

Abstract
Forkhead transcription factor C2 (FOXC2) is a transcription factor regulating vascular and lymphatic development, and its mutations are linked to lymphedema-distichiasis syndrome. FOXC2 is also a crucial regulator of the epithelial–mesenchymal transition processes essential for tumor metastasis. Here, we report the crystal structure of the FOXC2–DNA-binding domain in complex with its cognate DNA. The crystal structure provides the basis of DNA sequence recognition by FOXC2 for the T/CAAAC motif. Helix 3 makes the majority of the DNA–protein interactions and confers the DNA sequence specificity. The computational energy calculation results also validate the structural observations. The FOXC2 and DNA complex structure provides a detailed picture of protein and DNA interactions, which allows us to predict its DNA recognition specificity and impaired functions in mutants identified in human patients.
Introduction
Forkhead transcription factor C2 (FOXC2, UniProtKB Q99958) plays essential roles during vascular endothelial and lymphatic development. Recent studies have revealed that FOXC2, previously named mesenchymal forkhead 1, is a crucial regulator of epithelial–mesenchymal transition (EMT) processes.1−3 Epithelial–mesenchymal transition is a transformative cellular event in which epithelial cells become mesenchymal cells, characterized by more migratory and multipotent behaviors. In cancer cells, EMT is closely linked to tumor invasion and metastasis.4 Hence, FOXC2 has gained much interest as a novel cancer therapeutic target because of its critical roles in EMT processes.1−3,5−7 Notably, FOXC2 has been demonstrated to be abnormally highly expressed in stem cell populations of breast, colon, esophageal, and prostate cancers, which are culprits of cancer recurrence, metastasis, and drug resistance.1,6−9 Knockout of FOXC2 has significantly reduced tumor sizes,8 minimized neoplasia,10 and restored epithelial phenotypes sensitive to drugs.9
Forkhead transcription factor C2 belongs to the forkhead box (FOX) transcription factor protein family.11 The FOX family proteins share the forkhead or winged helix structure in their evolutionary conserved DNA-binding domain (DBD). The FOX proteins are grouped into 19 subfamilies, with more than 50 FOX proteins having been identified in humans to date.3,12,13 They play vital roles in development, apoptosis, metabolism, migration, proliferation, differentiation, and longevity-related processes,14 and mutations in some FOX family proteins are linked to severe phenotypic deformity.15 For example, mutations in FOXC2 have been linked to lymphedema-distichiasis syndrome, a condition characterized by abnormal lymphatic functions and heart abnormality.16−18
The 501 amino acid long human FOXC2 protein is composed of N- and C-terminal regulatory domains and the DBD (Figure 1). The evolutionary conserved DBD recognizes a consensus DNA motif (5′-(G/A)(T/C)(A/C)AA(C/T)A-3′).19 The DBD also contains a predicted nuclear localization signal (NLS, residues 135–142), less conserved among FOX family proteins (Figure 1). Interestingly, the cytoplasmic retention of FOXC2 by nuclear transport inhibition can prevent the mesenchymal transition of the cells.20 Thus, FOXC2 inhibitors targeting the NLS that interfere with nuclear transport may serve as potential cancer therapeutics. The majority of the FOXC2 mutations linked to lymphedema-distichiasis syndrome are insertion, deletion, and nonsense mutations. Among them, six identified missense mutations are located in the DBD, underscoring the functional importance of the DBD (Figure 1).17
Figure 1.

Overall structure of the FOXC2 DBD–DNA complex and sequence alignment. (A) A schematic of the protein–DNA complex. The FOXC2 DBD is indicated in cyan (helices), red (sheets), and magenta (coils). The DNA containing dual binding sites of FOXC2 are also illustrated in cartoon representation. The secondary structure elements and N- and C-termini are labeled. The dotted lines represent the loops missing in the Mol B model. (B) C-terminal residues of FOXC2, 148–161, are depicted in the stick representation with a 2Fo – Fc map (1σ level). (C) Sequence alignment of the FOX family DNA-binding domains. The numbering is based on the FOXC2 residues. The secondary structure elements are indicated, and the missense mutations identified in FOXC2 are also denoted on top of the sequences.
To understand the molecular basis of the pathological mechanisms of the FOXC2 mutations and to provide the structural basis for cancer therapeutic development, we have determined a three-dimensional structure of the FOXC2 DBD in complex with its cognate double-stranded DNA fragment. Multiple structures of forkhead domains were determined using NMR and X-ray crystallography methods.21−32 The FOXC2 DBD structure was determined by NMR without the cognate DNA (PDB ID: 1D5V).27 Without the bound DNA, the structure exhibits a dynamic nature of the domain in which the C-terminal residues are disordered. The FOXC2 DBD and DNA complex structure reported here provides a detailed picture of the protein and DNA interactions. The structural data combined with quantitative data on the FOXC2 DBD and various DNA motifs presented here will provide the basis for next-generation cancer therapeutics targeting FOXC2.
Results and Discussion
Overview of the FOXC2 DBD–DNA Complex
The structural study of the FOXC2 bound to the target DNA-containing palindromic forkhead-binding sites (AAATTGTTTATAAACAGCCCG/TTCGGGCTGTTTATAAACAAT) was carried out using the human FOXC2 DBD. In solution, the FOXC2 DBD–DNA complexes were readily formed and purified. The expression construct encodes residues 60–198, but moderate protein degradation was observed during the purification. The purified complexes were used for crystallization and structure determination. The crystal structure of the FOXC2 DBD–DNA complex was solved at the 3.06 Å resolution. The data collection and final refinement statistics are summarized in Table 1.
Table 1. Crystallographic Data and Refinement Statistics.
| crystallographic data | |
|---|---|
| X-ray source | 19-ID, APS |
| wavelength (Å) | 0.97918 |
| space group | C121 |
| a, b, c (Å), β (deg) | 128.37, 41.82, 82.65, 98.30 |
| resolution (Å) | 36.3–3.06 (3.15–3.06) |
| total no. of reflections | 23 999 |
| no. of unique reflections | 8013 (389) |
| multiplicity | 3.0 (3.1) |
| completeness (%) | 96.8 (98.0) |
| mean I/σ(I) | 12.3 (2.8) |
| Rmeas | 0.118 (0.427) |
| Rpim | 0.065 (0.233) |
| Rsym | 0.097 (0.356) |
| refinement statistics of the current model | |
|---|---|
| reflections used in the refinement | 7889 (602) |
| reflections used for Rfree | 389 (25) |
| Rwork/Rfree (%) | 23.3/26.8 |
| RMSD bond lengths (Å) | 0.011 |
| RMSD bond angles (deg) | 1.48 |
| no. of macromolecules | 2197 |
| average B-factor (Å2) | 79 |
| Ramachandran analysis (%) | |
| favored | 90.8 |
| allowed | 9.2 |
| outliers | 0.0 |
| rotamer outliers (%) | 0.0 |
One 21 base-pair DNA and 2 molecules of FOXC2 DBD are present in an asymmetric unit. The amino acid residues 70–164 in one of the two FOXC2 DBDs (Mol A) and 73–149 in the second molecule (Mol B) are modeled into the electron densities (Figure 1). Mol A is stabilized by multiple crystallographic contacts and displays well-ordered densities. However, Mol B is involved in fewer crystallographic contacts and is less ordered, resulting in a poorly defined electron density map (data not shown). Because of this, the quality of the Mol B model is worse than that of the Mol A model, and the overall discussion of the FOXC2 DBD is based on the structure of Mol A. The two FOXC2 DBDs bind to their respective recognition motifs on the 21-mer DNA double helix and are related by a 180° rotation symmetry (Figure 1A). However, no direct contacts between the two protein molecules were observed.
The FOXC2 DBD displays a canonical winged helix fold composed of three α-helices (α1–3), three β-strands (β1–3), and two winglike loops (W1, W2) (Figure 1A). The second winged loop (W2) is disordered in the previously reported forkhead structures, including the FOXC2 NMR structure (PDB ID: 1D5V).27 In our model, the second winged loop forms a pseudo-helical conformation with hydrophobic residues Leu148, Met155, Phe156, and Leu162 packing against the hydrophobic residues on helix 1 (Figure 1B). The W2 loop in Mol A is involved in multiple crystallographic contacts, which help to stabilize the conformation. In Mol B, the C-terminal residues 150–162 are disordered and not included in the model. This suggests that the second winged motif in the FOXC2 DBD has a flexible nature. The predicted NLS signal (residues 135–142) is located at the tip of the W1 loop and is highly flexible, making it easily accessible by the nuclear transport machinery and therapeutic agents.
While this manuscript was being prepared, crystal structures of FOXC2 DBD in complex with DNA motifs, ACAAATA and GTACACA, were reported.33 The protein structure exhibits a similar structural arrangement as our FOXC2 structure, with the root-mean-square distances among the Cα atoms ranging between 0.5 and 0.75 Å.
FOXC2 DBD–DNA Interactions
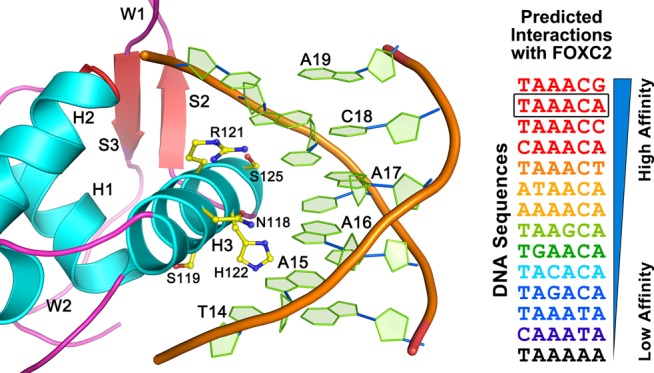
The DNA used for this structure study contains two forkhead-binding motifs (5′-TATAAACA-3′) in a palindromic arrangement. Both motifs are bound by the FOXC2 protein in an identical manner, and interactions between one of the proteins (Mol A) and the DNA motif are illustrated in Figure 2. Based on the NuProPlot diagram in Figure 2A, Mol A interacts with residues 12–19 of chain D, TATAAACA, and their complementary residues.34 Overall, the protein and DNA interactions are mediated through residues from the helices α1−α3 and wings 1 and 2 of the FOXC2 DBD. Interactions with the DNA bases were mostly mediated by the residues of helix 3 conferring specificity for the forkhead-binding motif. The residues from α1–2 and wings 1–2 bind to the backbone of the DNA, stabilizing the protein–DNA interactions. Lys72, Ser76, and Tyr77 interact with the DNA backbone near the 5′ region of the TATAAACA motif, while Asn96 and Tyr99 of α2 contact the DNA backbone in the 3′ end. Residues from the wing 1 region, Lys132, Ser144, Gly143, and Trp146, also interact with the DNA backbone in the 3′ region. Arg164 from W2 forms a hydrogen bond with the phosphate group at the 5′ section of the TATAAACA motif (Figure 2A).
Figure 2.

FOXC2 DBD and DNA interaction. (A) NuProPlot diagram of FOXC2 DBD–DNA interactions. The DNA segment interacting with the Mol A is illustrated. Hydrogen bonds and van der Waals interactions are represented by dotted blue and orange lines, respectively. (B) Detailed view of the FOXC2 DBD and its cognate DNA segment shown in a stereo diagram. The side chains of amino acid residues involved in the DNA base interactions are illustrated with the carbons in yellow, the oxygens in red, and the nitrogens in blue.
The most extensive interactions between the protein and DNA occur through helix 3, which sits perpendicularly on a major groove of the DNA (Figure 2B). Asn118, Ser119, Arg121, His122, and Ser125 from α3 form protein–base interactions. The residues are absolutely conserved across the forkhead family proteins, except at residue Ser119 (Ala in FOXP3) (Figure 1C), and interact with the canonical forkhead-binding motif. The FOXC2 DBD interacts with the DNA bases of residues 14–18 (TAAAC) and their complementary residues. The T14 interacts with Ser119 and His122. The O4 of the T14 hydrogen-bonds with the Nε2 of the His122 indole ring, forming a favorable interaction, while the Oγ of the Ser119 is in the range of the van der Waals interaction with the C7 of the T14. If the T14 is substituted by cytosine, the O4 would be changed to N4, forming an unfavorable bond with the His122 Nε2. In addition, the C7 would be missing, and interactions with the Ser119 would be abolished. The His122 also interacts with A10′ complementary to the T14, and substitution to guanine at this position will switch the N6 to O6. This change may be favorable for the interaction with the histidine imidazole ring. Changes to a purine of the T14, however, will induce conformational changes, losing any interactions with the Ser119 and His122. Therefore, T or C at position 14 seems to be favored for interactions with the FOXC2 DBD.
The His122 also forms van der Waals interactions with A15 and its complementary residue. The Nδ1 of the indole ring interacts with the N6 group of A15 and the O4 of the complementary thymine (T9′). If the A–T base pair changes to G–C, the interactions will still be maintained, but the change of the O4 to N4 in cytosine makes it less favorable for interactions with the Nδ1. Changes from purine to pyrimidine at this position are likely to abolish interactions with the H122. The His122 also determines DNA specificity for A16. The Nδ1 of the His122 forms a hydrogen bond with the O4 of T8′, complementary to A16. Changes to cytosine will substitute the O4 with N4 and would again make the bond less favorable with H122 Nδ1. Asn118 forms bidentate hydrogen bonds with the A16, making adenine the most optimal base at this position for protein interaction.
Ser125 exhibits structural complementarity with the T7′ base-pairing with the A17, and changes to any other bases at this position will eliminate van der Waals interactions at this position. Arg121 forms a hydrogen bond with the G6′ complementary to C18. The terminal amide group of Arg121 interacts with the O6 group of the guanine. Substitution to adenine at this position will change the O6 to N6, which is less favorable for interaction with Arg121. In our structure, neither A19 nor the complementary T5′ is within the 3.8 Å range for interactions with the protein.
Our structural analyses predict that the FOXC2 DBD favors the DNA motif (T/C)AAACN. To evaluate this prediction quantitatively, we performed a theoretical energy calculation with base substitutions. We calculated protein–DNA interaction energies using the program FoldX and our FOXC2 DBD crystal structure (Table 2).35 The DNA sequences were mutated at each position, and changes in protein–DNA interaction energies were calculated after energy minimization of the models. Overall, the free energy differences with DNA sequence variations agree with our structural analyses. Of the TAAACA sequence, the first T to C change did not demonstrate notable differences. All of the other changes resulted in mostly unfavorable interactions, except for the last adenine, which can be changed to any other bases without a significant energy cost. Based on our analyses, the DNA motif (T/C)AAACN is predicted to be the optimal sequence for binding with FOXC2.
Table 2. Calculated Relative Interaction Energy of FOXC2 with Various DNA Sequences.
| DNA sequence | calculated relative ΔG protein–DNA interaction (kcal/mol) |
|---|---|
| TAAACA (current structure) | 0.0 |
| CAAACA | 0.0 |
| AAAACA | 0.4 |
| TGAACA | 1.0 |
| TAGACA | 1.5 |
| TAAGCA | 0.6 |
| TAAATA | 1.5 |
| TAAAAA | 6.1 |
| TAAACG | –0.2 |
| TAAACC | 0.0 |
| TAAACT | 0.1 |
| ATAACA | 0.3 |
| CAAATA | 1.6 |
| TACACA | 1.2 |
We measured the binding affinity of the FOXC2 DBD to the DNA-containing TATAAACA motif using electrophoretic mobility shift assay (EMSA). The FOXC2 DBD exhibits an overall low affinity to its cognate DNA with the Kd value of 26 μM (Figure 3). The Kd values between FOXC2 and various DNA motifs have previously been measured using isothermal calorimetry, and their ranges are 0.79 μM for GTAAACA, 2.22 μM for GTACACA, and 100 μM for ACAAATA.33 The measured Kd value indicates more than 30-fold differences between TATAAACA and GTAAACA, which contains the core TAAACA motif. This may be due to differences in the DNA sequences and the techniques employed in the binding affinity measurement. Nonetheless, our computational data predict that the FOXC2 prefers TAAACA over TACACA, and CAAATA seems to be the least favored (Table 2). This result is in agreement with the isothermal calorimetry measurement data.33 This indicates that crystal structures of transcription factors combined with computational approaches can be a credible tool for DNA specificity determination.
Figure 3.

Electrophoretic mobility shift assay of FOXC2 and DNA. (A) FOXC2 and DNA binding by EMSA. The purified FOXC2 DBD was mixed with the DNA containing TAAACA motif, and the protein–DNA complexes were separated on a 6% acrylamide gel. Increasing concentrations of FOXC2 DBD, 0, 2.5, 5, 10, 25, 50, 75, 150, and 300 μM, were used (lanes 1–9). (B) Linear scale saturation binding curve of FOXC2 DBD measured by EMSA. The Kd and R2 were estimated as 26.4 ± 3.9 μM (95% CI: 18.7–37 μM) and 0.94, respectively. The error bar indicates a standard deviation of measurements from triplicate experiments.
FOXC2 Mutations in Lymphedema-Distichiasis Syndrome
Several missense mutations in the FOXC2 DBD are associated with pathogenic conditions in humans (Figure 1C). They are linked to the familial and sporadic lymphedema-distichiasis syndrome, a condition characterized by abnormal lymphatic functions, spinal cysts, and heart abnormality.16−18,36−38 To understand the effect of these mutations on DNA binding and protein stability, free energy differences of missense mutants from the wild type were calculated and compared.39 The mutant models were generated based on our FOXC2 DBD–DNA crystal structure, energy-minimized, and used for the calculation. Free energies of intraprotein interaction and protein–DNA interaction were computed, and the ΔGs from the wild type were calculated. Solvent molecules were not included in the calculation for the sake of simplicity. Table 3 presents the calculated ΔGs in the mutants.
Table 3. Calculated Relative Interaction Energy of FOXC2 Mutants.
| mutations | calculated relative ΔG intraprotein (kcal/mol) | calculated relative ΔG protein–DNA interaction (kcal/mol) |
|---|---|---|
| wild type | 0 | 0 |
| I85N | 1.9 | –0.1 |
| W116R | 10.0 | –0.2 |
| N118K | 1.8 | –2.7 |
| R121C | 0.7 | 2.4 |
| R121H | 4.1 | 4.0 |
| S125L | 1.0 | 3.9 |
As described previously, in the crystal structure, the side chains of Asn118, Arg121, and Ser125 make direct contacts with the DNA and are responsible for the DNA sequence specificity (Figure 4). Therefore, the mutations, N118K, R121C, R121H, and S125L, are expected to impact DNA binding. Figure 4 illustrates that the N118K mutation is likely to remove the hydrogen bonds to A16 (Figure 4A), and changes to cysteine or histidine from Arg121 are also predicted to remove multiple bonds to the DNA (Figure 4B). S125L mutation, however, seems to cause steric clashes with the DNA (Figure 4C). The energy calculation results also indicate an increase in the ΔGs of the protein–DNA interactions in the mutants (Table 3).
Figure 4.

Comparisons of wild type and FOXC2 missense mutants. Models of wild type, N118K (A), R121H (B), and S125L (C), are depicted in the stick representation. Dotted lines represent hydrogen bonds, and in panel (C), the clash among the atoms is illustrated.
The two other reported missense mutations in lymphedema-distichiasis patients, I85N and W116R, are not located at the DNA-binding interface. These residues seem to contribute to stabilizing the hydrophobic interactions in the protein core. Changes to polar amino acids at this position are likely to affect protein folding. Disruption of the intraprotein interactions results in higher free energy in these mutants compared to that in the wild type (Table 3).
Protein–Protein Interactions
The FOXC2 DBD exhibits low affinity to its cognate DNA, with a micromolar range of the Kd values (Figure 3).33 The FOXC2 was reported to interact with various transcription factors and co-occupy promotor and enhancer sequences.40 The FOXC2 alone exhibits low affinity, but interactions with other proteins are an essential driving force for DNA interaction. Among the binding partners, the E-twenty-six (ETS) family proteins were reported to physically interact with the FOXC2 and synergistically activate downstream targets.40 In zebrafish, FOXC2 together with ETS variant 2 (ETV2, UniProtKB Q30JB6) transcription factors control angiogenetic processes, and the two proteins are required for blood vessel formation.40 One of the downstream targets activated by FOXC2 and ETV2 is the myocyte-specific enhancer factor 2C (MEF2C, UniProtKB Q06413). MEF2C is an endothelial-specific transcription factor, and the enhancer sequences co-occupied by FOXC2 and ETV2 have been identified.40 We built a composite model of FOXC2 and ETV2 (UniProtKB O00321) DNA-binding domains bound to the FOX–ETS motif in the Mef2c enhancer sequence, based on the FOXO1 and ETS cocrystal structure (RCSB 4LG0).41 We generated the FOXC2–ETV2 complex model using the FOXC2 DBD crystal structure and an ETV2 model. The ETV2 model was generated by homology modeling, using the ETS1 as a template.41,42
Figure 5 illustrates the composite model of FOXC2 and ETV2 bound to the DNA. The ternary complex model indicates that no extensive interactions are present between the two proteins. However, the Lys113 of FOXC2 is positioned at the interface close to the Glu293 of ETV2, suggesting a potential interaction between the two factors. In our current composite model, the distance between the two residues is 5.6 Å. With that distance, direct contact would not be possible; however, the two residues are in the range for solvent-mediated interactions. The Glu293 is absolutely conserved among the ETS proteins, but its function remains unclear. The Lys113 of FOXC2 is located in the loop between the 310-helix and Helix 3. This loop is involved in neither DNA interactions nor protein core stabilization. The amino acid sequence of the loop is not highly conserved, but most forkhead proteins contain a basic amino acid in this loop. The conserved nature of the two binding partners implies a possible role for the two amino acids in protein–protein interactions. A similar intermolecular interaction has been observed between the cardiac transcription factors, NKX2.5 and TBX5.43 The ternary complex structure of NKX2.5 and TBX5 with the target DNA revealed physical interactions between the two proteins via salt bridges between the Lys and the Asp, highly conserved in TBX and NKX families across species. Except for the potential salt bridge, the FOXC2 and ETV2 models do not show extensive interfaces between the two DNA-binding domains. This implies that the major protein–protein interactions are mediated by the N- or C-terminal domains outside of the DNA-binding domains.
Figure 5.
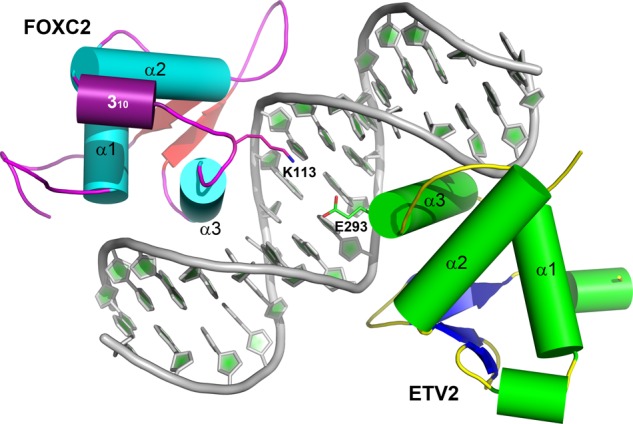
Composite model of FOXC2 and ETV2 bound to a DNA target. A ternary complex of FOXC2 and ETV2 bound to a FOX–ETS motif is illustrated. The FOXC2 follows the same color scheme as in Figure 1A, and ETV2 is indicated in green (helices), blue (sheets), and yellow (coils). The FOX–ETS DNA is illustrated in the cartoon representation. Two charged residues at the interface, Lys113 of FOXC2 and Glu293 of ETV2, are labeled.
Conclusions
We report the crystal structure of the FOXC2–DNA-binding domain in complex with its cognate DNA. The structure provides the basis for specificity of protein and DNA interactions. As in previously reported forkhead protein structures, the helix α3 forms the majority of the DNA–protein interactions and provides its DNA sequence specificity. Additional contacts to the DNA backbone by residues outside of helix 3 seem to stabilize the protein–DNA interactions. Furthermore, this structure provides the structural foundation that allows us to predict disruptions in the protein–DNA binding caused by pathological mutations identified in FOXC2 proteins.
Materials and Methods
Cloning of the FOXC2 DBD expression constructs and purification of the proteins were previously reported.10 The DNA encoding the human FOXC2 protein (amino acid residues 60–198) was inserted between the Nde I and EcoR I sites of a modified pET28b plasmid containing an N-terminal six histidine and a maltose-binding protein tag, cleavable using tobacco etch virus (TEV) protease. For protein expression, Escherichia coli BL21 (DE3)-RIPL cells were transformed with the FOXC2 DBD expression construct and grown in Luria broth. Protein expression was induced with 0.8 mM of β-d-1-thiogalactopyranoside for 4 h at 37 °C.
The FOXC2 DBD protein was purified using a standard affinity chromatography method. First, cells were lysed by sonicating for 4 min in 50 mM Tris–HCl pH 7.0, 150 mM NaCl, 20% glycerol (v/w), 2 mM MgCl2, and 2 mM β-mercaptoethanol (BME). A protease inhibitor cocktail tablet (Roche Life Science) and 0.2% polyethyleneimine were added to the lysed sample. The cell lysate was clarified by centrifugation at 35 000g for 30 min and applied to cobalt-charged sepharose beads (GE Healthcare). The protein was eluted with 750 mM imidazole. To remove the bulky fusion tag, the eluted fractions were incubated with TEV protease during an overnight dialysis step against a buffer composed of 50 mM Tris–HCl (pH 7.0), 100 mM NaCl, 12% glycerol (v/w), 2 mM MgCl2, and 2 mM BME at 4 °C. The protein was further purified by cation exchange chromatography using a Hi-Trap SP HP column (GE Healthcare). The cleaved proteins were eluted by a linear NaCl gradient from 50 mM to 1 M at pH 7.0.
The oligonucleotides for cocrystallization experiments were synthesized (Sigma Genosys) and annealed in 10 mM Tris pH 8.0, 100 mM NaCl, and 10 mM MgCl2, first by heating the solution to 95 °C for 10 min and then cooling down slowly to room temperature in a water bath.
For the protein–DNA complex purification, the double-stranded DNA and the purified FOXC2 DBD protein were mixed roughly in the molar ratio of 1:1.5. The protein–DNA complex was purified by size-exclusion chromatography using a Superdex 75 column (GE Healthcare) in a gel filtration buffer containing 50 mM Tris pH 7.0, 0.15 M NaCl, 2 mM BME, and 5% glycerol. The purified ternary complex was analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, concentrated to the final protein concentration of 10 mg/mL, and stored at −80 °C.
Crystallization, X-ray Data Collection, and Structure Determination
For crystallization, the purified complex of FOXC2–DNA was screened by the sparse matrix with crystallization drops containing 1 μL of protein–DNA sample and 1 μL of crystallization solution at room temperature. Plate-shaped crystals were obtained from well solutions containing 100 mM sodium acetate pH 4.6, 200 mM (NH4)2SO4, 25% w/v poly(ethylene glycol) (PEG) 2000 monomethyl ether (MME), and 10 mM BME. For data collection, the FOXC2 DBD–DNA crystals were cryoprotected in 100 mM sodium acetate pH 4.6, 200 mM (NH4)2SO4, 25% w/v PEG 2000 MME, 10 mM BME, and 30% glycerol and flash-frozen in liquid nitrogen.
The thin plate-shaped crystals of the FOXC2–DNA complex diffracted to 3.06 Å resolution at APS 19-ID beamline in Argonne National Laboratory synchrotron sources. The crystal belongs to the monoclinic C2 space group. Based on the unit cell dimensions and the molecular mass of the FOXC2 DBD/DNA complex, 2 DBD domains and a 21 bp double-stranded DNA were expected in an asymmetric unit with a solvent content of 60% (VM = 2.74 Å3/Da).44 The structure was determined by molecular replacement using the program Phaser45 and the crystal structure of the FOXA2 (PDB ID: 5X07) as a search model.46,47 The initial search revealed one unambiguous solution for the FOXC2–DNA complex. When the model was positioned in a unit cell, densities for the full DNA and the additional FOXC2 were visible. Subsequent iterative rounds of structural refinement were carried out using the PHENIX suite, followed by manual rebuilding using the program COOT.45,48,49 The final model was validated using Molprobity.50
Electrophoretic Mobility Shift Assay (EMSA)
The electrophoretic mobility shift assay was performed using gradient concentrations of purified FOXC2 DBD with a 16 bp DNA (caaggTAAACAaacca) containing the consensus motif on a 5% native polyacrylamide gel. The FOXC2 DBD was first diluted with a gel filtration buffer to the final concentrations of 0, 5, 10, 20, 50, 100, 150, 300, and 600 μM. For the EMSA sample preparation, 1 μM DNA and EMSA buffer containing 50 mM Tris pH 7.0, 5 mM MgCl2, 150 mM NaCl, 2 mM BME, 5% glycerol, and 10 μg/mL BSA were used. Five microliters of the protein sample were first premixed with 1 μL of DNA and 4 μL of EMSA buffer and incubated on ice for 20 min. The mixed samples were electrophoresed on an equilibrated native gel for 35 min and stained with SYBR Green from the EMSA kit (Invitrogen). The intensity measurement of the signals was performed using ImageJ.51
Calculation of Intraprotein and Protein–DNA Interaction Energies
Six FOXC2 mutants, I85N, W116R, N118K, R121C, R121H, and S125L, were modeled based on the FOXC2–DNA structure using the program COOT.49 The wild type and the mutant models were energy-minimized, and the intraprotein and protein–DNA interaction energies were calculated using the program FoldX.35
Acknowledgments
This work was supported by the Research Enhancement Fund of the University of Texas at Dallas (H.-J.N.). The authors thank Dr. Brian Black for the expression constructs and advice; the staff at the ID19 beamline of Advanced Photon Source for assistance during the X-ray data collection; and Dr. Yi Li and Chance Nowak for critical reading of the manuscript. The atomic coordinates and the structural factors were deposited in the Protein Data Bank as entry 6O3T.
Glossary
- A
adenine
- C
cytosine
- EMSA
electrophoretic mobility shift assay
- G
guanine
- T
thymidine
- ETS
E-twenty-six
- ETV2
ETS variant 2
- DBD
DNA binding domain
- BME
β-mercaptoethanol
- PEG
poly(ethylene glycol)
- MME
monomethyl ether
Author Present Address
‡ Department of Chemistry and Biotechnology, Swinburne University of Technology, Hawthorn, VIC 3122, Australia (A.J.).
Author Present Address
† Orca Biosystems Inc, 3475 Edison way, Menlo Park, California 94025, United States (L.P.).
Author Contributions
The manuscript was written through the contributions of all authors. All authors approved the final version of the manuscript.
The authors declare no competing financial interest.
References
- Mani S. A.; Guo W.; Liao M.-J.; Eaton E. N.; Ayyanan A.; Zhou A. Y.; Brooks M.; Reinhard F.; Zhang C. C.; Shipitsin M.; Campbell L. L.; Polyak K.; Brisken C.; Yang J.; Weinberg R. A. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell 2008, 133, 704–715. 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani S. A.; Yang J.; Brooks M.; Schwaninger G.; Zhou A.; Miura N.; Kutok J. L.; Hartwell K.; Richardson A. L.; Weinberg R. A. Mesenchyme Forkhead 1 (FOXC2) plays a key role in metastasis and is associated with aggressive basal-like breast cancers. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 10069–10074. 10.1073/pnas.0703900104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollier B. G.; Tinnirello A. A.; Werden S. J.; Evans K. W.; Taube J. H.; Sarkar T. R.; Sphyris N.; Shariati M.; Kumar S. V.; Battula V. L.; Herschkowitz J. I.; Guerra R.; Chang J. T.; Miura N.; Rosen J. M.; Mani S. A. FOXC2 expression links epithelial-mesenchymal transition and stem cell properties in breast cancer. Cancer Res. 2013, 73, 1981–1992. 10.1158/0008-5472.CAN-12-2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Craene B.; Berx G. Regulatory networks defining EMT during cancer initiation and progression. Nat. Rev. Cancer 2013, 13, 97–110. 10.1038/nrc3447. [DOI] [PubMed] [Google Scholar]
- Kume T. The Role of FoxC2 Transcription Factor in Tumor Angiogenesis. J. Oncol. 2012, 2012, 204593 10.1155/2012/204593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N.; Mimori K.; Yokobori T.; Sudo T.; Tanaka F.; Shibata K.; Ishii H.; Doki Y.; Mori M. FOXC2 is a Novel Prognostic Factor in Human Esophageal Squamous Cell Carcinoma. Ann. Surg. Oncol. 2011, 18, 535–542. 10.1245/s10434-010-1274-y. [DOI] [PubMed] [Google Scholar]
- Sano H.; LeBoeuf J. P.; Novitsky S. V.; Seo S.; Zaja-Milatovic S.; Dikov M. M.; Kume T. The Foxc2 transcription factor regulates tumor angiogenesis. Biochem. Biophys. Res. Commun. 2010, 392, 201. 10.1016/j.bbrc.2010.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kume T. The Role of FoxC2 Transcription Factor in Tumor Angiogenesis. J. Oncol. 2012, 2012, 204593 10.1155/2012/204593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paranjape A. N.; Soundararajan R.; Werden S. J.; Joseph R.; Taube J. H.; Liu H.; Rodriguez-Canales J.; Sphyris N.; Wistuba I.; Miura N.; Dhillon J.; Mahajan N.; Mahajan K.; Chang J. T.; Ittmann M.; Maity S. N.; Logothetis C.; Tang D. G.; Mani S. A. Inhibition of FOXC2 restores epithelial phenotype and drug sensitivity in prostate cancer cells with stem-cell properties. Oncogene 2016, 35, 5963. 10.1038/onc.2015.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castaneda M.; Chen L.; Pradhan L.; Li S.; Zein R.; Lee Y.; Lim H. S.; Nam H. J.; Lee J. A Forkhead Box Protein C2 Inhibitor: Targeting Epithelial-Mesenchymal Transition and Cancer Metastasis. ChemBioChem 2018, 19, 1359–1364. 10.1002/cbic.201800022. [DOI] [PubMed] [Google Scholar]
- Myatt S. S.; Lam E. W. The emerging roles of forkhead box (Fox) proteins in cancer. Nat. Rev. Cancer 2007, 7, 847–859. 10.1038/nrc2223. [DOI] [PubMed] [Google Scholar]
- Jackson B. C.; Carpenter C.; Nebert D. W.; Vasiliou V. Update of human and mouse forkhead box (FOX) gene families. Hum. Genomics 2010, 4, 345–352. 10.1186/1479-7364-4-5-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaestner K. H.; Knochel W.; Martinez D. E. Unified nomenclature for the winged helix/forkhead transcription factors. Genes Dev. 2000, 14, 142–146. 10.1101/gad.14.2.142. [DOI] [PubMed] [Google Scholar]
- Benayoun B. A.; Caburet S.; Veitia R. A. Forkhead transcription factors: key players in health and disease. Trends Genet. 2011, 27, 224–232. 10.1016/j.tig.2011.03.003. [DOI] [PubMed] [Google Scholar]
- Lehmann O. J.; Sowden J. C.; Carlsson P.; Jordan T.; Bhattacharya S. S. Fox’s in development and disease. Trends Genet. 2003, 19, 339–344. 10.1016/S0168-9525(03)00111-2. [DOI] [PubMed] [Google Scholar]
- Sargent C.; Bauer J.; Khalil M.; Filmore P.; Bernas M.; Witte M.; Pearson M. P.; Erickson R. P. A five generation family with a novel mutation in FOXC2 and lymphedema worsening to hydrops in the youngest generation. Am. J. Med. Genet., Part A 2014, 164, 2802–2807. 10.1002/ajmg.a.36736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellinger M. T.; Thome K.; Bernas M. J.; Erickson R. P.; Witte M. H. Novel FOXC2 missense mutation identified in patient with lymphedema-distichiasis syndrome and review. Lymphology 2008, 41, 98–102. [PubMed] [Google Scholar]
- Fang J.; Dagenais S. L.; Erickson R. P.; Arlt M. F.; Glynn M. W.; Gorski J. L.; Seaver L. H.; Glover T. W. Mutations in FOXC2 (MFH-1), a forkhead family transcription factor, are responsible for the hereditary lymphedema-distichiasis syndrome. Am. J. Hum. Genet. 2000, 67, 1382–1388. 10.1086/316915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson P.; Mahlapuu M. Forkhead transcription factors: key players in development and metabolism. Dev. Biol. 2002, 250, 1–23. 10.1006/dbio.2002.0780. [DOI] [PubMed] [Google Scholar]
- Golden D.; Cantley L. G. Casein kinase 2 prevents mesenchymal transformation by maintaining Foxc2 in the cytoplasm. Oncogene 2015, 34, 4702–4712. 10.1038/onc.2014.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu Y. P.; Chang C. H.; Shiu J. H.; Chang Y. T.; Chen C. Y.; Chuang W. J. Solution structure and backbone dynamics of the DNA-binding domain of FOXP1: insight into its domain swapping and DNA binding. Protein Sci. 2011, 20, 908–924. 10.1002/pro.626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin C.; Marsden I.; Chen X.; Liao X. Dynamic DNA contacts observed in the NMR structure of winged helix protein-DNA complex. J. Mol. Biol. 1999, 289, 683–690. 10.1006/jmbi.1999.2819. [DOI] [PubMed] [Google Scholar]
- Lee G. I.; Ding Z.; Walker J. C.; Van Doren S. R. NMR structure of the forkhead-associated domain from the Arabidopsis receptor kinase-associated protein phosphatase. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 11261–11266. 10.1073/pnas.2031918100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P. P.; Chen Y. C.; Li C.; Hsieh Y. H.; Chen S. W.; Chen S. H.; Jeng W. Y.; Chuang W. J. Solution structure of the DNA-binding domain of interleukin enhancer binding factor 1 (FOXK1a). Proteins 2002, 49, 543–553. 10.1002/prot.10227. [DOI] [PubMed] [Google Scholar]
- Sheng W.; Rance M.; Liao X. Structure comparison of two conserved HNF-3/fkh proteins HFH-1 and genesis indicates the existence of folding differences in their complexes with a DNA binding sequence. Biochemistry 2002, 41, 3286–3293. 10.1021/bi011908k. [DOI] [PubMed] [Google Scholar]
- Weigelt J.; Climent I.; Dahlman-Wright K.; Wikstrom M. Solution structure of the DNA binding domain of the human forkhead transcription factor AFX (FOXO4). Biochemistry 2001, 40, 5861–5869. 10.1021/bi001663w. [DOI] [PubMed] [Google Scholar]
- van Dongen M. J.; Cederberg A.; Carlsson P.; Enerback S.; Wikstrom M. Solution structure and dynamics of the DNA-binding domain of the adipocyte-transcription factor FREAC-11. J. Mol. Biol. 2000, 296, 351–359. 10.1006/jmbi.1999.3476. [DOI] [PubMed] [Google Scholar]
- Bandukwala H. S.; Wu Y.; Feuerer M.; Chen Y.; Barboza B.; Ghosh S.; Stroud J. C.; Benoist C.; Mathis D.; Rao A.; Chen L. Structure of a domain-swapped FOXP3 dimer on DNA and its function in regulatory T cells. Immunity 2011, 34, 479–491. 10.1016/j.immuni.2011.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boura E.; Rezabkova L.; Brynda J.; Obsilova V.; Obsil T. Structure of the human FOXO4-DBD-DNA complex at 1.9 A resolution reveals new details of FOXO binding to the DNA. Acta Crystallogr., Sect. D: Struct. Biol. 2010, 66, 1351–1357. 10.1107/S0907444910042228. [DOI] [PubMed] [Google Scholar]
- Littler D. R.; Alvarez-Fernandez M.; Stein A.; Hibbert R. G.; Heidebrecht T.; Aloy P.; Medema R. H.; Perrakis A. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 2010, 38, 4527–4538. 10.1093/nar/gkq194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud J. C.; Wu Y.; Bates D. L.; Han A.; Nowick K.; Paabo S.; Tong H.; Chen L. Structure of the forkhead domain of FOXP2 bound to DNA. Structure 2006, 14, 159–166. 10.1016/j.str.2005.10.005. [DOI] [PubMed] [Google Scholar]
- Tsai K. L.; Sun Y. J.; Huang C. Y.; Yang J. Y.; Hung M. C.; Hsiao C. D. Crystal structure of the human FOXO3a-DBD/DNA complex suggests the effects of post-translational modification. Nucleic Acids Res. 2007, 35, 6984–6994. 10.1093/nar/gkm703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Wei H.; Li J.; Liang X.; Dai S.; Jiang L.; Guo M.; Qu L.; Chen Z.; Chen L.; Chen Y. Structural basis for DNA recognition by FOXC2. Nucleic Acids Res. 2019, 47, 3752–3764. 10.1093/nar/gkz077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan L.; Nam H. J. NuProPlot: nucleic acid and protein interaction analysis and plotting program. Acta Crystallogr., Sect. D: Struct. Biol. 2015, 71, 667–674. 10.1107/S1399004715000139. [DOI] [PubMed] [Google Scholar]
- Schymkowitz J.; Borg J.; Stricher F.; Nys R.; Rousseau F.; Serrano L. The FoldX web server: an online force field. Nucleic Acids Res. 2005, 33, W382–W388. 10.1093/nar/gki387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogura Y.; Fujibayashi S.; Iida A.; Kou I.; Nakajima M.; Okada E.; Toyama Y.; Iwanami A.; Ishii K.; Nakamura M.; Matsumoto M.; Ikegawa S. A novel FOXC2 mutation in spinal extradural arachnoid cyst. Hum. Genome Var. 2015, 2, 15032 10.1038/hgv.2015.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Carpintero R.; Dominguez P.; Núñez M. T.; Patiño-García A. Spinal extradural arachnoid cysts in lymphedema-distichiasis syndrome. Genet. Med. 2010, 12, 532–535. 10.1097/GIM.0b013e3181e5c7ea. [DOI] [PubMed] [Google Scholar]
- van Steensel M. A.; Damstra R. J.; Heitink M. V.; Bladergroen R. S.; Veraart J.; Steijlen P. M.; van Geel M. Novel missense mutations in the FOXC2 gene alter transcriptional activity. Hum. Mutat. 2009, 30, E1002–E1009. 10.1002/humu.21127. [DOI] [PubMed] [Google Scholar]
- Siggers T. W.; Honig B. Structure-based prediction of C2H2 zinc-finger binding specificity: sensitivity to docking geometry. Nucleic Acids Res. 2007, 35, 1085–1097. 10.1093/nar/gkl1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Val S.; Chi N. C.; Meadows S. M.; Minovitsky S.; Anderson J. P.; Harris I. S.; Ehlers M. L.; Agarwal P.; Visel A.; Xu S. M.; Pennacchio L. A.; Dubchak I.; Krieg P. A.; Stainier D. Y.; Black B. L. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell 2008, 135, 1053–1064. 10.1016/j.cell.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choy W. W.; Datta D.; Geiger C. A.; Birrane G.; Grant M. A. Crystallization and preliminary X-ray analysis of a complex of the FOXO1 and Ets1 DNA-binding domains and DNA. Acta Crystallogr., Sect. F: Struct. Biol. Commun. 2014, 70, 44–48. 10.1107/S2053230X13024795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex N.; Peitsch M. C. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Pradhan L.; Gopal S.; Li S.; Ashur S.; Suryanarayanan S.; Kasahara H.; Nam H. J. Intermolecular Interactions of Cardiac Transcription Factors NKX2.5 and TBX5. Biochemistry 2016, 55, 1702–1710. 10.1021/acs.biochem.6b00171. [DOI] [PubMed] [Google Scholar]
- Matthews B. W. Solvent content of protein crystals. J. Mol. Biol. 1968, 33, 491–497. 10.1016/0022-2836(68)90205-2. [DOI] [PubMed] [Google Scholar]
- McCoy A. J.; Grosse-Kunstleve R. W.; Adams P. D.; Winn M. D.; Storoni L. C.; Read R. J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan L.; Genis C.; Scone P.; Weinberg E. O.; Kasahara H.; Nam H. J. Crystal structure of the human NKX2.5 homeodomain in complex with DNA target. Biochemistry 2012, 51, 6312–6319. 10.1021/bi300849c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnimann C. U.; Ptchelkine D.; Grimm C.; Muller C. W. Structural basis of TBX5-DNA recognition: the T-box domain in its DNA-bound and -unbound form. J. Mol. Biol. 2010, 400, 71–81. 10.1016/j.jmb.2010.04.052. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Struct. Biol. 2010, 66, 213–221. 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Struct. Biol. 2004, 60, 2126–2132. 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- Chen V. B.; Arendall W. B. 3rd; Headd J. J.; Keedy D. A.; Immormino R. M.; Kapral G. J.; Murray L. W.; Richardson J. S.; Richardson D. C. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr., Sect. D: Struct. Biol. 2010, 66, 12–21. 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A.; Rasband W. S.; Eliceiri K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]