

Abstract
Diagnostic tests based on proteomics analysis can have significant advantages over more traditional biochemical tests. However, low molecular weight (MW) protein biomarkers are difficult to identify by standard mass spectrometric analysis, as they are usually present at low concentrations and are masked by more abundant resident proteins. We have previously shown that mesoporous silica nanoparticles are able to capture a predominantly low MW protein fraction from the serum, as compared to the protein corona (PC) adsorbed onto dense silica nanoparticles. In this study, we begin by further investigating this effect using liquid chromatography–mass spectrometry (LC–MS)/MS and thermogravimetric analysis (TGA) to compare the MW of the proteins in the coronas of mesoporous silica nanoparticles with the same particle size but different pore diameters. Next, we examine the process by which two proteins, one small and one large, adsorb onto these mesoporous silica nanoparticles to establish a theory of why the corona becomes enriched in low MW proteins. Finally, we use this information to develop a novel system for the diagnosis of prostate cancer. An elastic net statistical model was applied to LC–MS/MS protein coronas from the serum of 22 cancer patients, identifying proteins specific to each patient group. These studies help to explain why low MW proteins predominate in the coronas of mesoporous silica nanoparticles, and they illustrate the ability of this information to supplement more traditional diagnostic tests.
Introduction
The early diagnosis of cancer is one of the most important goals of current medicine, and in the last two decades significant progress has been made in understanding the molecular processes involved in the origin and evolution of cancer.1 Recently, efforts have been made to use modern techniques such as proteomics and genomics to isolate, identify, and quantify specific molecules, that is, prognostic factors or biomarkers, that are closely related to a specific clinical outcome (in the absence of therapy).2 Many protein and peptide biomarkers belong to the low MW serum protein fraction (e.g. MW below 50 kDa) and their isolation by standard MS analysis is mostly unfeasible, as these low MW species are usually in low concentration and are masked by large quantities of resident proteins, such as immunoglobulins and albumin.3 Furthermore, most of these “small” proteins are especially sensitive to enzymatic degradation following collection, making their identification and determination difficult.4 Consequently, there is need for techniques that allow selective isolation, quantitative estimation, and statistical validation of the low MW proteins in the specific proteome assigned to a disease. In this context, we recently published several reports showing that the composition of the protein corona (PC) on porous nanoparticles is different from the corona adsorbed onto dense nanoparticles of the same size and composition.5,6 Specifically, porous nanoparticles retain significantly more low MW proteins (<50 kDa) from serum. Using stochastic optical reconstruction microscopy (STORM), we showed that small proteins have a better fit within the pores and are able to access the internal surface areas of the nanoparticles, while large proteins are mainly restricted to the external surface.7 Qualitative co-adsorption studies using one small and one large protein also showed that the presence of the small protein reduces the amount of large protein able to bind. We hypothesized that the small protein rapidly accesses and fills the pores, while the large protein must undergo restructuring (a high energy process) to fit into the pores, and consequently becomes rapidly excluded from them.
The ability to specifically capture the low MW fraction of serum proteins provides a unique opportunity to diagnose certain disease states using a simple blood draw and proteomics. Protein biomarker discovery and quantification by mass spectrometry (MS) is a powerful tool, but it is severely limited due to the intrinsic complexity of human serum samples.8,9 The isolation of low MW biomarkers by standard MS analysis is mostly unfeasible, as they are usually present at low concentrations and are masked by more abundant resident proteins, such as immunoglobulins and albumin.3 Furthermore, most of these small proteins are particularly sensitive to enzymatic degradation after collection, making their identification difficult.4 Albeit nanoparticles modified with different chemical groups have been used to selectively bind low-abundance proteins and peptides,10 it is optimistic to think that a single molecule is indicative of a pathologic state. Huo and co-workers developed a diagnostic test for early stage prostate cancer (PCa) based on the amount of human immunoglobulin G (IgG) retained in the PC of citrate-capped gold nanoparticles.11,12 This blood test is a significant advance over the currently clinically used prostate specific antigen (PSA) analysis, but it could be seriously affected by selectivity issues, as many pathological states can increase blood IgG concentration. Instead, analysis of the entire PC adsorbed onto porous nanoparticles by MS could reveal patterns of protein up and down regulation throughout the low MW proteome in the disease state versus the normal state. At this point, Mahmoudi and co-workers recently introduced the concept of a personalized PC (PPC) as a pattern linked to physiological conditions.13 This PPC could have a dramatic impact in cancer diagnosis, as many proteins change their plasmatic profile. In this sense, the sequestration of proteins in the nanoparticle PC could be used to detect low-abundant proteins related to tumorigenesis.14 In this line, Huo and co-workers also pointed out that nanoparticle corona proteomic analysis may reveal molecular profile differences between cancer and healthy serum samples,12 although no specific protocols and patterns were given to confirm such differences with statistical evidence. Moreover, Caracciolo and co-workers used lipid nanoparticles to isolate the PC from pancreatic cancer patient serum samples. Then, they characterized the different proteomic patterns by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and, using principal component analysis (PCA) and linear discriminant analysis, they obtained statistically significant differences between the healthy and the cancer groups.15
Our goal for the current set of experiments was threefold. First, we sought to compare the extent of protein adsorption on a set of nanoparticles with the same well-defined shapes and sizes but different pore diameters. Previously, we focused on materials with essentially the same pore diameter but with other properties that differed (particle size, surface modification, etc.). Second, we quantitatively compared the amount of a single small and large protein adsorbed onto the nanoparticle when both were present throughout the adsorption process versus when the large protein was given a “head start”, that is, the proteins were added sequentially with the small protein added last. Finally, we sought to develop a proteomic test based on the low MW proteins in porous nanoparticle coronas to show that the low MW proteomes of patients with various stages of PCa are statistically different from those of control patients, and that this information can be used to distinguish one patient group from another.
Results and Discussion
We previously showed that the nanoparticle size has an influence on protein adsorption, with small nanoparticles adsorbing more total protein than large ones.6 To minimize this effect and limit differences in protein adsorption to the porosity of each material, we prepared nanoparticles with diameters that were as similar as possible (see Table 1 and Figure S1). Dense, nonporous silica nanoparticles (“SNP”) with a particle size of ∼120 nm as determined by transmission electron microscopy (TEM) (Figure 1) were prepared by modifying the Stobër method.16 The particle size was confirmed by dynamic light scattering (DLS); the polydispersity of this material and of the mesoporous nanoparticles indicated reasonably consistent particle sizes throughout each sample. Standard mesoporous silica nanoparticles (“MSN”) with a hexagonally ordered pore structure similar to that of MCM-41, with dpore = 2.7 nm and approximately the same particle size as SNPs were prepared by polymerizing silicate monomers in the presence of surfactant micelles.17 Large-pore silica (“LPS”) nanoparticles with increasing dpore = 4.8, 6.2, 7.4, and 14 nm were prepared using a previously published procedure.18 These materials, with FDU-12 structure type narrow channels that connect larger cavities,19 were used because retaining the particle size of all materials used in this study was of primary importance. Cavity sizes in LPS nanoparticles are noted in Table 1 and were in the range 15.5–24.5 nm, significantly larger than the pore interconnections; thus, the limiting factor in protein adsorption onto these particles should be the pore diameter. In addition to the particle size, the zeta potentials for SNP, MSN, and LPS materials were also nearly the same, ranging from −12.0 to −16.4 mV. These values are mainly dependent on the synthesis method, and in the case of the porous nanoparticles, are related to the removal of the surfactant by extraction. Characterization of the nanoparticles using N2 physisorption showed that the surface areas (SBET) of the porous nanoparticles decreased as the pore diameter increased, as expected for these materials (Table 1, Figures S2 and S3). The pore volumes ranged from 0.60 to 0.92 cm3/g, indicating highly porous MSN and LPS nanoparticles.
Table 1. Physical Properties of MSN.
| particle diameter | N2 physisorption | ||||||||
| sample | phase | TEMb (nm) | DLSc (nm) | PdId | ζ-potential (mV) | dpore (nm) | dcavitya (nm) | SBET (m2/g) | Vpore (cm3/g) |
| SNP | dense SiO2 | 123.4 ± 19.8 | 164.2 ± 85.0 | 0.268 | –16.4 | 49.5 | |||
| MSN | MCM-41 | 113.7 ± 20.4 | 136.8 ± 56.6 | 0.171 | –14.1 | 2.7 | 1016.4 | 0.76 | |
| LPS-1 | FDU-12a | 121.4 ± 27.0 | 185.2 ± 94.1 | 0.316 | –15.2 | 4.8 | 15.5 | 680.5 | 0.92 |
| LPS-2 | FDU-12 | 107.8 ± 22.6 | 133.8 ± 48.1 | 0.129 | –14.8 | 6.2 | 20.5 | 421.2 | 0.82 |
| LPS-3 | FDU-12 | 111.0 ± 25.4 | 115.4 ± 44.2 | 0.147 | –12.0 | 7.4 | 20.4 | 360.7 | 0.74 |
| LPS-4 | FDU-12 | 108.4 ± 21.6 | 100.7 ± 48.3 | 0.230 | –12.5 | 14.0 | 24.5 | 176.3 | 0.60 |
FDU-12 consists of large cavities connected by much smaller pores.
As determined by the TEM study of single particles: mean ± SD (n ≥ 250).
Average hydrodynamic diameter: mean ± SD (n = 3).
PdI = polydispersity index from DLS measurements.
Figure 1.

Representative TEM images of silica nanoparticles: (a) dense nanoparticles; (b) MSN, with the MCM-41 structure; (c) LPS-1, with the FDU-12 structure consisting of large cavities interconnected by smaller pores. The cavities can be readily observed in this image.
Dependence of Serum Protein Adsorption on the Pore Diameter
Protein adsorption studies from whole serum were performed by separately suspending each type of nanoparticle in human serum for 1 h, followed by centrifugation to collect the nanoparticles and washing to remove any loosely adsorbed proteins. The total protein adsorbed was obtained by TGA (Table 2).
Table 2. Protein Adsorption Data from TGA.
| sample | dpore (nm) | SBET (m2/g) | mass of protein adsorbed (mg/g SiO2) | specific protein adsorption (mg/m2) |
|---|---|---|---|---|
| SNP | 49.5 | 38 | 0.77 | |
| MSN | 2.7 | 1016.4 | 212 | 0.21 |
| LPS-1 | 4.8 | 680.5 | 168 | 0.25 |
| LPS-2 | 6.2 | 421.2 | 134 | 0.32 |
| LPS-3 | 7.4 | 360.7 | 136 | 0.38 |
| LPS-4 | 14.0 | 176.3 | 147 | 0.83 |
As expected, the mass of the protein adsorbed was correlated with the surface area of the nanoparticles; materials with larger surface areas generally retained more protein. However, when the specific adsorption (i.e., the mass of protein per unit surface area) was calculated, the nanoparticles could be separated into two groups. Although SNP did not adsorb much protein, the specific adsorption was much higher than that for porous nanoparticles, which adsorbed nearly 4–7 times as much total protein but which had a much lower specific adsorption. When the pore diameter increased to 14 nm, the specific adsorption returned to a value similar to that for the dense materials (0.83 vs 0.77). This information supports a model in which a number of proteins present in serum are too large to access the internal surface area of mesoporous silica nanoparticles with pore diameters less than approximately 7 nm, leading to a PC that is significantly different from that on either dense nanoparticles or nanoparticles with large pores, the latter defined by our data as nanoparticles with pore diameters 14 nm or larger.
A preliminary assessment of the content of the PCs on the nanoparticles was obtained by gel electrophoresis (Figure S4). The low protein content of the dense nanoparticles is apparent from the lack of significant protein bands which, as it will be shown later on, correspond to a few proteins, mostly apolipoproteins and albumin. The porous nanoparticles all showed more proteins, and qualitatively, the bands associated with apolipoprotein A-1 and A-II are significantly more prominent in these samples than the bands associated with albumin. The most important difference between the coronas from dense and porous nanoparticles is the addition of proteins with MW < 50 kDa in the latter case. More detailed analysis of the content of the PCs of these particles is shown by proteomic analysis (Figure 2 and Table S1). Using the normalized spectral counts (NSpC) provided by liquid chromatography–MS (LC–MS) analysis gave the relative percent of each identified protein; multiplying this percentage by the mass of the total PC adsorbed onto each type of particle, as determined by TGA, gave the mass of each identified protein adsorbed onto the particles. These values (NSpC × TGA) were collected into four distinct MW ranges for clarity and plotted as a function of the pore diameter.
Figure 2.

Protein adsorption onto silica nanoparticles from human serum as a function of the pore diameter. (a) Percentage of different MW fractions in the PC. (b) Specific protein adsorption per unit surface area of the different MW fractions. The mass of each protein in the coronas of the nanoparticles was determined by multiplying the NSpC obtained from liquid chromatography tandem-mass spectrometry (LC–MS/MS) with the total mass of the PC obtained from TGA. For the sake of clarity, error bars have been omitted in the figure, but the information is provided in Table S1.
In the PCs of all nanoparticles, at least 90% of the protein mass consisted of proteins with MW < 70 kDa (Figure 2a). However, excluding serum albumin (MW = 69 kDa) and proteins with MW above 50 kDa highlighted the difference between dense and porous nanoparticles. The coronas of porous nanoparticles still consisted primarily of low MW proteins, with 80–85% of the total mass coming from proteins with MW < 50 kDa. Only 65% of the total PC mass of the dense nanoparticles came from proteins in this group. Within the group of porous nanoparticles, there was a rough trend that correlated to the pore diameter, with the smallest fraction of proteins <50 kDa associated with the material with the largest pores (LPS-4, 14 nm). Plotting the data in terms of specific adsorption (i.e., mass of proteins adsorbed in each category per unit surface area) highlighted the difference between LPS-4 and the other porous materials (Figure 2b). The total specific protein adsorption for LPS-4 and dense nanoparticles was similar to each other and different from that of the other porous nanoparticles, as shown in Table 2. However, the specific adsorption of proteins in the 50–70 kDa range for LPS-4 was also more similar to that of the dense nanoparticle sample than to that of the other porous samples. Thus, we can conclude that it was easier for proteins with masses above 50 kDa and particularly between 50 and 70 kDa to access the internal pore surfaces of porous nanoparticles with a very large pore diameter (e.g., 14 nm). This highlights the size discrimination effect due to the pore diameters of porous nanoparticles and provides some explanation for the predominance of low MW proteins in the coronas of porous nanoparticles. Finally, for additional specific details about the PCs adsorbed from total serum, we created a heat map comparing adsorption data for the 24 most abundant proteins (NSpC × TGA > 1 mg/g, Figure 3). The predominance of only a few types of proteins, namely apolipoproteins, is apparent. The complete proteomic profiles are shown in Table S2.
Figure 3.
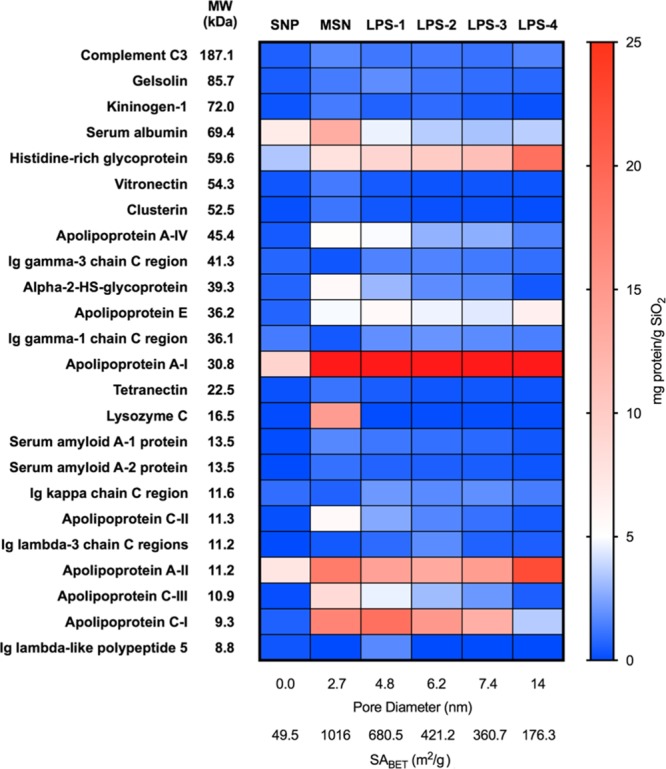
Relative abundance of human serum proteins in the coronas of silica nanoparticles. The 24 most abundant proteins (NSpC × TGA > 1 mg/g SiO2) across the entire data set of all identified proteins were organized according to the MW, and the data were presented as a heat map (scale shown at right of figure). The mass of apolipoprotein A-I on MSN through LPS-4 was 90.8, 78.9, 59.4, 65.1, and 71.7 mg/g SiO2 and is represented in red in the heat map.
Influence of the Pore Diameter on the Competitive Adsorption of Representative Small and Large Proteins
Having developed a model of protein adsorption onto porous nanoparticles, we next examined the effect of pore diameter on the adsorption of two specific proteins that are representative of “small” and “large” proteins: apolipoprotein A-II (abbreviated A-II) has a mass of 11.2 kDa and was one of the most prominent low MW proteins identified and complement C3 (abbreviated simply C3) was the largest protein of the group of 24 proteins identified in Figure 3. These two proteins were the subject of experiments we previously reported, where we used the STORM technique to provide qualitative observation of adsorption by fluorescence microscopy and calculating the relative penetration depths of each protein.7 Our results pointed out that when A-II and C3 were both present in the same solution, the adsorption of C3 was qualitatively much less than when C3 was the only protein in the solution. This may have occurred because when C3 was the only protein in the solution, some of it was able to bind near the pore entrances, but when A-II was present, A-II rapidly bound to the internal pore surfaces, blocking the pores and reducing the overall amount of C3 able to bind. According to this hypothesis, the pore diameter should have a significant effect on both the total amount of protein bound and the relative amounts of A-II and C3.
To study the effect of the pore diameter on the competitive adsorption of A-II and C3, we performed a series of protein adsorption experiments: C3 only, A-II only, C3 and A-II together in the same solution (represented as C3 + A-II), and finally C3 adsorbed first for 1 h followed by A-II (i.e., sequential adsorption of proteins that gave C3 a “head start”, represented as C3 → A-II). The results are summarized in Figure 4, where specific adsorption is plotted as a function of pore diameter. First, we found that the total specific protein adsorption increased with the pore diameter (Figure 4a), as expected from our previous experiments described above. In general, the total protein adsorption was the highest when C3 was given a head start, consistent with the hypothesis above because C3 binds to the pore entrances and external surfaces, and then A-II is able to bind to other surface sites to increase the overall mass of the adsorbed protein. In addition, whatever the pore size, it must be taken into account that both A-II and C3 present isoelectric points lower than 7 (respectively, 6.62 and 6.40), and negative effective charge at physiological pH (respectively −0.3 and −16.5), which means that electrostatic repulsion may happen even in the solution, limiting total adsorption. For materials with small pores, simultaneous adsorption of C3 and A-II did not result in as much adsorbed protein, because A-II prevented C3 from accessing pore entrance sites and because C3 weighs over 10 times as much as A-II, this significantly reduced the amount of adsorbed protein. The exception came for LPS-4, with the largest pore diameter, where the total amount of protein adsorbed was the same regardless of whether or not C3 and A-II were both present throughout the adsorption process. This can be rationalized by the fact that here, C3 is more fully able to access the internal porosity of the nanoparticles because the pores are much larger than those of the other materials.
Figure 4.

Comparison of specific protein adsorption of a representative heavy protein (complement C3) and light protein (apolipoprotein A-II) onto silica nanoparticles. “C3” = complement C3 alone; “A-II” = apolipoprotein A-II alone; “C3 + A-II” = complement C3 and apolipoprotein A-II in the same solution; “C3 → A-II” = sequential adsorption, i.e. complement C3 adsorbed first for 1 h, followed by apolipoprotein A-II. (a) Total specific adsorption of A-II and C3 proteins. (b) Specific adsorption of A-II or C-3 portions.
We also applied proteomic analysis to these samples to study the relative amounts of C3 and A-II adsorbed in each material as determined by NSpC × TGA (Figure 4b). For MSN, LPS-1, and LPS-2, with pore diameters of 2.7, 4.8, and 6.2 nm, respectively, the specific adsorption of C3 was very low regardless of whether the proteins were added simultaneously or sequentially. There was notably more C3 adsorbed in LPS-3 and LPS-4 (7.4 and 14 nm pore diameters), and for these samples, there was clearly more C3 adsorbed when it was added to the solution before A-II instead of being added simultaneously. This result indicated that C3 is able to adsorb more successfully when it does not compete with A-II for access to the internal surface area. A-II, on the other hand, was present in all samples regardless of the pore diameter although there was a notable switch in the amount of A-II adsorbed in the C3 + A-II experiment versus C3 → A-II. For materials with the smallest pores, that is, MSN, LPS-1, and LPS-2, more A-II adsorbed when it was added sequentially after C3 rather than simultaneously. The fact that there was any difference at all in these materials showed that even though there was very little C3 present, it still had an influence on the adsorption of A-II. When the pores were large enough to allow significant amounts of C3 to bind (LPS-3 and LPS-4), more A-II was adsorbed during simultaneous addition of proteins. This indicates that when C3 is able to enter the pores, it blocks them and decreases the diffusion of A-II within the pores to adsorption sites, promoting adsorption on their external surfaces; thus, less A-II binds when C3 is given a head start. On the other hand, when the pores are too small for appreciable amounts of C3 to enter, more A-II binds when it does not compete directly for adsorption sites near the pore entrances in the same solution. In addition, the latter result may also indicate that some exchange occurs between adsorbed C3 near the pore entrances and A-II. Overall, the results of these experiments showed that the sizes of the adsorbed proteins depended strongly on the pore diameter, and that they were also sensitive to the presence of other proteins in the solution.
Design of a Diagnostic Test for Prostate Cancer Development Using the Protein Corona of Mesoporous Silica Nanoparticles
It was apparent from the TGA data in Table 2 that the material with the largest surface area (MSN) adsorbed the most protein. As our other results above have shown, MSN also had the largest fraction of low MW proteins: more than 95% of the total protein mass in the corona consisted of proteins with MW < 70 kDa. Therefore, we chose this material as a “nano-concentrator” to demonstrate an application involving proteomic analysis of the PC to develop a diagnostic test for the progression of PCa. We began by optimizing the physical properties of the MSN required to obtain the highest possible fraction from human serum samples. MSN were prepared similar to the experiments mentioned above, and the particles’ surfaces were modified with different organic moieties (amine, carboxylate, or thiol). In all cases, highly porous nanoparticles were obtained (Table 3). After protein adsorption using human serum and quantitative analysis by LC–MS/MS and TGA, we clearly observed that thiol-derivatized MSN (“MSN-SH”) retained the largest amount of low MW proteins (Figure S5). This is consistent with previously published work, as the strongly hydrophobic character of thiol-modified silica materials favored interactions with the hydrophobic pockets present on protein surfaces as well as the formation of disulfide bridges.10,20 For this reason, we selected MSN-SH for subsequent studies of the PCs from serum samples of PCa patients.
Table 3. Physical Properties and Protein Adsorption Data of Functionalized MSN.
| N2 physisorption |
organic
group contenta |
||||||
|---|---|---|---|---|---|---|---|
| sample | dpore (nm) | SBET (m2/g) | Vpore (cm3/g) | (mmol/g) | (μmol/m2) | mass of protein adsorbed (mg/g SiO2)b | specific protein adsorption (mg/m2) |
| MSN-OH | 2.7 | 1016.4 | 0.76 | 212 | 0.21 | ||
| MSN-COOH | 2.2 | 822.6 | 0.36 | 0.67 | 0.81 | 201 | 0.24 |
| MSN-NH2 | 2.4 | 495.1 | 0.71 | 2.00 | 4.04 | 170 | 0.34 |
| MSN-SH | 2.5 | 882.5 | 0.72 | 2.57 | 2.91 | 368 | 0.42 |
As determined by elemental analysis.
As determined by TGA.
Since the late 1980s, PSA-based testing has been widely used for PCa detection. However, overdiagnosis and overtreatment occur frequently, involving serious side effects and unnecessary clinical expenses. Often, these problems are a result of the fact that PSA levels may also be elevated in healthy men, as PSA is prostate-specific but not PCa-specific. Indeed, meta-analyses show that in patients with PSA levels above 4 ng/mL, the positive predictive value of PSA is only about 25%.21 Moreover, second biopsies can detect cancer in 15–34% of patients whose first biopsy was negative.22 A recent U.S. preventive services task force recommended against PSA-based screening for PCa in all age groups, highlighting the need for new, clinically-useful, biomarkers.23,24
We sought to study the low MW proteomes of patients with various stages of PCa, using their PSA levels and biopsy status to divide them into groups and to provide a method of comparing the relative amounts of each protein from group to group. For this purpose, blood samples were taken from 22 patients from the following groups: metastatic PCa (“M” group, 4 patients); elevated PSA and positive biopsy for PCa (“B+” group, 6 patients); elevated PSA and negative biopsy (“B–” group, 7 patients); age-matched control patients with normal PSA levels (“C” group, 5 patients). Adsorption experiments were carried out over MSN-SH and subsequent analysis of the PC by LC–MS/MS and TGA showed a complex mixture of proteins with the MW ranging from 5 to 515 kDa, with 344 different protein assignments over the entire set of patient sera (Table S3). All proteins were organized by the mass of each protein contained in each patient’s corona, as calculated by NSpC × TGA, and statistical analysis using principal component analysis (PCA) was performed. According to PCA, the first three components of the proteomic distribution in different patient groups were represented in a tri-coordinate projection system (Figure S6). Then, to determine individual proteomes (which may correspond to the real PPC as described before),13,14 predictor variables associated with the four different patient groups were estimated by using an elastic net penalized multinomial regression model, and a total of 51 proteins were identified as model components. The combinations of the levels of these 51 proteins were sufficient to define the four groups; these data are presented in Figure 5a as a heat map.
Figure 5.

Statistical identification of proteins unique to each patient group after adsorbing serum proteins from patients onto thiol-modified mesoporous silica nanoparticles (MSN-SH). Patient groups are defined in the text. (a) Z-score differences (±4SD) corresponding to selected variables obtained by the elastic net penalized multinomial regression model for 51 identified proteins were clustered into groups based on correlation in their relative abundance (Pearson correlation coefficients). A heat map (scale at left of figure) was then generated from the Z-score data. Proteins present in a larger amount in a particular patient sample are in green; those in a smaller amount are in red. (b) 3D plot of the heat map, emphasizing groups of proteins unique to each patient group, based on Z-score data.
The data in this figure are presented as a “Z-score”, which is the number of standard deviations (SD) above (green) or below (red) the average value of the mass of a particular protein in each corona. A 3D representation of the heat map (Figure 5b) revealed that all PCa groups showed unique proteomic clusters, making it potentially feasible to quickly assign each patient to a particular diagnostic group. Many of the identified proteins belong to the low MW fraction, where a significant number of proteins whose serum concentration is severely affected by the pathological stage are expected to be found.8 Indeed, some of these proteins are strongly related to cancer development, such as plasma serine protease inhibitor in metastasis and positive biopsy patients (inhibits tissue- and urinary-type plasminogen activators, prostate-specific antigens, and kallikrein activities). Other detected up and down-regulated proteins with reported potential as cancer biomarkers are identified in Table S4.25
With proteomics data in hand, the predictive value of the model was tested by applying the elastic net model coefficients to the data of each patient.19 That is, each patient’s proteomics information was processed through the model without any bias as to their clinical diagnosis, and a prediction was made as to the probability of their assignment to a specific group. The results (Table 4) showed that patient assignments made from proteomics matched the clinical assignment quite well, with probabilities of matching assignments ranging from 49 to 78%. Importantly, the difference between the probability of being in the clinically assigned group versus the other groups was sizable. For example, patients with clinical assignments of B– (elevated PSA but negative biopsy) had a probability of 67% of being assigned to the B– group from proteomics data, but only a probability of 14% of being assigned to the B+ group; patients with B+ clinical assignments (elevated PSA and positive biopsy) had a probability of 59% of being assigned to the B+ group but only 17% probability of assignment to the B– group.
Table 4. Summary of Clinical Data for Prostate Cancer Patients and Prediction Values Obtained by Application of the Elastic Net Penalized Multinomial Regression Modela.
| probability
of patients in groupc |
||||||
|---|---|---|---|---|---|---|
| patient # | groupb | PSA (ng/mL) | M | B+ | B– | C |
| 1 | M | 235 | 0.78 | 0.08 | 0.05 | 0.09 |
| 2 | M | 426 | 0.66 | 0.16 | 0.09 | 0.08 |
| 3 | M | 26.7 | 0.73 | 0.11 | 0.07 | 0.10 |
| 4 | M | 530 | 0.61 | 0.14 | 0.08 | 0.17 |
| average | 0.70 | 0.12 | 0.07 | 0.11 | ||
| 5 | B+ | 7.6 | 0.09 | 0.57 | 0.17 | 0.18 |
| 6 | B+ | 9.7 | 0.05 | 0.69 | 0.10 | 0.16 |
| 7 | B+ | 5.1 | 0.09 | 0.55 | 0.17 | 0.19 |
| 8 | B+ | 8.1 | 0.08 | 0.57 | 0.23 | 0.12 |
| 9 | B+ | 7.4 | 0.06 | 0.63 | 0.18 | 0.13 |
| 10 | B+ | 6.5 | 0.06 | 0.56 | 0.19 | 0.18 |
| average | 0.07 | 0.59 | 0.17 | 0.16 | ||
| 11 | B– | 6.9 | 0.06 | 0.12 | 0.69 | 0.13 |
| 12 | B– | 5.8 | 0.04 | 0.13 | 0.72 | 0.10 |
| 13 | B– | 11.4 | 0.05 | 0.11 | 0.73 | 0.11 |
| 14 | B– | 8.7 | 0.07 | 0.15 | 0.65 | 0.13 |
| 15 | B– | 6.1 | 0.07 | 0.15 | 0.64 | 0.13 |
| 16 | B– | 5.3 | 0.06 | 0.13 | 0.64 | 0.17 |
| 17 | B– | 6.3 | 0.07 | 0.18 | 0.61 | 0.14 |
| average | 0.06 | 0.14 | 0.67 | 0.13 | ||
| 18 | C | 0.6 | 0.09 | 0.22 | 0.20 | 0.49 |
| 19 | C | 0.8 | 0.08 | 0.18 | 0.23 | 0.51 |
| 20 | C | 1.1 | 0.07 | 0.14 | 0.26 | 0.53 |
| 21 | C | 0.2 | 0.04 | 0.10 | 0.24 | 0.63 |
| 22 | C | 0.5 | 0.07 | 0.19 | 0.12 | 0.62 |
| average | 0.07 | 0.16 | 0.21 | 0.56 | ||
Best statistical matches for each patient group are indicated in bold.
Assigned after complete clinical examination and biopsy. M, metastatic PCa; B+, elevated PSA, positive PCa biopsy; B–, elevated PSA, negative PCa biopsy = negative biopsy; C, normal PSA (age-matched control). PSA = prostate-specific antigen.
Prediction based solely on proteomics data.
The distinction between B– and B+ patients is of special significance because the predictive value of the PSA test alone is only ∼25%. Instead, an initial screening with a PSA test followed by proteomics using MSN-SH, which only requires an additional blood sample, provides a simple predictive tool to avoid unnecessary, costly, and invasive biopsies. This is especially relevant to clearly discriminate patients with elevated PSA levels due to prostatitis or benign prostate hyperplasia from those with lethal forms of PCa. Conversely, continued monitoring of patients with elevated PSA levels using proteomics could also allow early intervention, for example, when assignment to the B+ group becomes indicated. The data set here is small and the information presented is preliminary; however, the probability difference between correct and incorrect assignments will only increase as more patients are added to the model and additional proteomics information is collected. This is the focus of our current studies.
Conclusions
We have shown that low MW proteins dominate the PCs of mesoporous silica nanoparticles, an effect that is more pronounced with small pore diameters. When the surface areas of each type of particle are taken into account by comparing the specific protein adsorption (protein adsorbed per unit surface area), silica nanoparticles with the largest pore diameter studied (14 nm) had PCs with very similar compositions to those of dense silica nanoparticles. This indicated that the protein size discrimination capability is most pronounced when the pore diameter is below 7.4 nm. Comparing the adsorption patterns of a representative low MW protein (apolipoprotein A-II) and a high MW protein (complement C3), we found that in silica nanoparticles with small pores, A-II diffuses easily within the pores while C3 does not. Interestingly, although only a small amount of C3 bound to small-pore silica nanoparticles, it still influenced the adsorbed quantity of A-II bound. In particles with larger pores, each protein was able to bind more successfully when the other was not present. These competitive adsorption experiments showed that in addition to the expected size exclusion effect on protein adsorption, there is still competition for surface adsorption sites. Finally, thiol-modified medium pore silica nanoparticles (MSN-SH) were used to adsorb proteins from sera of PCa patients, patients with elevated PSA but without PCa, and control patients. Application of a statistical model to the complete set of proteomics data allowed groups of proteins to be preliminarily identified as markers of each patient’s status. The proteomics and clinical assessments showed very good agreement, and the proteomics assignations had the added advantage of discriminating between patients with elevated PSA due to PCa and patients with elevated PSA for another reason. These experiments provide additional insight into the development of the PCs on MSNs, and illustrate how this information can be usefully applied to a diagnostic test.
Materials and Methods
Materials
All materials were purchased from Sigma-Aldrich unless otherwise noted, and used as received. Synthetic protocols corresponding to preparation of dense silica nanoparticles (SNPs), medium pore silica nanoparticles (MSNs), and large pore silica nanoparticles (LPSs), as well as surface modification with organic groups are fully detailed in the Supporting Information. Furthermore, the different techniques used for silica nanoparticle physico-chemical characterization are also described in the Supporting Information.
Human Serum Protein Adsorption Experiments
Prior to serum exposure, particles (1 mg) were suspended in 0.5 mL phosphate-buffered saline (PBS) (1×, pH 7.4). Subsequently, the serum sample (0.5 mL) was added and the mixture was incubated for 1 h at 37 °C and 1500 rpm in a ThermoMixer. Next, the protein-adsorbed particles were isolated through centrifugation at 14800 rpm at 4 °C for 5 min. Then, the supernatant was discarded and nanoparticles were resuspended in PBS (1 mL) and sonicated. This process was repeated three times in order to ensure that any free or loosely bound proteins were removed from the solution. After removal of the final PBS wash, nanoparticles containing the hard corona were suspended in digestion buffer (36 μL, 50 mM ammonium bicarbonate, 0.25 mM urea, 4% acetonitrile) and reducing buffer (6 μL, 100 mM dithiothreitol in H2O). These nanoparticle suspensions were allowed to incubate for 1 h at 70 °C. After 1 h, alkylation buffer (15 μL, 100 mM iodoacetamide) was added and the samples were incubated at room temperature for 20 min in a dark place. Protein digestion was achieved by adding trypsin (15 μL, 40 ng/μL) and incubating overnight at 37 °C. The following morning, formic acid (15 μL, 10%) was added to stop the digestion process. Prior to submitting the samples for proteomic analysis, the nanoparticles were centrifuged out of solution and the supernatant was submitted for analysis.
Single Protein Adsorption Experiments
Nanoparticles (1 mg) were suspended in 0.5 mL PBS (1×, pH 7.4). The particle suspension was then sonicated to disperse any aggregates. Subsequently, the suspension was diluted to a final concentration of 1 mg/mL with PBS containing apolipoprotein AII (40 μg/mL) and/or complement C3 protein (40 μg/mL). Proteins were supplied by Athens Research & Technologies. The adsorption proceeded for 1 h before isolating the particle–protein conjugates via centrifugation (1 min, 14 800 rpm). The protein adsorbed sample was then re-suspended in PBS (1 mL), sonicated, and centrifuged. This process was repeated three times in order to remove any loosely bound protein. In the case of sequential adsorption, C3 was adsorbed first for 1 h. Then, the supernatant was removed by centrifugation (1 min, 14 800 rpm) and the sample was washed three times with PBS as above. Afterwards, A-II adsorption was carried out for 1 h, and the protein–particle conjugate was separated and washed again three times with PBS. Subsequently, hard corona was extracted and analyzed as described above.
Proteomic Analysis
Prior to the MS/MS technique, preliminary characterization of the PC composition by one-dimensional SDS-PAGE was performed, and details are presented in the Supporting Information. Subsequently, for the proteomic analysis, samples were subjected to desalting and concentration by μC18 ZipTip (Millipore) according to the manufacturer protocol. 5 μL of each sample were loaded onto a trap column (NanoLC Column, 3μ C18-CL, 350 μm × 0.5 mm; Eksigen) and desalted with 0.1% trifluoroacetic acid at 3 μL/min for 5 min. The peptides were then loaded onto an analytical column (LC Column, 3 μC18-CL, 75 mm × 12 cm, Nikkyo) equilibrated in 5% acetonitrile and 0.1% formic acid. Elution was carried out with a linear gradient of 5–35% acetonitrile in 0.1% formic acid at a flow rate of 300 nL/min for 30 min. Peptides were analysed in a mass spectrometer nanoESI-qQTOF (6600 TripleTOF, AB SCIEX). A tripleTOF was operated in information-dependent acquisition mode, in which a 0.26 s TOF MS scan from 350 to 1250 m/z was performed, followed by 0.05 s product ion scans from 100 to 1600 m/z on the 50 most intense 2–5 charged ions. The system sensitivity was controlled with 2 fmol of 6 proteins (LC Packings). ProteinPilot 5.0 search engine (AB SCIEX) default parameters were used to generate a peak list directly from 6600 Triple TOF .wiff files. The paragon algorithm of ProteinPilot 5.0 was used to search the ExPASy protein database (616203 sequences; 181334896 residues) with the following parameters: trypsin specificity, cys-alkylation, taxonomy restricted to human, and the search effort set to through. To avoid using the same spectral evidence in more than one protein, the identified proteins are grouped based on MS/MS spectra by the ProteinPilot Pro Group algorithm. Thus, proteins sharing MS/MS spectra are grouped, regardless of the peptide sequence assigned. The protein within each group that can explain more spectral data with confidence is shown as the primary protein of the group. Only the proteins of the group for which there is individual evidence (unique peptides with enough confidence) are also listed, usually toward the end of the protein list. Protein identification was performed according to ProtScore units (eq 1). Here, proteins showing unused score >1.3 were identified with confidence ≥96%, and proteins showing identification confidence less than 96% were eliminated.
| 1 |
Calculation of Mass % of Individual Proteins
LC–MS/MS analysis of adsorbed proteins was carried in triplicate. In order to obtain a unique intensity signal for each identified protein, the signals obtained from all peptides that make up the protein were summed. Then, the NSpC for each protein, which represent the percentage of each protein identified in the proteomic analysis as a function of MW, were multiplied by the overall mass of adsorbed protein as determined by TGA. The result of this calculation (NSpC × TGA, eqs 2 and 3) is the contribution of each protein to the total adsorbed mass.26 SD were determined from these values.
| 2 |
| 3 |
Patient Selection and Clinical Sample Extraction
This study was conducted with the approval of the Ethics Committee of La Fe Hospital (Valencia, Spain). Informed consent was obtained from all cases for the examinations and experiments conducted. Clinical materials were used after written informed consent was obtained, according to protocols approved by the Institutional Review Board of La Fe Hospital.
Between January and February 2015, 17 patients were referred to the Department of Urology at La Fe Hospital for further evaluation of suspected PCa based on an abnormal digital rectal examination and/or an elevated serum PSA concentration.27 During the study period, 17 transrectal ultrasonography-guided biopsies were performed with an ultrasound system (Siemens Healthcare, Erlangen, Germany). An extended 10-core systematic random biopsy was performed using an automatic spring-loaded gun biopsy device with an 18-gauge needle, and the Gleason score and TNM adscription were established in all patients.27,28 Also, a computer tomography scan was performed in patients suspected of metastasis (PSA > 25 ng/mL). In parallel, 5 extra patients were selected as controls. Controls were defined as men who had been followed at least for 5 years on study with no PCa diagnosis. Blood samples from all patients were collected the same day that biopsies were scheduled in Vacutainer tubes (Becton Dickinson) and centrifuged at room temperature for 15 min at 3000 g. Serum was collected from the tubes, aliquoted, and stored at −80 °C.
Statistical Analysis
Statistical analysis of clinical data was performed using R software (version 3.2.2) and the R-package glmnet (version 2.0-2).
Data Pretreatment
Data were summarized by mean, SD, median, and first and third quartiles. Initial data exploration was performed using PCA.29 Moreover, identified proteins were clustered into groups based on correlation to their relative abundance (Pearson correlation coefficients), and variables with zero variance were excluded. A total of 187 proteins were clustered in a heat map.
Elastic Net Penalized Multinomial Regression Model
Association of the predictor variables with the four different patient groups was assessed using an elastic net penalized multinomial regression model.30 Elastic net penalization allows for variable selection by shrinking the coefficients of the variables not related to the response to zero. Thus, variables with non-zero coefficients are considered as important predictors. Selection of the shrinkage parameter (lambda) for the elastic net model was performed by twenty repetitions of 10-fold cross-validation. The one-standard-error rule was used. The results of the elastic net model were depicted in a heat map including only the selected variables. Finally, the probability (Pr) to ascribe the corresponding patient to a specific PCa group was calculated through the minimum error model.30
Model Validation
An internal model validation was performed using 1000 iterations of bootstrapping. Optimism-corrected kappa statistic was 0.58 and accuracy was 0.69.
Acknowledgments
Financial support from the University of Vermont, the Spanish Ministry of Economy and Competitiveness (projects TEC2016-80976-R and SEV-2016-0683), and the Generalitat Valenciana (project PROMETEO/2017/060), is gratefully acknowledged. We thank Dr. Jaime Font de Mora for his assistance in the clinical sample collection and Dr. David Hervás for the statistical study supervision. We also appreciate the assistance of the electron microscopy service of the Universitat Politècnica de València.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00460.
Full synthesis and characterization details of the nanoparticles used in these experiments, including DLS and N2 physisorption; characterization and identification of adsorbed proteins, including SDS-PAGE and LC-MS/MS; tables of NSpC × TGA for all proteins; and PCA of experimental data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Wulfkuhle J. D.; Liotta L. A.; Petricoin E. F. Proteomic Applications for the Early Detection of Cancer. Nat. Rev. Cancer 2003, 3, 267–275. 10.1038/nrc1043. [DOI] [PubMed] [Google Scholar]
- Tirumalai R. S.; Chan K. C.; Prieto D. A.; Issaq H. J.; Conrads T. P.; Veenstra T. D. Characterization of the Low Molecular Weight Human Serum Proteome. Mol. Cell. Proteomics 2003, 2, 1096–1103. 10.1074/mcp.m300031-mcp200. [DOI] [PubMed] [Google Scholar]
- Marshall J.; Kupchak P.; Zhu W.; Yantha J.; Vrees T.; Furesz S.; Jacks K.; Smith C.; Kireeva I.; Zhang R.; et al. Processing of Serum Proteins Underlies the Mass Spectral Fingerprinting of Myocardial Infarction. J. Proteome Res. 2003, 2, 361–372. 10.1021/pr030003l. [DOI] [PubMed] [Google Scholar]
- Clemments A. M.; Muniesa C.; Landry C. C.; Botella P. Effect of Surface Properties in Protein Corona Development on Mesoporous Silica Nanoparticles. RSC Adv. 2014, 4, 29134–29138. 10.1039/c4ra03277b. [DOI] [Google Scholar]
- Clemments A. M.; Botella P.; Landry C. C. Protein Adsorption From Biofluids on Silica Nanoparticles: Corona Analysis as a Function of Particle Diameter and Porosity. ACS Appl. Mater. Interfaces 2015, 7, 21682–21689. 10.1021/acsami.5b07631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemments A. M.; Botella P.; Landry C. C. Spatial Mapping of Protein Adsorption on Mesoporous Silica Nanoparticles by Stochastic Optical Reconstruction Microscopy. J. Am. Chem. Soc. 2017, 139, 3978–3981. 10.1021/jacs.7b01118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aebersold R.; Mann M. Mass Spectrometry-Based Proteomics. Nature 2003, 422, 198. 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- Gerszten R. E.; Accurso F.; Bernard G. R.; Caprioli R. M.; Klee E. W.; Klee G. G.; Kullo I.; Laguna T. A.; Roth F. P.; Sabatine M.; et al. Challenges in Translating Plasma Proteomics from Bench to Bedside: Update from the NHLBI Clinical Proteomics Programs. Am. J. Physiol. Lung Cell Mol. Physiol. 2008, 295, L16–L22. 10.1152/ajplung.00044.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamburro D.; Fredolini C.; Espina V.; Douglas T. A.; Ranganathan A.; Ilag L.; Zhou W.; Russo P.; Espina B. H.; Muto G.; et al. Multifunctional Core-Shell Nanoparticles: Discovery of Previously Invisible Biomarkers. J. Am. Chem. Soc. 2011, 133, 19178–19188. 10.1021/ja207515j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huo Q.; Colon J.; Cordero A.; Bogdanovic J.; Baker C. H.; Goodison S.; Pensky M. Y. A Facile Nanoparticle Immunoassay for Cancer Biomarker Discovery. J. Nanobiotechnol. 2011, 9, 20. 10.1186/1477-3155-9-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng T.; Pierre-Pierre N.; Yan X.; Huo Q.; Almodovar A. J. O.; Valerio F.; Rivera-Ramirez I.; Griffith E.; Decker D. D.; Chen S.; et al. Gold Nanoparticle-Enabled Blood Test for Early Stage Cancer Detection and Risk Assessment. ACS Appl. Mater. Interfaces 2015, 7, 6819–6827. 10.1021/acsami.5b00371. [DOI] [PubMed] [Google Scholar]
- Hajipour M. J.; Raheb J.; Akhavan O.; Arjmand S.; Mashinchian O.; Rahman M.; Abdolahad M.; Serpooshan V.; Laurent S.; Mahmoudi M. Personalized Disease-Specific Protein Corona Influences the Therapeutic Impact of Graphene Oxide. Nanoscale 2015, 7, 8978–8994. 10.1039/c5nr00520e. [DOI] [PubMed] [Google Scholar]
- Palchetti S.; Pozzi D.; Mahmoudi M.; Caracciolo G. Exploitation of nanoparticle-protein corona for emerging therapeutic and diagnostic applications. J. Mater. Chem. B 2016, 4, 4376–4381. 10.1039/c6tb01095d. [DOI] [PubMed] [Google Scholar]
- Caputo D.; Papi M.; Coppola R.; Palchetti S.; Digiacomo L.; Caracciolo G.; Pozzi D. A Protein Corona-Enabled Blood Test for Early Cancer Detection. Nanoscale 2017, 9, 349–354. 10.1039/c6nr05609a. [DOI] [PubMed] [Google Scholar]
- Nozawa K.; Gailhanou H.; Raison L.; Panizza P.; Ushiki H.; Sellier E.; Delville J. P.; Delville M. H. Smart Control of Monodisperse Stöber Silica Particles: Effect of Reactant Addition Rate on Growth Process. Langmuir 2005, 21, 1516–1523. 10.1021/la048569r. [DOI] [PubMed] [Google Scholar]
- Cai Q.; Luo Z.-S.; Pang W.-Q.; Fan Y.-W.; Chen X.-H.; Cui F.-Z. Dilute Solution Routes to Various Controllable Morphologies of MCM-41 Silica with a Basic Medium†. Chem. Mater. 2001, 13, 258–263. 10.1021/cm990661z. [DOI] [Google Scholar]
- Gao F.; Botella P.; Corma A.; Blesa J.; Dong L. Monodispersed Mesoporous Silica Nanoparticles with Very Large Pores for Enhanced Adsorption and Release of DNA. J. Phys. Chem. B 2009, 113, 1796–1804. 10.1021/jp807956r. [DOI] [PubMed] [Google Scholar]
- Fan J.; Yu C.; Gao F.; Lei J.; Tian B.; Wang L.; Luo Q.; Tu B.; Zhou W.; Zhao D. Cubic Mesoporous Silica with Large Controllable Entrance Sizes and Advanced Adsorption Properties. Angew. Chem. Int. Ed. 2003, 42, 3146–3150. 10.1002/anie.200351027. [DOI] [PubMed] [Google Scholar]
- Edwards R. A.; Woody R. W. Spectroscopic Studies of Cibacron Blue and Congo Red Bound to Dehydrogenases and Kinases. Evaluation of Dyes as Probes of the Dinucleotide Fold. Biochemistry 1979, 18, 5197–5204. 10.1021/bi00590a026. [DOI] [PubMed] [Google Scholar]
- Mistry K.; Cable G. Meta-Analysis of Prostate-Specific Antigen and Digital Rectal Examination as Screening Tests for Prostate Carcinoma. J. Am. Board Fam. Pract. 2003, 16, 95–101. 10.3122/jabfm.16.2.95. [DOI] [PubMed] [Google Scholar]
- Roehl K. A.; Antenor J. A. V.; Catalona W. J. Serial Biopsy Results in Prostate Cancer Screening Study. J. Urol. 2002, 167, 2435–2439. 10.1016/s0022-5347(05)64999-3. [DOI] [PubMed] [Google Scholar]
- Moyer V. A. Screening for Prostate Cancer: U.S. Preventive Services Task Force Recommendation Statement. Ann. Intern. Med. 2012, 157, 120–134. 10.7326/0003-4819-157-2-201207170-00459. [DOI] [PubMed] [Google Scholar]
- Prensner J. R.; Rubin M. A.; Wei J. T.; Chinnaiyan A. M. Beyond PSA: The next Generation of Prostate Cancer Biomarkers. Sci. Transl. Med. 2012, 4, 127rv3. 10.1126/scitranslmed.3003180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polanski M.; Anderson N. L. A list of candidate cancer biomarkers for targeted proteomics. Biomark. Insights 2007, 1, 1–48. 10.1177/117727190600100001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monopoli M. P.; Walczyk D.; Campbell A.; Elia G.; Lynch I.; Bombelli F. B.; Dawson K. A. Physical–Chemical Aspects of Protein Corona: Relevance to in Vitro and in Vivo Biological Impacts of Nanoparticles. J. Am. Chem. Soc. 2011, 133, 2525–2534. 10.1021/ja107583h. [DOI] [PubMed] [Google Scholar]
- Heidenreich A.; Aus G.; Bolla M.; Joniau S.; Matveev V. B.; Schmid H. P.; Zattoni F. EAU Guidelines on Prostate Cancer. Eur. Urol. 2008, 53, 68. 10.1016/j.eururo.2007.09.002. [DOI] [PubMed] [Google Scholar]
- American Joint Committee . American Join Committee on Cancer Staging Manual, 5th ed., Philadelphia, 1997; pp 219–222.
- Jolliffe I. T.Principal Component Analysis, 2nd ed.; Springer-Verlag: New York, 2002. [Google Scholar]
- Friedman J.; Hastie T.; Tibshirani R. Regularization Paths for Generalized Linear Models via Coordinate Descent. J. Stat. Softw. 2010, 33, 1–22. 10.18637/jss.v033.i01. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.