Rac1 approach described above19, 20, 21, and control of the green fluorescent protein DRONPA40. The kinetics of activation and inactivation for different photo-proteins can also be modulated according to the goals of the experiment. The LOV2 domain used here, which is activated in under a millisecond, can be mutated to alter the half-life for return to the dark state from 2 to 496 seconds37, 41.
An important advantage of the method described here is that it does not control protein activity by controlling localization. Rather an intact analog of the target protein can be produced, with modifications away from the sites of interaction with endogenous ligands. Furthermore, light activation is rapid (< 1ms) and the kinetics of return to the dark state can be adjusted using specific point mutations37. The uniRapR and PI approaches require the identification of insertion sites, surface exposed loops where the inserted domains will be allosterically coupled to the active site. Here we describe an approach to identify insertion sites through computational screening, but sometimes there are only a few appropriate surface loops, so testing empirically can suffice. A comprehensive empirical design strategy can also be pursued by creating random domain insertion libraries with nuclease digestion, multiplex inverse PCR, and circular permutation42. Such an approach may identify additional sites not found by computational approaches, and is especially useful for proteins that can be tested in bacteria or yeast.
Limitations
For any given target protein, a robust approach to assay protein activity must be available. The protocol includes structure design, so a protein structure must be available or accessible via homology modeling. If a homology model has to be built, and there is no close homolog structure available, the computational protocol described in Steps 1–8 cannot be applied. In that case, one can identify potential loop sites using sequence-based algorithms43. Finally, the approach requires a certain stability in the target protein. For some exceptionally unstable proteins, domain insertion will not be feasible.
The strategy described here is most effective for structures that include tight, short, surface exposed loops connecting interacting structural units. These features may not be present in some proteins, such as transmembrane proteins without any intra- and extra-cellular structured domains, intrinsically-disordered proteins, or unstable proteins (proteins with the folding free energy difference (ΔGU) less than ~ 5 kcal/mol). However, for many proteins, solvent accessible, evolutionarily non-conserved, and tight loops should be permissive for domain insertion, as the termini of uniRapR and LOV2 domains are in close proximity. For transmembrane proteins, the only practical option might be to insert the sensory domain into a non-transmembrane portion of the host protein. There are inherent limitations of using light for protein control, including phototoxicity and the inaccessibility of some targets to light. For photo-activation studies, it is important to measure the activity of the inactivated protein, and be sure that expression levels do not produce undesired biological effects prior to activation.
Experimental design
The PROCEDURE has four sections: Section 1 describes a computational strategy to identify the sites for domain insertion (steps 1–8). Section 2 describes the molecular cloning (steps 9–14). Sections 3 and 4 describe the testing of engineered proteins using in vitro assays (step 15, options A-C) and live cell imaging (steps 16–19). As proof of principle systems, we focus on kinases, Rho guanine exchange factors (GEF), and Rho GTPases. Src kinase and Vav2 were used as examples.
Identification of domain insertion sites.
Insertion sites must fulfill two important criteria. The domain insertion should not interfere with the folding and function of the target protein, and the perturbation induced by the uniRapR or LOV2 domains must effectively alter the active site. To this end, we recommend selecting “tight loops”1 that control the proximity of two internal structured units (e.g. connect two parallel/antiparallel helices or strands, Fig. 3a). These internal units can directly span from the loop to the active site, or can contact a second structured unit that reaches the active site. This positioning of the insertion sites can lead to effective active site distortion by the uniRapR or LOV2 domains. The surface exposed residues at the insertion site should not be functionally important, but should only play a role in holding together internal secondary structures. The placement of the external sensory domains LOV2 or uniRapR should not sterically block important interactions. Insertion into tight surface loops is possible owing to the short distances between the termini of uniRapR and LOV2 (Fig. 3a). Tight loops can often be simply selected by visual inspection of the protein structure, which can be obtained from the protein data bank. When it is available, we use data from the literature to eliminate protein regions that are important for interaction with endogenous ligands.
Figure 3. An approach to design allosteric protein switches.
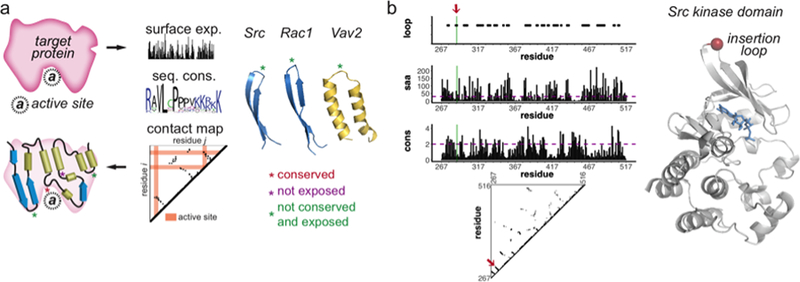
a) The three dimensional structure or a structural homolog can be used to identify secondary structures, surface exposure of each residue, and the contact map. By collecting sequences from different species and homologue domain sequences, sequence conservation is calculated and used to identify surface sequences less likely to be important for function. The insertion sites (shown with green asterisks) are tight surface loops connecting interacting elements of interior secondary structure (e.g. two parallel strands or helices) b) Src kinase domain was used as an example. The loop sites along with other parameters, including solvent accessible area (saa) and sequence conservation (cons), contact map were used to identify insertion sites. The red arrow on the plots and red sphere on the structure indicates the site that was selected.
Identification of domain insertion sites from homology models.
If no X-ray or NMR structure is available, a homology model of the structure can be built using tools such as I-Tasser44, Modeller45, Rosetta46. Due to its automatic pipeline algorithm, accuracy and speed, we prefer I-Tasser for homology modeling. We identify the surface exposure of amino acids by computing the solvent accessible area (SAA) using Stride47, which also can provide the secondary structure information to select short loops. To identify sites where surface residues do not play important roles, we perform evolutionary sequence conservation analysis for each residue by collecting sequences of the target domain from all the available proteins and species in Pfam48. While there are multiple ways to select the sites, we recommend obtaining the domain sequences from Pfam48, and feeding the Pfam-derived sequence alignments to the online MISTIC server49. This server can perform multiple tasks including sequence conservations, mutual information to infer coevolution mapped on the protein structure, and a sequence-based approach to detect the allosteric sites. By using a multiple sequence alignment matrix, MISTIC provides Kullback-Liebler49 conservation calculated from the frequency of a partition of amino acids in a position and the background frequency of the amino acid in nature, obtained from the UniProt database.
Once the insertion loops are identified, multiple designs can be generated using alternate linkers to connect the inducible domain to the target domain. We generally insert the inducible domain into the middle of the insertion loop without any linker, with a linker consisting of glycine, proline, glycine (GPG), or with glycine, serine, glycine (GSG) residues. In initial designs, we recommend testing several possible linkers. If the protein is not fully active in the on-state, the linker should be elongated with flexible residues (e.g. glycine and serine). If the protein shows some activity in the off-state, the linker should be shortened.
Picking the right strategy.
There are several critical points to be considered when deciding which protein control strategy to use. With analogs that can be inhibited, one must be aware of potential overexpression artifacts. Before irradiation, transfection of photo-inhibitable analogs results in overexpression and inhibition can restore endogenous protein levels. It is possible that in some cases the cell will compensate for expression of the analog by downregulating the endogenous protein, but this is not always the case. Therefore, one might have to use a knockdown and rescue approach or replace the native gene. For analogs that are activatable, ideally the protein analog will have little effect until it is triggered. It can therefore be overexpressed and used to study effects of activation with exact localization and/or timing. For PI-Vav2 and PI-Rac1, we compared overexpression of the analog to replacement of the endogenous protein; in both cases photoinhibition produced retraction or protrusion changes, but the approach used affected the magnitude of the effects1.
Triggering with a ligand versus with light has distinct advantages and disadvantages. In our experience, rapamycin-induced activation has been very effective for inducing robust phenotypes, but activation is irreversible and cannot be focused on one subcellular region. Triggering with light provides much more rapid changes that can be localized, but it is harder to implement, especially deep within tissues. uniRapR can be photoactivated using a version of rapamycin caged with a photocleavable protecting group50. For protein analogs based on insertion of LOV2, it is possible to introduce mutations to tune the recovery kinetics of LOV220. LOV2 can be activated rapidly, within less than a second. The t1/2 is 1.7s for the I427T mutant and 496 s for the V416L mutant, providing fast and slow recovery options37. The latter are useful for long time scale processes, where the specimen need only be irradiated for less than a second every eight minutes.
Testing proteins engineered for allosteric control.
One must be able to reliably test the activity and response of the engineered protein. For kinases, we have used immunoprecipitation from cell extracts (step 15, option A). For Rho GEFs and GTPases we have used co-immunoprecipitation assays51 and assays (step 15, options B and C) based on Förster resonance energy transfer (FRET) biosensors52. We quantify expression and the extent of pulldown using peptide tags such as myc and Flag fused to the target protein. In some cases, terminus of a protein may be functionally important, so the tag location should be chosen carefully. To increase the dynamic range and ensure that protein activity is under the control of the experimentalist, constitutively active mutant analogs and wild type protein should be used as positive controls. As negative controls, cells expressing catalytically inactive mutants and cells not expressing engineered proteins should be included. As a positive control, cells expressing non-engineered proteins should be included. The effect of induction itself on cellular signaling and cell behavior should be tested with cells that do not express any of the constructs.
For imaging studies, we recommend an open chamber or a perfusion system to deliver rapamycin into the cell medium. The microscope should be capable of irradiating at the excitation wavelengths of the fluorophores described, and capable of switching between wavelengths used for LOV2 activation and fluorescence visualization. For LOV2-based optogenetic control, the excitation and emission wavelengths used for imaging fluorophores should be selected carefully, so that they do not to overlap with the absorption wavelengths of LOV2. Because LOV2 is excited at 300–500 nm, we recommend using red fluorescent proteins, e.g. mCherry, or yellow fluorescent proteins, e.g. YPet or mVenus. For the yellow proteins, a 510-nm excitation filter with a sufficiently narrow bandwidth (for example Semrock FF-510/10) can avoid LOV2 excitation. LOV2 is extremely sensitive to the blue light that is present in ambient light or from computer screens, so all the light sources nearby should be covered with high-pass yellow or red filters.
Levels of expertise needed to implement the protocol.
The protocols described here require basic knowledge and facilities for mammalian tissue culture, and fluorescence microscopy of living cells.
MATERIALS
REAGENTS
Molecular cloning
DNA encoding residues 1–198 of the uniRapR domain (Addgene Plasmid #45381) This plasmid is used to design single-chain rapamycin-induced allosteric proteins.
DNA encoding residues 1–87 of the iFKBP domain (Addgene Plasmid #25933) and residues 1–96 of FRB (Addgene Plasmid #25920).
This plasmid is used to design dual-chain rapamycin-induced allosteric proteins.
DNA encoding residues 1–143 of the LOV2 domain (Addgene Plasmid #86974). This plasmid is used to design photo-activatable (PA) or photo-inhibitable (PI) proteins.
Plasmid vectors for transient mammalian cell expression: e.g., pTriex (Addgene Plasmid # 66110), pcDNA (Addgene Plasmid # 52535)
Plasmid vectors for viral infection: e.g., pBabe (Addgene Plasmid #91876), pLenti (Addgene Plasmid # 39481), FUW (Addgene Plasmid # 84008)
Q5 Hot Start High-Fidelity 2X Master Mix (New England Biolabs, cat. no. M0494S)
Pfu Turbo enzyme (Agilent, cat. no. 600250) CRITICAL A high fidelity DNA polymerase is required when amplifying long pieces of DNA using PCR. Alternatively, Gibson assembly mix (New England Biolabs, cat. no. E2611S) can be used for isothermal assembly of inserted and host plasmid DNA fragments.
DNA amplification kit (Fischer Scientific, cat. no. FERK0503 and Qiagen, cat. no. 12643) CAUTION Buffers in the kit can cause serious eye and skin irritation. The experimenter should use gloves and goggles.
Transient or stable expression of constructs in mammalian cell lines
CAUTION The cell lines used in your research should be regularly checked to ensure they are authentic and are not infected with mycoplasma.
HEK293T cell line (ATCC, cat. no. CRL-3216 or cat. no. CRL-11268)
Engineered mouse embryonic fibroblasts (Clontech, cat. no. 630914) that express a tetracycline element for doxycycline-off (tet-off) stable expression
DMEM medium (Invitrogen, cat. no. 12-604Q) with GlutaMAX supplement (Thermo-Fisher, cat. no. 10566016)
FBS (Corning, cat. no. 35015CV)
Transfection reagents such as FuGENE 6 (Promega, cat. no. E2691) or Lipofectamine (Thermo Fisher Scientific, cat. no. 11668-019 or L3000-015)
Kinase assay
HEPES (Sigma, cat. no. 54457-50G-F)
EGTA (Sigma, cat. no. E0396)
NP-40 (abcam, cat. no ab142227)
Sodium fluoride (NaF) (Sigma-Aldrich, cat. no. S7920)
Sodium orthovanadate (Na3VO4) (Sigma-Aldrich, cat. no. 450243)
MgCl2 (Sigma-Aldrich, cat. no. M8266)
MnCl2 (Sigma-Aldrich, cat. no. 244589)
Brij-35 (Thermo-Fischer, cat. no. 20150)
KCl (Sigma-Aldrich, cat. no. P9541)
NaCl (Sigma-Aldrich, cat. no. S9888)
Protein G-coupled agarose beads (Millipore, cat. no. 16-266) for kinase assay
Purified kinase substrate (see Reagent setup)
ATP (New England Biolabs, cat. no. P0756S)
Bovine serum albumin (New England Biolabs, cat. no. B9000S)
1 mM rapamycin (LC Laboratories, R-5000) stock solution in ethanol
2× Laemmli SDS-PAGE protein sample buffer (Sigma-Aldrich, cat. no. S3401)
Protease inhibitor cocktail tablets (Sigma-Aldrich, cat. no. 11697498001)
Anti-myc antibody (Millipore, clone 4A6, cat. no. 05-724)
Anti-flag antibody (Sigma-Aldrich, cat. no. F3165-1MG)
Biochemical GEF and GTPase activation assays
Bacterial expression plasmid to express mutant Rho GTPase such as pGEX-4T1-Rac1 G15A (Addgene Plasmid # 69355), pGEX-4T1-Cdc42 G15A (Addgene Plasmid # 69356), and pGEX-4T1-RhoA G17A (Addgene Plasmid # 69357)
Active GTPase pull-down assay kit for Rac1 (Cytoskeleton, cat. no. BK035), Cdc42 pull-down assay kit (Cytoskeleton, cat. no. BK034), and RhoA pull-down assay kit (Cytoskeleton, cat. no. BK036)
LB bacterial medium (see Reagent Setup)
IPTG (Life Technologies, cat. no. 15529019)
E. coli. BL21 competent cells (New England Biolabs, cat. no. C2527I)
Triton X-100 (Sigma-Aldrich, cat. no. X100-100ML)
Protease inhibitor cocktail tablets (Sigma-Aldrich, cat. no. 11697498001)
Dithiothreitol (DTT) (Life Technologies, cat. no. D1532)
Glutathione sepharose slurry (abcam, cat. no. ab193267)
Coomassie Plus (Bradford) protein reagent (Thermo Fisher Scientific, cat. no. 23236).
Live cell imaging
Ham’s F-12 Kaighn’s Modification with L-glutamine and without phenol red (Caisson, cat. No. HFL12-500ML). CRITICAL Phenol red fluorescence can interfere with imaging observations
1 M HEPES (Gibco, cat. No. 15630-106)
100 μg/ml poly-L-lysine (PLL; mol. wt. 150,000 to 300,000; Sigma-Aldrich, cat. no. P4832)
Fibronectin (Sigma, cat. no. F0635)
EQUIPMENT
Molecular cloning
Thermocycler
Gel electrophoresis unit
37° C bacterial incubator
37° C bacterial shaker
Kinase, GEF and GTPase immunoprecipitation assays
6-well tissue culture plates (Corning, 07-200-83)
37° C 5% CO2 mammalian cell incubator
37° C heater with shaking capability
Cell scraper
High-content imaging
96-well microplates (Corning, cat. no. 3904)
A fluorescence microscope with 10X or 20X objective and capable of imaging 96-well plates.
Live cell imaging
25 mm round glass coverslips, 0.17 mm thick (Fisher Scientific) CRITICAL The coating reagent of the coverslip may depend on the cellular process of interest. For migration studies, we use 5 μg/ml fibronectin (see Reagent Setup).
Culture chambers (e.g. Attofluor Cell Chamber, Thermo Fisher Scientific, cat. No. A78167).
A fluorescence microscope with a 40X, 1.3. N.A. oil-immersion objective, and the ability to switch excitation and emission wavelengths. CRITICAL When using a mercury arc lamp, the microscope can be equipped with the following band pass filters and dichroic mirrors: ET430/24X (for LOV2 illumination), ET572/35X (RFP excitation), HQ620/60 M (RFP emission) from Chroma Technology Corporation, FF-520/15 (YFP excitation) and FF-565/24 (YFP emission) from Semrock Inc. 440/500/580 dichroic mirror (for RFP) and T545LP dichroic mirror (for YFP) from Chroma Technology Corporation. The microscope should also be capable of adjusting irradiation intensity, in our case using neutral density filters, e.g. with 1% transmittance.
Computational identification of insertion sites
A three-dimensional structure of the target protein domain (e.g. a crystal structure or high resolution NMR structure). We show Src kinase (pdb id: 6f3f) as an example here. If a structure is not available, a homology modeling suite, for example I-Tasser (https://zhanglab.ccmb.med.umich.edu/I-TASSER/), is required. Detailed procedures on how to create an homology model using I-Tasser can be found in Roy et al.53
Online Pfam (https://pfam.xfam.org/) database to collect domain sequences.
MISTIC web server (http://mistic.leloir.org.ar/index.php) to calculate conservation of amino acids.
Stride package (http://webclu.bio.wzw.tum.de/stride/) to calculate the secondary structure and solvent accessible area.
A protein structure molecular visualization package to view the proposed insertions. Recommended packages include PyMOL (http://pymol.org/) and UCSF Chimera (http://www.cgl.ucsf.edu/chimera).
A tool to calculate contact maps. We recommend the CMView module of PyMOL (http://www.bioinformatics.org/cmview/download.html).
REAGENT SETUP
Lysis buffer 20 mM HEPES-KOH, pH 7.8, 50 mM KCl, 1 mM EGTA, 1% (v/v) NP-40, 1mM NaF, 0.2 mM Na3VO4, and protease inhibitor tablet. The buffer without the inhibitors can be stored at 4° C up to six months. NaF, Na3VO4, and protease inhibitors should be added immediately before use.
Kinase reaction buffer 25 mM HEPES-KOH, pH 7.5, 5 mM MgCl2, 5 mM MnCl2, 0.5 mM EGTA, 0.005% (v/v) Brij-35 detergent, 1mM NaF and 0.1 mM Na3VO4.
Kinase wash buffer 20 mM HEPES-KOH, pH 7.8, 50 mM KCl, 100 mM NaCl, 1 mM EGTA, 1% (v/v) NP-40.
GEF bacterial lysis buffer 20 mM HEPES pH 7.5; 150 mM NaCl; 5 mM MgCl2; 1% (v/v) Triton X-100; 1 mM DTT with protease inhibitors. DTT and protease inhibitors should be added immediately before use.
GEF bacterial wash buffer HBS 20 mM HEPES, pH 7.5; 150 mM NaCl.
Purified paxillin /ATP mix: 0.1 mM ATP and 0.05 mg/mL Paxillin in Kinase Buffer. The paxilin/ATP mix should be freshly prepared. GST fused N-terminal domain of paxilin is purified as described in Lyons et al 54.
5 μg/ml fibronectin (Sigma, cat. no. F0635) in phosphate-buffered saline (PBS)
LB medium (Thermodisher, cat. no. 12780052) with the appropriate antibiotic for selection (for example 50 μg/ml carbenicillin)
PROCEDURE
Computational identification of insertion sites. TIMING 1–2 hours
-
1
Go to UniProt (http://www.uniprot.org/) and obtain the UniProt ID and amino acid sequence of the target protein in FASTA format. For example, search “Src” and click on “P05480” for SRC_MOUSE.
-
2
Go to the Protein Data Bank (https://www.rcsb.org) to check the availability of the target domain structure. For example, search for “Src” and find a structure for kinase domain, e.g. “6F3F” for mouse Src. Download PDB Format. CRITICAL STEP: If the structure is not available, build a homology model for the target domain using I-Tasser as described in Roy et al.53 (http://zhanglab.ccmb.med.umich.edu/I-TASSER/), which requires the FASTA sequence and subscription to the web site.
-
3
Minimize the structural clashes using Chiron55 at http://chiron.dokhlab.org by providing the pdb file. This step is required if the protein is to be simulated. Chiron identifies steric clashes in a protein structure based on a clash-score (derived from Van der Waals repulsion energy distributions of high-resolution structures), and removes the clashes through minimization by discrete molecular dynamics.
-
4
Go to Pfam (http://pfam.xfam.org) and enter the UniProt ID from step 1. Select the target domain (For example, P05480 for Src and click on “Pkinase_Tyr”, which will provide Pfam ID PF07714 for tyrosine kinase domains). Click on the domain name, and Pfam will provide a Pfam ID for the domain.
-
5
Go to the MISTIC web server (http://mistic.leloir.org.ar/index.php). Enter the Pfam ID (for example PF07714), select a reference sequence (for example SRC_MOUSE) to upload the multiple sequence alignment (MSA). Next, upload the pdb file from (either a pdb structure such as 6f3f.pdb for mouse Src tyrosine kinase domain downloaded from PDB or a homology model). This server will provide the sequence conservation (Kullback-Liebler or KL conservation) of each amino acid as a table or mapped on the protein structure. Mark all residues with a KL value less than 2 as non-conserved. An example for Src kinase domain (residues 267–516) is provided (Fig. 3b).
-
6
Run Stride (download at http://webclu.bio.wzw.tum.de/stride/) using the command from the terminal: stride protein_file.pdb. Use the kinase domain (residues 267–516) saving the structure of this region only in PyMOL. Stride will generate an output that includes detailed secondary structure assignment, phi and psi angles of the tertiary structure, and solvent accessible area. The output can be written as stride protein_file.pdb > output_file.txt. Mark the residues that have SAA values (the column name is –Area-, which are provided in the last column of the output, higher than 30 Å2. An example for Src kinase domain is provided (Fig. 3b).
-
7
Install and start CMView (http://www.bioinformatics.org/cmview), load the pdb file and select 7 Å (distance between two α-carbons) as a threshold to compute the contact map. Investigate the resulting 2-D plot and select the tight loops that extend perpendicular to the diagonal line (Fig. 3). An example for Src kinase domain is provided (Fig. 3b). CMView will start also PyMOL. Visualize the tight loops using the guidelines above by selecting the relevant residues on the protein sequence to confirm that the loops are surface exposed.
Molecular cloning of the designed constructs TIMING 3–4 d
CRITICAL: The chemogenetic or optogenetic strategy to be pursued should be selected carefully (see Experimental Design). For synthesis of photoactivatable rapamycin, detailed protocols are published in Karginov et al52.
-
8
Design primers to amplify by PCR the sequence to be inserted into the designated site. Primers should be designed so that the resulting megaprimer (amplified insert) should have flanking ends that align to both sides of the insertion site of the target cDNA 25–30 bp homology (Fig. 4).
CRITICAL STEP: We usually insert the inducible domain into the middle of the insertion loop without any linker, with a linker consisting of glycine, proline, glycine (GPG), or with glycine, serine, glycine (GSG) residues.
-
9Perform the following PCR master mix.
Component Amount (µl) Final concentration Nuclease-free water 22 2x Q5 High-fidelity master mix 25 1X 5’ F primer (10 µM) 1 0.2 µM 3’ R primer (10 µM) 1 0.2 µM Template DNA (50ng /µL) 1 1 ng/ µL Total 50 -
10Amplify insert using the following conditions
Step Temperature Time Initial denaturation 98 °C 30 sec 32 cycles 98 °C 15 sec 55–65 °C 30 sec 72 °C 30–60 sec/kb Final extension 72 °C 2 min -
11
Gel purify resulting megaprimer using Gel Extraction kit (GENEJET, cat. no. FERK0692).
-
12
Use the purified megaprimer (300–500 ng) and target plasmid template to carry out site-directed mutagenesis as described in Agilent kit (reference kit).
-
13
Sequence the cDNA of the constructs and amplify them using midi- or maxi-prep kits (Qiagen) for transient transfection or viral delivery. The purified cDNA should be endotoxin free and the OD 260/280 ratio should be between 1.7–1.9. PAUSE POINT DNA can be stored at −20° C indefinitely.
Figure 4. Quikchange strategy to clone iFKBP, uniRapR, or LOV2 into a target gene.

In Step 1, forward and reverse primers are used in PCR to generate megaprimers consisting of a fragment aligning to the target gene and a flanking region aligned to the inserted sequence. In Step 2, Quikchange PCR is performed using the megaprimers and target plasmid. F and R denote forward and reverse primers. The blue region indicates the target gene, and the blue dots indicate synthesized DNA with Quikchange PCR. The insertion sites of kinases, GEFs and GTPases are at the bottom.
Mammalian expression and activity tests TIMING 3–4 d
-
14
Proceed with Option A to perform an in vitro kinase assay, Option B for a Rho GEF or GTPase activity assay using co-immunoprecipitation, or Option C for using high-content microscopy.
A). Testing inducible kinase activity in vitro TIMING 4 d
CRITICAL. This assay to test the activity of immunoprecipitated engineered or wild type kinases using its purified substrate. Here we provide an assay for Src kinase, but this procedure should be applied to other kinases.
Coat 6-well plates by coating with 100 μg/ml poly-L-Lys overnight. Wash the wells with PBS three times, and seed 1–1.5 million cells of HEK 293T cells. Seed fewer cells (10000–20000) on fibronectin-coated coverslips to monitor single cell motility in imaging experiments. Return cells to the incubator.
When the cells reach 70–80% confluency the next day, transfect in the constructs with Fugene or Lipofectamine reagents following the manufacturer’s recommendations. Because overexpression of kinases can be toxic for cells, use Fugene, which results in lower expression and less toxicity. For high-content imaging experiments, use Lipofectamine, for higher transfection efficiency. For lipofectamine transfection, add the transfection mix with serum free media to the cells and add medium with 20% FBS 4–6 hours after the transfection. For fugene-based transfection, add the transfection mix without changing the medium. CRITICAL STEP: If a photo-sensitive construct is tested, keep the plates in the dark (e.g. by covering them with aluminum foil).
Wash Protein G-coupled agarose beads (10 μl per sample) with cold lysis buffer three times and incubate with 0.5–1 μl (per sample) of tag antibody (4A6 anti-myc or M2A anti-flag antibody) by mixing them with 10 μl of bead suspension (for each sample) in 1.5-ml tube at least 2 hours at 4 °C.
Wash the beads three times by resuspending in 1 ml of lysis buffer containing 1mg/ml BSA and centrifuge 1 min at 4000g at 4 °C.
Resuspend the beads in 50 μl of lysis buffer.
Treat the cells from Step 15A(ii) with the inducer or vehicle (250 nM of rapamycin or equal volume ethanol, blue light or equal period darkness) in a 37 °C incubator for at least 30 min. A blue light LED panel can be used to illuminate the specimens (~ 0.02 nW/µm2 power density at λ=445 nm, with a pulse protocol of 10 s light on and 10 s light off).
Remove the medium, and wash the cells with PBS.
-
Remove PBS, add 400 μl of lysis buffer to each well, and scrape the cells using a cell scraper.
CRITICAL STEP: For rapamycin-induced samples, add rapamycin (500 nM of final concentration) to the lysis buffer.
Collect the cell lysates in 1.5-ml tubes. Clear the lysate with by centrifuging 10 min at 3000g at 4 °C. Transfer 20 μl of supernatant to a new tube for Western blotting, and use the other ~380 μl for the next steps.
Incubate the lysates with the conjugated beads at 4 °C for 1.5 to 2 hr.
Centrifuge the beads at 3000g for 2 minutes, at 4 °C. Resuspend the beads in 500 μl of wash buffer. Repeat twice.
Centrifuge the beads at 3000g, at 4 °C. Resuspend the beads in 500 μl of kinase buffer. Repeat twice.
Resuspend the beads in 40 μl of kinase buffer, keep 20 μl for the kinase reaction, and keep 20 μl for Western Blot at −20 °C.
Add 10 μl of saturated amount of substrate/ATP mix (e.g. 0.05 mg/mL Paxillin with 0.1 mM of ATP to test Src) to the kinase reaction tube and immediately shake at 1000 rpm for 10 min at 37 °C using a heating 1.5 ml tube shaker.
Stop the reaction by adding 30 µl of 2X Laemmli protein sample buffer equal to the sample volume. Denature the proteins at 95° C for 5 min. Cool down.
Perform western blot analysis using antibodies for phosphorylated and total substrate to test the activity, anti-tag antibody to evaluate the amount of protein, a cellular marker as a loading control (e.g. vinculin, Gadph). Assess whether the on/off activity of the designed construct is dependent on induction based on the western blot results.
?TROUBLESHOOTING
| Step | Problem | Possible Reason | Solution |
|---|---|---|---|
| 2 | The predicted structure of my domain by I-Tasser does not resemble other domains in the same family. | There are no close structural homologs. | Predict the portions of the proteins that have structural homologs. If there are none, predict the secondary structures and target the loops 59. |
| 13, BOX 4 xii | No activity seen after triggering by rapamycin or for PI constructs in the dark. | The domain insertion overly perturbs the target protein function. | Change the flexible linkers between the host and the inserted domain. A set of flexible linkers with different lengths can be tested (see text). |
| 13, BOX 4 xii | “Leakiness”, e.g. activity without rapamycin, or for PI constructs in the light. | The perturbation from the inserted domain to the target domain is not efficiently transmitted. | Remove residues iteratively from the insertion loop (Fig. 6). |
| BOX 5,v | No expression is observed. | Problem in the DNA, or the protein is not properly folded. | Sequence the entire plasmid to check potential frame-shifting mutations in the coding region, or mutations and deletions in the promoter region. Alternatively, the inserted domain may irreversibly unfold the target protein. In this case, elongate the linkers with flexible residues or test a different insertion site. |
| 15 | Cells are not healthy. | Multiple factors such as the amount of the engineered protein in the cell, presence of transfection reagent, media components, pH and/or temperature of the imaging medium. | Use different concentrations of the transfected plasmid and transfection reagent, remove the transfection reagent next day, double check the presence of HEPES in the medium, make sure the temperature of the medium and the imaging stage is 37°. |
| 18 | In imaging experiments, there are phenotypes indicating leaky activity. | Potential light leaking into the cells before induction, or the engineered protein shows activity in the off state. | Grow the cells in total darkness and keep the samples from computer light during imaging sessions. If the engineered protein is leaky, remove residues iteratively between the target domain and the inserted domain. |
B). Testing GEFs and GTPases with pull-down assays TIMING 4 d
CRITICAL This assay to test the activity of immunoprecipitated engineered or wild type kinases using its purified substrate. Here we provide an assay for Src kinase, but this procedure should be applied to other kinases.
Transform BL21 cells with either a GST-tagged mutant GTPase for a GEF assay, or a GTPase effector domain for a GTPase assay. Use Rac1(G15A)-GST, RhoA(T17A)-GST, and Cdc42(G15A)-GST), which interact with only the active form of GEFs51. Use GST-PDB, which can interact with active Rac1 and Cdc42, or GST-Rhotekin-RBD, which can interact with active RhoA.
Pick a colony and grow in 0.5 L of LB media overnight at 37° C to full density (O.D = 0.8–1.0), using induction with 100 μM of IPTG. Then incubate the culture at room temperature (~25° C) overnight (~16 h).
Harvest the bacteria by centrifugation at 6000 rpm for 20 minutes. Lyse the bacteria with 10 ml of cold GEF bacterial lysis buffer (see Reagent Setup) on ice, sonicate for 1 min on ice (4 pulses, 30 sec each on ice with 1 min rests), and clear the lysate by centrifugation at 15000g for 15 min at 4° C.
Transfer the cleared lysate to a new tube, add 300 µl of glutathione sepharose slurry and rotate at 4° C for an hour.
Wash the beads with GEF bacterial wash buffer HBS.
Estimate the protein concentration using BCA protein assay kit (Pierce) according to manufacturer’s instructions. PAUSE POINT Proteins attached to beads can be stored at −80° C for at least a month with 0.5 volume glycerol.
Seed 1.2 millions of HEK293T cells (per well) in 6-well plates. When cells are approximately 70 % confluent, transfect with the designed uniRapR or PI GEF constructs or control (WT, constitutively active, and inactive mutant) constructs using Lipofectamine. In 100 µl serum free media (per sample), mix the plasmids and lipofectamine, and incubate for 15–20 minutes at room temperature. Then add 900 µl of serum free medium (makes 1 ml total) on top of the transfection mix. Remove the medium from the cells, and add 1 ml of the mix gently to the cells. After 4–6 hours of incubation, add 1 ml serum with 20% FBS to make the final FBS concentration 10%. Incubate the cell overnight and replace the medium next day with fresh serum with 10% FBS.
Next day, remove the media, wash the cells with PBS, and lyse the cells with 300 μl of lysis buffer. Freeze 20 μL of lysate to check the expression with a Western Blot.
Incubate the lysate from the previous step with 10 μg of GST-tagged protein from step vi on a rotator for an hour at 4° C.
Wash the beads with 500–750 μL of lysis buffer (see Reagent Setup) three times, by centrifugation (3000 rpm for 2 minutes at 4° C) and resuspend them in 50 μl of lysis buffer.
Perform the Western blot as described in Steps 17B xii-xiii (use all the pulled-down material mixed with sample dye and β-mercaptoethanol) and use the results to analyse the dependence of the tested construct on induction as shown on Figure 5.
Figure 5. An expected result for PI-Vav2.
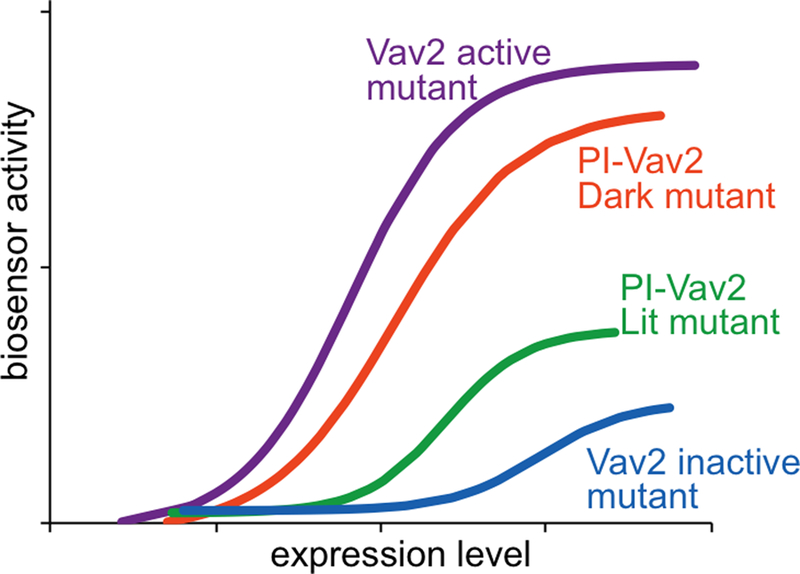
Vav2 active and inactive mutants were used as positive and negative controls. PI-Vav2 in the dark state or with dark-state mutation should have an activity similar to Vav2 activity. Conversely, Vav2 in the lit-state or with lit-state mutation should have an activity similar to the catalytically-dead Vav2 activity.
C). High-content live cell imaging TIMING 3 d
CRITICAL: Here we provide an assay for GEFs and GTPases. When testing an engineered construct with biosensors, titration is essential for accurate evaluation. This high-content assay enables rapid testing of biosensor activity in live cells transfected with increasing amounts of biosensor or engineered constructs. The precise assay used will of course depend on the protein being targeted. A detailed methods article regarding the assay use for GEFs and GTPases here can be found at Slattery et al52.
Coat black walled MicroClear 96-well microplates treated for tissue culture with 100 μg/ml poly-L-Lys. Wash the wells with PBS three times, and seed 30,000–40,000 cells on each well.
When the cells reach 70–80% confluency the next day, transfect the cells using the detailed protocol published by Slattery et al52.
Warm L15 Leibovitz medium supplemented with 5% (vol/vol) FBS in a tissue culture incubator so that the medium is warmed by the time it is used for imaging.
Image the plates using a microscope enabling imaging of 96-well plates as described in the published, detailed protocol by Slattery et al52.
Analyze the images using the information described in BOX 1. An expected result for PI-Vav2 is shown in Fig. 5.
BOX 1. Image analysis TIMING 1–2 hours.
Multiple programs including MATLAB modules56, 57 and ImageJ plugins58 can be used for image analysis. The analysis of high-content biosensor imaging, which includes background subtractions, bleed-through corrections, and normalized FRET calculations for each pixel, is provided in Slattery et al52. For single cell imaging analysis, MovThresh 56, 57 can be used for masking. The ImageJ plugin ADAPT58 can be used to analyze the morphological parameters cell area, roundness, circularity, and solidity. The Proactive 56, 57 MATLAB module provides quantification of protrusive and retractive activity. MATLAB modules are available at http://www.hahnlab.com/tools/software.html
Live cell imaging and image processing TIMING 3 d
-
15
Seed cells sparsely onto coverlips coated with 5 μg/ml fibronectin in DMEM medium supplemented with 10% (vol/vol) FBS. The number of cells depend on the cell line. For example, we recommend 200,000 HeLa cells for a 35-mm culture dish. Grow the cells in an incubator overnight. CRITICAL STEP: If there will be a transfection the next day, photo-sensitive samples should be covered with aluminum foil. To image transiently transfected HeLa cells, uniRapR-Src (Addgene, #45381) with a cell membrane marker (Addgene, # 54491) can be used.
-
16
Prior to imaging, warm L15 Leibovitz medium supplemented with 5% (vol/vol) FBS and 10 mM of HEPES in a tissue culture incubator. Mount the coverslips in a chamber suitable for live cell imaging. CRITICAL: Work under red light for blue light-sensitive samples. Add the amount (2 mL for 35 mm imaging dish) of warm imaging medium. CRITICAL STEP Keep the samples away from any light sources that include blue light wavelengths, such as computer screens or ambient light. A filter on the computer screen and ambient light source to block the light at wavelength less than 500 nm can be used, but simply using a red light bulb can suffice. The computer can be kept far from the microscope stage, or a ‘tent’ of foil or paper can be built over the sample on the stage. CRITICAL STEP: Addition of HEPES is important to maintain the approporate pH of the medium which is essential for cell health. ?TROUBLESHOOTING
-
17
Image cells at RFP or YFP wavelengths and illuminate LOV2 using GFP excitation settings (400–500 nm, see Live cell imaging in Equipments). Take images every 10 to 30 seconds for 90 minutes to monitor spontaneous motility of fibroblasts. For rapamycin-sensitive samples, add 100–500 nM of rapamycin or its analog (non-immunosuppresive analogs such as iRap7 and AP21967 (Takara, cat. no. 635055) require micromolar doses) after obtaining sufficient images to establish a baseline. CRITICAL STEP: If transmitted light is used for imaging, a long-pass red filter should be used to avoid LOV2 activation.
-
18
Analyze the images using the information described in BOX 1.
Anticipated Results
We have provided guidelines for the development of rapamycin and blue light regulated proteins. The guidelines should help to identify sites for insertion of iFKBP, uniRapR and LOV2 domains. If the target protein is a kinase or a Rho GEF or a Rho GTPase, these guidelines should also provide an approach to assess regulation of the target protein. Both in vitro and in living cells, the stimulus (rapamycin or light) should induce no change in the activity of the constructs when no domain has been inserted. The constructs that have the inserted domain, but also bear an inactivating mutation, should also not respond to rapamycin or blue light. With some constructs, one might find “leakiness” (activity in the off form) or reduced activation when full activity is desired. To overcome this, optimization of linkers is usually important, and some insertion sites will be better than others (Fig. 6). For activatable analogs, reduced maximal activation is more desirable than leakiness, as one can express more of the protein to achieve the desired level of activity in the on state. For photo-inhibited proteins, the tradeoff is less clear and will depend on the experiment. Effects on motility can be readily monitored using the many published tools designed to quantify motile behavior 56, 57, 58.
Figure 6. Anticipated results.

(A) The control with uniRapR should provide full activity with rapamycin and no activity without rapamycin. Conversely, in photo-inhibitable (PI) constructs, the protein should be deactivated with light. UniRapR-Src is deactivated without rapamycin, and the activity is rescued with rapamycin. Wild type or constitutively active (Src Y527F or YF) proteins should be used as positive controls, whereas catalytically inactive (kinase dead or KD) mutants can be used as negative controls. The expression of the constructs should be compared. Paxilin and focal adhesion kinase (FAK) are Src substrates. An alternative method should be also used to test the engineered constructs. In this case, the motility of mouse embryonic fibroblasts (MEFs) and translocation of PI-Src to focal adhesions are monitored upon photo-inhibition of PI-Src. (B) If significant activity is observed in the off state of a uniRapR-protein or in a PI-protein upon light treatment, the linkers between the sensory and host domain should be shortened. If insufficient activity is observed in the on state of uniRapR-protein or in the dark state of PI-protein, the linkers between the sensory and host domain should be elongated, or another insertion site should be tested. The same strategy should be pursued if there is lower expression of the constructs compared to controls. Some level of leakiness can be tolerated, depending on the biology and expression levels of the endogenous proteins. UniRapR-PAK1 insertion site is optimized by inserting uniRapR in between different residues of the loop. In another case uniRapR-PAK1 was generated by inserting uniRapR domain into a different loop (uniRapR-PAK1-L2). These figures were taken from published work1, 11, 13.
TIMING
Steps 1–8, computational identification of insertion sites: several hours
Steps 9–14, molecular cloning of the designed constructs: 4 d
Step 15, options A-C; mammalian expression and activity tests: 3–4 d
Steps 16–19; live cell imaging and image processing: 3 d
COVER TEASER.
Engineering proteins for allosteric control
Please indicate up to four primary research articles where the protocol has been used and/or developed.
Dagliyan, O. et al. Engineering extrinsic disorder to control protein activity in living cells. Science 354, 1441–1444 (2016)
Karginov, A.V. et al. Dissecting motility signaling through activation of specific Src-effector complexes. Nature Chem. Biol. 10(4): 286–290 (2014)
Dagliyan, O. et al. Rational design of a ligand-controlled protein conformational switch. Proc Natl Acad Sci U S A. 110(17):6800–4 (2013)
Karginov, A.V. et al. Engineered allosteric activation of kinases in living cells. Nature Biotechnology; 28(7): 743–7. (2010)
ACKNOWLEDGMENETS
This work was supported by NIH Grants R35GM122596 and P41-EB002025 (K.M.H), and R01-GM114015, R01-GM064803, and R01-GM123247 (N.V.D).
Footnotes
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
REFERENCES
- 1.Dagliyan O, et al. Engineering extrinsic disorder to control protein activity in living cells. Science 354, 1441–1444 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Karginov AV, et al. Dissecting motility signaling through activation of specific Src-effector complexes. Nat Chem Biol 10, 286–290 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chu PH, et al. Engineered kinase activation reveals unique morphodynamic phenotypes and associated trafficking for Src family isoforms. Proc Natl Acad Sci U S A 111, 12420–12425 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ostermeier M Designing switchable enzymes. Current Opinion in Structural Biology 19, 442–448 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oakes BL, et al. Profiling of engineering hotspots identifies an allosteric CRISPR-Cas9 switch. Nat Biotechnol 34, 646–651 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Edwards WR, Busse K, Allemann RK, Jones DD. Linking the functions of unrelated proteins using a novel directed evolution domain insertion method. Nucleic Acids Res 36, e78 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Inoue T, Do Heo W, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nature Methods 2, 415–418 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bayle JH, Grimley JS, Stankunas K, Gestwicki JE, Wandless TJ, Crabtree GR. Rapamycin analogs with differential binding specificity permit orthogonal control of protein activity. Chem Biol 13, 99–107 (2006). [DOI] [PubMed] [Google Scholar]
- 9.Spencer DM, Wandless TJ, Schreiber SL, Crabtree GR. Controlling Signal-Transduction with Synthetic Ligands. Science 262, 1019–1024 (1993). [DOI] [PubMed] [Google Scholar]
- 10.Karginov AV, Ding F, Kota P, Dokholyan NV, Hahn KM. Engineered allosteric activation of kinases in living cells. Nature Biotechnology 28, 743–U1756 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dagliyan O, et al. Engineering Pak1 Allosteric Switches. ACS Synth Biol 6, 1257–1262 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tucker CL, Fields S. A yeast sensor of ligand binding. Nature Biotechnology 19, 1042–1046 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Dagliyan O, et al. Rational design of a ligand-controlled protein conformational switch. Proc Natl Acad Sci U S A 110, 6800–6804 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Banaszynski LA, Chen LC, Maynard-Smith LA, Ooi AGL, Wandless TJ. A rapid, reversible, and tunable method to regulate protein function in living cells using synthetic small molecules. Cell 126, 995–1004 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bonger KM, Chen LC, Liu CW, Wandless TJ. Small-molecule displacement of a cryptic degron causes conditional protein degradation. Nature Chemical Biology 7, 531–537 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kennis JT, Crosson S, Gauden M, van Stokkum IH, Moffat K, van Grondelle R. Primary reactions of the LOV2 domain of phototropin, a plant blue-light photoreceptor. Biochemistry 42, 3385–3392 (2003). [DOI] [PubMed] [Google Scholar]
- 17.Peter E, Dick B, Baeurle SA. Mechanism of signal transduction of the LOV2-Jalpha photosensor from Avena sativa. Nat Commun 1, 122 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Harper SM, Neil LC, Gardner KH. Structural basis of a phototropin light switch. Science 301, 1541–1544 (2003). [DOI] [PubMed] [Google Scholar]
- 19.Wu YI, et al. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 461, 104–U111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang XB, He L, Wu YI, Hahn KM, Montell DJ. Light-mediated activation reveals a key role for Rac in collective guidance of cell movement in vivo. Nature Cell Biology 12, 591–U154 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayashi-Takagi A, et al. Labelling and optical erasure of synaptic memory traces in the motor cortex. Nature 525, 333–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guo XM, Liao ZH, Tao YZ, Wang FF, Ma L. [Optogenetic activation of dorsal hippocampal astrocytic Rac1 blocks the learning of associative memory]. Sheng Li Xue Bao 69, 241–251 (2017). [PubMed] [Google Scholar]
- 23.Das A, Dines M, Alapin JM, Lamprecht R. Affecting long-term fear memory formation through optical control of Rac1 GTPase and PAK activity in lateral amygdala. Sci Rep 7, 13930 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mills E, Chen X, Pham E, Wong S, Truong K. Engineering a Photoactivated Caspase-7 for Rapid induction of Apoptosis. Acs Synthetic Biology 1, 75–82 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Pham E, Mills E, Truong K. A Synthetic Photoactivated Protein to Generate Local or Global Ca2+ Signals. Chemistry & Biology 18, 880–890 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Schierling B, Pingoud A. Controlling the DNA Cleavage Activity of Light-Inducible Chimeric Endonucleases by Bidirectional Photoactivation. Bioconjugate Chemistry 23, 1105–1109 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Renicke C, Schuster D, Usherenko S, Essen LO, Taxis C. A LOV2 Domain-Based Optogenetic Tool to Control Protein Degradation and Cellular Function. Chemistry & Biology 20, 619–626 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Baarlink C, Wang HC, Grosse R. Nuclear Actin Network Assembly by Formins Regulates the SRF Coactivator MAL. Science 340, 864–867 (2013). [DOI] [PubMed] [Google Scholar]
- 29.Rao MV, Chu PH, Hahn KM, Zaidel-Bar R. An Optogenetic Tool for the Activation of Endogenous Diaphanous-related Formins Induces Thickening of Stress Fibers Without An Increase in Contractility. Cytoskeleton 70, 394–407 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rivera-Cancel G, Motta-Mena LB, Gardner KH. Identification of natural and artificial DNA substrates for light-activated LOV-HTH transcription factor EL222. Biochemistry 51, 10024–10034 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang X, Chen X, Yang Y. Spatiotemporal control of gene expression by a light-switchable transgene system. Nat Methods 9, 266–269 (2012). [DOI] [PubMed] [Google Scholar]
- 32.Liang FS, Ho WQ, Crabtree GR. Engineering the ABA Plant Stress Pathway for Regulation of Induced Proximity. Science Signaling 4, (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miyamoto T, et al. Rapid and orthogonal logic gating with a gibberellin-induced dimerization system. Nature Chemical Biology 8, 465–470 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yazawa M, Sadaghiani AM, Hsueh B, Dolmetsch RE. Induction of protein-protein interactions in live cells using light. Nature Biotechnology 27, 941–U105 (2009). [DOI] [PubMed] [Google Scholar]
- 35.Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, Tucker CL. Rapid blue-light-mediated induction of protein interactions in living cells. Nature Methods 7, 973–U948 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levskaya A, Weiner OD, Lim WA, Voigt CA. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 461, 997–1001 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang H, et al. LOVTRAP: an optogenetic system for photoinduced protein dissociation. Nat Methods, (2016). [DOI] [PMC free article] [PubMed]
- 38.Lee S, et al. Reversible protein inactivation by optogenetic trapping in cells. Nat Methods 11, 633–636 (2014). [DOI] [PubMed] [Google Scholar]
- 39.Taslimi A, et al. An optimized optogenetic clustering tool for probing protein interaction and function. Nat Commun 5, 4925 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou XX, Chung HK, Lam AJ, Lin MZ. Optical Control of Protein Activity by Fluorescent Protein Domains. Science 338, 810–814 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zayner JP, Sosnick TR. Factors that control the chemistry of the LOV domain photocycle. PLoS One 9, e87074 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanwar M, Wright RC, Date A, Tullman J, Ostermeier M. Protein switch engineering by domain insertion. Methods Enzymol 523, 369–388 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Aydin Z, Altunbasak Y, Borodovsky M. Protein secondary structure prediction for a single-sequence using hidden semi-Markov models. BMC Bioinformatics 7, 178 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y. The I-TASSER Suite: protein structure and function prediction. Nat Methods 12, 7–8 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Webb B, Sali A. Comparative Protein Structure Modeling Using MODELLER. Curr Protoc Protein Sci 86, 2 9 1–2 9 37 (2016). [DOI] [PubMed] [Google Scholar]
- 46.Alford RF, et al. The Rosetta All-Atom Energy Function for Macromolecular Modeling and Design. J Chem Theory Comput 13, 3031–3048 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Heinig M, Frishman D. STRIDE: a web server for secondary structure assignment from known atomic coordinates of proteins. Nucleic Acids Res 32, W500–502 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Finn RD, et al. Pfam: the protein families database. Nucleic Acids Res 42, D222–230 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simonetti FL, Teppa E, Chernomoretz A, Nielsen M, Marino Buslje C. MISTIC: Mutual information server to infer coevolution. Nucleic Acids Res 41, W8–14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Karginov AV, et al. Light Regulation of Protein Dimerization and Kinase Activity in Living Cells Using Photocaged Rapamycin and Engineered FKBP. Journal of the American Chemical Society 133, 420–423 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Garcia-Mata R, Wennerberg K, Arthur WT, Noren NK, Ellerbroek SM, Burridge K. Analysis of activated GAPs and GEFs in cell lysates. Methods Enzymol 406, 425–437 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Slattery SD, Hahn KM. A High-Content Assay for Biosensor Validation and for Examining Stimuli that Affect Biosensor Activity. Curr Protoc Cell Biol 65, 14.15.11–14.15.31 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5, 725–738 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lyons PD, Dunty JM, Schaefer EM, Schaller MD. Inhibition of the catalytic activity of cell adhesion kinase beta by protein-tyrosine phosphatase-PEST-mediated dephosphorylation. J Biol Chem 276, 24422–24431 (2001). [DOI] [PubMed] [Google Scholar]
- 55.Ramachandran S, Kota P, Ding F, Dokholyan NV. Automated minimization of steric clashes in protein structures. Proteins 79, 261–270 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Tsygankov D, Bilancia CG, Vitriol EA, Hahn KM, Peifer M, Elston TC. CellGeo: a computational platform for the analysis of shape changes in cells with complex geometries. J Cell Biol 204, 443–460 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tsygankov D, Chu PH, Chen H, Elston TC, Hahn KM. User-friendly tools for quantifying the dynamics of cellular morphology and intracellular protein clusters. Methods Cell Biol 123, 409–427 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Barry DJ, Durkin CH, Abella JV, Way M. Open source software for quantification of cell migration, protrusions, and fluorescence intensities. J Cell Biol 209, 163–180 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yan R, Xu D, Yang J, Walker S, Zhang Y. A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Sci Rep 3, 2619 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]