

Abstract

Intrinsically disordered proteins (IDPs) lack an ordered 3D structure. These proteins contain one or more intrinsically disordered protein regions (IDPRs). IDPRs interact promiscuously with other proteins, which leads to their structural transition from a disordered to an ordered state. Such interaction-prone regions of IDPs are known as molecular recognition features. Recent studies suggest that IDPs provide structural plasticity and functional diversity to viral proteins that are involved in rapid replication and immune evasion within the host cells. In the present study, we evaluated the prevalence of IDPs and IDPRs in human T lymphotropic virus type 1 (HTLV-1) proteome. We also investigated the presence of MoRF regions in the structural and nonstructural proteins of HTLV-1. We found abundant IDPRs in HTLV-1 bZIP factor, p30, Rex, and structural nucleocapsid p15 proteins, which are involved in diverse functions such as virus proliferation, mRNA export, and genomic RNA binding. Our study analyzed the HTLV-1 proteome with the perspective of intrinsic disorder identification. We propose that the intrinsic disorder analysis of HTLV-1 proteins may form the basis for the development of protein disorder-based drugs.
1. Introduction
Structurally, proteins are heterogeneous entities consisting of folded regions with variable conformational dynamics and disordered regions that do not have well-folded structures. Intrinsically disordered proteins (IDPs) are proteins that lack an ordered 3D structure. They contain one or more intrinsically disordered protein regions (IDPRs).1−3 Several studies have indicated that IDPs and proteins containing IDPRs are abundant in nature with ∼25–30% of eukaryotic proteins being mostly disordered and >50% of the disordered eukaryotic proteins possessing long-disordered regions.2,4 Functions of IDPs and IDPRs typically complement the functions of ordered proteins and domains. IDPs and IDPRs have been identified as key players in many critical cellular processes, such as being the component of macromolecular complexes wherein their conformational flexibility helps mediate interactions with binding partners.5 Because of the conformational plasticity, IDPs can act as hubs in the interaction networks and accomplish several functions in cell signaling and regulation.2,3 IDPRs are relatively long (>30 residues) stretches of disordered regions that flank globular domains on one or both ends.1,3,6 IDPs and IDPRs are also being implicated in various human diseases.7 It has been recognized that disordered regions can be a new and attractive target for drug design.8 IDPs typically interact with binding partners through short sequence motifs called molecular recognition features (MoRFs), making them capable of undergoing a disorder-to-order transition.9−12 IDPRs are enriched in amino acids that have a high mean net charge (that promote a strong electrostatic repulsion) and a low mean hydropathy (that lower the compaction of the polypeptide). Such amino acids are termed as “disorder-promoting amino acids” and include Gln, Ser, Pro, Glu, and Lys. Similarly, amino acids that have a low mean net charge and high mean hydropathy are termed as “order-promoting amino acids”, such as Cys, Trp, Ile, Tyr, Phe, Leu, Val, Asn, His, and Met. However, there are some amino acids that are intermediate between disorder- or order-promoting amino acids, such as Ala, Gly, Thr, Arg, and Asp.1 Because IDPs lack structural conformation under physiological conditions, they exist as the heterogeneous ensemble and are capable of rapidly changing conformation upon binding. In addition to this, IDPs are unable to crystallize; therefore, computational tools have provided an advantageous approach for analyzing IDPs/IDPRs.
Various studies have indicated that the prevalence of IDPs in the viral proteome is similar to eukaryotes; however, the prevalence of IDPs is higher in RNA viruses.13 The disordered nature of viral proteins is well suited for viral life-cycle and physiology. A large number of viruses have a small genome that encodes for only a few proteins, and the intrinsically disordered nature of viral protein enables them to interact with and modulate a wide range of host proteins and pathways. The IDPRs also reduce the drastic effect of the mutation on the structure and function of IDPs, which are highly prevalent in RNA viruses.14 Most of the IDPs from viruses contain MoRF regions that regulate the functionality of the viruses. These MoRFs are very crucial for the viral proteins that interact with host proteins to take over the cell for viral replication.15,16
The intrinsically disordered nature of the proteome of human T lymphotropic virus type 1 virus (HTLV-1) has not been studied till date. In the present study, we analyzed the prevalence of IDPs in HTLV-1 proteome and evaluated the MoRF regions. HTLV-1 was the first virus found to be linked with human malignancy and it has evolved to persist in the host for decades despite a vigorous virus–specific host immune response. A better understanding of IDPs/IDPRs and MoRF-based interactions of HTLV-1 proteins might assist in the development of novel compounds that could alter the protein–protein interactions and, therefore, can be implicated for the treatment of HTLV-1 infestation. In this study, we evaluated the prevalence of intrinsic disorder in HTLV-1 proteome using well-established computational methods as used earlier by several groups.15−23 Further, we have correlated the degree of structural disorder with functionality of HTLV-1 proteins. This study provides an unconventional direction that might help elucidating the molecular mechanisms of virus–host interaction.
2. Results and Discussion
HTLV-1 is a single-stranded retrovirus, about 100 nm in diameter, which expresses various proteins capable of inducing oncogenicity.24 According to an estimate, about 1 out of 25 HTLV-1 infected individuals develop malignancy, once the proviral DNA is integrated into the host genome upon transmission.25 The complex retroviral genome codes for viral structural proteins (Gag, Pro, Pol, and Env glycoproteins), regulatory proteins (Rex and Tax), and accessory proteins (p12, p13, and p30).24 The negative strand of the proviral mRNA encodes for basic leucine-zipper factor (HBZ).24,26 The complete genome structure of the HTLV-1 virus is depicted in Figure 1.
Figure 1.

Gene structure of HTLV-1 virus.
Despite its obvious threat, no disorder analysis has been reported for HTLV-1 proteins till date. Additionally, complete structures of only protease (PDB ID: 4YDF; protease in complex with 3,4-disubstituted pyrrolidines inhibitor), capsid p24 (PDB ID: 1QRJ), and Env (PDB ID: 1MG1) have been solved so far. Only partial structures for matrix p19 (PDB ID: 4ZNY; residue 119–129 in complex the human TSG101-UEV domain), rex (PDB ID: 1EXY; residues 1–16 bound to it RNA aptamer), and tax (PDB ID: 2AV1; residue 11–19 bound to human class I MHC HLA-A2 mutated at E63Q and K66A) are available. Therefore, computational tools may provide more advantage for analyzing the disorder propensities of the HTLV-1 proteins.
2.1. Compositional Profiling
The IDPs tend to differ in the composition of amino acids when compared with structured/ordered proteins. Hence, a simple protein sequence analysis termed as “compositional profiling” can provide a lot of information about the nature of a protein. Table 1 shows that matrix protein p19 (MA), nucleocapsid (NC), HTLV-1 bZIP factor (HBZ), p30, and Rex proteins were found to be fairly enriched in disorder-promoting amino acids compared with order-promoting amino acids, suggesting that they are likely to form IDPRs. In addition to this, the compositional profiles of all HTLV-1 structural proteins and nonstructural proteins were compared with a set of disordered proteins obtained from DisProt database (Figure 2A,B respectively). Individual comparative analysis is discussed below.
Table 1. Percentage of Order-Promoting, Neutral-Promoting, and Disorder-Promoting Amino Acids within the HTLV-1 Proteome.
| protein name | % order-promoting amino-acids (%) | % neutral amino acids (%) | % disorder-promoting amino acids (%) |
|---|---|---|---|
| MA | 35.62 | 24.01 | 40.29 |
| CA | 36.49 | 29.42 | 34.09 |
| NC | 30.60 | 25.88 | 43.52 |
| protease | 40.00 | 32.80 | 27.20 |
| RTp49 | 42.80 | 25.72 | 31.48 |
| RTp62 | 43.29 | 26.13 | 30.58 |
| IN | 40.72 | 30.49 | 28.79 |
| SU | 42.93 | 22.50 | 34.57 |
| TM | 45.50 | 28.39 | 26.11 |
| HTLV-1 basic leucine-zipper factor (HBZ) | 23.33 | 33.96 | 42.71 |
| P12 | 62.63 | 18.18 | 19.19 |
| P13 | 41.39 | 28.70 | 29.91 |
| P30 | 31.64 | 33.12 | 35.24 |
| Rex | 25.41 | 29.61 | 44.98 |
| Tax | 44.80 | 25.19 | 30.01 |
Figure 2.

Compositional profiling analysis. (A) Structural proteins present within HTLV-1. (B) Nonstructural proteins present within HTLV-1. The amino acid composition of HTLV-1 proteins is compared with a set of disorder proteins (black bars) that was obtained from DisProt database. Positive values correspond to amino acid residues that are found abundantly in specific HTLV-1 proteins. The negative values correspond to amino acid residues that are depleted within the specific HTLV-1 protein. Amino acids were placed from most order-promoting to most disorder-promoting, respectively.
2.2. Per-Residue Disordered Propensity and MoRF Analysis
The per-residue disorder propensity analysis of HTLV-1 proteins was carried out by using predictors, such as PONDR VSL2, PONDR VL-XT, PONDR-FIT, IUPred, and RONN, all of which have been assessed by CASP.27−29 The overall output of each predictor used was taken into consideration to determine the overall propensity of forming an IDPRs. Tables 2 and 3 show the physiochemical properties and the per-residue disorder profile of each HTLV-1 protein, respectively. The molecular weight (MW) and pI shown in Table 2 were predicted using the ProtParam tool available in ExPASy Bioinformatics Resource Portal (https://web.expasy.org/protparam/). Consistent with the compositional profiling analysis, per-residue propensity analysis also shows that the proteins—MA, NC, HBZ, p30, and Rex had several IDPRs within their sequence. Also, the p13 protein and the C-terminal domain of the Tax regulatory protein were predicted to contain IDPRs. The PONDR-FIT versus JRONN plot further showed the likelihood of forming disordered regions for NC, HBZ, Rex, p13, p30, and MA proteins (Figure 3A).
Table 2. Some Physicochemical and Intrinsic Disorder Properties of HTLV-1 Proteinsa.
| protein | length (MW) | pI | PPIDVLXT (%) | PPIDVSL2 (%) | PPIDFIT (%) | PPIDIUPred (long) (%) | PPIDIUPred (short) (%) | PPIDmean (%) |
|---|---|---|---|---|---|---|---|---|
| Gag-Pro-Pol | 1462 (162.9 kDa) | 9.31 | 25.58 | 33.38 | 13.54 | 21.25 | 14.56 | 21.66 |
| MA | 129 (14.2 kDa) | 9.25 | 38.76 | 50.39 | 41.08 | 31.00 | 38.76 | 42.63 |
| CA | 214 (23.8 kDa) | 8.19 | 34.11 | 40.19 | 17.28 | 30.84 | 24.29 | 24.76 |
| NC | 85 (9.42 kDa) | 7.78 | 57.64 | 100 | 100 | 64.70 | 51.76 | 91.76 |
| Protease | 125 (13.6 kDa) | 9.18 | 27.20 | 27.20 | 20.8 | 8.8 | 17.6 | 20.00 |
| RTp49 | 439 (49.2 kDa) | 8.98 | 18.68 | 21.18 | 8.42 | 8.68 | 8.68 | 9.13 |
| RTp62 | 585 (65.5 kDa) | 9.50 | 13.85 | 18.97 | 6.32 | 6.67 | 6.67 | 6.49 |
| IN | 295(33.0 kDa) | 9.62 | 26.44 | 37.29 | 17.28 | 17.28 | 11.52 | 17.96 |
| Env | 488 (53.9 kDa) | 8.45 | 16.80 | 15.98 | 4.71 | 0.28 | 1.63 | 4.30 |
| SU | 292 (32.36 kDa) | 7.63 | 21.57 | 27.05 | 7.53 | 1.02 | 5.13 | 8.21 |
| TM | 176 (19.31 kDa) | 9.55 | 15.90 | 11.93 | 3.40 | 0 | 3.40 | 2.84 |
| HBZ | 206 (24.9 kDa) | 6.83 | 84.95 | 89.80 | 83.49 | 64.07 | 48.05 | 81.06 |
| P12 | 99 (11.1 kDa) | 12 | 9.09 | 29.29 | 26.26 | 0 | 5.05 | 8.08 |
| P13 | 87 (10.1 kDa) | 11.76 | 55.17 | 59.77 | 71.26 | 13.79 | 25.28 | 51.72 |
| P30 | 241 (26.8 kDa) | 11.84 | 53.53 | 86.31 | 86.30 | 39.00 | 36.51 | 52.69 |
| Rex | 189 (20.6 kDa) | 9.54 | 69.31 | 81.48 | 82.01 | 81.48 | 64.55 | 79.36 |
| Tax | 353 (39.5 kDa) | 6.75 | 13.03 | 26.91 | 11.04 | 7.36 | 13.31 | 11.33 |
The length, pI, and predicted percentile of intrinsic disorder of the protein were calculated and shown.
Table 3. Disorder Regions and MoRF of Individual HTLV-1 Proteinsa.
| protein name | disorder regions | MORF regions |
|---|---|---|
| MA | 1–14 | 63–70 |
| 89–129 | 82–90 | |
| 123–129 | ||
| CA | 1–33 | 18–33 |
| 93 | 57–68 | |
| 95–106 | 116–122 | |
| 166 | ||
| 214 | ||
| NC | 1–16 | 64–75 |
| 24–85 | 80–85 | |
| protease | 1–19 | |
| 120–125 | ||
| RTp49 | 1–17 | 21–32 |
| 72–89 | ||
| 203–205 | ||
| 437–439 | ||
| RTp62 | 1–17 | 21–32 |
| 72–89 | ||
| 204–206 | ||
| IN | 1–10 | 227–232 |
| 41–49 | 253–276 | |
| 91–98 | ||
| 148–152 | ||
| 214 | ||
| 246–295 | ||
| SU | 165–172 | |
| 283–292 | ||
| TM | 172–176 | |
| HBZ | 32–189 | 1–30 |
| 45–51 | ||
| 200–206 | ||
| P12 | 92–99 | |
| P13 | 43–87 | |
| P30 | 72 | 152–163 |
| 75–155 | 172–190 | |
| 197–241 | ||
| Rex | 1–28 | 25–65 |
| 68–189 | 80–92 | |
| 106–117 | ||
| 143–155 | ||
| 168–174 | ||
| Tax | 1–3 | |
| 89–94 | ||
| 261 | ||
| 324–353 |
Regions highlighted in bold correspond to disordered regions having more than 30 amino acids at a stretch.
Figure 3.

Abundance of intrinsic disorder in various HTLV-1 proteins. (A) PONDR-fit vs JRONN plot analysis represents the co-relation of disorder content between PONDR-FIT (y-axis) and JRONN (x-axis) predictors. The plot distinguishes proteins as highly ordered [IDP score <10% (and <0.1), yellow field], moderately [IDP score between 10 and 30% (and between 0.1 and 0.3), light blue field], and highly disordered [IDP score >30% (and >0.3), purple area]. (B) CH–CDF analysis of structural and nonstructural proteins present within HTLV-1. From the corresponding figure, the co-ordinates for each protein were calculated using the output of two binary classifiers, where the x-axis co-ordinates represent the average vertical distance of the corresponding protein from the boundary line in the CH-plot and the y-axis represents the average vertical distance of the CDF curve from the CDF boundary in the CDF plot. Additionally, four quadrants were depicted: quadrant 1 (Q1), proteins predicted to be disordered by CH plot, but ordered by CDF plot; quadrant 2 (Q2), proteins predicted to be ordered by both methods; quadrant 3 (Q3), proteins predicted to be disordered by CDF plot, but ordered by CH plot (e.g., putative molten globules and mixed proteins); and quadrant 4 (Q4), proteins predicted to be disordered by both methods, including proteins with extended disorders.
MoRF regions are short molecular sites present within a long-disordered region capable of undergoing a transition from partial disordered to ordered state.9−11,30 Such transitions occur upon binding to protein interactors or other molecular interactors that are important for cellular recognition, signaling, and regulation.9−12,30 In our study, MoRF regions were predicted by using the ANCHOR algorithm that is based on a pair-wise estimation energy approach and developed from IUPred disorder predictors.31Table 3 shows the MoRF regions predicted by the ANCHOR algorithm. In-depth analysis of individual proteins is discussed below.
The intractability of HTLV-1 proteins was evaluated by using Viruses.STRING database that provides both experimental and predicted protein–protein interaction information.32 The result of the Viruses.STRING analysis is shown in Supporting Information Figure S1.
2.3. Evaluation of Disorder Propensities by Binary Classifiers
With respect to the per-residue propensity analysis, the overall status of the proteins as “ordered” or “disordered” was predicted by using binary classifiers. A combination of the outputs of two classifiers, that is, CH (charged-hydropathy) and CDF (cumulative distribution frequency), was used to generate the CH–CDF plot. The CH-plot analysis can differentiate proteins with random coils and pre-molten globules from molten globules and well-structured proteins.33,34 On the other hand, the CDF-plot analysis can differentiate between all disordered conformations, that is, distinguishing molten globules and mixed proteins from well-structured proteins.34,35 The CDF-plot analysis is more sensitive to protein disorders compared with the CH-plot analysis due to the inclusion of various protein attributes other than the charge and hydropathic properties.34 The location of a given protein on the CH–CDF plot gives information about its overall physical and structural characteristics. The CH–CDF plot depicted in Figure 3B shows that the HTLV-1 proteins—HBZ, NC, Rex, and p30 fall in quadrant 4, indicating that they are likely to be IDPs as a whole. On the other hand, the HTLV-1 proteins—p13 and MA fall in quadrant 3, indicating that they are mixed proteins and are likely to contain both disordered regions as well as ordered regions. Additionally, these proteins have a characteristic low net charge and low hydropathy, the features that are similar to those of unstructured proteins. The remaining HTLV-1 proteins fall in quadrant 2, indicating that they are mostly structured/ordered proteins.
2.4. Analysis of Structural Proteins of HTLV-1
The complex HTLV-1 genome codes for several structural proteins include Gag, Pro, Pol (UniProt ID P14078), and the Env glycoproteins gp62 (UniProt ID P14075).24 The Gag-Pro-Pol polyprotein is further classified into a gene cluster that includes numerous proteins. The Gag protein functions in the assembly of the virus and takes part in the infectivity on the host.36 The Gag protein is made up of three independent proteins: matrix protein (MA; responsible for anchoring of the Gag polyprotein to the plasma membrane), capsid protein (CA; important for the oligomerization of Gag during assembly and core formation during virus maturation), and nucleocapsid protein (NC; interacts with the genomic RNA signals and encapsulates the viral genomic RNA). All of the three proteins are cleaved by a retroviral protease protein, which is encoded by the pro gene.37−40 Also, the Pol polyprotein is cleaved by a protease into reverse transcriptase (RT)/RNase H (p49 and p62 subunit) and integrase (IN), which are required for the replication and integration of the viral genome.24,41,42Figure 4 shows that per-residue disorder propensity analysis of gag-pro-pol polyprotein and its cleavage into its respective products.
Figure 4.
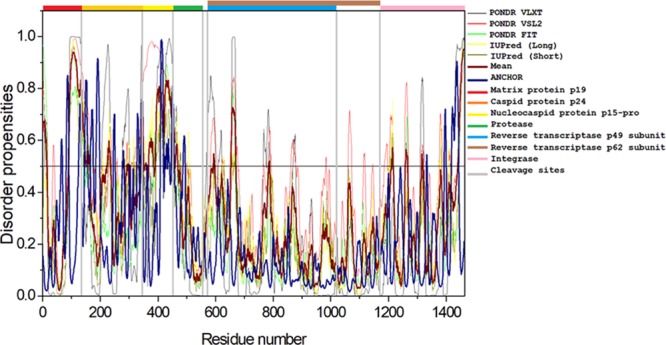
Intrinsic disorder predisposition of Gag-Pro-Pol polyprotein. Disorder propensities were generated by PONDR VLXT, PONDR VSL2, PONDR-FIT, IUPred (long), and IUPred (short) and are depicted by black, red, green yellow, and dark yellow lines, respectively. Dark red dashed lines represent the average of all disorder predictor’s scores used in this analysis. Blue dotted lines represent MoRF binding regions predicted using ANCHOR. Colored bars at the top of the plot show the localization of the individual proteins. Corresponding cleavage sites leading to the generation of mature individual proteins are shown in gray bars.
2.4.1. Matrix Protein p19 (MA)
HTLV-1 is an encapsulated virus whose envelope contains the viral MA within the inner membrane. The MA is crucial for anchoring of the Gag polyprotein to the plasma membrane38 and plays a role in the genomic RNA packaging.43 Its ability to anchor relies on the N-terminal myristoylation and the presence of basic amino acids, like Lys and Arg that stabilize the binding of the Gag polyprotein to the acidic plasma membrane surface.38,40 The basic amino acids are probably clustered in one region, even though distributed throughout the primary sequence of the MA protein. The HTLV-1 MA contains eight basic amino acids that individually do not have any role in the attachment but as a whole, and they help in the anchoring of the Gag polyprotein to the plasma membrane.38,40 In fact, it has been reported that none of the basic residues is important for Gag targeting but nearly all are crucial for subsequent stages of production, events leading to budding of the virus particles, and infectivity.38 Thus, such an association is required for the final assembly and budding of the virus particles.
The HTLV-1 MA, consisting of 129 amino acid residues, has a MW of ∼14.2 kDa and mean predicted percent of intrinsic disorder (PPIDmean) of 42.63% (Table 2). The PPIDmean was obtained from the output of five predictors used in our analysis (PONDR VLXT, PONDR VSL2, PONDR FIT, IUPred Long, and IUPred short). The compositional profiling analysis of MA, when compared with a set of disordered proteins, showed that MA is enriched in several order-promoting amino acids (e.g., Ile, Tyr, Leu, and His) but depleted in two disorder-promoting amino acids (i.e., Glu and Lys). Additionally, MA was found to be highly depleted in Val, Asn, Met, Thr, Asp, and Gly but highly enriched in Gln, Pro, and Ser when compared with DisProt proteins (Figure 2A). A detailed amino acid composition of HTLV-1 MA is shown in Figure 5A.
Figure 5.

Detailed amino acids composition for structural HTLV-1 proteins. (A) Matrix p19, (B) capsid p24, (C) nucleocapsid p15, (D) protease, (E). RNase H/RTp49 sub-unit, (F) RNase H/RTp62 subunit, (G) IN, (H) SU, and (I) TM. Red color represents disorder-promoting amino acids, blue color represents order-promoting amino acids, and yellow color represents amino acids that are indifferent to disorder or order.
The disorder propensity analysis of HTLV-1 MA is illustrated in Figure 6A. In the figure, on combining the output scores of all the predictors, the mean analysis showed the presence of two disordered regions—[1–14] and [89–129], where the longest disordered region was 41 residues long [89–129]. Interestingly, the CDF-plot analysis (Figure 3B) predicted this protein to be disordered, whereas the CH-plot analysis predicted this protein to be ordered. Thus, an agreement can be made that this protein is characterized by low mean net charge and low hydropathy but it might contain other characteristics that favor unstructured assembly.
Figure 6.

Disorder predisposition of structural proteins within the HTLV-1 genome. (A) Matrix p19, (B) capsid p24, (C) nucleocapsid p15, (D) protease, (E) RNase H/RTp49 sub-unit, (F) RNase H/RTp62 subunit, and (G) IN. Disorder profiles were generated by PONDR VLXT, PONDR VSL2, PONDR-FIT, IUPred (long), and IUPred (short) and are depicted by black, red, green yellow, and dark yellow lines, respectively. Dark red dashed lines represent the average of all disorder predictor’s scores used. Blue dotted lines represent MoRF binding regions predicted using ANCHOR.
The ANCHOR analysis showed the presence of three ANCHOR indicated binding sites (AIBS): [63–70], [82–90], and [123–129] (Figure 6A). One region was filtered out due to insufficient criteria [48–48]. Interestingly, AIBS—[82–90] partially coincides with the longest disordered region—[89–129] predicted by the mean analysis. Also, it is interesting to note that this region is the MA–CA cleavage site, which suggests that the flexibility in this region is required for protease binding and cleavage. A recent cryo-EM analysis indicated that the HTLV-1 Gag virus-like particles (VLPs) lattice is less ordered compared with HIV Gag VLPs, which could be due to disordered regions in HTLV-1 MA.44
2.4.2. Capsid Protein p24 (CA)
Like any other retroviruses, HTLV-1 is enveloped and packaged with two genomic RNA within a spherical core structure. This core structure is formed by the ∼24 kDa CA. The HTLV-1 CA protein functions in the Gag oligomerization during assembly and in the formation of the spherical core that encapsulates the genomic RNA-nucleocapsid complex.39 Initially, the HTLV-1 CA is synthesized as a domain of the structural Gag-polyprotein. As the Gag assembles on the plasma membrane along with other viral proteins and RNA, acquiring the outer membrane for budding from the infected cells, the CA protein is cleaved into a newly folded mature virion by a viral protease protein.37 The mature CA that forms the core folds into two independent domains, N-terminal domain (NTD; stretching between 15 and 127 amino acid position) and the C-terminal domain (CTD; stretching between 131 and 206 amino acid position), which are joined by a linker peptide (128AKD130 residues).37,39 The NTD residues form a β-hairpin upon cleavage, which is further stabilized by the formation of salt bridges between the conserved P1 and a Asp/Glu residue from helix 3.37 This refolded CA protein was postulated to be important for the CA–CA interface, crucial for the formation of the spherical core, and is required for initiating replication in the next infectivity.37 Mutational analysis showed that the CTD region is not involved in the Gag–Gag interaction, whereas NTD mutation impairs virion morphogenesis, and thus considered essential for VLP formation.37
The HTLV-1 CA consists of 214 amino acids and has a MW of ∼24 kDa and PPIDmean of 24.76% (Table 2). The compositional profiling analysis of HTLV-1 CA, when compared with a set of disorder proteins, showed that it is enriched in several order-promoting amino acids (i.e., Trp, Leu, and His) and depleted in several disorder-promoting amino acids (i.e., Ser, Glu, and Lys) (Figure 2A). However, HTLV-1 CA was found to be highly enriched in Pro and Gln that are disorder-promoting amino acids. A detailed amino acid composition of HTLV-1 CA is shown in Figure 5B.
Mean disorder propensity analysis of HTLV-1 CA showed the presence of five disordered regions—[1–33], [93], [95–106], [166], and [209–214], with the longest disordered region being 33 residues long [1–33] (Figure 6B). With respect to the CH–CDF plot analysis, HTLV-1 CA was predicted to be fully ordered protein (Figure 3B). This prediction is supported by an NMR study that predicted HTLV-1 CA to be more rigid compared with HIV CA.45
The ANCHOR analysis of HTLV-1 CA (Figure 6B) showed the presence of three AIBS—[18–23], [57–68], and [116–122]. Also, three regions were filtered out due to their small sizes, [41–44], [148–148], and [150–152]. The AIBS—[18–23] is present within the longest disordered region—[1–33] as predicted by the mean analysis. The presence of disordered regions and AIBS suggests that CA might exhibit some degree of flexibility. As a polyprotein, during cleavage and after cleavage, this flexibility might help it to form a rigid structure using the Pro1-Asp54 salt bridge.46
2.4.3. Nucleocapsid Protein p15 (NC)
The retroviral particle, NC, is an important component of the capsid as it encloses the dimeric genomic RNA. It is critical for recognition and packaging of the viral genome and hence considered a suitable target for antiretroviral therapy. The HTLV-1 NC is a neutral protein characterized by a basic NTD and a negatively charged CTD.47 Two zinc-finger binding domains (i.e., ZF1 and ZF2) are present at positions 11–28 and 34–51.47 The NC of HIV plays a critical role in the binding and packing of RNA and also has strong chaperone activity that helps in reverse transcription and integration of the viral genome. However, the HTLV-1 NC nonspecifically binds to genomic RNA and exhibits very poor chaperone activity.48 It has been reported that zinc-finger binding domains can interact with each other, while NTD can interact with the CTD via electrostatic interactions when NC is not bound to RNA. Such an intramolecular interaction is postulated to control the binding of this protein to DNA and impairing chaperone properties of this protein.47
The HTLV-1 NC consists of 85 amino acids with a MW of ∼9.4 kDa. PPIDmean predicted an average of 91.76% disordered region in the protein (Table 2). The compositional profiling analysis of the HTLV-1 NC, when compared with a set of disorder proteins, showed that NC is enriched in several order-promoting amino acids (e.g., Cys, Trp, Leu, and His) and depleted in several disorder-promoting amino acids (e.g., Ser and Glu) (Figure 2A). Additionally, the HTLV-1 NC was found to be depleted in Ile, Tyr, Phe, Val, Asn, Met, and Ala and highly enriched in Pro when compared with DisProt proteins. A detailed amino acid composition of the HTLV-1 NC is shown in Figure 5C.
Disorder propensity analysis of the HTLV-1 NC shows the presence of two disordered regions, that is, [1–16] and [24–85], with the longest disordered region being 62 residues long [24–85] (Figure 6C). As a whole, the CH–CDF plot (Figure 3B) analysis predicted this protein to be fully disordered. The disordered and flexible nature of the NC might enable the intramolecular interaction of the protein to preclude it from functioning as a better chaperone. The disordered nature of the NC might also be responsible for its inability to bind with the packing signal on the HTLV-1 genomic RNA.
The ANCHOR analysis showed the presence of two AIBS—[64–75] and [80–85] (Figure 6C). One region was filtered out due to insufficient criteria [19–19]. Interestingly, both the AIBS coincide with the longest disordered region predicted [47–105] by the mean analysis of the predictors used.
2.4.4. Protease
HTLV-1 protease is a homodimeric aspartic protease that consists of 125 amino acid residues and has a high sequence similarity with other retroviral proteases. It is encoded in a reading frame spanning from the 3′-end of the gag region to the 5′-end of the pol region (Figure 1), where its synthesis occurs by ribosomal frameshifting. Structurally, this protein has an active site motif similar to other retroviral proteases and has homology in sequences flanking the active region as well.49 Upon maturation, this protease cleaves the Gag precursor into MA, CA, and NC proteins, as well as Pol precursor into RT and IN proteins.41 This indicates the crucial role of HTLV-1 protease in viral replication.
Reports suggest that the C-terminal extension of this protein is key to the dimerization in the absence of a ligand.50 The flexibility of the C-terminal tail allows the protease to switch between a compact structure and a more freely opened structure.50 The HTLV-1 protease consists of 125 amino acids and has a MW of ∼13.6 kDa. The average percentile of disorder region is quite low with a PPIDmean of 20%.
The compositional profiling analysis of the HTLV-1 protease showed that it is enriched in several order-promoting amino acids (i.e., Ile, Leu, and Val) and depleted in several disorder-promoting amino acids (i.e., Ser, Glu, and Lys) (Figure 2A). Additionally, the HTLV-1 protease was found to be depleted in Trp, Tyr, Phe, His, Asn, Met, and Gly and highly enriched in Gln and Pro when compared with DisProt proteins. A detailed amino acid composition of protease is shown in Figure 5D.
Disorder propensity analysis of the HTLV-1 protease showed the presence of two disordered regions, that is, [1–19] and [120–125], with the longest disordered region being 19 residues long [1–19] (Figure 6D). As a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed no AIBS present whatsoever.
2.4.5. Reverse Transcriptase
The HTLV-1 RT protein is essential for the replication of the viral genome, making it a potential drug target. This protein has not been fully characterized yet; however, a recent study has suggested that Pol encodes for IN, a large subunit of reverse transcriptase p62 (RTp62), and a small subunit of reverse transcriptase p49 (RTp49).51
RTp49, the small subunit, consists of 439 amino acids with a MW of ∼49 kDa. The disordered region present within this protein is predicted to be fairly low with a PPIDmean of 9.13% (Table 2). The compositional profiling analysis of the HTLV-1 RTp49 showed that it is enriched in several order-promoting amino acids (e.g., Trp, Leu, and His) and depleted in two disorder-promoting amino acids (i.e., Lys and Glu) (Figure 2A). Additionally, the protein was depleted in Val, Asn, Met, Arg, Asp, Gly, and Ala and highly enriched in Gln and Pro when compared with DisProt proteins. A detailed amino acid composition of the HTLV-1 RTp49 is shown in Figure 5E. Disorder propensity analysis of the protein is illustrated in Figure 6E. The mean analysis showed the presence of four disordered regions, that is, [1–17], [72–89], [203–205], and [437–439], with the longest disordered region being 17 residues long [1–17] and [72–89]. However, as a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed the presence of one AIBS, that is, [21–32] (Figure 6E).
RTp62, the large subunit, consists of 585 amino acid residues with a MW of ∼65.5 kDa. The disorder regions present within this protein are predicted to be fairly low with a PPIDmean of 6.49% (Table 2). The compositional profiling analysis of the HTLV-1 RTp62 shows that it is enriched in several order-promoting amino acids (i.e., Trp, Leu, and His) and depleted in two disorder-promoting amino acids (i.e., Lys and Glu) (Figure 2A). Additionally, the protein was found to be depleted in Val, Asn, Met, Asp, Gly, and Ala and highly enriched in Gln and Pro when compared with DisProt proteins. A detailed amino acid composition of the HTLV-1 RTp62 is shown in Figure 5F. Disorder propensity analysis, illustrated in Figure 6F, showed the presence of three disordered regions, that is, [1–17], [72–89], and [204–206], with the longest disordered region being 18 residues long [72–89]. However, as a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed the presence of one AIBS, that is, [21–32] (Figure 6F).
2.4.6. Integrase
The Pol also encodes for IN that consists of 295 amino acid residues and has a MW of ∼33 kDa. In general, IN is a retrovirus enzyme that mediates the integration of the viral DNA into the host’s genomic DNA.52 However, currently, limited information is available for HTLV-1 IN. The disordered propensity of this protein was predicted to be fairly low with a PPIDmean of 17.96% (Table 2). The compositional profiling analysis of the HTLV-1 IN showed that it is enriched in several order-promoting amino acids (e.g., Cys, Trp, Leu, His, Asn, and Arg) and depleted in two disorder-promoting amino acids (i.e., Glu and Lys) (Figure 2A). Additionally, the protein was found to be depleted in Phe, Val, Met, Asp, and Gly and moderately enriched in Gln, Ser, and Pro when compared with DisProt proteins (Figure 2A). A detailed amino acid composition of the protein is shown in Figure 5G.
Disorder propensity analysis of the HTLV-1 IN showed the presence of six disordered regions, that is, [1–10], [41–49], [91–98], [148–152], [214], and [276–295], with the longest disordered region being 20 residues long [276–295] (Figure 6G). As a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed the presence of two AIBS, that is, [227–232] and [253–276] (Figure 6G). Three regions—[31–33], [72–76], and [165–167] were filtered out as they did not meet the criteria. The AIBS—[276–295] partially coincides with the disordered region [276–295], as predicted by the mean analysis.
2.4.7. Envelope Glycoprotein (Env)
The HTLV-1 Env is encoded by the gene env (gp62) and is synthesized in the endoplasmic reticulum, whereupon maturation it is cleaved to a ∼46 kDa surface membrane protein (SU) and a ∼21 kDa transmembrane protein (TM).40Figure 7A shows the disorder propensity of Env glycoprotein and its cleavage products—SU and TM, respectively. Colored bars at the top of the plot represent the location of various proteins encoded within the Env.
Figure 7.

Intrinsic disorder predisposition of envelope glycoprotein. Disorder propensities were generated by PONDR VLXT, PONDR VSL2, PONDR-FIT, IUPred (long), and IUPred (short) and are depicted by black, red, green yellow, and dark yellow lines, respectively. Dark red dashed lines represent the average of all disorder predictor’s scores used. Blue dotted lines represent MoRF binding regions predicted using ANCHOR. Colored bars at the top of the plot show the localization of the individual proteins. Corresponding cleavage sites leading to the generation of mature individual proteins are shown in gray bars. (A) Disorder predisposition of Env glycoprotein precursors, (B) SU, and (C) TM.
2.4.8. Surface Membrane Protein
The HTLV-1 SU protein consists of 292 amino acid residues and has a MW of ∼32 kDa. It contains three domains: an NTD (characterized by a α-helix and a highly variable β-sheet), a central domain (characterized by a Pro-rich region followed by a helix-turn-helix domain), and a C-terminal domain (characterized by a conserved-PPI/FS-motif) that are separated by hinges.40,53 The percentage of disorder region within SU was predicted to be fairly low with a PPIDmean of 8.21% (Table 2). The compositional profiling analysis of the SU protein showed that it is enriched in several order-promoting amino acids (i.e., Cys, Trp, Tyr, Leu, and His) and depleted in three disorder-promoting amino acids (i.e., Glu, Lys, and Gln) (Figure 2A). Additionally, the SU protein was found to be depleted in Met, Arg, Asp, Gly, and Ala and highly enriched in Ser and Pro when compared with DisProt proteins. A detailed amino acid composition of the SU protein is shown in Figure 5H. Disorder propensity analysis showed the presence of two disordered regions, that is, [165–172] and [283–292], with the longest disordered region being 10 residues long [283–292] (Figure 7B). As a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed no AIBS whatsoever (Figure 7B).
2.4.9. Transmembrane Protein
The HTLV-1 TM protein contains two domains, an N-terminal trimeric coiled-coil formed by the leucine-zipper-like region and a C-terminal domain that is structurally distinct from other retroviruses.54 Studies have suggested that the leucine-zipper-like region is important for the fusion process, whereas the C-terminal domain is crucial for the postfusion steps required for the virus’s infectivity.53 The TM protein consists of 176 amino acid residues with a MW of ∼19 kDa. The disorder propensity of TM protein was predicted to be fairly low with a PPIDmean of 2.84% (Table 2). The compositional profiling analysis of the TM protein, when compared with a set of disorder proteins, showed that it is enriched in several order-promoting amino acids (i.e., Cys, Trp, Leu, His, and Arg) and depleted in three disorder-promoting amino acids (i.e., Glu, Lys, and Pro) (Figure 2A). Additionally, the TM protein was found to be depleted in Phe, Leu, Met, Thr, and Asp and enriched in Gln and Ser when compared with DisProt proteins. A detailed amino acid composition of the TM protein is shown in Figure 5I. Disorder propensity analysis showed the presence of one disordered region of 5 residues long [172–176] (Figure 7C). As a whole, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis showed no AIBS whatsoever (Figure 7C).
2.5. Analysis of Nonstructural HTLV-1 Proteins
Besides the structural proteins (Gag, Pol, and Env), the complex retrovirus HTLV-1 also encodes for nonstructural proteins that typically constitute the regulatory and accessory proteins. HTLV-1 uses alternative splicing and internal initiation codons to generate these nonstructural proteins, which lies within the pX region of the viral genome, that is, between the env and the 3′LTR (Figure 1).24,55
The HTLV-1 nonstructural proteins are encoded within four ORFs present within the pX region. The ORF I and ORF II produce an alternative spliced form of mRNA that encodes four accessory proteins, p12I, p27I, p13II, and p30II.56−58 These accessory proteins have been reported to be critical for the viral infectivity and for maintaining a high viral load, activation of the host cell, and regulation of gene transcription.24,55,56 HTLV-1 genome encodes for two regulatory proteins—Rex and Tax within the pX ORF III and ORF IV that share a common doubly spliced transcript.59−61 The ORF III encodes for Rex protein that is primarily responsible for the nuclear export of unspliced or singly spliced viral RNA.24 The ORF IV encodes for Tax protein that potentially activates transcription, not only from the viral promoter (Tax-responsive elements) but also from the enhancer elements of many cellular genes involved in the host cell proliferation.24,62 The Tax protein has oncogenic capabilities essential for HTLV-1 transformation of the primary human T-cells.62,63
The minus strand (−) of the HTLV-1 proviral genome also encodes for several isoforms (generated from unspliced and spliced mRNA) of the HTLV-1 basic leucine-zipper factor (bZIP), denoted as HBZ. Reports suggest that HBZ interacts with cellular factors like Jun B, c-Jun, Jun D, cAMP response element binding (CREB), and CREB binding protein (CBP)/p300 to modulate both the viral and cellular gene expression.26,64 It was also reported that HBZ plays a crucial role in T-cell proliferation.64
2.5.1. HTLV-1 Basic Leucine-Zipper Factor (HBZ)
The minus strand of HTLV-1 genome encodes for spliced HBZ (206 amino acid residues) and unspliced HBZ (209 amino acid residues) proteins, denoted as sHBZ and usHBZ, respectively. The only difference between sHBZ and usHBZ is of seven amino acids present within the NTD, which are responsible for the short life span of usHBZ compared with sHBZ in Adult T-cell Lymphoma cells.64 Both forms of HBZ contain three domains: an N-terminal activation domain (AD), a central domain, and a basic ZIP domain.64 Two −LXXLL– like motifs are present within the N-terminal AD, which are known to bind to CBP/p300, thereby inhibiting the recruitment of HTLV-1 promoter.65 The HBZ bZIP binds to CREB/CREB2, which abolishes the Tax-responsive elements and results in the suppression of Tax-mediated transcription forms.26 It has been reported that sHBZ can induce proliferation via the first exon of the sHBZ transcript, whereas usHBZ is unable to induce proliferation.66 Additionally, HBZ can interact with c-Jun/JunB and suppress c-Jun/JunB-mediated signaling by abrogating its DNA binding capability.67 On the other hand, HBZ can activate transcription by forming heterodimers with JunD that binds to Sp1 transcription factor and enhances the transcription of telomerase RT.68 Another important finding was the inhibition of the classical NF-κβ pathway, critical for innate immunity and inflammatory responses, by HBZ. The HBZ selectively suppresses p65-mediated classical NF-κβ pathway without inhibiting alternative NF-κβ signaling by (i) binding to p65 (abolishing its DNA binding capability) and (ii) increasing the production of PDLIM2, an E3 ubiquitin ligase for p65.69 This is crucial for HTLV-1 virulence, survival, and infectivity. Recent reports suggest that transgenic expression of HBZ increases Foxp3+ Treg cells and effector/memory T cells, leading to the development of T cell lymphoma and systematic inflammatory diseases.70 HBZ can also enhance TGF-β signaling, critical for the development of Treg. These findings suggest that HBZ is crucial for HTLV-1 proliferation, virulence, survival, and infectivity.71
The sHBZ protein consists of 206 amino acids residues and has a MW of ∼6.5 kDa. The average disorder propensity of the protein was found to be very high with a PPIDmean of 81.06% (Table 2). The compositional profiling analysis of the protein shows that it is enriched in two order-promoting amino acids (i.e., Leu and Arg) and depleted in two disorder-promoting amino acids (i.e., Ser and Pro) (Figure 2B). Additionally, the sHBZ protein was found to be depleted in Ile, Tyr, Phe, His, Val, Asn, Thr, Asp, and Ala and highly enriched in disorder-promoting amino acids (Lys, Gln, and Glu) when compared with DisProt proteins. A detailed amino acid composition of the sHBZ protein is shown in Figure 8A.
Figure 8.

Detailed amino acids composition for nonstructural HTLV-1 proteins. (A) HBZ, (B) P12I, (C) p13, (D) p30II, (E) Rex, and (F) Tax. Red color represents disorder-promoting amino acids, blue color represents order-promoting amino acids, and yellow color represents amino acids that are indifferent to disorder or order.
Detailed disorder propensity analysis of the sHBZ protein showed the presence of one disordered region, that is, [32–189] that was 158 residues long (Figure 9A). As a whole, the CH–CDF analysis predicted this protein to be fully disordered (Figure 3B). It is interesting to note that the disordered region encompasses the NTD of HBZ that through its tandem (−LXXLL−) domains interacts with transcription factor—CREB and also binds to KIX-domain of coactivator—p300/CBP.26,72,73
Figure 9.

Disorder predisposition of nonstructural proteins within the HTLV-1 genome. (A) HBZ, (B) P12I, (C) p13, (D) p30II, (E) Rex, and (F) Tax. Disorder profiles were generated by PONDR VLXT, PONDR VSL2, PONDR-FIT, IUPred (long), and IUPred (short) and are depicted by black, red, green yellow, and dark yellow lines, respectively. Dark red dashed lines represent the average of all disorder predictor’s scores used in this analysis. Blue dotted lines represent MoRF binding regions predicted using ANCHOR.
The ANCHOR analysis showed the presence of three AIBS, that is, [1–30], [43–51], and [200–206] (Figure 9A). Three regions were filtered out [99–103], [121–124], and [161–162] due to their small size. The binding region [43–51] coincides with the disordered region predicted by the mean analysis and also coincides with second −LXXL– (44–53) motif. Because most of the interactions and functions of HBZ are mediated through its NTD, it was expected for this region to be intrinsically disordered. A recent study has also supported these results, which provided evidence of the intrinsically flexible nature of the HBZ NTD.74
2.5.2. P12
The p12 protein is generally localized in cellular membranes, particularly the endoplasmic reticulum and the Golgi bodies.75 The amino acid analysis suggested the p12 to be a highly hydrophobic structural protein, which is characterized by at least 32% Leu and 17% Pro.75 The p12 protein is highly conserved and contains four Pro-rich Src homology 3 (SH3) binding domains, a motif critical for interaction with proteins involved in intracellular signaling. A di-Leu motif (−DXXXLL−) at amino acid position 26–31 and two transmembrane domains at positions 12–30 and 48–67 have also been identified. The p12 protein interacts with interleukin-2 receptor (IL-2R), drastically affects its surface expression, and increases transcriptional activity of the signal transducers and activators of transcription-5 (STAT5). The ability of p12 to lower the threshold of T-cell activation and IL-2 production and the ability to increase responsiveness to IL-2 by STAT5 activation suggests that p12 amplifies physiological stimulations and it is involved in the proliferation of infected cells in a microenvironment depleted in antigens and growth factors.76,77 The p12 protein has been reported to bind to free MHC class I heavy chains that subsequently fail to bind to β2-microglobulin and translocated back to the cytosol, where they are degraded by the proteasome. This binding contributes to evasive measures of HTLV-1 from the host’s immune response.
The p12 protein consists of 99 amino residues and has a MW of ∼9.9 kDa. The disorder propensity of the p12 protein was found to be fairly low with a PPIDmean of 8.08% (Table 2). The compositional profiling analysis of the protein showed that it is enriched in three order-promoting amino acids (i.e., Trp, Phe, and Leu) but depleted in three disorder-promoting amino acids (i.e., Lys, Gln, and Glu) (Figure 2B). Additionally, the protein was depleted in Ile, Tyr, His, Val, Asn, Asp, Thr, Gly, and Ala and highly enriched in Ser and Pro when compared with DisProt proteins. A detailed amino acid composition of the p12 protein is shown in Figure 8B. The disorder propensity analysis of p12 is depicted in Figure 9B.
Mean analysis of the disorder predictors showed only one disordered region that was 8 residues long [92–99] (Figure 9B). In addition, the CH–CDF analysis predicted this protein to be fully ordered, as a whole (Figure 3B). The ANCHOR analysis showed no AIBS whatsoever.
2.5.3. P13
The p13 protein consists of 87 amino acid residues and has a MW of ∼10 kDa. It consists of a short leading sequence (between 1 and 5 amino acid position), a positively charged amphipathic α-helix with an N-terminal mitochondrial targeting signal between 22 and 31, a transmembrane domain at position 30–40, a flexible hinge region between 42 and 48, and a C-terminus β-hairpin homologous to an α-bungarotoxin binding peptide at position 65–75.76,78 The C-terminus of the p13 protein also contains multiple −PXXP– motifs. Though p13 protein is usually localized in the inner membrane of mitochondria, at a high level it might be localized even in the nucleus, especially when co-expressed with Tax, suggesting its potential role in modulating the apoptosis and transcriptional regulation.78 Because of its mitochondrial localization, p13 was proposed to function in mediating K+ influx, inner mitochondrial membrane potential, and electron transport chain activity, which subsequently affects Ca2+ and ROS production.78 Modulation of ROS level has been proposed to provide life-long persistence to HTLV-1 within the host cells.78 At a high level, the p13 protein is ubiquitinated by Tax, leading to its translocation into the nucleus, thereby hampering Tax-mediated viral gene transcription.78,79
The disorder propensity of the p13 protein was found to be quite high with a PPIDmean of 51.72% (Table 2). The compositional profiling analysis of the p13 protein showed that it is enriched in several order-promoting amino acids (i.e., Trp, Leu, His, and Arg) but depleted in three disorder-promoting amino acids (i.e., Lys, Gln, and Glu) (Figure 2B). Additionally, the protein was depleted in Phe, Asn, Asp, Gly, and Ala and highly enriched in Thr, Ser, and Pro when compared with DisProt proteins. A detailed amino acid composition of the p13 protein is shown in Figure 8C.
The mean analysis of the p13 protein showed only one long disordered region that was 45 residues long [43–87] (Figure 9C). Interestingly, the CH–CDF analysis predicted this protein to be disordered by CDF analysis, whereas the CH analysis predicted this protein to be ordered (Figure 3B), suggesting that although this protein is characterized by low mean net charge and low hydropathy, this protein might contain other characteristics that favor disordered structure. The ANCHOR analysis showed no AIBS whatsoever.
2.5.4. P30
The P30 protein consists of 241 amino acid residues and has a MW of ∼26.8 kDa. The p30 protein is highly basic with a net positive charge. It contains three nuclear localization signals (NLS1 between 66 and 73; NLS2 between 91 and 98; and NLS3 between 200 and 241 amino acid position) along with an Arg-rich nucleolar localization/retention (NoRS) domain between 73 and 78 position. The P30 protein also contains a Rex-binding domain between 131 and 164, a p300-binding domain between 131 and 161, and a DNA-binding domain between 100 and 179, which represses LTR-mediated transcription.24,75,76 Although the exact role of p30 is still unknown, few reports have postulated its function in cellular processes like cell cycle, DNA repair, and mRNA export.24 It has also been reported that the p30 protein promotes virus latency to escape host immune responses by retaining tax/rex mRNA and preventing their export into the cytoplasm, which subsequently decreases the expression of these two positive regulators of HTLV-1 replication.80 This, in turn, favors cell proliferation and clonal expansion of the infected cells. The P30 protein is also known to interact with many host proteins, such as ATM, REG-gamma, p300, TIP60, PU1, and MRN complex, to regulate viral and cellular gene expression, to modulate the cellular environment, and to facilitate viral persistence and transformation.81
The average disorder propensity of the p30 protein was found to be quite high with a PPIDmean of 52.69% (Table 2). The compositional profiling analysis of the p30 protein, when compared with a set of disorder proteins, showed that the protein is enriched in several order-promoting amino acids (e.g. Cys, Trp, Phe, Leu, His, and Arg) but depleted in three disorder-promoting amino acids (e.g., Lys, Gln, and Glu) (Figure 2B). Additionally, the protein was depleted in Ile, Tyr, Val, Asn, Met, Asp, Gly, and Ala and highly enriched in Ser and Pro when compared with DisProt proteins. A detailed amino acid composition of the p30 protein is shown in Figure 8D. The mean analysis of the p30 protein showed two disordered regions, that is, [75–155] and [197–241], with the longest disordered region being 81 residues long [75–155] and the second longest disordered region being 45 residues long [197–241] (Figure 9D). Additionally, the CH–CDF analysis predicted this protein to be fully disordered, as a whole (Figure 3B). The disordered nature of the p30 protein might enable it to interact with various host proteins and viral RNA at the same time and act as a multifunctional protein required for HTLV-1 persistence. The ANCHOR analysis showed two AIBS, that is, [152–163] and [172–190]. One region was filtered out [116–117] (Figure 9D). Region [152–163] was partially present within the disordered region as predicted by the mean analysis.
2.5.5. Rex
HTLV-1 encodes a regulatory protein—Rex that actively transports unspliced and singly spliced mRNA from the nucleus into the cytoplasm.24,82 The HTLV-1 Rex is a positive regulator phosphoprotein that consists of 189 amino acid residues and has a MW of ∼21 kDa. It is found predominantly within the nucleus, nucleoli, and nucleolar speckles of infected HTLV-1 cell lines.24,82,83 The Rex protein contains multiple functional domains that help in the HTLV-1 replication and pathogenesis. The protein constitutes an NLS corresponding to the N-terminal residues (1–19), which is highly rich in Arg.82 This stretch of positive residues also acts as the RNA binding domain, where it binds to the Rex responsive elements (RxRE) of the viral unspliced mRNA.82,84,85 This binding is a crucial step for transactivation of mRNA export. The Rex protein also contains two flanking multimerization domains corresponding to positions [57–66] and [106–124]. Multiple Rex proteins can bind to a single mRNA via protein–protein interactions and protein–RNA interaction prior to export.82 An AD, Leu-rich domain containing a nuclear export signal (NES), is also present within the Rex protein at the central region. NES is a Leu-rich motif that can interact with numerous other proteins via protein–protein interactions.82
The disorder propensity of the Rex protein was found to be rather high with a PPIDmean of 79.36% (Table 2). The compositional profiling analysis of the Rex protein showed that the protein is enriched in two order-promoting amino acids (e.g., Trp and Arg) but depleted in two disorder-promoting amino acids (i.e., Lys and Glu) (Figure 2B). Additionally, the protein was found to be depleted in Ile, His, Val, Asn, Asp, Gly, and Ala and highly enriched in Gln, Ser, and Pro when compared with DisProt proteins. A detailed amino acid composition of the Rex protein is shown in Figure 8E. Disorder propensity analysis of the Rex protein is illustrated in Figure 9E. The mean of disorder predictors used showed the presence of two disordered regions, that is, [1–28] and [68–189], with the longest disordered region being 122 residues long [68–189]. As a whole, the CH–CDF analysis predicted this protein to be fully disordered (Figure 3B). The ANCHOR analysis showed the presence of five AIBS, that is, [25–65], [80–92], [106–117], [143–155], and [168–174] (Figure 9E). Region [25–65] partially coincides with the smaller disordered region as predicted by the mean analysis, whereas [80–92], [106–117], [143–155], and [168–174] coincide with the longer disordered region predicted by the mean analysis. The data presented here are supported by a computation analysis of the phosphorylated and unphosphorylated Rex protein, which predicted unphosphorylated Rex protein to be fully disordered protein.86
2.5.6. Tax
The HTLV-1 Tax oncoprotein is the major detrimental protein, which aids in the activation of viral and cellular gene expression and triggers uncontrolled cell proliferation and transformation.24 The HTLV-1 Tax protein consists of 353 amino acid residues and has a MW of ∼39.5 kDa. It is predominantly found in the nucleus due to the presence of NLS within its N-termini. It can be present in the cytoplasm, from where it gets translocated to the nucleus and activates viral LTR-mediated transcription via binding to the Tax-responsive elements (TxRE).24,62 The Tax protein mediates viral gene transcription by interacting with CREB/activating transcription factor and forming a complex with TxRE, where it regulates LTR’s transactivation both positively as well as negatively. Such interactions repress expressions of the cellular genes, such as p53, cyclin A, and c-my.87 Additionally, Tax also interacts with activators involved in the SRF pathway such as activator protein 1 (Sap1). Upon stabilization of these complexes, Tax recruits transcription initiation co-activators–CBP/p300 and P-CAF. Tax is involved in various cellular functions within the HTLV-1 infected cells, rendering viral persistence and neoplastic transformation.87,88 Upon ubiquitination, Tax binds to NF-κβ essential modulators (NEMO and IKKγ), TAK1, and MAP3K, thereby activating and stimulating IKK activity and subsequently resulting in the activation of NF-κβ pathway.62 Tax stimulates cell proliferation from G1 phase to S phase through Rb hyperphosphorylation and activation of E2F transcription activators. Additionally, it activates CDK6 by interacting with CDK4, CDK6, and CDK inhibitors.62,87 It is also known that Tax promotes DNA damage by suppressing the expression of DNA polymerase β and human telomerase RT, essential for DNA repair.87 Interestingly, Tax contains a PBZ binding motif (PBM, consisting of four amino acid sequences) at the C-terminus, which is thought to be important for transformation and proliferation. Tax PBM has been demonstrated with three PDZ containing proteins (hDLG, MAGUK, and pro-IL16) in humans, all of which are involved in cell cycle regulation and tumor suppression.87
The disorder propensity analysis showed that the Tax protein contains a limited number of disordered regions with a PPIDmean of 11.33% (Table 2). The compositional profiling analysis of the Tax protein showed that Tax is enriched in several order-promoting amino acids (e.g., Cys, Trp, Phe, Leu, and His) but depleted in two disorder-promoting amino acids (i.e., Lys and Glu) (Figure 3B). Additionally, Tax was found to be depleted in Val, Asn, Met, Arg, Asp, and Ala and highly enriched in Pro when compared with DisProt proteins. A detailed amino acid composition of the Tax protein is shown in Figure 8F. The mean analysis of the disorder predictors used showed three disordered regions to be present in the Tax protein, that is, [1–3], [89–94], and [324–353], with the longest disordered region being 30 residues long [324–353] (Figure 9F). However, the CH–CDF analysis predicted this protein to be fully ordered (Figure 3B). The ANCHOR analysis predicted no AIBS whatsoever.
3. Conclusions
Most of the HTLV-1 proteins analyzed in this study contained disordered region and are known to involve in diverse mechanisms of virus survival and immune evasion. Specifically, IDPRs, having more than 30 amino acids at a stretch, were predicted in HTLV-1 bZIP factor, p30, Rex, and structural nucleocapsid p15 proteins, which are involved in diverse functions such as virus proliferation, mRNA export, and genomic RNA binding. Additionally, as a whole, these proteins were predicted and classified as IDPs, where disorder percentile was not below 50 percent. Very limited study has been carried out concerning the mechanisms of the HTLV-1 pathogenesis. The present study provides novel insights from the unique perspective of intrinsically disordered regions of HTLV-1 proteins that might help elucidating the molecular mechanisms of virus–host interaction. The disordered proteins or the proteins containing IDPRs, which have highly dynamic nature and flexibility, play a significant role in disease development and progression. As disordered regions are attractive and challenging drug targets, strategies should be developed to find small inhibitors against IDPRs that could serve as effective antivirals. The occurrence of intrinsic disorder in HTLV-1 proteins strongly suggests that disorder needs to be seriously considered in the drug discovery process against HTLV-1 infection.8,89,90 Also, a detailed biophysical study of the HTLV-1 disordered proteins could provide a platform to develop novel antiviral therapeutics.
4. Methods
4.1. Retrieval of Sequences
A total of eight protein sequences of HTLV-1 were retrieved from UniProt database for disorder analysis (namely, P14075—envelope glycoprotein gp62, P14078—Gag-Pro-Pol polyprotein, P0C745—HTLV-1 basic zipper factor, P0CK16—accessory protein p12I, P0CK17—accessory protein p30II, P0C206—Rex, P14079—Tax and C6L855—p13 protein). All the sequences belong to HTLV-1 virus isolate Caribbean HS-35 subtype-A.
4.2. Compositional Profiling
Compositional profiling, performed using Composition Profiler, was used to study the disordered nature of the HTLV-1 proteins. Compositional profiling was performed by calculating the fractional difference in the composition, (CX – Corder)/(Corder), for each amino acid within the query protein set (i.e., HTLV-1 proteins) and a set of reference proteins (i.e., disordered proteins obtained from DisProt Database).91 In (CX – Corder)/(Corder), CX represents the content of a given amino acid in the query protein or the DisProt protein and Corder represents the standard value for a set of ordered proteins from PDB Select 25.91,92
4.3. Analysis of Per-Residue Disorder Propensity
Several disorder predictors are available online, out of which, the predictors of the PONDR family (e.g. PONDR VLXT, PONDR VSL2B, and meta-predictor PONDR FIT), IUPred, and RONN were used to evaluate the disorder propensities of the HTLV-1 proteins.28,29,93−95 Among these, PONDR VSL2B, which is based on two predictors (one optimized for long disordered regions whereas another optimized for short disorder regions) combined by an independent meta-predictor, has the best accuracy for the prediction of disordered regions of various lengths.29 PONDR VLXT, although not known to be the most accurate predictor, is very sensitive to local sequence peculiarity where disordered binding sites are usually associated.27 PONDR FIT meta-predictor consists of six individual predictors (PONDR VLXT, PONDR VSL2, PONDR VL3, IUPred, FoldIndex, and TopIDP) and its accuracy is better than each of its component predictors.95 IUPred prediction is based on the ab initio approach that uses statistical interaction potential. IUPred short has a comparatively better prediction accuracy due to its ability to predict missing residues in X-ray structures.93 The outputs of the following predictors were in the form of score value between 0 and 1 for individual residues of a particular protein. Residues with output values above 0.5 were considered as disordered. Per-residue disorder propensity analysis for each individual protein was interpreted.
Additionally, for further illustration of the disorder propensities of the HTLV-1 proteins, the PONDR FIT–JRONN plot was constructed.94,95 In the corresponding plot, HTLV-1 proteins were segregated as highly ordered (disorder score < 0.1, percentage of disordered < 10%), moderately disordered (disorder score between 0.1 and 0.3, percentage of disordered between 10 and 30%), and highly disordered (disorder score > 0.3, percentage of disordered > 30%).
4.4. Disorder Propensity Analysis by Binary Classifiers
Additionally, binary classifiers were used to determine the overall status of a protein as “ordered” or “disordered” as a whole. A combination of the outputs of two classifiers was used, that is, CH and CDF, to obtain the CH–CDF plot. In the CH–CDF plot, the X-axis represents the average vertical distance of the CDF curve from the CDF boundary line and the Y-axis represents the vertical distance of the corresponding protein in CH plot from the CH boundary line.33−35
4.5. MoRF Analysis
The MoRF regions were predicted by using the ANCHOR algorithm31 that is based on the pair-wise estimation energy approach developed for IUPred disorder predictors. Binding sites predicted by this approach are generally termed as AIBS. This analysis was performed for all HTLV-1 proteins.
Acknowledgments
The study was partly supported by a research grant from Department of Biotechnology, India (grant no. BT/PR16871/NER/95/329/2015). D.L.L. thanks UGC for providing fellowship.
Glossary
Abbreviations
- HTLV-1
human T lymphotropic virus type-1
- IDPs
intrinsically disordered proteins
- IDPRs
intrinsically disordered protein regions
- MoRF
molecular recognition features
- AIBS
ANCHOR indicated binding sites
- CH
charged hydropathy
- CDF
cumulative distribution frequency
- PPID
predicted percentile of intrinsic disorder
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b01017.
Protein–protein interaction network of entire HTLV-1 proteome with Homo sapiens as generated by STRING database (PDF)
Author Contributions
D.L.L. performed and generated all the data. D.L.L., H.S., A.S., and T.T. conceived the study. D.L.L., H.S., R.A., and T.T. analyzed the data and drafted the manuscript. All authors read and approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Oldfield C. J.; Dunker A. K. Intrinsically disordered proteins and intrinsically disordered protein regions. Annu. Rev. Biochem. 2014, 83, 553–584. 10.1146/annurev-biochem-072711-164947. [DOI] [PubMed] [Google Scholar]
- Liu Z.; Huang Y. Advantages of proteins being disordered. Protein Sci. 2014, 23, 539–550. 10.1002/pro.2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa P. Intrinsically disordered proteins: a 10-year recap. Trends Biochem. Sci. 2012, 37, 509–516. 10.1016/j.tibs.2012.08.004. [DOI] [PubMed] [Google Scholar]
- Fuxreiter M.; Tóth-Petróczy Á.; Kraut D. A.; Matouschek A. T.; Lim R. Y. H.; Xue B.; Kurgan L.; Uversky V. N. Disordered proteinaceous machines. Chem. Rev. 2014, 114, 6806–6843. 10.1021/cr4007329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright P. E.; Dyson H. J. Intrinsically disordered proteins in cellular signalling and regulation. Nat. Rev. Mol. Cell Biol. 2015, 16, 18–29. 10.1038/nrm3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N. A decade and a half of protein intrinsic disorder: biology still waits for physics. Protein Sci. 2013, 22, 693–724. 10.1002/pro.2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Oldfield C. J.; Dunker A. K. Intrinsically disordered proteins in human diseases: introducing the D2 concept. Annu. Rev. Biophys. 2008, 37, 215–246. 10.1146/annurev.biophys.37.032807.125924. [DOI] [PubMed] [Google Scholar]
- Joshi P.; Vendruscolo M.. Druggability of Intrinsically Disordered Proteins. Advances in Experimental Medicine and Biology; Springer, 2015; Vol. 870, pp 383–400. [DOI] [PubMed] [Google Scholar]
- Yan J.; Dunker A. K.; Uversky V. N.; Kurgan L. Molecular recognition features (MoRFs) in three domains of life. Mol. Biosyst. 2016, 12, 697–710. 10.1039/c5mb00640f. [DOI] [PubMed] [Google Scholar]
- Malhis N.; Wong E. T. C.; Nassar R.; Gsponer J. Computational Identification of MoRFs in Protein Sequences Using Hierarchical Application of Bayes Rule. PLoS One 2015, 10, e0141603 10.1371/journal.pone.0141603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhis N.; Gsponer J. Computational identification of MoRFs in protein sequences. Bioinformatics 2015, 31, 1738–1744. 10.1093/bioinformatics/btv060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V.; Oldfield C. J.; Mohan A.; Radivojac P.; Cortese M. S.; Uversky V. N.; Dunker A. K. Characterization of molecular recognition features, MoRFs, and their binding partners. J. Proteome Res. 2007, 6, 2351–2366. 10.1021/pr0701411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokuriki N.; Oldfield C. J.; Uversky V. N.; Berezovsky I. N.; Tawfik D. S. Do viral proteins possess unique biophysical features?. Trends Biochem. Sci. 2009, 34, 53–59. 10.1016/j.tibs.2008.10.009. [DOI] [PubMed] [Google Scholar]
- Xue B.; Blocquel D.; Habchi J.; Uversky A. V.; Kurgan L.; Uversky V. N.; Longhi S. Structural disorder in viral proteins. Chem. Rev. 2014, 114, 6880–6911. 10.1021/cr4005692. [DOI] [PubMed] [Google Scholar]
- Mishra P. M.; Uversky V. N.; Giri R. Molecular Recognition Features in Zika Virus Proteome. J. Mol. Biol. 2018, 430, 2372–2388. 10.1016/j.jmb.2017.10.018. [DOI] [PubMed] [Google Scholar]
- Singh A.; Kumar A.; Yadav R.; Uversky V. N.; Giri R. Deciphering the dark proteome of Chikungunya virus. Sci. Rep. 2018, 8, 5822. 10.1038/s41598-018-23969-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giri R.; Kumar D.; Sharma N.; Uversky V. N. Intrinsically Disordered Side of the Zika Virus Proteome. Front. Cell. Infect. Microbiol. 2016, 6, 144. 10.3389/fcimb.2016.00144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uversky V. N.; Roman A.; Oldfield C. J.; Dunker A. K. Protein intrinsic disorder and human papillomaviruses: increased amount of disorder in E6 and E7 oncoproteins from high risk HPVs. J. Proteome Res. 2006, 5, 1829. 10.1021/pr0602388. [DOI] [PubMed] [Google Scholar]
- Xue B.; Mizianty M. J.; Kurgan L.; Uversky V. N. Protein intrinsic disorder as a flexible armor and a weapon of HIV-1. Cell. Mol. Life Sci. 2012, 69, 1211–1259. 10.1007/s00018-011-0859-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X.; Xue B.; Dolan P. T.; LaCount D. J.; Kurgan L.; Uversky V. N. The intrinsic disorder status of the human hepatitis C virus proteome. Mol. Biosyst. 2014, 10, 1345–1363. 10.1039/c4mb00027g. [DOI] [PubMed] [Google Scholar]
- Meng F.; Badierah R. A.; Almehdar H. A.; Redwan E. M.; Kurgan L.; Uversky V. N. Unstructural biology of the Dengue virus proteins. FEBS J. 2015, 282, 3368–3394. 10.1111/febs.13349. [DOI] [PubMed] [Google Scholar]
- Whelan J. N.; Reddy K. D.; Uversky V. N.; Teng M. N. Functional correlations of respiratory syncytial virus proteins to intrinsic disorder. Mol. Biosyst. 2016, 12, 1507–1526. 10.1039/c6mb00122j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redwan E. M.; AlJaddawi A. A.; Uversky V. N. Structural disorder in the proteome and interactome of Alkhurma virus (ALKV). Cell. Mol. Life Sci. 2019, 76, 577–608. 10.1007/s00018-018-2968-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannian P.; Green P. L. Human T Lymphotropic Virus Type 1 (HTLV-1): Molecular Biology and Oncogenesis. Viruses 2010, 2, 2037–2077. 10.3390/v2092037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gessain A.; Cassar O. Epidemiological Aspects and World Distribution of HTLV-1 Infection. Front. Microbiol. 2012, 3, 388. 10.3389/fmicb.2012.00388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemasson I.; Lewis M. R.; Polakowski N.; Hivin P.; Cavanagh M.-H.; Thebault S.; Barbeau B.; Nyborg J. K.; Mesnard J.-M. Human T-cell leukemia virus type 1 (HTLV-1) bZIP protein interacts with the cellular transcription factor CREB to inhibit HTLV-1 transcription. J. Virol. 2007, 81, 1543–1553. 10.1128/jvi.00480-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunker A. K.; Lawson J. D.; Brown C. J.; Williams R. M.; Romero P.; Oh J. S.; Oldfield C. J.; Campen A. M.; Ratliff C. M.; Hipps K. W.; Ausio J.; Nissen M. S.; Reeves R.; Kang C.; Kissinger C. R.; Bailey R. W.; Griswold M. D.; Chiu W.; Garner E. C.; Obradovic Z. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. 10.1016/s1093-3263(00)00138-8. [DOI] [PubMed] [Google Scholar]
- Deng X.; Eickholt J.; Cheng J. A comprehensive overview of computational protein disorder prediction methods. Mol. Biosyst. 2012, 8, 114–121. 10.1039/c1mb05207a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z.; Meszaros B.; Simon I. Bioinformatical approaches to characterize intrinsically disordered/unstructured proteins. Briefings Bioinf. 2010, 11, 225–243. 10.1093/bib/bbp061. [DOI] [PubMed] [Google Scholar]
- Disfani F. M.; Hsu W.-L.; Mizianty M. J.; Oldfield C. J.; Xue B.; Dunker A. K.; Uversky V. N.; Kurgan L. MoRFpred, a computational tool for sequence-based prediction and characterization of short disorder-to-order transitioning binding regions in proteins. Bioinformatics 2012, 28, i75–i83. 10.1093/bioinformatics/bts209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z.; Meszaros B.; Simon I. ANCHOR: web server for predicting protein binding regions in disordered proteins. Bioinformatics 2009, 25, 2745–2746. 10.1093/bioinformatics/btp518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook H.; Doncheva N. T.; Szklarczyk D.; von Mering C.; Jensen L. J., Viruses.STRING: A Virus-Host Protein-Protein Interaction Database. Viruses 2018,10 (). 10.3390/v10100519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Oldfield C. J.; Xue B.; Hsu W. L.; Meng J.; Liu X.; Shen L.; Romero P.; Uversky V. N.; Dunker A. Improving protein order-disorder classification using charge-hydropathy plots. BMC Bioinf. 2014, 15, S4. 10.1186/1471-2105-15-s17-s4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F.; Oldfield C.; Meng J.; Hsu W. L.; Xue B.; Uversky V. N.; Romero P.; Dunker A. K., Subclassifying disordered proteins by the CH-CDF plot method. Biocomputing, 2012; pp 128–139. [PubMed]
- Xue B.; Oldfield C. J.; Dunker A. K.; Uversky V. N. CDF it all: consensus prediction of intrinsically disordered proteins based on various cumulative distribution functions. FEBS Lett. 2009, 583, 1469–1474. 10.1016/j.febslet.2009.03.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazurov D.; Heidecker G.; Derse D. HTLV-1 Gag protein associates with CD82 tetraspanin microdomains at the plasma membrane. Virology 2006, 346, 194–204. 10.1016/j.virol.2005.10.033. [DOI] [PubMed] [Google Scholar]
- Rayne F.; Bouamr F.; Lalanne J.; Mamoun R. Z. The NH2-terminal domain of the human T-cell leukemia virus type 1 capsid protein is involved in particle formation. J. Virol. 2001, 75, 5277–5287. 10.1128/jvi.75.11.5277-5287.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Blanc I.; R A. R.; Dokhelar M. C. Multiple functions for the basic amino acids of the human T-cell leukemia virus type 1 matrix protein in viral transmission. J. Virol. 1999, 73, 1860–1867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu C. C.; Bouamr F.; Yao X.; Carter C.; Tjandra N. Structural analysis of the N-terminal domain of the human T-cell leukemia virus capsid protein. J. Mol. Biol. 2001, 306, 783–797. 10.1006/jmbi.2000.4395. [DOI] [PubMed] [Google Scholar]
- Le Blanc I.; Grange M. P.; Delamarre L.; Rosenberg A. R.; Blot V.; Pique C.; Dokhélar M. C. HTLV-1 structural proteins. Virus Res. 2001, 78, 5–16. 10.1016/s0168-1702(01)00278-7. [DOI] [PubMed] [Google Scholar]
- Shuker S. B.; Mariani V. L.; Herger B. E.; Dennison K. J. Understanding HTLV-I protease. Chem. Biol. 2003, 10, 373–380. 10.1016/s1074-5521(03)00104-2. [DOI] [PubMed] [Google Scholar]
- Trentin B.; Rebeyrotte N.; Mamoun R. Z. Human T-cell leukemia virus type 1 reverse transcriptase (RT) originates from the pro and pol open reading frames and requires the presence of RT-RNase H (RH) and RT-RH-integrase proteins for its activity. J. Virol. 1998, 72, 6504–6510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M.; Grigsby I. F.; Gorelick R. J.; Mansky L. M.; Musier-Forsyth K. Retrovirus-specific differences in matrix and nucleocapsid protein-nucleic acid interactions: implications for genomic RNA packaging. J. Virol. 2014, 88, 1271–1280. 10.1128/jvi.02151-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigsby I. F.; Zhang W.; Johnson J. L.; Fogarty K. H.; Chen Y.; Rawson J. M.; Crosby A. J.; Mueller J. D.; Mansky L. M. Biophysical analysis of HTLV-1 particles reveals novel insights into particle morphology and Gag stochiometry. Retrovirology 2010, 7, 75. 10.1186/1742-4690-7-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornilescu C. C.; Bouamr F.; Carter C.; Tjandra N. Backbone15N relaxation analysis of the N-terminal domain of the HTLV-I capsid protein and comparison with the capsid protein of HIV-1. Protein Sci. 2003, 12, 973–981. 10.1110/ps.0235903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouamr F.; Cornilescu C. C.; Goff S. P.; Tjandra N.; Carter C. A. Structural and dynamics studies of the D54A mutant of human T cell leukemia virus-1 capsid protein. J. Biol. Chem. 2005, 280, 6792–6801. 10.1074/jbc.m408119200. [DOI] [PubMed] [Google Scholar]
- Morcock D. R.; Kane B. P.; Casas-Finet J. R. Fluorescence and nucleic acid binding properties of the human T-cell leukemia virus-type 1 nucleocapsid protein. Biochim. Biophys. Acta 2000, 1481, 381–394. 10.1016/s0167-4838(00)00181-3. [DOI] [PubMed] [Google Scholar]
- Wu W.; Hatterschide J.; Syu Y.-C.; Cantara W. A.; Blower R. J.; Hanson H. M.; Mansky L. M.; Musier-Forsyth K. Human T-cell leukemia virus type 1 Gag domains have distinct RNA-binding specificities with implications for RNA packaging and dimerization. J. Biol. Chem. 2018, 293, 16261–16276. 10.1074/jbc.ra118.005531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhnert M.; Steuber H.; Diederich W. E. Structural basis for HTLV-1 protease inhibition by the HIV-1 protease inhibitor indinavir. J. Med. Chem. 2014, 57, 6266–6272. 10.1021/jm500402c. [DOI] [PubMed] [Google Scholar]
- Kádas J.; Boross P.; Weber I. T.; Bagossi P.; Matúz K.; Tözsér J. C-terminal residues of mature human T-lymphotropic virus type 1 protease are critical for dimerization and catalytic activity. Biochem. J. 2008, 416, 357–364. 10.1042/bj20071132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell M. S.; Tozser J.; Princler G.; Lloyd P. A.; Auth A.; Derse D. Synthesis, processing, and composition of the virion-associated HTLV-1 reverse transcriptase. J. Biol. Chem. 2006, 281, 3964–3971. 10.1074/jbc.m507660200. [DOI] [PubMed] [Google Scholar]
- Seegulam M. E.; Ratner L. Integrase inhibitors effective against human T-cell leukemia virus type 1. Antimicrob. Agents Chemother. 2011, 55, 2011–2017. 10.1128/aac.01413-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg A. R.; Delamarre L.; Preira A.; Dokhélar M. C. Analysis of functional conservation in the surface and transmembrane glycoprotein subunits of human T-cell leukemia virus type 1 (HTLV-1) and HTLV-2. J. Virol. 1998, 72, 7609–7614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobe B.; Center R. J.; Kemp B. E.; Poumbourios P. Crystal structure of human T cell leukemia virus type 1 gp21 ectodomain crystallized as a maltose-binding protein chimera reveals structural evolution of retroviral transmembrane proteins. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 4319–4324. 10.1073/pnas.96.8.4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M.; Jeang K.-T. Human T-cell leukaemia virus type 1 (HTLV-1) infectivity and cellular transformation. Nat. Rev. Cancer 2007, 7, 270–280. 10.1038/nrc2111. [DOI] [PubMed] [Google Scholar]
- Bartoe J. T.; Albrecht B.; Collins N. D.; Robek M. D.; Ratner L.; Green P. L.; Lairmore M. D. Functional role of pX open reading frame II of human T-lymphotropic virus type 1 in maintenance of viral loads in vivo. J. Virol. 2000, 74, 1094–1100. 10.1128/jvi.74.3.1094-1100.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derse D.; Mikovits J.; Ruscetti F. X-I and X-II open reading frames of HTLV-I are not required for virus replication or for immortalization of primary T-cells in vitro. Virology 1997, 237, 123–128. 10.1006/viro.1997.8781. [DOI] [PubMed] [Google Scholar]
- Fukumoto R. Human T-lymphotropic virus type 1 non-structural proteins: Requirements for latent infection. Cancer Sci. 2013, 104, 983–988. 10.1111/cas.12190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cann A. J.; Rosenblatt J. D.; Wachsman W.; Shah N. P.; Chen I. S. Y. Identification of the gene responsible for human T-cell leukaemia virus transcriptional regulation. Nature 1985, 318, 571–574. 10.1038/318571a0. [DOI] [PubMed] [Google Scholar]
- Felber B.; Paskalis H.; Kleinman-Ewing C.; Wong-Staal F.; Pavlakis G. The pX protein of HTLV-I is a transcriptional activator of its long terminal repeats. Science 1985, 229, 675–679. 10.1126/science.2992082. [DOI] [PubMed] [Google Scholar]
- Inoue J.; Yoshida M.; Seiki M. Transcriptional (p40x) and post-transcriptional (p27x-III) regulators are required for the expression and replication of human T-cell leukemia virus type I genes. Proc. Natl. Acad. Sci. U.S.A. 1987, 84, 3653–3657. 10.1073/pnas.84.11.3653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azran I.; Schavinsky-Khrapunsky Y.; Aboud M. Role of Tax protein in human T-cell leukemia virus type-I leukemogenicity. Retrovirology 2004, 1, 20. 10.1186/1742-4690-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grassmann R.; Aboud M.; Jeang K.-T. Molecular mechanisms of cellular transformation by HTLV-1 Tax. Oncogene 2005, 24, 5976–5985. 10.1038/sj.onc.1208978. [DOI] [PubMed] [Google Scholar]
- Zhao T.; Matsuoka M. HBZ and its roles in HTLV-1 oncogenesis. Front. Microbiol. 2012, 3, 247. 10.3389/fmicb.2012.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc I.; Polakowski N.; André-Arpin C.; Cook P.; Barbeau B.; Mesnard J.-M.; Lemasson I. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J. Biol. Chem. 2008, 283, 23903–23913. 10.1074/jbc.m803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usui T.; Yanagihara K.; Tsukasaki K.; Murata K.; Hasegawa H.; Yamada Y.; Kamihira S. Characteristic expression of HTLV-1 basic zipper factor (HBZ) transcripts in HTLV-1 provirus-positive cells. Retrovirology 2008, 5, 34. 10.1186/1742-4690-5-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbous J.; Arpin C.; Gaudray G.; Piechaczyk M.; Devaux C.; Mesnard J.-M. The HBZ factor of human T-cell leukemia virus type I dimerizes with transcription factors JunB and c-Jun and modulates their transcriptional activity. J. Biol. Chem. 2003, 278, 43620–43627. 10.1074/jbc.m307275200. [DOI] [PubMed] [Google Scholar]
- Thébault S.; Basbous J.; Hivin P.; Devaux C.; Mesnard J.-M. HBZ interacts with JunD and stimulates its transcriptional activity. FEBS Lett. 2004, 562, 165–170. 10.1016/s0014-5793(04)00225-x. [DOI] [PubMed] [Google Scholar]
- Zhao T.; Yasunaga J.-i.; Satou Y.; Nakao M.; Takahashi M.; Fujii M.; Matsuoka M. Human T-cell leukemia virus type 1 bZIP factor selectively suppresses the classical pathway of NF- B. Blood 2009, 113, 2755–2764. 10.1182/blood-2008-06-161729. [DOI] [PubMed] [Google Scholar]
- Satou Y.; Yasunaga J.-i.; Zhao T.; Yoshida M.; Miyazato P.; Takai K.; Shimizu K.; Ohshima K.; Green P. L.; Ohkura N.; Yamaguchi T.; Ono M.; Sakaguchi S.; Matsuoka M. HTLV-1 bZIP factor induces T-cell lymphoma and systemic inflammation in vivo. PLoS Pathog. 2011, 7, e1001274 10.1371/journal.ppat.1001274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T.; Satou Y.; Sugata K.; Miyazato P.; Green P. L.; Imamura T.; Matsuoka M. HTLV-1 bZIP factor enhances TGF- signaling through p300 coactivator. Blood 2011, 118, 1865–1876. 10.1182/blood-2010-12-326199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook P. R.; Polakowski N.; Lemasson I. HTLV-1 HBZ protein deregulates interactions between cellular factors and the KIX domain of p300/CBP. J. Mol. Biol. 2011, 409, 384–398. 10.1016/j.jmb.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerc I.; Polakowski N.; André-Arpin C.; Cook P.; Barbeau B.; Mesnard J.-M.; Lemasson I. An interaction between the human T cell leukemia virus type 1 basic leucine zipper factor (HBZ) and the KIX domain of p300/CBP contributes to the down-regulation of tax-dependent viral transcription by HBZ. J. Biol. Chem. 2008, 283, 23903–23913. 10.1074/jbc.m803116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang K.; Stanfield R. L.; Martinez-Yamout M. A.; Dyson H. J.; Wilson I. A.; Wright P. E. Structural basis for cooperative regulation of KIX-mediated transcription pathways by the HTLV-1 HBZ activation domain. Proc. Natl. Acad. Sci. U.S.A. 2018, 115, 10040–10045. 10.1073/pnas.1810397115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrecht B.; Lairmore M. D. Critical role of human T-lymphotropic virus type 1 accessory proteins in viral replication and pathogenesis. Microbiol. Mol. Biol. Rev. 2002, 66, 396–406. 10.1128/mmbr.66.3.396-406.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C.; Harrod R. L.; Ciminale V.; Franchini G. Human T-cell leukemia/lymphoma virus type 1 nonstructural genes and their functions. Oncogene 2005, 24, 6026–6034. 10.1038/sj.onc.1208977. [DOI] [PubMed] [Google Scholar]
- Michael M.; Nair A.; Lairmore M. D. Role of accessory proteins of HTLV-1 in viral replication, T cell activation, and cellular gene expression. Front Biosci 2004, 9, 2556–2576. 10.2741/1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards D.; Fenizia C.; Gold H.; Castro-Amarante M. F. d.; Buchmann C.; Pise-Masison C. A.; Franchini G. Orf-I and orf-II-encoded proteins in HTLV-1 infection and persistence. Viruses 2011, 3, 861–885. 10.3390/v3060861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andresen V.; Pise-Masison C. A.; Sinha-Datta U.; Bellon M.; Valeri V.; Washington Parks R.; Cecchinato V.; Fukumoto R.; Nicot C.; Franchini G. Suppression of HTLV-1 replication by Tax-mediated rerouting of the p13 viral protein to nuclear speckles. Blood 2011, 118, 1549–1559. 10.1182/blood-2010-06-293340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicot C.; Dundr M.; Johnson J. M.; Fullen J. R.; Alonzo N.; Fukumoto R.; Princler G. L.; Derse D.; Misteli T.; Franchini G. HTLV-1-encoded p30II is a post-transcriptional negative regulator of viral replication. Nat. Med. 2004, 10, 197–201. 10.1038/nm984. [DOI] [PubMed] [Google Scholar]
- Anupam R.; Doueiri R Fau - Green P. L.; Green P. L. The need to accessorize: molecular roles of HTLV-1 p30 and HTLV-2 p28 accessory proteins in the viral life cycle. Front. Microbiol. 2013, 4, 275. 10.3389/fmicb.2013.00275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younis I.; Green P. L. The human T-cell leukemia virus Rex protein. Front. Biosci. 2005, 10, 431–445. 10.2741/1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota S.; Hatanaka M.; Pomerantz R. J. Nucleo-cytoplasmic redistribution of the HTLV-I Rex protein: alterations by coexpression of the HTLV-I p21x protein. Virology 1996, 220, 502–507. 10.1006/viro.1996.0339. [DOI] [PubMed] [Google Scholar]
- Bai X. T.; Sinha-Datta U.; Ko N. L.; Bellon M.; Nicot C. Nuclear export and expression of human T-cell leukemia virus type 1 tax/rex mRNA are RxRE/Rex dependent. J. Virol. 2012, 86, 4559–4565. 10.1128/jvi.06361-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogerd H. P.; Tiley L. S.; Cullen B. R. Specific binding of the human T-cell leukemia virus type I Rex protein to a short RNA sequence located within the Rex-response element. J. Virol. 1992, 66, 7572–7575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kheirabadi M.; Taghdir M. Is unphosphorylated Rex, as multifunctional protein of HTLV-1, a fully intrinsically disordered protein? An in silico study. Biochem. Biophys. Rep. 2016, 8, 14–22. 10.1016/j.bbrep.2016.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxus M.; Twizere J. C.; Legros S.; Dewulf J. F.; Kettmann R.; Willems L. The HTLV-1 Tax interactome. Retrovirology 2008, 5, 76. 10.1186/1742-4690-5-76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuh M.; Derse D. Ternary complex factors and cofactors are essential for human T-cell leukemia virus type 1 tax transactivation of the serum response element. J. Virol. 2000, 74, 11394–11397. 10.1128/jvi.74.23.11394-11397.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo A.; Aiello C.; Grieco P.; Marasco D. Targeting “Undruggable” Proteins: Design of Synthetic Cyclopeptides. Curr. Med. Chem. 2016, 23, 748–762. 10.2174/0929867323666160112122540. [DOI] [PubMed] [Google Scholar]
- Russo A.; Manna S. L.; Novellino E.; Malfitano A. M.; Marasco D. Molecular signaling involving intrinsically disordered proteins in prostate cancer. Asian J. Androl. 2016, 18, 673–681. 10.4103/1008-682X.181817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacic V.; Uversky V. N.; Dunker A. K.; Lonardi S. Composition Profiler: a tool for discovery and visualization of amino acid composition differences. BMC Bioinf. 2007, 8, 211. 10.1186/1471-2105-8-211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. 10.1093/nar/28.1.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dosztanyi Z.; Csizmok V.; Tompa P.; Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005, 21, 3433–3434. 10.1093/bioinformatics/bti541. [DOI] [PubMed] [Google Scholar]
- Yang Z. R.; Thomson R.; McNeil P.; Esnouf R. M. RONN: the bio-basis function neural network technique applied to the detection of natively disordered regions in proteins. Bioinformatics 2005, 21, 3369–3376. 10.1093/bioinformatics/bti534. [DOI] [PubMed] [Google Scholar]
- Xue B.; Dunbrack R. L.; Williams R. W.; Dunker A. K.; Uversky V. N. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. 10.1016/j.bbapap.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.