

Abstract
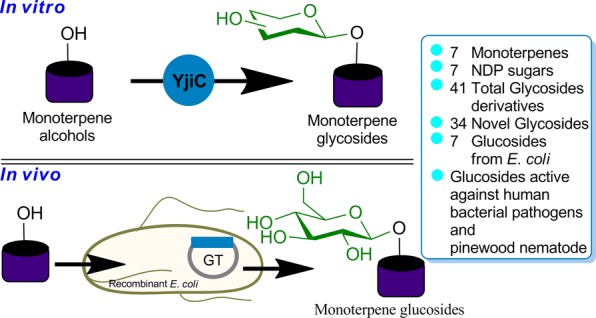
A promiscuous Bacillus glycosyltransferase (YjiC) was explored for the enzymatic synthesis of monoterpene O-glycosides in vitro and in vivo. YjiC converted seven monoterpenes into 41 different sugar-conjugated novel glycoside derivatives. The whole-cell biotransformation of the same set of monoterpenes exhibited robust enzyme activity to synthesize O-glucosyl derivatives from Escherichia coli. These newly synthesized selected monoterpene-O-glucosyl derivatives exhibited enhanced antibacterial activities against human pathogenic bacteria and antinematodal activities against pine wood nematode Bursaphelenchus xylophilus.
Introduction
Approximately 55,000 terpenoids have been identified from diverse sources. They are well known to exhibit anticancer, antimicrobial, antifungal, antiviral, antiparasitic, anti-inflammatory, antioxidative, antihyperglycemic, and skin permeation enhancing activities.1 Monoterpenes that contain 10 carbon atoms in various branched and often cyclic structures sharing p-methane are increasingly getting popular as flavoring agents and perfumery products, spices and seasoning of foods, and additives in bath, oil lamps, massages, and aromatherapy.2 They are major constituents of essential oils and are nontoxic and nonmutagenic to human. Thus, they are generally recognized as safe (GRAS) for use as drug permeation enhancers.3 Using formulations containing menthol, thymol, carvacrol, linalool, anethole, and limonene in humans does not lead to skin irritation or sensitization.4 However, these compounds have extremely low water solubility that their use in the industry is limited.5 The major hurdle in modifying these molecules to other derivatives is associated with loss of inherent flavor and certain pharmacological properties. However, glycosides of monoterpenes have high water solubility. Sugar-conjugated terpenes can retain their inherent flavor after hydrolysis of the compound, which releases terpene and sugar. Moreover, sugar-conjugated natural products are known to exhibit different biological activities.6 Although a small number of monoterpene glycosides have been isolated from natural sources, few glycosides have been produced using the whole-cell biotransformation of microbial and plant cells.7 Selected monoterpene glycosides chemically synthesized by Higashiyama and Sakata have several limitations associated with their regio- and stereoselectivities, final yields, purification steps, and protection/deprotection of functional groups.8
In recent years, we have explored the catalytic promiscuity of glycosyltransferases (GTs; EC 2.4) for the synthesis of diverse sugar-conjugated natural products. YjiC, one of the Bacillus licheniformis DSM 13 glycosyltransferases belonging to GT1 family proteins in CAZy classification (www.cazy.org), has been discovered to possess highly flexible activity toward a wide range of plant and microbial polyketides.9 To investigate the potential of using this recombinant enzyme to synthesize glycodiverisified monoterpenes, we examined its activity with selected commercially available medicinally important monoterpene alcohols and different nucleotide diphosphate sugars (NDP-sugars).
Results and Discussion
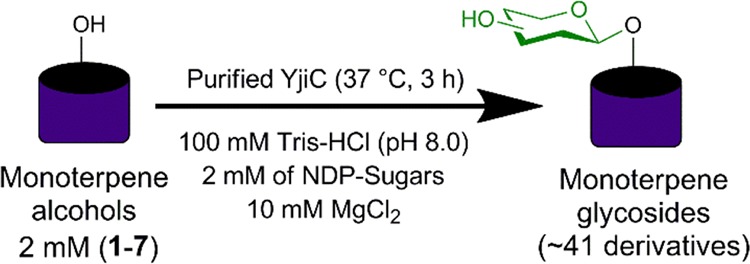
To study the promiscuity of YjiC toward monoterpenes, eugenol (1) was initially used as an acceptor substrate, while UDP-α-d-glucose (UDP-Glc) was used as a glucose donor. Then, Ni-affinity purified His6-tagged fusion protein produced in Escherichia coli BL21 (DE3) was allowed to react with 2 mM 1 in 200 mM Tris-HCl buffer (pH 8.0) containing 20 μg/mL YjiC and 2 mM UDP-Glc at 37 °C for 3 h (Scheme 1). After adding methanol as a quenching reagent, the reaction was analyzed by high-performance liquid chromatography–photodiode array (HPLC-PDA) and high-resolution quadruple time-of-flight electrospray ionization mass spectrometry (HRqTOF-ESI/MS) in positive mode. The reaction lacking eugenol served as a control reaction. MS analysis of 1a showed a spectrum of an ion peak at m/z [M + Na]+ 349.1270, which resembled glucose (162 amu greater)-conjugated 1 for which the calculated exact mass was 349.1263 Da. The sister fragment of 1 with an ion peak at m/z [M + H]+ 165.0914 confirmed 1 (Figure S1). Under identical conditions, six other monoterpene alcohols thymol (2), isoeugenol (3), carvacrol (4), α-terpineol (5), (+)-menthol (6), and (−)-borneol (7) were assessed as acceptor substrates. HPLC-PDA and HRqTOF-ESI/MS analyses confirmed the glucose-conjugated monoterpene alcohols (Figures S2–S7 and Table S1). Conversion rates for most of these compounds were more than 40% except for 5 and 7, which had a conversion rate of only 30 and 16%, respectively. The maximum conversion rate was achieved for 3 (∼78%), followed by that for 1 (65%) and 4 (51%).
Scheme 1. YjiC Mediated In Vitro Glycodiversification of Monoterpene Alcohols.
Among the seven monoterpenes (1–7) used as acceptor substrates, four molecules (1–4) contain a benzene ring in their structure (Figure 1A). The conversion percentage of these molecules to glucosides (1a–4a) is fairly high when UDP-Glc was used as the glucose donor substrate (Figure 1B). This results provide the evidence to support that YjiC has more substrate specificity toward a phenolic ring containing the acceptor substrates. A previous report has shown that YjiC converted phenolic compounds such as flavonoids, stilbenes, and chalcones to multiple glucosides by conjugating glucose moieties to multiple available phenolic hydroxyl groups.9 Similarly, the conversion of 7 to 7a was the least among all. Structurally, compound 7 is a bicyclic monoterpene, while the remaining compounds are monocyclic. This structural difference could be a possible reason for less preference by YjiC over other acceptor substrates for glycosylation.
Figure 1.

Catalytic promiscuity of YjiC toward selected monoterpenols and diverse NDP-d/l-sugars. (A) Structures of the monoterpenols and glycosides generated (2b, 3b, 4b, 5b, 5f, 5g, 6f, and 7b were not detected). (B) Percent conversion of glycodiversified monoterpenol derivatives catalyzed by YjiC. (C) Structures of different sugar moieties conjugated to monoterpenols. Asterisk (*) represents the glucosylated products that are identified by 1H and 13C NMR analyses. ND indicates no detection of the product.
All products (1a–7a) were prepared in large scale under identical reaction conditions and purified by prep-HPLC (Figure S8) for structural characterization. 1H and 13C NMR spectroscopic analyses of 1a–7a were in good agreement with glucose-conjugated 1–7 (Figures S9–S15). The appearance of a distinct doublet spectrum between δ 4.1 and 5.0 ppm with a coupling constant of ∼J = 7.5 Hz (Table S2) represented the anomeric proton (1′) of the glucose moiety in β configuration. Other spectra for glucose were in the range of 3 to 5 ppm. In 13C NMR, anomeric carbon was distinctly observed at around 100 ppm, while other spectra of glucose were present between 60 and 80 ppm (Figures S9–S15).
To further expand the chemical diversity of monoterpene alcohol glycosides, in addition to UDP-Glc, six other nucleotide diphosphate (NDP)-sugars [UDP-galactose (UDP-Gal), UDP-glucuronic acid (UDP-GlcA), UDP-N-acetylgalactosamine (UDP-GalNAc), UDP-N-acetylglucosamine (UDP-GluNAc), TDP-rhamnose (TDP-Rhm), and GDP-fucose (GDP-Fuc)] were reacted with all 1–7 acceptor substrates. HPLC-PDA and HRqTOF-ESI/MS analyses (Figures S17–S22) of all these reaction mixtures showed the capacity of YjiC to tolerate a wide range of NDP-sugars to transfer most of these sugars to acceptor molecules, generating a total of 41 glycoside derivatives (1b–g to 7b–g) (Figure 1). However, the enzyme had a relatively small catalytic activity for other sugars. With most of these sugar donors, the conversion rate was limited (below 5–10%). One of the sugars used in the reaction, GalNAc, was conjugated with only 1 and 6 with detectable conversion percentage. Thus, we did not scale up these reactions to preparative scale for structural elucidation or biological activities.
The conversion of 1–7 to 1a–7a was higher when UDP-Glc was used as a sugar donor. However, the conversion of the same set of molecules with other NDP-sugars (b–g) to glycosides (1b–g to 7b–g) was comparatively very low (Figure 1). This result demonstrated that UDP-Glc is one of the preferred substrates among seven different NDP-sugars. In nature, UDP-Glc is abundantly present in all living cells, and many GTs accept UDP-Glc to transfer to a range of acceptor substrates within the cell.10 YjiC, though found as a promiscuous enzyme to accept wide ranges of substrates and NDP-sugars,9 preferred UDP-Glc over other NDP-sugars. However, this statement warrants further studies such as the crystal structure of YjiC and molecular docking of different acceptors and donor NDP-sugars to provide a clear understanding of substrates preferences.
Experiments were also carried out to check the possible synthesis of diverse sugar-conjugated monoterpenes using YjiC and cheap synthetic sugar donors such as p-nitrophenyl-β-d-xyloside (8) and p-nitrophenyl-β-d-galactoside (9) in one-pot transglycosylation reactions. Most GTs are capable of reversing the glycosylation reaction in the presence of NDPs.11 This property of GT helps to use alternative cheap sugar donors instead of relatively expensive NDP-sugars for the cost-effective production of natural product glycosides.12 Purified YjiC was used along with other reaction ingredients under aforementioned reaction conditions, and 1 was used as the sugar acceptor in Tris-HCl buffer at 37 °C. The reaction was carried out for 13 h. As shown in Figure 2, various NDPs such as ADP, TDP, CDP, GDP, and UDP were used at a concentration of 4 mM to transfer sugar moieties xylose (Xyl) and galactose (Gal) from 8 and 9, respectively. In the presence of high concentrations of NDPs, YjiC drove the reaction in the opposite direction to produce NDP-Xyl and NDP-Gal eventually utilized by the same enzyme to conjugate Xyl and Gal moieties to 1 in the coupled one-pot reaction. Byproduct 10 was released in the reaction medium, which developed a yellow color representing the progress of the reaction. Samples were taken at different time points to check the production of 1e and 1h. HPLC-PDA analysis showed that all five NDPs could be equally used to carry out transglycosylation with a conversion rate of ∼60% from 1 to 1e using 9 as the Gal donor. The conversion reached almost the maximum within 30 min. It was increased slightly until 13 h. However, the synthesis of 1h was lower under identical conditions when 8 was used as the Xyl sugar donor. The conversion rate of 1 to 1h was below 5% in ADP, GDP, and CDP. However, it reached ∼35% with UDP and 20% with TDP after 13 h of reaction (Figure 2).
Figure 2.

Transglycosylation by YjiC. (A) Scheme of trans-glycosylation via p-nitrophenyl β-d-galactoside (8) and p-nitrophenyl β-d-xyloside (9) using the enzyme YjiC and eugenol as the substrate in the presence of five different nucleotide diphosphates (UDP, TDP, ADP, GDP, and CDP). (B) Conversion percentage of 1 to eugenol β-d-galactoside (1e) and (C) eugenol β-d-xyloside (1h) at different time intervals in the presence of different nucleotides.
Many organic compounds with limited water solubility precipitate during in vitro enzymatic reaction. Thus, the use of organic co-solvents is considered to be useful for the enhancement of enzymatic reactions. We also studied the effect of co-solvent such as dimethyl sulfoxide (DMSO) for the conversion of monoterpenes to their glucosides. Compound 2 was randomly selected to check its conversion to 2a in the presence of DMSO, which is generally considered as a beneficial co-solvent for the enzymatic reactions. However, the result showed no increment in the conversion percentage of 2 to 2a in the presence of DMSO. The conversion percentage was somehow similar when there was no DMSO in the reaction mixture and in the presence of 5 and 10% DMSO. Instead, the conversion was decreased when the DMSO concentration was increased from 15 to 25% to the final concentration of the reaction mixture (Figure S23). DMSO is a good organic solvent that solubilizes organic molecules and makes them available for enzymes to react in the mixture. However, in the case of compound 2, the water solubility is 0.98 g/L (∼6.6 mM). The reaction mixture contained only 2 mM 2, which is completely soluble in water or reaction buffer condition. Thus, there was no additional catalytic enhancement in the conversion of 2 to 2a while using DMSO as a co-solvent in the reaction.
The stability of YjiC under reaction conditions was also studied using compound 2 as an acceptor substrate for glycosylation. Four sets of glycosylation reactions were set up in triplet and placed in a 37 °C water bath. These reaction mixtures contained all reaction ingredients except the donor and acceptor substrates. Each set of reaction was activated after 0, 2, 5, and 10 h of incubating at a 37 °C water bath by adding UDP-Glc and compound 2. The relative conversion percentage of 2 to its glucoside 2a was determined using HPLC-PDA analysis at different time intervals from 2 to 180 min. When the reaction was activated at 0 h, the enzyme reached the maximum conversion within 15 min and started to reverse the reaction as the accumulation of compound 2 was seen. This decrease in product concentration is because of the deglycosylation property of YjiC. YjiC drove the reaction in the opposite direction in the presence of high concentrations of nucleotide diphosphates such as UDP and glucosides. This behavior of YjiC was also observed with other acceptor substrates in previous studies.9 In a similar manner, when the reaction was activated after 2 h, the enzyme achieved the maximum conversion considerably late after 60 min and started to decrease in product concentration due to deglycosylation by YjiC. However, when the reaction mixture was activated after 5 and 10 h, the enzyme activity was dramatically decreased and did not achieve the maximum conversion percentage (Figure S24). These results clearly showed that the enzyme’s activity was decreased over time under the reaction condition at 37 °C.
Since the enzymatic synthesis of natural product glycosides requires purified enzymes and large quantities of NDP-sugars are costly, the process is difficult to scale up for practical quantity biosynthesis. Thus, we shifted the biosynthesis of monoterpene glucosides into the microbial system using recombinant E. coli BL21 (DE3) harboring YjiC plasmid (Figure 3A). The recombinant strain can utilize indigenous cytosolic UDP-Glc as a glucose donor and biotransform exogenously supplemented monoterpene alcohols to respective glucoside derivatives in Luria–Bertani broth medium.13 These growing recombinant cells were induced by isopropyl β-d-1-thiogalactopyranoside at a final concentration of 0.5 mM and incubated at 20 °C for 12 h for protein expression. These induced cells were then supplemented with exogenous 0.5 mM monoterpene alcohols (dissolved in DMSO) and further incubated at 20 °C for biotransformation. Time-dependent HPLC-PDA and ELSD analysis of 1–7 revealed the conversion of all substrates into respective O-glucosides and excreted out of the cells into the growth medium (Figure S25). Interestingly, the conversion reached the maximum within 4 h and remained almost constant or slightly increased until 68 h. The in vitro conversion profile was different from in vivo conversion. The conversion of 5 was elevated to 80% in the in vivo reaction. However, it was limited to 30% in the in vitro reaction system. Conversions of 2 and 4 were decreased, while those of 1 and 3 remained almost the same as those in the in vitro conversion system. Cell lysates were also subjected to HPLC-PDA and ELSD analyses for the possible accumulation of glucosides inside the cells. However, the concentrated cell lysate methanol extract did not show any glucoside peak in considerable abundance (Figure S26). When the same strain was supplemented with additional 5% sterile glucose in the culture medium at 0, 12, and 24 h of substrate feeding, it showed elevated conversion for most monoterpenes. Conversions of 3a and 6a reached ∼100%, while those of 1a and 5a reached 90% in 36 h. Those of 2a, 4a, and 7a reached 60% or more (Figure 3B). The in vivo conversion of 1–7 established the cost-effective biosynthesis of 1a–7a in a regiospecific manner through a sustainable and green approach.
Figure 3.
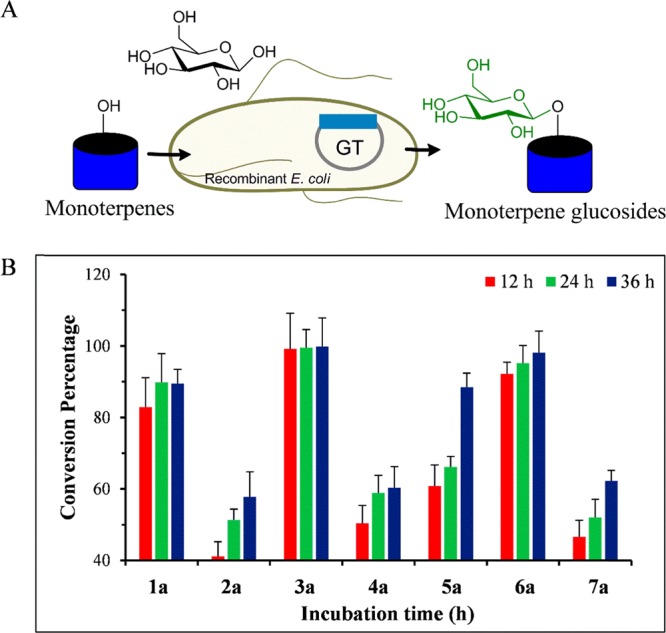
(A) Schematic representation of the whole-cell biotransformation of monoterpenes into glucosides using recombinant E. coli cells. (B) In vivo conversion percentage of monoterpenes into respective glucosides using whole cells of E. coli BL21 (DE3) harboring pET28-YjiC at different time intervals. The conversion of all monoterpenes was checked at a 0.5 mM final concentration. A 5% final concentration of sterile glucose was also supplemented at 0, 12, and 24 h.
All these newly synthesized glucoside derivatives (1a–7a) were produced using the in vivo system [isolated yields: 1a (93.3%), 2a (47.6%), 3a (91.3%), 4a (51.5%), 5a (75.4%), 6a (68%), and 7a (43%)]. They were purified using prep-HPLC and used for antibacterial and antinematodal assays. All seven O-glucoside derivatives of monoterpene alcohols were accessed for their antibacterial activities against 16 different Gram-positive and Gram-negative human pathogens (Table S4). Most Staphylococcus aureus strains used in this study were methicillin-resistant or methicillin-susceptible (MRSA/MSSA) superbugs. Since most of the glucosides tested showed antibacterial activities in disc-diffusion assay (Table S5), we further determined the minimum inhibitory concentration (MIC) values of these compounds (1–7 and 1a–7a) in liquid culture. Results showed the loss of antibacterial activity of glucoside derivatives 1a, 2a, 3a, and 4a for most pathogens tested, although they retained similar MIC values for selected MRSA, Kocuria rhizophila, and Proteus hauseri strains. However, 5a retained the antibacterial activity similar to its parent molecule against most pathogens tested. Moreover, it showed 2-fold lower MIC values against K. rhizophila and Klebsiella pneumonia. Interestingly, 6a and 7a not only retained the antibacterial potential of their respective parent molecules but also gained antibacterial properties (2- to 4-fold higher than their parent molecules against MRSA and MSSA strains). 6a also exhibited a 4-fold lower MIC value than 6 against P. hauseri. It had a 2-fold lower MIC value against several pathogens, including MSSA, MRSA, Salmonella enterica, and K. rhizophila (Table 1).
Table 1. MIC Values of Compounds against Selected Pathogensa.

ND, not determined based on disc-diffusion assay; sky blue color, 4-fold lower in MIC values; brown color, 2-fold lower; light golden color, activity retained as the parent compound.
The same set of molecules were also used to study their potential application for pinewood nematode control. In vitro antinematodal activity was determined using Bursaphelenchus xylophilus, a pinewood nematode known to infect pine trees, resulting in pine wilt and finally death of the plant.14 Pine wilt epidemics widely occurs in eastern Asian countries such as Japan, Korea, China, and Taiwan, and in North and South American countries. This nematode is also threatening wild ecosystem worldwide.15 Nematode infection can be controlled by applying organophosphorus compounds such as thiacloprid, acetamiprid, fenitrothion, and malathion. However, concerns for the use of such toxic molecules have increased due to their toxicity via food chain and environmental contamination issues (www.fao.org).16 Alternative nematocides including levamisole, thionazin, and avermectin analogs such as emamectin and milbemactin are also used to prevent pine wilt disease including other plant wilt diseases.17 In this context, we assayed plant volatile metabolites with GRAS status having strong odor such as monoterpenes (1–7) and their glucoside derivatives synthesized in this study (1a–7a) against pinewood nematode B. xylophilus through in vitro experiment. Most of these glucoside derivatives of monoterpenes (1, 1a, 3, 3a, 4, 4a, 6, 6a, 7, and 7a) lost their antinematodal properties. However, compounds 2a and 5a exhibited significantly higher antinematodal activities, with lethal concentration 50% (LC50%) values of 1.002 and 0.246 mM, respectively (Table 2). The LC50% concentration of 2a was approximately 2-fold lower than its parent aglycone, whereas the value of 5a was 16-fold lower than compound 5. Previously, it has been reported that selected monoterpenes possess antinematodal activities.18 This preliminary evidence of superior antinematodal activities of 2a and 5a opens up opportunities to develop these safe-to-human molecules as future lead compounds for the control of pine wilt disease.
Table 2. LC50 Values of Compounds against B. xylophilus.
| compounds | LC50 (%) (× 10–3 M) |
|---|---|
| 1 | 0.95005 |
| 1a | 3.83283 |
| 2 | 2.08364 |
| 2a | 1.00205 |
| 3 | 3.80631 |
| 3a | 7.66565 |
| 4 | 1.03849 |
| 4a | 16.00727 |
| 5 | 4.05188 |
| 5a | 0.24653 |
| 6 | 0.49915 |
| 6a | 7.85162 |
| 7 | 0.50567 |
| 7a | 7.90164 |
The mode of action of glycoconjugates synthesized in this study against nematode and bacteria is unknown. There are chances of deglycosylation of these molecules prior to acting on the target. Nevertheless, we also cannot rule out the fact that many natural product glycosides such as doxorubicin, erythromycin, avermectins, and amphotericin B are active only when sugars are conjugated on them. Thus, there are equal chances of acting on target either in aglycon form after deglycosylation or as it is in the glycoside form in in vivo systems. Thus, investigation of the exact behavior of these compounds in in vivo systems could be the subject of the next study.
Although YjiC is well known as a promiscuous GT to glycosylate different classes of natural products, biosynthesis of glycodiversified monoterpenoids is the first report. In nature, a significant number of terpenes exist as glycosides while conjugation of sugar unit is crucial for their activity.19 Very few reports are available on the biosynthesis of monoterpenoid glycosides. Selected monoterpenes glycosides were reported to be produced by plant whole-cell biotransformation.7 Many monoterpene glycosides are chemically synthesized in industrial level. In this context, the enzyme has shown broad applicability in the biotech industries to synthesize human beneficial monoterpene glycosides either in a cell-free system or using engineered microbial cells. Since both enzymatic and microbial synthesis methods are eco-friendly, sustainable, cost-effective, robust, and easy to scale up at the industrial level, this study provides a broad path in the field of monoterpene glycosides synthesis using green approach.
Experimental Part
General Experimental Procedures
A reversed-phase high-performance liquid chromatography–photodiode array (HPLC-PDA) was performed with a C18 column (YMC-Pack ODS-AQ; 4.6 mm internal diameter (I.D.), 250 mm long, 5 μm particle size) connected to a PDA detector (SPD-M20A) (Shimadzu, Japan) for the analysis of 1–5 and their derivatives using the binary condition of H2O (0.01% trifluoroacetic acid buffer) and 100% acetonitrile (ACN) at a flow rate of 1 mL/min for 30 min. The ACN concentrations were 15% (0–12 min), 75% (12–18 min), and 15% (18–30 min) and stopped at 30 min. Compounds 6 and 7 and their derivatives are UV inactive. So, they were analyzed by reversed-phase HPLC connected to a evaporative light scattering detector (ELSD) (ELSD-LT II) (Shimadzu, Japan) under the same binary condition of H2O (0.01% trifluoroacetic acid (TFA) buffer) and acetonitrile (ACN) at a flow rate of 1 mL/min for 30 min. The temperature of the ELSD detector was set at 60 °C with a N2 gas flow pressure with 350 psi. Since 6 and 7 were highly volatile, we were unable to detect them; however, their glucosylated derivatives were detected in ELSD. High-resolution quadruple time-of-flight electrospray ionization mass spectrometry (HRQTOF-ESIMS) was carried out in positive ion mode on an Acquity mass spectrometer (with UPLC; Waters, Milford, MA, USA) coupled with a Synapt G2-S system (Waters).
The compounds were extracted using a double volume of ethyl acetate (2:1, v/v) using a Soxhlet extractor. The Soxhlet extractor was kept still to separate the mixture for 4–6 h in room temperature after shaking. The organic fraction of the extract was evaporated using a rotary evaporator. The extract was subjected to preparative-HPLC (prep-HPLC) for purification. Each glucosylated compounds were purified using prep-HPLC (Shimadzu, Tokyo, Japan) with a C18 column (YMC–Pack ODS-AQ (250 × 20 mm I.D., 10 μm) connected to a UV detector under the binary condition of H2O (0.05% TFA buffer) and 100% ACN at a flow rate of 10 mL/min for 35 min. The ACN concentrations were 20% (0–5 min), 50% (5–10 min), 70% (10–15% min), 90% (15–25 min), 50% (25–30 min), and 20% (30–35 min). To purify the glucosylated derivatives of 6 and 7, the eluted solvent was collected in a separate tube for every 1 min interval. Each fraction was analyzed by HPLC-ELSD, and the pure fraction was concentrated for further use.
The pure fraction of glucosylated compounds was first dried using a rotary evaporator and then lyophilized (HyperVAC-Max, HyperCOOL). The dried sample was taken and dissolved in DMSO-d6 (Sigma-Aldrich) and further analyzed with a 300 MHz nuclear magnetic resonance (NMR) spectrometer. One-dimensional NMR (1H NMR and 13C NMR) was performed to confirm the structures of the glycosylated monoterpenes. All the raw data were processed using TopSpin 3.1 software (Bruker) and further analyzed by using MestReNova 8.0 software (Mestrelab Research S.L., Spain).
The standard curve of 6a and 7a was prepared to determine the conversion percentage in in vitro and in vivo reactions. A standard calibration curve was created using the integrated HPLC-ELSD peak area of both compounds at different concentrations (312.5, 156.25, 78.125, 39.66, and 19.5 μg/mL).
In Vitro Glucosylation Assay
The reaction was carried out in a total volume of 450 μL by mixing the purified YjiC enzyme (20 μg/mL) with 200 mM Tris-HCl (pH 8.0) containing 10 mM MgCl2, 2 mM monoterpenes (1–7), and 2 mM UDP-α-d-glucose as a sugar donor. The reaction mixture was then incubated at 37 °C for 3 h. Three parallel assays were routinely carried out. A double volume of chilled methanol was used for quenching the reaction. Aliquots was removed by centrifugation at 13,475g for 20 min and subjected to HPLC-PDA or HPLC-ELSD.
Use of Co-Solvent in the Assay
To study the effect of co-solvent in product formation, the reaction was carried out using DMSO as a co-solvent. The reaction was performed in a total volume of 200 μL by mixing YjiC enzyme (20 μg/mL) with 200 mM Tris-HCl (pH 8.0), 10 mM MgCl2, 2 mM 2, and 2 mM UDP-Glc as a sugar donor. Six sets of reactions were carried out using 0, 5, 10, 15, 20, and 25% of the final concentration of DMSO in the reaction mixture. Reactions were incubated at 37 °C for 3 h. After 3 h, reactions were quenched with a double volume of chilled methanol. Aliquots were removed by centrifugation and analyzed by HPLC-PDA.
Stability of Glycosyltransferase under Reaction Conditions
To study the stability of glycosyltransferase under reaction conditions, reactions were carried out in four different tubes under aforementioned in vitro reaction conditions. All the reaction ingredients were added in the microcentrifuge tubes except the donor and acceptor substrates. All the tubes containing reaction ingredients were incubated at a 37 °C water bath. Substrates 2 and UDP-Glc were added in the tubes and activated the reaction mixture at 0, 2, 5, and 10 h. After activation of the reaction mixture, 50 μL of the sample was taken from the reaction mixture at different time intervals (2, 15, 30, 60, 90, 120, 150, and 180 min) and quenched by 450 μL of chilled methanol. The aliquots were centrifuged, and each sample was analyzed by using HPLC-PDA.
Biosynthesis of Diverse Monoterpene Glycosides
To synthesize diverse sugar-conjugated monoterpenes, reactions were carried out with six different nucleotide diphosphate sugars including d and l sugars. For the NDP-d-sugars, four different types of sugars were used. In correlation to glucose, C-2 position-modified sugars (UDP-α-d-N-acetyl galactosamine and UDP-α-d-N-acetyl glucosamine), a C-4 position-modified (UDP-α-d-galactose), and a C-6 position-modified sugar (UDP-α-d-glucuronic acid) were used. Similarly, two NDP-l-sugars, TDP-l-rhamnose and GDP-l-fucose, were also used for the in vitro reactions. The reaction mixture contents were added in an identical concentration to aforementioned methods. Each substrate (1 to 7) was reacted with all six different NDP-sugars. Altogether, 42 different reactions were carried out and incubated at 37 °C for 3 h. Three parallel assays were routinely carried out. The reactions were quenched with a double volume of chilled methanol. The aliquots after centrifugation was analyzed by either HPLC-PDA or HPLC-ELSD. All the in vitro reaction samples were further analyzed by HRQTOF-ESI-MS analysis.
One-Pot Transglycosylation Reaction
The one-pot transglycosylation reaction was carried out using various commercially available synthetic p-nitrophenyl glycosides such as p-nitrophenyl-β-d-galactoside, p-nitrophenyl-β-d-xyloside, and p-nitrophenyl-α-d-galactoside in the presence of five different nucleotide diphosphates (NDPs) such as UDP, ADP, CDP, GDP, and TDP. The reaction mixture containing 4 mM p-nitrophenyl glycosides, 4 mM NDP, 2 mM 1 (as an acceptor substrate), 10 mM MgCl2, 200 mM Tris-HCl buffer (pH 8.0), and 20 μg/mL pure YjiC in a total volume of 300 μL was incubated at 37 °C for 13 h. The sample was collected at different time intervals (15, 30, 60, 180, and 780 min). Each sample was analyzed by HPLC-PDA. Three parallel assays were routinely carried out.
Whole-Cell Bioconversion of Monoterpenes
E. coli BL21 (DE3) harboring pET28a-YjiC was prepared in a 5 mL culture volume in LB broth containing kanamycin antibiotic for seed culture. The culture was incubated at 37 °C and kept for overnight growth. The following day, 100 μL of pre-inoculum was transferred to the 50 mL LB broth medium containing kanamycin antibiotic. This was kept at 37 °C until the OD600nm reached 0.6. Then, the culture was induced by 0.5 mM isopropyl β-d-thiogalactoside (IPTG) and kept at 20 °C for 12 h. Standard monoterpenes prepared in dimethyl sulfoxide (DMSO) was added to a final concentration of 0.5 mM. Additionally, 2% sterile glucose was supplemented to each flask and again kept at 20 °C for 60 h. The sample was taken every 12 h and analyzed by HPLC-PDA or HPLC-ELSD. The same set of additional experiments were carried out in which, instead of 2% glucose, 5% glucose was supplemented while feeding monoterpenes and every 12 h. The conversion profile of each substrate was monitored every 12 h interval. Three parallel assays were routinely carried out. The conversion percentage of each substrate was calculated using the integrated peak area of the substrate and product peak.
Eugenol β-d-Glucoside (1a)
White amorphous powder; HR-ESI-MS m/z 349.1270 [M + Na]+ (calcd for C16H22NaO7, 349.1263 Da); λmax: 276 nm; 1H NMR (300 MHz, DMSO-d6): δH 7.00 (d, J = 8.3 Hz, 1H), 6.79 (d, J = 2.0 Hz, 1H), 6.67 (dd, J = 8.2, 2.0 Hz, 1H), 5.94 (ddt, J = 16.8, 10.0, 6.7 Hz, 1H), 5.12–4.98 (m, 2H), 4.84 (d, J = 7.3 Hz, 1H), 3.74 (s, 3H), 3.66 (dd, J = 11.8, 1.9 Hz, 1H), 3.44 (dd, J = 11.8, 5.5 Hz, 1H), 3.34–3.19 (m, 5H), 3.19–3.11 (m, 1H). 13C NMR (75 MHz, DMSO-d6): δ 149.40, 145.38, 138.38, 133.97, 120.81, 116.09, 116.01, 113.50, 100.80, 77.47, 77.34, 73.74, 70.21, 61.19, 56.17, 39.54.
Thymol β-d-Glucoside (2a)
White amorphous powder; HR-ESI-MS m/z 335.1464 [M + Na]+ (calcd for C16H24NaO6, 335.1471 Da); λmax: 276 nm; 1H NMR (300 MHz, DMSO-d6): δH 7.05 (d, J = 7.7 Hz, 1H), 6.89 (d, J = 1.6 Hz, 1H), 6.76 (dd, J = 8.3, 1.4 Hz, 1H), 4.75 (d, J = 7.6 Hz, 1H), 3.70 (dd, J = 11.8, 2.0 Hz, 1H), 3.46 (dd, J = 11.8, 5.8 Hz, 1H), 3.38–3.23 (m, 3H), 3.21–3.10 (m, 1H), 2.23 (s, 3H), 1.13 (t, J = 6.5 Hz, 6H). 13C NMR (75 MHz, DMSO-d6): δ 155.11, 136.16, 134.62, 125.91, 122.92, 116.34, 101.77, 77.51, 77.32, 73.94, 70.34, 61.27, 26.12, 23.49, 23.22, 21.43.
Isoeugenol β-d-Glucoside (3a)
White amorphous powder; HR-ESI-MS m/z 349.1264 [M + Na]+ (calcd for C16H22NaO7, 349.1263 Da); λmax: 259 nm; 1H NMR (300 MHz, DMSO-d6): δH 7.04–6.97 (m, 1H), 6.98 (s, 1H), 6.83 (dd, J = 8.5, 2.0 Hz, 1H), 6.33 (dd, J = 15.8, 1.6 Hz, 1H), 6.18 (dq, J = 15.8, 6.3 Hz, 1H), 4.87 (d, J = 7.3 Hz, 1H), 3.77 (s, 3H), 3.66 (dd, J = 11.8, 2.0), 3.44 (dd, J = 11.8, 5.5 Hz, 1H), 3.35–3.17 (m, 3H), 3.21–3.09 (m, 1H), 1.82 (dd, J = 6.4, 1.4 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 149.52, 146.19, 132.12, 131.02, 124.14, 118.92, 115.87, 110.14, 100.61, 77.50, 77.33, 73.70, 70.17, 61.17, 56.14, 18.63.
Carvacrol β-d-Glucoside (4a)
White amorphous powder; HR-ESI-MS m/z 335.1471 [M + Na]+ (calcd for C16H24NaO6, 335.1471 Da); λmax: 270 nm; 1H NMR (300 MHz, DMSO-d6): δH 7.02 (dd, J = 7.6, 0.8 Hz, 1H), 6.94 (d, J = 1.7 Hz, 1H), 6.75 (dd, J = 7.7, 1.7 Hz, 1H), 4.77 (d, J = 7.7 Hz, 1H), 3.69 (dd, J = 11.7, 2.0 Hz, 1H), 3.46 (dd, J = 11.7, 5.9 Hz, 1H), 3.39–3.21 (m, 3H), 3.21–3.09 (m, 1H), 2.80 (p, J = 6.9 Hz, 1H), 2.15 (d, J = 0.7 Hz, 3H), 1.18 (s, 3H), 1.15 (s, 3H). 13C NMR (75 MHz, DMSO-d6): δ 156.06, 147.73, 130.47, 124.45, 119.84, 113.49, 101.55, 77.55, 77.27, 73.85, 70.40, 61.27, 33.74, 24.42, 24.32, 16.14.
α-Terpineol β-d-Glucoside (5a)
Powder; HR-ESI-MS m/z 339.1783 [M + Na]+ (calcd for C16H28NaO6, 349.1784 Da); λmax: 200 nm; 1H NMR (300 MHz, DMSO-d6): δH 5.32 (d, J = 5.1 Hz, 1H), 4.28 (d, J = 7.7 Hz, 1H), 3.61 (dt, J = 11.6, 1.7 Hz, 1H), 3.42–3.31 (m, 1H), 3.22–2.95 (m, 3H), 2.88 (dd, J = 8.8, 7.7 Hz, 1H), 2.15–1.63 (m, 5H), 1.60 (d, J = 3.1 Hz, 4H), 1.51 (d, J = 12.1 Hz, 1H), 1.10 (d, J = 8.5 Hz, 6H). 13C NMR (75 MHz, DMSO-d6): δ 133.69, 121.40, 97.49, 77.49, 76.90, 74.11, 70.79, 70.76, 61.75, 43.58, 31.05, 26.88, 25.10, 24.14, 23.73, 22.94.
(+)-Menthol β-d-Glucoside (6a)
Powder; HR-ESI-MS m/z 341.1938 [M + Na]+ (calcd for C16H30NaO6, 341.1940 Da); 1H NMR (300 MHz, DMSO-d6): δH 4.14 (d, J = 7.7 Hz, 1H), 3.64 (dd, J = 11.7, 1.7 Hz, 1H), 3.42 (dd, J = 11.6, 5.1 Hz, 1H), 3.26 (td, J = 10.5, 4.2 Hz, 1H), 3.19–2.95 (m, 1H), 2.91 (t, J = 8.1 Hz, 1H), 2.42 (td, J = 7.1, 2.3 Hz, 1H), 2.19 (d, J = 12.6 Hz, 1H), 1.57 (t, J = 12.0 Hz, 1H), 1.34 (s, 1H), 1.14 (t, J = 11.2 Hz, 1H), 0.92 (d, J = 12.1 Hz, 2H), 0.84 (dd, J = 6.7, 5.3 Hz, 6H), 0.77 (s, 1H), 0.72 (d, J = 6.9 Hz, 3H). 13C NMR (75 MHz, DMSO-d6): δ 104.86, 80.38, 77.22, 77.06, 74.24, 70.50, 61.57, 48.92, 43.88, 34.51, 31.61, 24.43, 23.01, 22.78, 21.68, 16.51.
(−)-Borneol β-d-Glucoside (7a)
White amorphous powder; HR-ESI-MS m/z 339.1770 [M + Na]+ (calcd for C16H30NaO6, 339.1784 Da); 1H NMR (300 MHz, DMSO-d6): δH 4.07 (d, J = 7.8 Hz, 1H), 4.01–3.90 (m, 1H), 3.64 (d, J = 11.4 Hz, 1H), 3.53–3.34 (m, 2H), 3.19–2.99 (m, 3H), 2.91 (t, J = 8.2 Hz, 1H), 2.05 (tdd, J = 12.8, 6.6, 3.0 Hz, 2H), 1.75–1.50 (m, 2H), 1.13 (ddt, J = 31.9, 13.0, 4.0 Hz, 3H), 0.81 (d, J = 1.7 Hz, 9H). 13C NMR (75 MHz, DMSO-d6): δ 102.38, 82.85, 77.24, 77.20, 73.94, 70.53, 61.57, 49.09, 48.11, 44.70, 36.02, 28.33, 26.80, 20.21, 19.25, 14.06.
Acknowledgments
This work was supported by a grant from the Next-Generation BioGreen 21 Program (SSAC, grant# PJ013137), Rural Development Administration, Republic of Korea.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00535.
Experimental procedures, chromatographic (HPLC-PDA, HPLC-ELSD), spectrometric (ESI/MS), and spectroscopic (UV-VIS, NMR) analyses of reaction products, and results of biological assays (PDF)
Author Contributions
∥ P.B. and R.P.P. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- a Chouhan S.; Sharma K.; Guleria S. Antimicrobial activity of some essential oils-present status and future perspectives. Medicines 2017, 4, 58. 10.3390/medicines4030058. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ludwiczuk A.; Skalicka-Woźniak K.; Georgiev M. I.. Terpenoids. In Pharmacognosy Fundamentals, Applications and Strategies; Badal S., Delgoda R., Eds.; Elsevier: Amsterdam, 2017; 233–266. [Google Scholar]
- a Bohlmann J.; Keeling C. I. Terpenoid biomaterials. Plant J. 2008, 54, 656–669. 10.1111/j.1365-313X.2008.03449.x. [DOI] [PubMed] [Google Scholar]; b Caputi L.; Aprea E. Use of terpenoids as natural flavouring compounds in food industry. Recent. Pat. Food, Nutr. Agric. 2011, 3, 9–16. 10.2174/2212798411103010009. [DOI] [PubMed] [Google Scholar]; c Abbas F.; Ke Y.; Yu R.; Yue Y.; Amanullah S.; Jahangir M. M.; Fan Y. Volatile terpenoids: multiple functions, biosynthesis, modulation and manipulation by genetic engineering. Planta 2017, 246, 803–816. 10.1007/s00425-017-2749-x. [DOI] [PubMed] [Google Scholar]
- Tassou C. C.; Nychas G.-J. E.; Skandamis P. N.. Herbs and spices and antimicrobials. In Handbook of Herbs and Spices; Peter K. V. Ed.; CRC Press: Washington, DC, 2004; 2, 22–40. [Google Scholar]
- a Krishnaiah Y. S. R.; Bhaskar P.; Satyanarayana V. Penetration-enhancing effect of ethanol-water solvent system and ethanolic solution of carvone on transdermal permeability of nimodipine from HPMC gel across rat abdominal skin. Pharm. Dev. Technol. 2004, 9, 63–74. 10.1081/PDT-120027419. [DOI] [PubMed] [Google Scholar]; b Kararli T. T.; Kirchhoff C. F.; Penzotti S. C. Jr. Enhancement of transdermal transport of azidothymidine (AZT) with novel terpene and terpene-like enhancers: In vivo-in vitro correlations. J. Controlled Release 1995, 34, 43–51. 10.1016/0168-3659(94)00128-H. [DOI] [Google Scholar]; c Krishnaiah Y. S. R.; Satyanarayana V.; Bhaskar P. Enhanced percutaneous permeability of nicardipine hydrochloride by carvone across the rat abdominal skin. Drug Dev. Ind. Pharm. 2003, 29, 191–202. 10.1081/DDC-120016727. [DOI] [PubMed] [Google Scholar]
- Martins M. A. R.; Silva L. P.; Ferreira O.; Schröder B.; Coutinho J. A. P.; Pinho S. P. Terpenes solubility in water and their environmental distribution. J. Mol. Liq. 2017, 241, 996–1002. 10.1016/j.molliq.2017.06.099. [DOI] [Google Scholar]
- a Weymouth-Wilson A. C. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 1997, 14, 99–110. 10.1039/np9971400099. [DOI] [PubMed] [Google Scholar]; b Méndez C.; Salas J. A. Altering the glycosylation pattern of bioactive compounds. Trends Biotechnol. 2001, 19, 449–456. 10.1016/S0167-7799(01)01765-6. [DOI] [PubMed] [Google Scholar]; c Griffith B. R.; Langenhan J. M.; Thorson J. S. ’Sweetening’ natural products via glycorandomization. Curr. Opin. Biotechnol. 2005, 16, 622–630. 10.1016/j.copbio.2005.10.002. [DOI] [PubMed] [Google Scholar]
- a Orihara Y.; Furuya T.; Hashimoto N.; Deguchi Y.; Tokoro K.; Kanisawa T. Biotransformation of isoeugenol and eugenol by cultured cells of Eucalyptus perriniana. Phytochemistry 1992, 31, 827–831. 10.1016/0031-9422(92)80022-7. [DOI] [PubMed] [Google Scholar]; b Nakagawa H.; Dobashi Y.; Sato T.; Yoshida K.; Tsugane T.; Shimura S.; Kirimura K.; Kino K.; Usami S. α-Anomer-selective glucosylation of menthol with high yield through a crystal accumulation reaction using lyophilized cells of Xanthomonas campestris WU-9701. J. Biosci. Bioeng. 2000, 89, 138–144. 10.1016/S1389-1723(00)88727-7. [DOI] [PubMed] [Google Scholar]; c Sato T.; Takeuchi H.; Takahashi K.; Kurosu J.; Yoshida K.; Tsugane T.; Shimura S.; Kino K.; Kirimura K. Selective alpha-glucosylation of eugenol by alpha-glucosyl transfer enzyme of Xanthomonas campestris WU-9701. J. Biosci. Bioeng. 2003, 96, 199–202. 10.1016/S1389-1723(03)90127-7. [DOI] [PubMed] [Google Scholar]; d Shimoda K.; Kondo Y.; Nishida T.; Hamada H.; Nakajima N.; Hamada H. Biotransformation of thymol, carvacrol, and eugenol by cultured cells of Eucalyptus perriniana. Phytochemistry 2006, 67, 2256–2261. 10.1016/j.phytochem.2006.07.006. [DOI] [PubMed] [Google Scholar]
- Higashiyama T.; Sakata I.. Menthol glycoside, process for preparing the same and method for releasing menthol therefrom. U. S. Patent 4038270A, 1977.
- a Pandey R. P.; Parajuli P.; Koirala N.; Park J. W.; Sohng J. K. Probing 3-hydroxyflavone for in vitro glycorandomization of flavonols by YjiC. Appl. Environ. Microbiol. 2013, 79, 6833. 10.1128/AEM.02057-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Pandey R. P.; Gurung R. B.; Parajuli P.; Koirala N.; Tuoi L. T.; Sohng J. K. Assessing acceptor substrate promiscuity of YjiC-mediated glycosylation toward flavonoids. Carbohydr. Res. 2014, 393, 26. 10.1016/j.carres.2014.03.011. [DOI] [PubMed] [Google Scholar]; c Parajuli P.; Pandey R. P.; Koirala N.; Yoon Y. J.; Kim B. G.; Sohng J. K. Enzymatic synthesis of epothilone A glycosides. AMB Express. 2014, 4, 31. 10.1186/s13568-014-0031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Pandey R. P.; Li T. F.; Kim E. H.; Yamaguchi T.; Park Y. I.; Kim J. S.; Sohng J. K. Enzymatic synthesis of novel phloretin glucosides. Appl. Environ. Microbiol. 2013, 79, 3516. 10.1128/AEM.00409-13. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Pandey R. P.; Parajuli P.; Koirala N.; Lee J. H.; Park Y. I.; Sohng J. K. Glucosylation of isoflavonoids in engineered Escherichia coli. Mol. Cells 2014, 37, 172. 10.14348/molcells.2014.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Pandey R. P.; Parajuli P.; Shin J. Y.; Lee J.; Lee S.; Hong Y. S.; Park Y. I.; Kim J. S.; Sohng J. K. Enzymatic biosynthesis of novel resveratrol glucoside and glycoside derivatives. Appl. Environ. Microbiol. 2014, 80, 7235. 10.1128/AEM.02076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Pandey R. P.; Chu L. L.; Kim T. S.; Sohng J. K. Bioconversion of tetracycline antibiotics to novel glucoside derivatives by single-vessel multienzymatic glycosylation. J. Microbiol. Biotechnol. 2018, 28, 298. 10.4014/jmb.1709.09072. [DOI] [PubMed] [Google Scholar]; h Nguyen T. T. H.; Pandey R. P.; Parajuli P.; Han J. M.; Jung H. J.; Park Y. I.; Sohng J. K. Microbial synthesis of non-natural anthraquinone glucosides displaying superior antiproliferative properties. Molecules 2018, 23, 2171. 10.3390/molecules23092171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Lairson L. L.; Henrissat B.; Davies G. J.; Withers S. G. Glycosyltransferases: structures, functions, and mechanisms. Annu. Rev. Biochem. 2008, 77, 521. 10.1146/annurev.biochem.76.061005.092322. [DOI] [PubMed] [Google Scholar]; b Liang D. M.; Liu J. H.; Wu H.; Wang B. B.; Zhu H. J.; Qiao J. J. Glycosyltransferases: mechanisms and applications in natural product development. Chem. Soc. Rev. 2015, 44, 8350. 10.1039/C5CS00600G. [DOI] [PubMed] [Google Scholar]; c Gloster T. M. Advances in understanding glycosyltransferases from a structural perspective. Curr. Opin. Struct. Biol. 2014, 28, 131. 10.1016/j.sbi.2014.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zhang C.; Griffith B. R.; Fu Q.; Albermann C.; Fu X.; Lee I. K.; Li L.; Thorson J. S. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 2006, 313, 1291. 10.1126/science.1130028. [DOI] [PubMed] [Google Scholar]; b Modolo L. V.; Li L.; Pan H.; Blount J. W.; Dixon R. A.; Wang X. Crystal structures of glycosyltransferase UGT78G1 reveal the molecular basis for glycosylation and deglycosylation of (iso)flavonoids. J. Mol. Biol. 2009, 392, 1292. 10.1016/j.jmb.2009.08.017. [DOI] [PubMed] [Google Scholar]
- a Thibodeaux C. J.; Melançon C. E. III; Liu H. W. L. Natural-product sugar biosynthesis and enzymatic glycodiversification. Angew. Chem. Int. Ed. Engl. 2008, 47, 9814. 10.1002/anie.200801204. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Härle J.; Bechthold A.. Chapter 12. The power of glycosyltransferases to generate bioactive natural compounds. In Methods in Enzymology; Hopwood D. A. Ed.; Elsevier: Amsterdam, 2009; 458, 309–333. [DOI] [PubMed] [Google Scholar]; c Pandey R. P. Diversifying natural products with promiscuous glycosyltransferase enzymes via a sustainable microbial fermentation approach. Front. Chem. 2017, 5, 110. 10.3389/fchem.2017.00110. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Gantt R. W.; Peltier-Pain P.; Singh S.; Zhou M.; Thorson J. S. Broadening the scope of glycosyltransferase-catalyzed sugar nucleotide synthesis. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 7648–7653. 10.1073/pnas.1220220110. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Erb A.; Weiss H.; Härle J.; Bechthold A. A bacterial glycosyltransferase gene toolbox: generation and applications. Phytochemistry 2009, 70, 1812–1821. 10.1016/j.phytochem.2009.05.019. [DOI] [PubMed] [Google Scholar]
- a Zhou W.; Bi H.; Zhuang Y.; He Q.; Yin H.; Liu T.; Ma Y. Production of cinnamyl alcohol glucoside from glucose in Escherichia coli. J. Agric. Food Chem. 2017, 65, 2129–2135. 10.1021/acs.jafc.7b00076. [DOI] [PubMed] [Google Scholar]; b Lim C. G.; Wong L.; Bhan N.; Dvora H.; Xu P.; Venkiteswaran S.; Koffas M. A. G. Development of a recombinant Escherichia coli strain for overproduction of the plant pigment anthocyanin. Appl. Environ. Microbiol. 2015, 81, 6276–6284. 10.1128/AEM.01448-15. [DOI] [PMC free article] [PubMed] [Google Scholar]; c He Q.; Yin H.; Jiang J.; Bai Y.; Chen N.; Liu S.; Zhuang Y.; Liu T. Fermentative production of phenolic glucosides by escherichia coli with an engineered glucosyltransferase from Rhodiola sachalinensis. J. Agric. Food Chem. 2017, 65, 4691–4697. 10.1021/acs.jafc.7b00981. [DOI] [PubMed] [Google Scholar]; d Parajuli P.; Pandey R. P.; Trang N. T. H.; Chaudhary A. K.; Sohng J. K. Synthetic sugar cassettes for the efficient production of flavonol glycosides in Escherichia coli. Microb. Cell Fact. 2015, 14, 76. 10.1186/s12934-015-0261-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tóth A. Bursaphelenchus xylophilus, the pinewood nematode: its significance and a historical review. Acta Biol. Szeged. 2011, 55, 213. [Google Scholar]; b Futai K. Pine wood nematode, Bursaphelenchus xylophilus. Annu. Rev. Phytopathol. 2013, 51, 61. 10.1146/annurev-phyto-081211-172910. [DOI] [PubMed] [Google Scholar]
- Rodrigues J. M.National eradication programme for the pinewood nematode. In Pine Wilt Disease: A Worldwide Threat to Forest Ecosystems; Mota M. M.; Vieira P. R. Eds.; Springer Verlag GmbH: Heidelberg, 2008, 5–14. [Google Scholar]
- a Eddleston M.; Buckley N. A.; Eyer P.; Dawson A. H. Management of acute organophosphorus pesticide poisoning. Lancet. 2008, 371, 597. 10.1016/S0140-6736(07)61202-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Priyadharshini U. K.; Latha R.; Kavitha U.; Nirmala N. Effects of organophosphorus pesticides on cardiorespiratory parameters among the farmers. J. Clin. Diagn. Res. 2017, 11, CC01–CC04. 10.7860/JCDR/2017/26724.10590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marasigan K.; Toews M.; Kemerait R. Jr.; Abney M. R.; Culbreath A.; Srinivasan R. Evaluation of alternatives to carbamate and organophosphate insecticides against thrips and tomato spotted wilt virus in peanut production. J. Econ. Entomol. 2016, 109, 544. 10.1093/jee/tov336. [DOI] [PubMed] [Google Scholar]
- a Zhou L.; Wang J.; Wang K.; Xu J.; Zhao J.; Shan T.; Luo C.. Secondary metabolites with antinematodal activity from higher plants. In Studies in Natural Products Chemistry; Rahman A. Ed. Elsevier: Oxford, UK, 2012, 37, 67–114. [Google Scholar]; b Barbosa P.; Lima A. S.; Vieira P.; Dias L. S.; Tinoco M. T.; Barroso J. G.; Pedro L. G.; Figueiredo A. C.; Mota M. Nematicidal activity of essential oils and volatiles derived from Portuguese aromatic flora against the pinewood nematode, Bursaphelenchus xylophilus. J. Nematol. 2010, 42, 8–16. [PMC free article] [PubMed] [Google Scholar]; c Li H. Q.; Liu Q. Z.; Liu Z. L.; Du S. S.; Deng Z. W. Chemical Composition and nematicidal activity of essential oil of Agastache rugosa against Meloidogyne incognita. Molecules 2013, 18, 4170–4180. 10.3390/molecules18044170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivas F.; Parra A.; Martinez A.; Garcia-Granados A. Enzymatic glycosylation of terpenoids. Phytochem. Rev. 2013, 12, 327. 10.1007/s11101-013-9301-9. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.