

Abstract
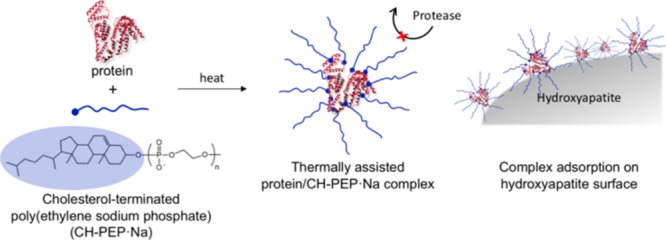
Protein therapeutics has recently attracted interest in various medical treatments. However, the structure and function preservation in proteins under physiological conditions is still an important issue and reliable immobilization techniques are required. In this study, the thermally assisted complexation of proteins with amphiphilic polyphosphoesters is proposed as a new methodology for their durability improvement. Amphiphilic cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na) was synthesized via the organocatalytic ring-opening polymerization of 2-methoxy-2-oxo-1,3,2-dioxaphospholane initiated by cholesterol as the hydrophobic molecule and followed by demethylation and neutralization. For the protein nanocarrier preparation, a complex of the amphiphilic CH-PEP·Na with bovine serum albumin (BSA) was formed through the hydrophobic interactions between the lipophilic moieties of the protein and the cholesteryl groups of the CH-PEP·Na chains, which were induced by thermal treatment at 90 °C. The resulting complex size ranged between 27 and 51 nm, as confirmed by dynamic light scattering. The complexes dispersed in an aqueous medium exhibited a high stability in size for up to 1 month of storage. CH-PEP·Na efficiently inhibited the thermal aggregation and sedimentation of BSA, unlike poly(ethylene sodium phosphate) (PEP·Na) and cholesterol-terminated poly(ethylene glycol) (CH-PEG). In addition, CH-PEP·Na was able to protect the complexed BSA against proteolytic digestion and the BSA–CH-PEP·Na complexes well adsorbed onto hydroxyapatite even in the presence of BSA (5.5 g/dL). Hence, thermally induced protein–CH-PEP·Na complexes can be a potential tool for the development of bone and dental applications.
Introduction
The demand for the medical use and commercialization of therapeutic proteins is forecasted to grow dramatically over the next few years. The specificity and bioactivity of such proteins have had a positive impact on the treatments for autoimmune diseases, cancers, and other human disorders,1−5 but they lack in proteolytic stability and have a short blood circulation time.6−8 To enhance the pharmacokinetics of the delivered proteins, several chemical modification techniques, such as the poly(ethylene glycol) (PEG) immobilization, called PEGylation,9,10 have been developed. However, despite the improved intravascular half-life of the proteins, the PEG non-biodegradability and decreased bioactivity because of a permanent conjugation are a serious concern.7,11 As an alternative, Wurm and co-workers have proposed degradable protein–polyphosphoester (PPE) conjugates with a relatively high bioactivity retention to address such PEG shortcomings.12,13 Nonetheless, protein–polymer conjugations require multistep preparations and purification. Noncovalent binding is an interesting alternative formation route involving electrostatic forces, hydrogen bondings, and hydrophobic attractions. The thermally assisted complexation of proteins with amphiphilic polymers is a facile and timesaving approach to create protein carriers, but its challenge lies in suppressing the protein aggregation and preventing the protein bioactivity from thermal denaturation. Akiyoshi et al. have designed a cholesterol-bearing pullulan (CHP) nanogel having a chaperone-like activity for the thermal stabilization of carbonic anhydrase B:14 the self-assembly of the cholesteryl groups provided physical cross-linking to form stable CHP nanogels in water. Hence, the use of amphiphilic CHP for the thermostabilization of proteins can be a significant strategy.
On the other hand, PPEs are promising polymers in the biomedical field because of their versatility, biocompatibility, enzymatic degradability, and biomimetic building blocks of natural molecules, that is, nucleic acids.15−17 In contrast to biodegradable polycarboxylic acid esters such as poly(lactic acid) and poly(ε-caprolactone), diverse functional groups can be introduced to PPEs.18,19 Poly(ethylene sodium phosphate) (PEP·Na) is a type of water-soluble PPE possessing a phosphodiester backbone and negative charges. It has exhibited strong bone affinity in both in vitro and in vivo and high cell viability against bone cells.20−22 PEP·Na can be simply synthesized via the ring-opening polymerization (ROP) of cyclic phosphoester monomers by using organocatalysts and alcohols as initiators,21−26 whereas its polarity can be conveniently adjusted to be hydrophilic or amphiphilic by using suitable hydroxyl-containing initiators. Cholesterol, which is a ubiquitous lipid molecule present in body systems, was used as an initiator to prepare amphiphilic cholesterol-terminated PEP·Na (CH-PEP·Na). In this study, amphiphilic CH-PEP·Na was newly introduced to form complexes with proteins assisted by thermal treatment. Once the temperature rose, the interior hydrophobic amino acids of the proteins became exposed. The hydrophobic interactions between the nonpolar segments of the amphiphilic polymer and the exposed lipophilic proteins spontaneously occurred, whereas the hydrophilic PEP·Na chains stabilized such self-assembled complexes at the periphery because of the electrostatic repulsive force of the phosphate anions, whereas the polymers also hampered the aggregation of the unfolded proteins.27 The bovine serum albumin (BSA) formed a complex with CH-PEP·Na without the need for protein modification and organic solvents. After the thermally assisted complexation, CH-PEP·Na exhibited a great potential to form complexes with BSA at high temperature without BSA thermal aggregation and the peripheral CH-PEP·Na protected the complexed BSA well under protease conditions. In addition, the obtained complexes showed affinity for hydroxyapatite (HAp) by adsorbing onto its surface.
Results and Discussion
Preparation and Characterization of PPEs
Three types of PPEs, cholesterol-terminated poly(2-methoxy-2-oxo-1,3,2-dioxaphospholane) (CH-PMP), CH-PEP·Na, and PEP·Na, were successfully synthesized via the ROP of 2-methoxy-2-oxo-1,3,2-dioxaphospholane (MP), as shown in Scheme 1. These polymers were examined for a targeted degree of polymerization (DP) of 70. Their molecular weights and polydispersity indexes (PDIs) were evaluated by gel permeation chromatography (GPC), as summarized in Table 1 (GPC traces of the PPEs are displayed in Figure S1). The proton nuclear magnetic resonance (1H NMR) spectra (Figure 1) revealed the characteristic peaks of CH-PMP at 4.3, 3.8, and 0.7–1.0 ppm, which correlate respectively to the methylene and methyl protons of the PMP chain and the methyl protons of the cholesteryl group. After demethylation and cation exchange, the triester PMP was transformed into diester PEP·Na, demonstrating the upfield shift (3.9 ppm) of the methylene protons from the polymeric backbone and the disappearance of the methoxy signal. However, slight methoxyl groups still remained. The percentage of demethylation calculated from an NMR spectrum is reported in Table 1.
Scheme 1. Synthetic Route of Polyphosphoesters.

Table 1. Molecular Characteristics of Polyphosphoesters.
| polymer | Mn,NMRa (kDa) | Mn,GPCb (kDa) | DPa | PDIb | demethylationa (%) | cmc (g/L) |
|---|---|---|---|---|---|---|
| CH-PEP·Na | 11.1 | 9.5 | 73 | 1.48 | 98 | 0.04 |
| PEP·Na | 8.1 | 1.25 | 97 | |||
| CH-PMP | 12.2 | 8.8 | 86 | 1.46 | 0.01 |
Estimated by proton nuclear magnetic resonance.
Determined by gel permeation chromatography.
Figure 1.
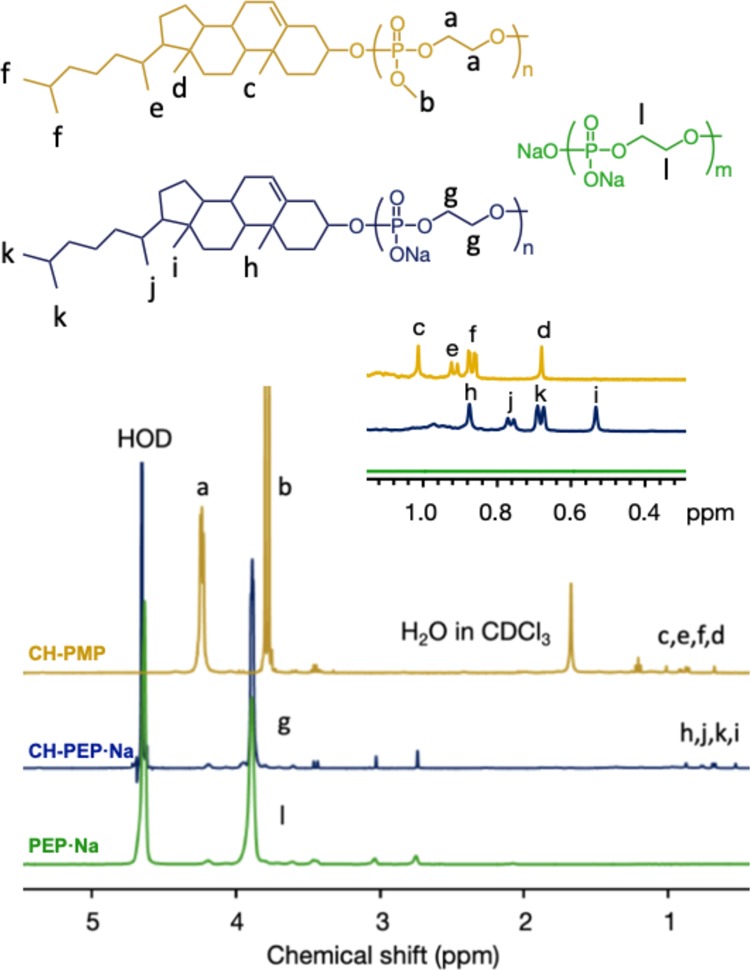
Proton nuclear magnetic resonance spectra of cholesterol-terminated poly(2-methoxy-2-oxo-1,3,2-dioxaphosphoranedioxaphospholane) (CH-PMP), cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na), and poly(ethylene sodium phosphate) (PEP·Na).
To suppress the protein aggregation under thermal conditions, the self-assembly of amphiphilic polymers is necessary to stabilize the proteins. Hence, the critical micelle concentration (cmc) of the amphiphilic cholesterol-terminated polymers was determined using a pyrene fluorescence probe in an aqueous medium. The cmc’s of CH-PEP·Na and CH-PMP were 0.04 and 0.01 g/L, respectively, and the higher value of CH-PEP·Na was probably due to the higher hydrophilicity of the anionic PEP·Na compared with the nonionic PMP and to its anionic repulsion.28
Formation and Characterization of BSA–PPE Complexes
At 90 °C, BSA was easily aggregated because of thermal denaturation, showing white turbidity (Figure 2A). In contrast, the BSA suspensions exhibited transparency in the presence of amphiphilic CH-PEP·Na. The complex suspensions became more transparent with increases in the CH-PEP·Na content, reaching a transmittance of almost 100% as unheated BSA. During the heat treatment, BSA basically started to unfold and the exposed hydrophobic parts of denatured BSA led to protein aggregation. After the addition of CH-PEP·Na, the hydrophobic regions of the denaturing protein bonded with the cholesteryl appendages at the polymeric chain ends, whereas the anionic PEP·Na chains, which were exposed to the surrounding aqueous medium, prevented protein aggregation because of electrostatic repulsion and steric stabilization.14,27 The other two polymers, CH-PMP and PEP·Na, did not exhibit any thermal stabilization property for the protein; for the case of PEP·Na, in particular, the complexes showed the lowest transmittance because of the absence of nonpolar molecules. Although CH-PMP is an amphiphilic PPE as CH-PEP·Na, it could not efficiently inhibit the protein aggregation because of the lower hydrophilicity of PMP compared with PEP·Na and the absence of electrostatic repulsions. Another hydrophilic polymer, PEG, containing cholesteryl groups at the polymeric chain ends (CH-PEG) was also used to form a complex with BSA (BSA/CH-PEG ratios of 1:2, 1:5, and 1:10). After heat treatment, the BSA–PEG complexes exhibited a turbid appearance (see Figure S2), revealing the thermal aggregation of BSA at 90 °C. According to the phase separation of PEG in phosphate-buffered saline (PBS),29 hydrophobic PEG aggregates at high temperatures.30 Therefore, CH-PEG (MW 5008) lost the hydrophilicity to stabilize BSA during heating. Circular dichroism (CD) measurements were also performed (see Figure S3) to confirm that CH-PEG does not suppress the thermal denaturation of the protein.
Figure 2.
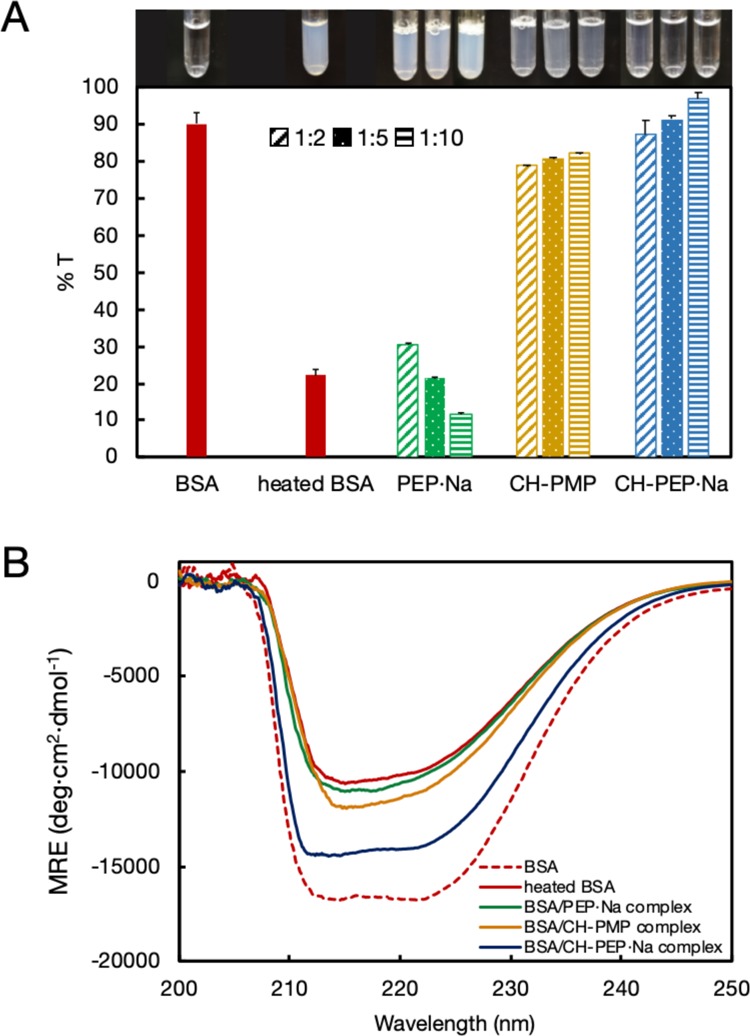
(A) Transmittance (%) and observation images (inset) of native BSA, heated BSA, and BSA–polyphosphoester complexes with different BSA/polymer molar ratios (1:2, 1:5, and 1:10) (CH-PEP·Na: cholesterol-terminated poly(ethylene sodium phosphate); PEP·Na: poly(ethylene sodium phosphate); CH-PMP: cholesterol-terminated poly(2-methoxy-2-oxo-1,3,2-dioxaphosphoranedioxaphospholane)). (B) Circular dichroic spectra of native BSA, heated BSA, and BSA–polyphosphoester complexes with a molar ratio of 1:10. Mean residual ellipticities were plotted as a function of wavelength.
To elucidate the effect of different PPEs, the secondary structural alteration of complexed BSA was performed using CD measurements (Figure 2B). The CD spectrum of native BSA showed two negative signals, at 212 and 222 nm, at 37 °C, and the heated BSA exhibited a huge reduction in the CD intensity, indicating a decrease of the α-helical structure and a denaturation of BSA upon the thermal treatment.31 The signals of BSA–CH-PMP and BSA–PEP·Na did not significantly differ from that of the heated BSA, suggesting that neither CH-PMP nor PEP·Na could preserve the conformational structure of the protein. In the presence of CH-PEP·Na (BSA/CH-PEP·Na ratio of 1:10; the other ratios are shown in Figure S4), the loss in the negative band was interestingly much smaller compared to that observed for the heated BSA and the other complexes, suggesting that only CH-PEP·Na could stabilize the complexed BSA, for the same reason mentioned before. Thus, amphiphilic CH-PEP·Na can be considered an effective excipient for suppressing thermal denaturation and aggregation.
The complexation between BSA and CH-PEP·Na was further confirmed by GPC (Figure 3A); native BSA, the BSA–CH-PEP·Na (1:10) complex, and the unheated mixture of BSA and CH-PEP·Na (1:10) were eluted with PBS. The GPC traces revealed a decrease in the retention time and a peak broadening was observed after thermal treatment with the polymer, indicating the complex formation. In our study it is notable that the complexation could not occur without thermal inducement, that is, CH-PEP·Na did not interact with BSA at ambient temperature. As shown in Figure 3B, BSA and CH-PEP·Na formed spherical-like complexes under thermal treatment in an aqueous medium. However, a small amount of free BSA still remained in the complex solution observed from a tiny peak of the complex trace at the retention time of 14 min (solid line, Figure 3A). 94% of complex purity was calculated from the area under its GPC trace. In addition, according to the bicinchoninic acid (BCA) assay, the BSA/CH-PEP·Na ratio in the complexes was 1:12.7 by mol, resembling feeding ratios of 1:10.0 equiv. The BSA–CH-PEP·Na (1:10) complex with weight-average Mw of 1.9 × 105 g/mol was obtained as evaluated by size exclusion chromatography with multiangle light scattering (SEC-MALS) measurement (see Figure S5). On the basis of the data derived from the BCA assay and SEC-MALS, we could estimate that a complex comprising one BSA molecule complexed with 13 CH-PEP·Na chains. This result verified that CH-PEP·Na had an excellent ability to protect aggregation of protein molecules.
Figure 3.

(A) Gel permeation chromatography elution profiles of native BSA, heated BSA–cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na) (1:10) complex, and a mixture of BSA and CH-PEP·Na at 25 °C. (B) Transmission electron microscopy image of the BSA–CH-PEP·Na (1:10) complex (scale bar 200 nm).
To further confirm the complex formation by heat treatment, the sizes of the complexes obtained with different feeding polymeric ratios were characterized by dynamic light scattering (DLS) (Figure 4). By increasing the amphiphilic CH-PEP·Na content, the complex size gradually decreased from 51 to 27 nm because, at higher CH-PEP·Na fractions, the polymeric chains probably stabilized the entire surface of the heat-denatured proteins, resulting in large protein aggregation blocking and the formation of smaller complexes. Inhibition of protein aggregation by high polymeric content was also described, that thermal aggregation of lysozyme was suppressed with increasing polymeric concentrations by functioning as a molecular shield.32 This is consistent with the previous result showing the inhibition of the protein thermal aggregation by CH-PEP·Na. As PPE analogs are biodegradable, the stability during long-term storage was examined. Steinbach et al. reported a good stability of degradable poly(ethyl ethylene phosphate)–protein conjugates during storage at 4 °C for 1 year.33 In our study, the size and PDI values derived from the DLS data did not significantly change over 1 month of storage at room temperature and undesirable aggregates were not found. Hence, BSA–CH-PEP·Na complexes can ensure a high stability for long-term storage.
Figure 4.
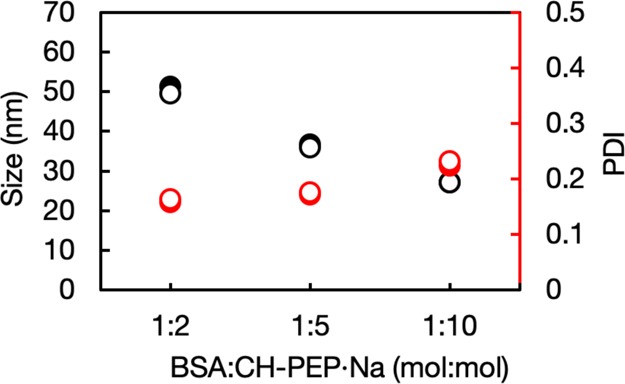
Size (black) and PDI (red) of BSA–cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na) complex suspended in PBS before (closed circles) and after (open circles) storage for 1 month.
The BSA–CH-PEP·Na complexes and native BSA were subjected to trypsin degradation using a stealth polymer to protect the susceptible protein cargos from proteolytic attacks. The matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectra (Figure 5) confirmed that native BSA was easily digested by trypsin, generating peptide fragments (see Table S1); interestingly, the BSA–CH-PEP·Na (1:10) complex did not exhibit any discernible signals, demonstrating an excellent stability in the presence of trypsin. The protection against trypsin digestion was further evaluated by DLS technique as exhibited in Figure S6. Several polymers, such as poly(ethylene glycol)monomethyl ether methacrylate-co-poly(nitrophenylcarbonate) (PEGMA-co-PNPC),34 poly(ethylene glycol)-b-poly(N-2-hydroxypropylmethacrylamide) (PEG-b-PHPMA),6 and carbohydrate-b-poly(propylene glycol),35 have the potential ability to protect encapsulated proteins. Hence, CH-PEP·Na can be considered as a promising polymeric carrier for protein delivery and protection under proteolytic conditions.
Figure 5.

Matrix-assisted laser desorption/ionization time-of-flight mass spectra of (A) native BSA and (B) BSA–cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na) (1:10) complex under trypsin digestion.
HAp, a formula of calcium phosphate (Ca10(PO4)6OH2), is the main mineral portion in teeth and bones.36 It contains two species of binding sites, positively charged calcium and negatively charged phosphate groups. In our previous works, PEP·Na exhibited excellent affinity for both HAp substrates20,37 and in vivo bones.21 Accordingly, BSA-fluoresce in isothiocyanate conjugate (FITC-BSA)–CH-PEP·Na complexes were anticipated to adsorb onto HAp to continue this complex platform for bone-targeting nanomedicine. HAp was immersed into a FITC-BSA–CH-PEP·Na suspension for 3 h. As the reference range for albumin concentrations in human serum is approximately 3.3–5.2 g/dL,38 values between 2.5 and 5.5 g/dL were used in our experiment to investigate the effect of albumin on the affinity of the complexes for HAp. The coverage ratio and quantity of the adsorbed complexes per unit amount of HAp was estimated by comparing the final complex concentration in the supernatant with that in the original solution before immersion with HAp. As shown in Figure 6A, both plateau plots of coverage ratio (1.8 to 2.0 × 10–3) and the amount of adsorbed complex (26.6–28.4 μg/mg HAp) revealed no significant changes in the values upon increasing the albumin concentration, thus implying that the FITC-BSA–CH-PEP·Na complexes strongly deposited on the HAp surface and the albumin could not block the binding avidity between the complexes and the HAp particles. The uncomplexed FITC-BSA (Figure 6B), however, exhibited a decrease in affinity for HAp as the albumin concentration increased because of the competitive adsorption of that protein. This phenomenon could be confirmed by the fluorescence micrographs of HAp treated with FITC-BSA and FITC-BSA–CH-PEP·Na complexes. The fluorescence signal of FITC-BSA-treated HAp disappeared in the presence of free albumin, whereas that of the complex-treated HAp with and without albumin remained steady (see Figure S7). Therapeutic proteins have been recently developed for bone34,39 and dental40,41 treatments; because of the various advantages of CH-PEP·Na, our protein–CH-PEP·Na complexes could be potentially used as biotherapeutic delivery vehicles with high proteolytic stability for such applications.
Figure 6.
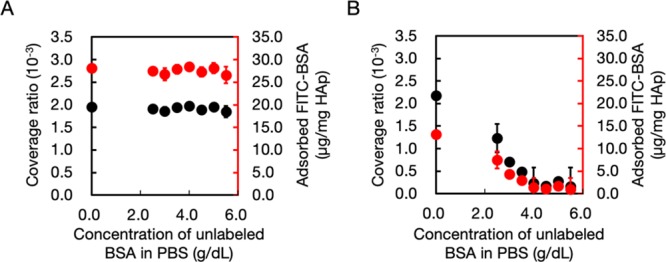
Surface coverage ratio (black circles) and quantity (red circles) of (A) complex and (B) BSA–fluorescein isothiocyanate conjugate (FITC-BSA) adsorbed on HAp, treated, respectively, with FITC-BSA–cholesterol-terminated poly(ethylene sodium phosphate) (CH-PEP·Na) (1:10) complex and FITC-BSA, in the absence and presence of free albumin at different concentrations.
Conclusions
In summary, amphiphilic CH-PEP·Na was used to fabricate protein–polymer complexes via a method as simple as thermal treatment. This noncovalent strategy was created by utilizing the hydrophobic attraction between proteins and cholesteryl appendages. Compared with the other two polyphosphoesters tested, PEP·Na and CH-PMP, CH-PEP·Na provided a considerably greater protein stabilization to heat because of its amphiphilicity and negative charges. The BSA–CH-PEP·Na complexes were obtained at 90 °C, as verified by DLS and transmission electron microscopy (TEM). The CH-PEP·Na chains were able to protect the complexed proteins in the presence of proteolytic cleavage and excellently maintain the complex size stability over 1 month. Besides, FITC-BSA–CH-PEP·Na complexes have a binding affinity for HAp surfaces even in the presence of free albumin. This work should be further extended on the complexation of other proteins with CH-PEP·Na for bone and dental treatments.
Experiments
Materials
2-Chloro-2-oxo-1,3,2-dioxaphospholane (COP) was kindly supplied by NOF Corporation (Japan) and cholesterol was obtained from Kanto Chemical (Japan). Macro-prep ceramic HAp type II 40 μm was purchased from Bio-Rad. BSA, FITC-BSA, and Dulbecco’s PBS was purchased from Sigma-Aldrich and used as received. 1,8-Diazabicyclo[5.4.0]-undec-7-ene (DBU) was purified by distillation under reduced pressure using a fraction of bp 130 °C/3.0 mmHg (lit.: bp 80 °C/0.6 mmHg). 2,6-Lutidine was purified by vacuum distillation. The ion exchanger Amberlite IR-120 (Merck KGaA) was washed with deionized (DI) water before use. Trimethylamine (TMA) was purchased from Nacalai Tesque. Water was purified using a Millipore Milli-Q system (resistivity 18.2 MΩ·cm at 25 °C), involving ultraviolet (UV) irradiation, ion exchange, and filtration. The BSA amount was quantified using the Pierce BCA Protein Assay Kit (Thermo Scientific). The other reagents were purchased from Wako Pure Chemical and used without further purification. MP was synthesized as reported in a previous work.22
Methods
The PPE structure was confirmed by 1H NMR using a JEOL (Japan) operating at 400 MHz. The transmittance at 550 nm of the complex suspensions was recorded with a JASCO V-650 UV–visible light (vis) spectroscope (Japan). The fluorescence measurements were performed using an F-2500 spectrophotometer (Hitachi, Japan) to investigate the cmc. The molecular weight of the polymers was measured with a gel permeation chromatographer and anRI-2031 refractive index detector (JASCO, Japan). For the analysis of CH-PEP·Na and PEP·Na, acetate buffer was selected as eluent and PEG standards (7.5 × 102 to 1.4 × 105 Da) were used to build a calibration curve, whereas for CH-PMP, chloroform as eluent and polystyrene standards (2.8 × 103 to 1.3 × 106 Da) were used. The complex formation was performed using a UV-2075 UV–vis detector (JASCO, Japan) recorded at 280 nm. A phosphate buffer containing NaCl (0.3 M) was used as the eluent. DLS was also carried out with a Zetasizer Nano-ZS (Malvern Instruments Ltd., UK) to analyze the average diameter and PDI of the complexes based on intensity. CD spectra were recorded on a J-1500 CD spectrometer (JASCO, Japan). The secondary structures of the proteins were investigated in the 200–250 nm wavelength range using a quartz cuvette; the samples were prepared with PBS (pH 7.4) to obtain a protein concentration of 2 μM and a scan rate of 100 nm/min at 37 °C, with 8 accumulation scans, was adopted. The molecular weight of a complex was carried out by SEC-MALS (TOSOH) with column (WTC-050S5, Wyatt technology) (Mw range = 15 000–5 000 000 Da) using two detectors, a refractive index detector (Optilab T-rEX, Wyatt technology) and MALS detector (DAWN HELEOS-II, Wyatt technology). Dulbecco’s PBS was used as a solvent for the complexes and as an eluent for SEC analysis with a flow rate of 0.5 mL/min at 30 °C.
Preparation of PPEs
As shown in Scheme 1, CH-PMP was synthesized via the ROP of MP (60 mmol, 70 equiv) using cholesterol (0.86 mmol, 1 equiv) and DBU (0.86 mmol, 1 equiv) as initiator and catalyst, respectively.20,22 The mixture was dissolved in dichloromethane (5 mL) and the reaction was allowed to proceed under an argon atmosphere in an ice bath for 1 h and subsequently stirred at ambient temperature for 2 h. Then, CH-PMP was purified by reprecipitation in toluene followed by diethyl ether and dried under vacuum, resulting in a viscous colorless liquid that was stored in a refrigerator at 4 °C. To prepare CH-PEP·Na, the purified CH-PMP was further demethylated with TMA (120 mmol, 140 equiv) over 24 h, and then the mixture was stirred with cation exchange resins twice to replace the quaternized ammonium ions with H+ ones. The polymer was subsequently purified using dialysis against DI water using a dialysis membrane with a molecular weight cut-off (MWCO) of 3500 Da. The so-obtained polymer was neutralized with an NaOH solution, dialyzed against DI water (MWCO = 3500 Da), and lyophilized to obtain CH-PEP·Na. PEP·Na was also synthesized as described for CH-PEP·Na, starting from MP (36 mmol, 70 equiv), methanol (0.51 mmol, 1 equiv), and DBU (0.51 mmol, 1 equiv). Both pure CH-PEP·Na and PEP·Na were finally obtained as white solids and stored at 4 °C.
The cmc’s of the cholesterol-containing PPEs were determined using pyrene as a fluorescence probe, according to a previously published method.42,43 The concentrations of the polymer solutions were varied from 2 × 10–5 to 5 mg/mL. A pyrene solution in acetone was added to each sample and the resulting mixtures were dissolved in PBS. The spectra were recorded with a fluorescence spectrophotometer using an excitation wavelength of 336 nm.
Preparation of BSA–PPE Complexes
BSA (120 μmol, 1 equiv) was dissolved in PBS (1 mL). Each PPE (0 μmol, 0 equiv; 240 μmol, 2 equiv; 600 μmol, 5 equiv; 1200 μmol, 10 equiv) in PBS (1 mL) was prepared separately and then mixed with the BSA solution, obtaining final BSA concentrations of 60 μM. The resulting mixtures were heated at 90 °C under stirring for 15 min. The free polymers were removed by centrifugal filtration (MWCO: 50k, Merck Millipore) and the complexes were then re-dispersed in PBS.
BSA/CH-PEP·Na Ratio Determination in the Complexes
SEC-MALS was conducted to determine the weight-average molecular weight of the complexes in PBS. The dn/dc value of 0.185 mL/g was used in the molecular weight calculations processed by ASTRA software. The BSA amount in the complex solution was then quantified via the BCA assay using the albumin standard curve (concentration range = 125–2000 μg/mL). The content of the complexed CH-PEP·Na was calculated according to the following equation. Both the complex weight and BSA/CH-PEP·Na ratio (by mol) data were used for estimating the number of protein molecules to the CH-PEP·Na chain in a complex.
* from the BCA assay.
Stability against Trypsin Digestion
The enzymatic degradation study of native and complexed BSA was performed according to a modified version of a previously published method.34 The samples were 10-fold diluted with acetonitrile (10%) and NaHCO3 (90%) buffer at pH = 8, with a final protein concentration of 0.40 mg/mL, and then digested with trypsin (trypsin/protein = 1:25) at 37 °C for 17 h. The MALDI matrix was prepared from a mixture of acetonitrile, Milli-Q water, and trifluoroacetic acid (50:47.5:2.5) containing α-cyano-hydroxycinnamic acid (10 mg/mL). The digested samples and the matrix were mixed at a 1:1 ratio and spotted on an Anchor Chip target to undergo fragmental analysis operated by Flex Control software.
Binding Affinity of FITC-BSA–CH-PEP·Na Complexes for Ceramic HAp Surfaces
HAp microparticles (30 mg) were treated with the FITC-BSA–CH-PEP·Na (1:10, 500 μL) complex suspension containing different amounts of albumin (0, 2.5, 3.0, 3.5, 4.0, 4.5, 5.0, and 5.5 g/dL), as a protein model in human serum, for 3 h. The fluorescence intensity of the centrifuged supernatants was measured with a spectrophotometer and derived from the initial value to determine the amount of complex adsorbed onto HAp; the excitation and emission wavelengths were 495 and 520 nm, respectively. The surface area coverage ratio is defined as the ratio between the total surface area of the adsorbed complexes and the HAp surface area.
Acknowledgments
The authors would like to thank Dr. Kazunari Akiyoshi and Dr. Shin-ichi Sawada for their valuable advice and technical support. This study was supported by the MEXT Private University Research Branding Project and KAKENHI from the Japan Society for the Promotion of Science (JSPS), Japan (#16H03185).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03585.
Photographs of heated BSA solution with CH-PEG, CD spectra of heated BSA with CH-PEG, CD spectra of heated BSA with CH-PEP·Na, SEC-MALS chromatogram of fluorescence micrographs of HAp microparticles in contact with FITC-BSA–CH-PEP·Na complex, main peptide fragments of BSA (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ma X.; Zhao Y.; Liang X.-J. Theranostic nanoparticles engineered for clinic and pharmaceutics. Acc. Chem. Res. 2011, 44, 1114–1122. 10.1021/ar2000056. [DOI] [PubMed] [Google Scholar]
- Riehemann K.; Schneider S. W.; Luger T. A.; Godin B.; Ferrari M.; Fuchs H. Nanomedicine-Challenge and Perspectives. Angew. Chem., Int. Ed. 2009, 48, 872–897. 10.1002/anie.200802585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R.; Lillard J. W. Nanoparticle-based targeted drug delivery. Exp. Mol. Pathol. 2009, 86, 215–223. 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. Multifunctional nanocarriers. Adv. Drug Deliv. Rev. 2006, 58, 1532–1555. 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Yan Y.; Such G. K.; Johnston A. P. R.; Best J. P.; Caruso F. Engineering particles for therapeutic delivery: prospects and challenges. ACS Nano 2012, 6, 3663–3669. 10.1021/nn3016162. [DOI] [PubMed] [Google Scholar]
- Blackman L. D.; Varlas S.; Arno M. C.; Houston Z. H.; Fletcher N. L.; Thurecht K. J.; Hasan M.; Gibson M. I.; O’Reilly R. K. Confinement of therapeutic enzymes in selectively permeable polymer vesicles by polymerization-induced self-assembly (PISA) reduces antibody binding and proteolytic susceptibility. ACS Cent. Sci. 2018, 4, 718–723. 10.1021/acscentsci.8b00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisal D. S.; Kosloski M. P.; Balu-Iyer S. V. Delivery of therapeutic proteins. J. Pharm. Sci. 2010, 99, 2557–2575. 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchilin V. P.; Lukyanov A. N. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov. Today 2003, 8, 259–266. 10.1016/s1359-6446(03)02623-0. [DOI] [PubMed] [Google Scholar]
- Alconcel S. N. S.; Baas A. S.; Maynard H. D. FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym. Chem. 2011, 2, 1442–1448. 10.1039/c1py00034a. [DOI] [Google Scholar]
- Chen J.; Zhao M.; Feng F.; Sizovs A.; Wang J. Tunable Thioesters as ″Reduction″ Responsive Functionality for Traceless Reversible Protein PEGylation. J. Am. Chem. Soc. 2013, 135, 10938–10941. 10.1021/ja405261u. [DOI] [PubMed] [Google Scholar]
- Duncan R.; Spreafico F. Polymer Conjugates. Clin. Pharmacokinet. 1994, 27, 290–306. 10.2165/00003088-199427040-00004. [DOI] [PubMed] [Google Scholar]
- Pelosi C.; Duce C.; Russo D.; Tiné M. R.; Wurm F. R. PPEylation of proteins: synthesis, activity, and stability of myoglobin-polyphosphoester conjugates. Eur. Polym. J. 2018, 108, 357–363. 10.1016/j.eurpolymj.2018.09.019. [DOI] [Google Scholar]
- Steinbach T.; Wurm F. R. Degradable Polyphosphoester-Protein Conjugates: ″PPEylation″ of Proteins. Biomacromolecules 2016, 17, 3338–3346. 10.1021/acs.biomac.6b01107. [DOI] [PubMed] [Google Scholar]
- Akiyoshi K.; Sasaki Y.; Sunamoto J. Molecular Chaperone-Like Activity of Hydrogel Nanoparticles of Hydrophobized Pullulan: Thermal Stabilization with Refolding of Carbonic Anhydrase B. Bioconjugate Chem. 1999, 10, 321–324. 10.1021/bc9801272. [DOI] [PubMed] [Google Scholar]
- Li R.; Elsabahy M.; Song Y.; Wang H.; Su L.; Letteri R. A.; Khan S.; Heo G. S.; Sun G.; Liu Y.; et al. Functional, degradable zwitterionic polyphosphoesters as biocompatible coating materials for metal nanostructures. Langmuir 2018, 35, 1503. 10.1021/acs.langmuir.8b02033. [DOI] [PubMed] [Google Scholar]
- Pei P.; Sun C.; Tao W.; Li J.; Yang X.; Wang J. ROS-sensitive thioketal-linked polyphosphoester-doxorubicin conjugate for precise phototriggered locoregional chemotherapy. Biomaterials 2019, 188, 74–82. 10.1016/j.biomaterials.2018.10.010. [DOI] [PubMed] [Google Scholar]
- Simon J.; Wolf T.; Klein K.; Landfester K.; Wurm F. R.; Mailänder V. Hydrophilicity regulates the stealth properties of polyphosphoester-coated nanocarriers. Angew. Chem., Int. Ed. 2018, 57, 5548–5553. 10.1002/anie.201800272. [DOI] [PubMed] [Google Scholar]
- Bauer K. N.; Tee H. T.; Velencoso M. M.; Wurm F. R. Main-chain poly(phosphoester)s: history, syntheses, degradation, bio-and flame-retardant applications. Prog. Polym. Sci. 2017, 73, 61–122. 10.1016/j.progpolymsci.2017.05.004. [DOI] [Google Scholar]
- Steinbach T.; Wurm F. R. Poly(phosphoester)s: a new platform for degradable polymers. Angew. Chem., Int. Ed. 2015, 54, 6098–6108. 10.1002/anie.201500147. [DOI] [PubMed] [Google Scholar]
- Hirano Y.; Iwasaki Y. Bone-specific poly(ethylene sodium phosphate)-bearing biodegradable nanoparticles. Colloids Surf. B: Biointerfaces 2017, 153, 104–110. 10.1016/j.colsurfb.2017.02.015. [DOI] [PubMed] [Google Scholar]
- Iwasaki Y.; Yokota A.; Otaka A.; Inoue N.; Yamaguchi A.; Yoshitomi T.; Yoshimoto K.; Neo M. Bone-targeting poly(ethylene sodium phosphate). Biomater. Sci. 2018, 6, 91–95. 10.1039/c7bm00930e. [DOI] [PubMed] [Google Scholar]
- Kootala S.; Tokunaga M.; Hilborn J.; Iwasaki Y. Anti-resorptive functions of poly(ethylene sodium phosphate) on human osteoclasts. Macromol. Biosci. 2015, 15, 1634–1640. 10.1002/mabi.201500166. [DOI] [PubMed] [Google Scholar]
- Libiszowski J.; Kałużynski K.; Penczek S. Polymerization of cyclic esters of phosphoric acid. VI. Poly(alkyl ethylene phosphates). Polymerization of 2-alkoxy-2-oxo-1,3,2-dioxaphospholans and structure of polymers. J. Polym. Sci. Polym. Chem. Ed. 1978, 16, 1275–1283. 10.1002/pol.1978.170160610. [DOI] [Google Scholar]
- Zhang S.; Zou J.; Zhang F.; Elsabahy M.; Felder S. E.; Zhu J.; Pochan D. J.; Wooley K. L. Rapid and versatile construction of diverse and functional nanostructures derived from a polyphosphoester-based biomimetic block copolymer system. J. Am. Chem. Soc. 2012, 134, 18467–18474. 10.1021/ja309037m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi R.; Iwasaki Y. High mineral affinity of polyphosphoester ionomer-phospholipid vesicles. J. Biomed. Mater. Res. 2013, 101, 318–325. 10.1002/jbm.a.34321. [DOI] [PubMed] [Google Scholar]
- Zhang S.; Wang H.; Shen Y.; Zhang F.; Seetho K.; Zou J.; Taylor J.-S. A.; Dove A. P.; Wooley K. L. A simple and efficient synthesis of an acid-labile polyphosphoramidate by organobase-catalyzed ring-opening polymerization and transformation to polyphosphoester ionomers by acid treatment. Macromolecules 2013, 46, 5141–5149. 10.1021/ma400675m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadhukhan N.; Muraoka T.; Ui M.; Nagatoishi S.; Tsumoto K.; Kinbara K. Protein stabilization by an amphiphilic short monodisperse oligo(ethylene glycol). Chem. Commun. 2015, 51, 8457–8460. 10.1039/c4cc10301g. [DOI] [PubMed] [Google Scholar]
- Moroi Y.; Nishikido N.; Saito M.; Matuura R. The critical micelle concentration of ionic-nonionic detergent mixtures in aqueous solutions. III. J. Colloid and Interface Sci. 1975, 52, 356–363. 10.1016/0021-9797(75)90210-6. [DOI] [Google Scholar]
- Silva L. H. M. d.; Coimbra J. S. R.; Meirelles A. J. d. A. Equilibrium phase behavior of poly(ethylene glycol) + potassium phosphate + water two-phase systems at various pH and temperatures. J. Chem. Eng. Data 1997, 42, 398–401. 10.1021/je9602677. [DOI] [Google Scholar]
- Chua G. B. H.; Roth P. J.; Duong H. T. T.; Davis T. P.; Lowe A. B. Synthesis and thermoresponsive solution properties of poly[oligo(ethylene glycol) (meth)acrylamide]s: biocompatible PEG analogues. Macromolecules 2012, 45, 1362–1374. 10.1021/ma202700y. [DOI] [Google Scholar]
- Borzova V. A.; Markossian K. A.; Chebotareva N. A.; Kleymenov S. Y.; Poliansky N. B.; Muranov K. O.; Stein-Margolina V. A.; Shubin V. V.; Markov D. I.; Kurganov B. I.. Kinetics of thermal denaturation and aggregation of bovine serum albumin. PLoS One 2016,11, -e0153495. 10.1371/journal.pone.0153495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan R.; Matsumura K. A zwitterionic polymer as a novel inhibitor of protein aggregation. J. Mater. Chem. B 2015, 3, 5683–5689. 10.1039/c5tb01021g. [DOI] [PubMed] [Google Scholar]
- Steinbach T.; Becker G.; Spiegel A.; Figueiredo T.; Russo D.; Wurm F. R. Reversible bioconjugation: biodegradable poly(phosphate)-protein conjugates. Macromol. Biosci. 2017, 17, 1600377. 10.1002/mabi.201600377. [DOI] [PubMed] [Google Scholar]
- Dutta K.; Hu D.; Zhao B.; Ribbe A. E.; Zhuang J.; Thayumanavan S. Templated self-assembly of a covalent polymer network for intracellular protein delivery and traceless release. J. Am. Chem. Soc. 2017, 139, 5676–5679. 10.1021/jacs.7b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura T.; Sasaki Y.; Akiyoshi K. Biotransporting self-assembled nanofactories using polymer vesicles with molecular permeability for enzyme prodrug cancer therapy. Adv. Mater. 2017, 29, 36. 10.1002/adma.201770258. [DOI] [PubMed] [Google Scholar]
- Low S. A.; Kopeček J. Targeting polymer therapeutics to bone. Adv. Drug Deliv. Rev. 2012, 64, 1189–1204. 10.1016/j.addr.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki Y.; Katayama K.; Yoshida M.; Yamamoto M.; Tabata Y. Comparative physicochemical properties and cytotoxicity of polyphosphoester ionomers with bisphosphonates. J. Biomater. Sci. Polym. Ed. 2012, 24, 882–895. 10.1080/09205063.2012.710823. [DOI] [PubMed] [Google Scholar]
- Jang Y.; Lee S.-T.; Kim T.-J.; Jun J.-S.; Moon J.; Jung K.-H.; Park K.-I.; Chu K.; Lee S. K. High albumin level is a predictor of favorable response to immunotherapy in autoimmune encephalitis. Sci. Rep. 2018, 8, 1012. 10.1038/s41598-018-19490-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Newman M. R.; Benoit D. S. W. Development of controlled drug delivery systems for bone fracture-targeted therapeutic delivery: A review. Eur. J. Pharm. Biopharm. 2018, 127, 223. 10.1016/j.ejpb.2018.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakashima M.; Reddi A. H. The application of bone morphogenetic proteins to dental tissue engineering. Nat. Biotechnol. 2003, 21, 1025. 10.1038/nbt864. [DOI] [PubMed] [Google Scholar]
- Huq N. L.; Cross K. J.; Ung M.; Myroforidis H.; Veith P. D.; Chen D.; Stanton D.; He H.; Ward B. R.; Reynolds E. C. A review of the salivary proteome and peptidome and saliva-derived peptide therapeutics. Int. J. Pept. Res. Ther. 2007, 13, 547. 10.1007/s10989-007-9109-9. [DOI] [Google Scholar]
- McRae Page S.; Martorella M.; Parelkar S.; Kosif I.; Emrick T. Disulfide cross-linked phosphorylcholine micelles for triggered release of camptothecin. Mol. Pharm. 2013, 10, 2684–2692. 10.1021/mp400114n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topel Ö.; Çakır B. A.; Budama L.; Hoda N. Determination of critical micelle concentration of polybutadiene-block-poly(ethyleneoxide) diblock copolymer by fluorescence spectroscopy and dynamic light scattering. J. Mol. Liq. 2013, 177, 40–43. 10.1016/j.molliq.2012.10.013. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.