

Abstract
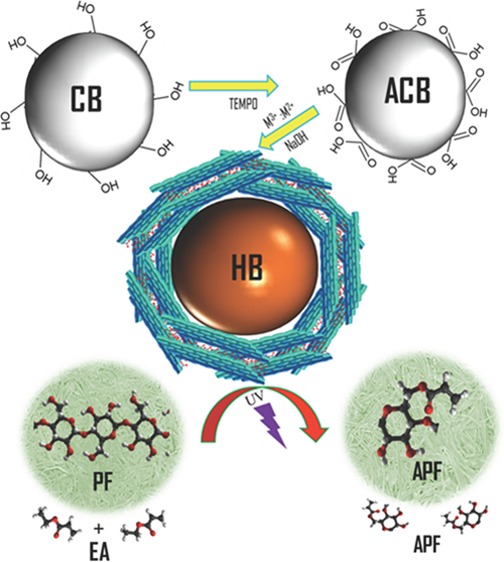
Cellulose-based materials are very attractive for emerging bioeconomy as they are renewable, inexpensive, and environmentally friendly. Cellulose beads are spherical and porous and can be highly engineered to be used as catalyst support material. This type of inorganic catalysts is cost-effective and suitable for multiple re-usage and has been rarely explored in cellulose reaction research. In this work, NiFe-layered double hydroxide (LDH) was tailor-made in situ on anionic cellulose beads to form a hybrid, supported photocatalyst for the first time. The hybrid beads were prepared in a size larger than the pulp fibers in order to make the catalysis reaction heterogeneous in nature. Hydrophilic pulp fibers were converted into hydrophobic pulp by photocatalytic topochemical grafting of ethyl acrylate using the LDH-cellulose bead catalyst. The approach identified for the modification of the pulp fibers is the “hydrogen abstraction–UV photografting” because the low-energy, UV radiation-induced grafting offers advantages, such as a reduced degradation of the backbone polymer and a control over the grafting reaction. After grafting, the pulp fibers showed increased water repellency and unaltered thermal stability, indicating the hydrophobic, plasticizing nature of the pulp, which in turn accounts for its thermoformable behavior. These acrylated pulp fibers can be further designed/customized for waterproof or oil absorption applications.
Introduction
Because of rising environment concerns and society demands, bio-based materials are of great interest to the scientists and industries. Especially cellulose fiber-based products are much attractive owing to their properties and abundance in nature. Many industries convert cellulose into value-added consumer products such as absorbents, paper, textiles, and fillers for pharmaceutical tablets. Nevertheless, all these employ cellulose in its natural hydrophilic form. However, when cellulose is made hydrophobic by chemical treatments, its range of applicability widens. This brings more opportunity in research and development, production followed by consumer supply. Hydrophobic cellulose has very big potential in the food packaging industry in the near future and it could replace “fossil-derived chemicals”-based packaging polymers.1−3 Pulp fibers (PFs) in the form of translucent see-through thin films for covering and wrapping of food products is one of the desired target applications. Also, this type of bioplastic material can be extruded and molded into container bottles or vessels to store liquids, food, water-resistant building insulation material, stationary phase in column or thin layer chromatography employing nonaqueous (organic) solvents, oil-absorbent sponges, water-repellent papers, and nonwoven fabrics.
There are literature reports where hydrophobic cellulose composites are derived from physical grafting/mixture interactions on cellulose by water-repelling organic moieties.4−6 However, these composites are made by using either expensive, time-consuming synthetic routes, or environment polluting stoichiometric reagents. In addition, their technical specifications require further improvement in order to compete with other nonrenewable materials used for the same applications. Hence, there is a need to produce these functional materials by simple, inexpensive, eco-friendly chemical routes with high turnover and target specifications. Our research meets this need as our protocol utilizes (i) water as solvent medium, (ii) kraft PFs without any treatment, (iii) abundantly available bio–geo mineral-based catalyst, to yield target hydrophobicity and thermal stability specification values. Further, our novel catalyst system was heterogenized in order to separate the catalyst particles from the reaction mixture after grafting. This means that the physical size/shape of the catalyst particles was considerably larger for easy filtration from the solvent medium. Hence, our catalyst material can be regenerated and recycled and our synthesis and the final product exhibits promising qualities that might fulfill the needs of industry.
In the present study, ethyl acrylate (EA) was coupled with PFs via dissociative esterification to add value to cellulose/pulp in terms of its induced hydrophobic properties. EA or acrylic acid ethyl ester was technically used as a precursor in many polymers, textiles, nonwoven fabric and as additives in paints. EA is a clear, colorless liquid with relatively low vapor pressure, readily miscible with most of the organic solvents and sparingly soluble in water. Structure-wise, EA has a five carbon chain with an olefinic π bond in one end and two paraffinic carbons at the other end with a connecting functional group, −COO– (ester).7 This makes its hydrogen bond acceptor count 2 and topological polar surface area (PSA) of 23 Å2. The hydrogen bond acceptor count indicates the number of electronegative centers with a lone pair of electrons readily available to form bonding with the hydrogen of another molecule. The surface area covering the polar atoms (O, N, S, etc.) in the molecule gives the PSA in angstrom units. If a molecule possesses more polarizable groups, then it would have more PSA. EA possess the “O”-induced polar surface. These properties are thoroughly exploited in topochemical modification of PFs to bring hydrophobicity in fibers as against its intrinsic hydrophilic nature bestowed by −OH groups. The topochemical modification of PFs was carried out in both noncatalyzed and catalyzed routes under irradiated and nonirradiated conditions.
The catalyst we aimed to design to facilitate EA grafting on pulp was an inorganic material, named layered double hydroxide (LDH). Naturally occurring mineral hydrotalcite [Mg6Al2(OH)16]CO3·4H2O] is the best example for a LDH compound. It forms by charge neutralizing the cationic hydroxide layers by mobile anions in interlayer galleries. LDH belongs to the family of lamellar ionic solids possessing superior properties like crystallinity, tunable surface area–size–porosity, cation–anion exchangeability, swelling, and memory effect. These properties make LDH an interesting candidate for exploring in the area of catalysis, photocatalysis, adsorbents, composites, and medicine. LDH is identified for its efficient and selective heterogeneous/photocatalytic application in chemical syntheses, decomposition of pollutants, and production of energy.8−17 Sobhana and coworkers31 synthesized NiFe LDH by the co-precipitation method and carried out decomposition of orange II dye, a water pollutant, by using NiFe LDH as a photocatalyst. Gong et al. employed Ni–Fe LDH nanoplates in electrocatalysis to oxidize water, as oxygen evolution is an important side pathway in water splitting and metal–air batteries.18 NiFe LDH composed with carbon nitride has been proven an efficient photocatalyst for water oxidation and reduction reactions,19 whereas NiFe LDH fabricated with CdS liberated hydrogen from aqueous methanol solution photocatalytically.20 Ni nanoparticles decorated NiFe LDH performed as a bifunctional catalyst in oxygen evolution reaction and urea oxidation reaction under electrochemical conditions.21
In the present work, LDH was chosen to be employed as a photocatalyst for the following reasons: (i) semiconducting, (ii) ability to initiate hydrogen abstraction for radical formation, and (iii) potential to transfer photogenerated electrons to the reactant surface. Photocatalysis can be done in either heterogeneous condition or homogenous environment. In homogeneous phase reactions, separation of the catalyst (i.e., LDH) from the product is often impossible, whereas under heterogeneous conditions, the solid catalyst particles can be separated by simple filtration. Therefore, modification of the catalyst turned to be very important and a challenging step as both the reactant (PFs) and catalyst (LDH particles) existed in the solid phase. Heterogeneous catalysts have advantages owing to their ease of separation, reusability, and cost-effectiveness. In order to make these types of catalysts, catalytically active nano-/microcomponent should be anchored/supported on an inert bulk solid matrix. In the present case, the size difference between the supported catalyst and the reactant PFs must be high, as both are in the solid state.
Cellulose has gained increasing interest in materials research because of its ecological properties, such as its natural abundance, biodegradability, and renewability. Cellulose dissolution and regeneration into definite physical forms/shapes has been a focus in recent years.22,23 The bead form of cellulose is one of the physical forms, which could show numerous applications such as stationary phase in chromatographic column, pharmaceutical fillers, absorbent, structure-directing agent, and so forth.24 We realized that the cellulose bead (CB) could act as catalyst support or catalyst anchoring site matrix so that the fine particles of LDH can be immobilized on it and used in heterogeneous catalysis.
The most important feature in our study is that cellulose in PFs has been derivatized using an organic moiety (EA) through proper chemical bonding as opposed with widely reported weak physical bonding/interaction (van der Waals force). The chemistry between host cellulosic −OH groups and guest organic moiety in functional composites is not well understood and reported. Thus, our investigation paves way to address the intricacies of bonding, need of having proper chemical composites, and robust properties in material applications. Further, we have attempted to plasticize/hydrophobize cellulose in its original form (PF in water medium) instead of first derivatizing it into some other form, for example, ether, before functionalizing with EA. There are many reports in which acrylic acid, acrylonitrile, EA was grafted on cellulose ethers. However, the bonding is not of chemical type and it is merely a physical mixture of cellulose and the grafted molecule.25−27 We believe that super-hydrophobic cellulose acrylate derivative material will be a potential renewable substitute for petroleum-based nonrenewable poly (-olefin, ester) plastics.
Results and Discussion
Hybrid Bead Catalysts
An eco-friendly solvent system (alkali/urea aqueous solution) was used to dissolve microcrystalline cellulose, and the dissolved cellulose was extruded through a needle nozzle into a regenerative acid medium for coagulation shaping into spherical beads (CB). Further, the beads were subjected to (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) oxidation.
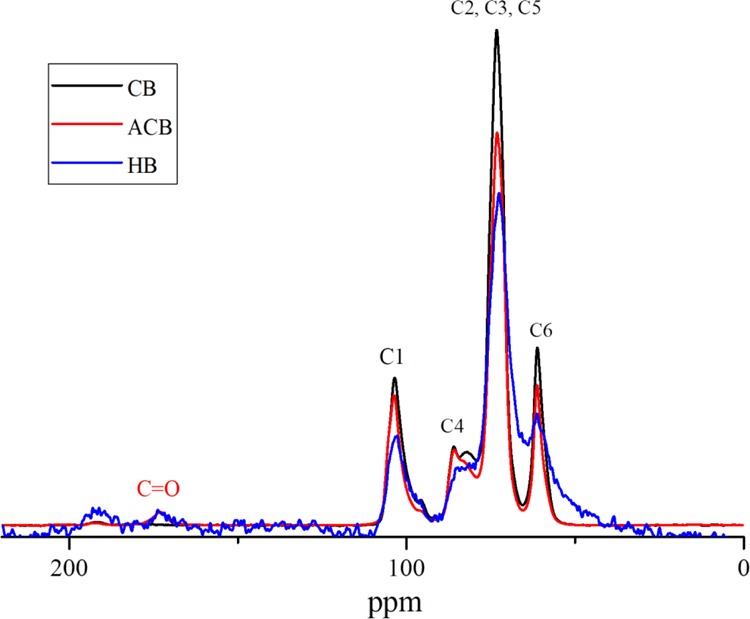
Oxidation brings anionic charges on the beads via the introduction of the −COOH group in place of −CH2OH in cellulose moiety. Oxidation of the beads anionic CB (ACB) was confirmed with reference to the unoxidized CBs via Raman spectroscopy. The oxidized CBs exhibit peaks at 1614 cm–1 corresponding to −COOH out of phase stretching frequency28,29 (Figure 1). These ACB were used as support material to anchor catalytically active LDH nanoparticles, which was prepared from mixed metal salt solution of Ni/Fe ratio 3:1. Heterogenization of the LDH particle catalyst is vital for better separation of catalyst particles from the PF solution at the end of the reaction (Figure S1). Hence, we chose large-sized CB material as a matrix to immobilize the photoactive NiFe LDH platelets. This type of novel hybrid structures was used as heterogeneous catalysts for the first time. Powder X-ray diffraction (PXRD), attenuated total reflectance (ATR)–Fourier transform infrared (FTIR), NMR, and scanning electron microscopy (SEM)–energy-dispersive X-ray spectroscopy (EDX) measurements proved the successful synthesis and growth of LDH on ACB.
Figure 1.
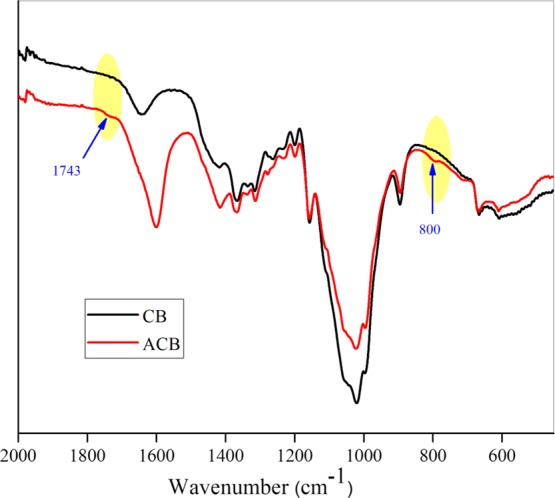
Raman spectra of CBs.
XRD measurements were studied to ensure the authenticity of the structure and purity of the synthesized materials. Figure 2 shows the powder XRD patterns of the CBs obtained from regenerated cellulose (CB), ACBs, LDH–ACB hybrid beads (HBs). The extruded beads (CB) and oxidized CBs (ACB) showed three different peaks at 11.22°, 19.17°, and 20.24°, which were indexed as the (101), (101̅), and (002) planes of the cellulose II structure. This confirmed the crystallinity of the bead-shaped regenerated and oxidized cellulose.30 The unsupported LDH material usually displays the reflections at 11.9°, 24°, 33°, 38°, and 42° corresponding to (003), (006), (012), (015), and (018) planes, respectively, which suggested the formation of NiFe LDH.31 These strong reflections arising from the layers of LDH demonstrated the crystalline nature of LDH particles.
Figure 2.
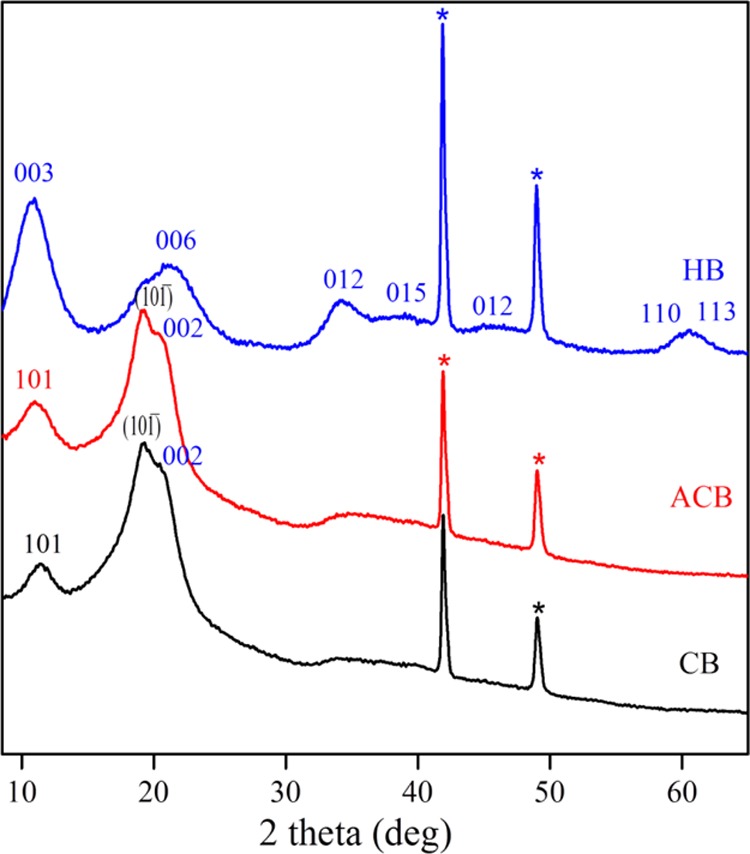
XRD of CBs. * Peaks from the copper sample holder (to be ignored).
The PXRD of the HB material exhibited plane reflections attributable to both ACB and LDH discussed above. The diffraction peak appeared at 10.9° (003 planes) was the peaks overlap aroused from both ACB and LDH. Thus, the original peak for cellulose at 11.22° merged with the LDH diffraction peak (Figure 2), suggesting enhanced LDH coverage on CBs. The other peaks from LDH showed a layered structure of rhombohedral symmetry in agreement with the hydrotalcite-like structure reference, JCPDS no. 40-0215. This indicated the co-existence of NiFe LDHs and cellulose in the hybrid material. Between these two components, cellulose was obtained commercially as microcrystalline cellulose, whereas NiFe LDH was synthesized in situ (in ACB–water dispersion medium) as well as an individual control sample. Therefore, the purity of the synthesized LDH should be vindicated.
As elaborated in the experimental part, LDH was synthesized from its precursors, nickel(II) nitrate hexahydrate Ni(NO3)2·6H2O and iron(III) nitrate nonahydrate Fe(NO3)3·9H2O in the presence of Na2CO3, NaOH in ACB-dispersed water solution. There is always a possibility of anion exchange in LDH intergalleries initiated by foreign anionic species; in the present case, it is anionic cellulose. The XRD data suggested that there was no such intercalation, as the (003) reflection from HB did not shift to the lower angle side (Figure 2). Hence, anionic cellulose was not dissolved in the metal salt medium and subsequently not displaced the already existing anions (CO32–, NO3–) in nascent LDH. The reason could be the steric hindrance between the brucite layers of LDH, which made the bulgy cellulose polymer unable to penetrate inside and undergo anion exchange. Thus, electrostatic/ionic type chemical bonding was avoided between LDH and ACB. This implied only the formation of physical bonding between LDH–ACB, facilitated by van der Waals forces. These forces were the outcome of hydrogen bonding between the −OH groups of LDH and −COOH groups of ACB. Another thing in regard with purity is that, the co-precipitation reaction might also form undesirable byproducts like Ni(OH)3, Fe(OH)2–3, Ni(CO3)3, Fe(CO3)3, Ni2O3, FeO, and Fe2O3. However, their characteristic diffraction peaks were not found in XRD spectra. Hence, no other detectable impurity phases were evidenced during the characterization of control LDH and HB materials.
The sharp narrow peaks/peak heights of 003 and 006 reflections revealed the smaller crystallite size of LDH particles.32 The basal spacing value for NiFe LDH in HB was calculated as 8.10 Å (Table S1), and this value was in good agreement with the literature.31 This higher value is due to the perpendicular orientation of the interlayer anion to the cationic lamellar sheets and their weaker electrostatic interaction.33,34 The hybrid formation from LDH and ACB was allowed under air atmosphere and therefore, the majority of the anions occupying the interlayer will be carbonates with the inclusion of some nitrate anions. The interference of nitrate anions is inevitable from the precursor solution that contributes to an increase in the basal spacing between the layers.31 The distance between the metals in the cationic layer was calculated to be 3.02 Å using the formula “a = 2 × d(110)” (Table S1). The XRD reflections from HB were slightly broader in comparison to the highly ordered structure of LDH that we reported earlier.31 This is because of the higher octahedral ionic radius of Ni2+ (0.72 Å) and Fe3+ (0.69 Å) occupying the brucite-like sheets. The peaks at 110 and 113 were merged into one peak, signifying the arrangement of metal cations in the lamellar layers.34 The modification of the CBs with TEMPO oxidation has thus apparently resulted in the hybrid formation between LDH and cellulose.
The ATR–FTIR studies (Figure 3) conveyed the information on the nature of bonding in CB, ACB, and LDH synthesized in the presence of ACBs (LDH–ACB), that is, HBs. As a known fact, the cellulose polymer possesses primary and secondary hydroxyl groups (1°, 2° −OH), and the repetitive monomer units of carbon rings are connected via an ether bond (−O−). The TEMPO oxidation yields −COOH groups in place of 1°–OH groups and rest of the 2°–OH remain unaffected. The anionic CBs showed broadband in the region of 3500–3000 cm–1 corresponding to stretching of cellulosic −OH bonds and intramolecular hydrogen bonds (X–H). It is also a known fact that water molecules vibrate in the same region, hence the peak overlaps between the vibration frequencies of cellulosic −OH, X–H, and H2O molecules. The peaks at 2893 and 1422 cm–1 asserted the presence of amorphous as well as crystalline nature in ACBs. A narrow peak at and 1601 cm–1 was due to bending of −OH groups in adsorbed water. The typical 1155, 102, and 888 cm–1 vibrational frequencies were results of stretching of C–O–C glycosidic ether bond, ring C–C bond, and C–O–C stretching of β-(1 → 4)-glycosidic linkages in cellulose molecule, respectively.35 The LDH-hybridized ACBs show similar vibrational frequencies, which are already observed at 3400 and 1611 cm–1 with plain ACBs. The intensity boost was offered by stretching vibration of free −OH groups in brucite layers and bending vibration of interlayer H2O molecules of LDH material. The unique peak at 1356 cm–1 was emerged by the asymmetric stretching of carbonate (CO3–) anions bridging between brucite layers. The vibration pattern at lower wavelengths 775, 573,, 479 cm–1 were characterized as O–M–O, M–OH, M–O–M bonds (M—Ni, Fe).31
Figure 3.
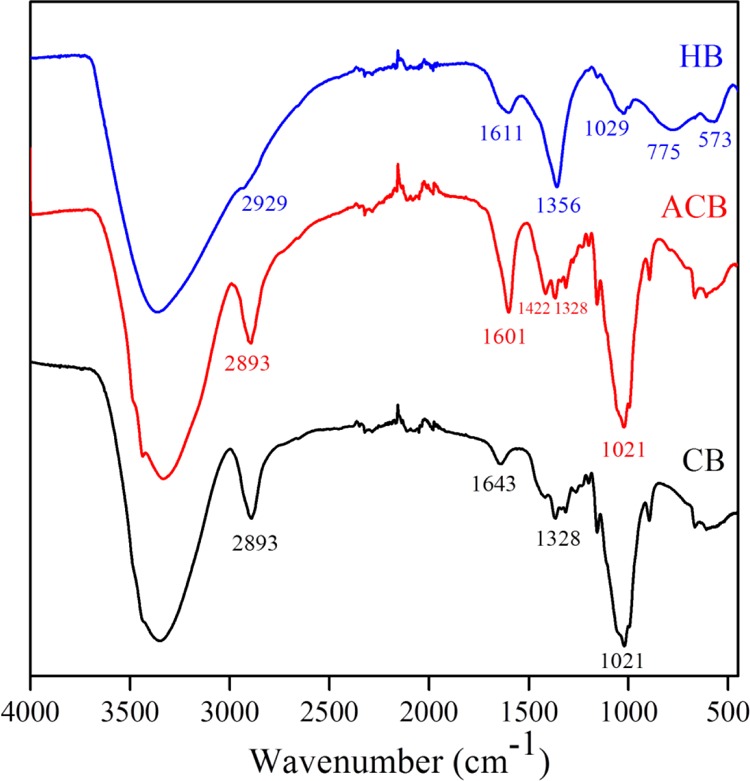
FTIR of the CBs.
The cross-polarization magic angle spinning Carbon-13 nuclear magnetic resonance (CP MAS 13C NMR) supplied the information that CB contains mainly amorphous cellulose. ACB has disclosed the carbonyl carbon signal from the carboxylic acid groups, which appeared at 173 ppm35 (Figure 4). The exact level of oxidation cannot be determined with CP MAS. The HB catalyst sample seemed to contain mainly inorganics, as the signals were very weak. Despite accumulating 30 000 scans, the spectrum looked noisy, and it was almost impossible to tell if the carbonyl signal had shifted at all.
Figure 4.
NMR spectra of CBs.
Figure 5 shows the SEM images of the surfaces of CBs and LDH-hybridized ACB (HB). The freshly prepared CBs show smooth, uniform, and bubble-like or cauliflower-like surface without any contaminant particles (Figure 5a,b). It was expected that when microcrystalline cellulose was dissolved in an alkali solution (NaOH, urea) and then neutralized in acidic medium (HNO3), it would have formed salts. Nevertheless, there was no sign of salt lumps/crystal formation across the scanned surfaces. The cross section of the CBs also revealed a clean uniform surface, which resembled the surface of scooped ice cream, whereas the morphology of the LDH-precipitated surface of ACBs (HB) looked rough and grainy (Figure 5d) as against CBs, for the air-dried sample. The high magnification image (Figure 5e) exhibited the formation of LDH crystals in definite shapes (e.g., blood platelets) on the surface of ACBs covering them completely. On the contrary, the cross section of HBs did not show any presence of LDH crystals (Figure 5f) and it looked similar to the cross section of CBs (Figure 5c). It should be noted here that the oxidation condition chosen for CBs was controlled in such a way that it can only oxidize the −CH2OH located on the external/outer surface of CBs. This way, the LDH deposition was tuned to be only on the outer surface of ACB via hydrogen bonding. Thus, the photocatalytically active LDH was made richly available on the outer surface of the HBs for catalysis. The morphology of the freeze-dried HB was rough (Figure 5g) at low magnification, whereas at high magnification, it looked like a porous weblike structure in plant walls and the pattern was uniform across the surface of the HBs (Figure 5h,i)
Figure 5.

SEM images of CBs.
EDX analysis was performed in parallel to scanning of morphology on the beads (CBs, HBs), in which HBs show a clear presence of Ni and Fe, that was absent on CBs (Figure 6).
Figure 6.

EDX spectra of CBs.
Topochemical Grafting on PFs
Cellulose-based hybrid materials comprising the inorganic component LDH were used as photon harvesting catalysts (HB) in order to bring novel functionalities (acrylate groups) in the cellulose chain. The empirical formula of LDH is written as [M1–x2+Nx3+(HO–)2]x+[(Xn–)x/n·yH2O]x−. The catalyst, as named, possesses a layered structure of repetitive positively charged brucite layers and interconnecting anionic lamellar layers in between. These layers contain −OH groups and interstitial crystalline H2O molecules which has the potential to interact with −OH-rich PFs and electronegatively (O=C–O−) polarizable EA.
A catalyst is a foreign substance, which increases the rate of the reaction when it is added to the reaction medium. A catalyst can either participate in a reaction through chemical bonds or just act as an active site, to reduce the activation energy barrier of a given reaction. Heterogeneous catalysts possess active sites in which the reactants bind/adsorb and are activated to undergo chemical transformations (bond breaking/making) followed by product desorption. Here, LDH facilitates the adsorption of PFs and EA on its surface through weak hydrogen bonding. The bonding was between LDH −OH groups and cellulosic −CH2OH, >CHOH or −O–, >C=O (PSA) of EA. In addition, LDH is a well-known photocatalyst, which can harvest photons from irradiated energy to its surface. Thus, LDH −OH groups and its surface utilized as active sites to induce PF and EA radicalization followed by esterification reaction.
The generation of radicals could occur by hydrogen abstraction from the −CH2OH group in the cellulose chain and [O=C)···(OC2H5] cleavage in EA as shown in Figure 7. The photogenerated cellulose radical and acryloyl radical on the LDH surface recombined to form the cellulose acrylate. This molecular route allows the newly designed catalyst to graft the hydrocarbon/organic moieties on the cellulose chain to achieve hydrophobicity and thermoformability by disrupting hydrogen bonds between the cellulose polymer chains.
Figure 7.

Scheme for the photocatalytic esterification of PFs.
The technique adopted for topochemical functionalization of PFs by EA was “hydrogen abstraction–UV photografting”.36 Here, UV radiation is the limiting factor for the reaction. Therefore, the reaction was controlled mostly like “stop and go” by switching on/off UV lamp. The reaction steps were carried out in dark, so that only the irradiated area undergone reaction and the dark areas remain unreacted. As in most cases, we believe that hydrogen abstraction is the rate-determining step. The abstraction of hydrogen preferred at the −OH on the C6 position is due to limited hindrance around the cellulose chain. In this case, the cellulose polymer was excited to become a macroradical. This was induced by the addition of LDH particles as a catalyst, which played their role as a binding active site and photosensitizer. The enhanced catalytic efficiency was achieved because of the superficial growth of LDH platelets over the anionic CBs, which offered more surface-exposed catalytic active sites for grafting of EA on pulp. In addition, there was no metal (Ni/Fe) leaching from LDH (catalyst) and LDH leaching from its anchoring support, ACBs. Hence, the catalyst was stable.
This photografting method showed advantages in several points such as (i) easy and controllable introduction of the graft moiety as the radiation is the limiting reagent for the reaction to happen, (ii) the bulk properties of the parent material (PFs) remains undamaged, (iii) ensures covalent attachment of the graft moiety and gives long-term chemical stability of introduced functional group in contrast to the physically mixed/coated (composite) material, (iv) the cost of radiation energy spent for UV radiation was lower than the ionizing radiation, (v) UV-mediated photocatalysis was environmentally friendly as the stoichiometric esterification reagents cause waste generation and pollution, and (vi) above all, the procedure itself is simple and facile.
Various factors affecting the grafting efficiency on the PFs was considered viz., homopolymerization, partitioning effect, and screening effect. Homopolymerization occurs when the repeating EA monomers chain grow without attaching to the PFs, that is, −EA to EA–. The partitioning effect occurs when there is a decreased concentration of EA available at the grafting site, which makes cellulose and EA remain unreacted. The screening effect arises when the PFs are not exposed to UV irradiation due to the increased concentration of PFs followed by congestion of fibers in solution medium. This was studied by optimizing the operation concentration as 500 mg/100 mL. Acidic treatment enhances grafting and hinders homopolymer formation in the case of homopolymerization and partitioning effect. However, the nature and choice of the acids are also important. Among many mineral acids, sulfuric acid is effective, but it also depends on the polymer backbone. The choice of nitric acid has the possibility or tendency to degrade cellulose during irradiation. Hydrochloric acid could exert a negative effect in grafting efficiency, as chlorine could be incorporated in the final product acrylated PF (APF). Therefore, sulfuric acid was used in our experiments to enhance the grafting efficiency. The grafting was carried out in a slightly acidic condition under UV irradiation for 15–60 min, with a definite amount of catalyst (air-dried HB). Control experiments for EA grafting on PFs were also performed; (i) in the absence of the catalyst and UV irradiation unacrylated PF (UAPF 1), (ii) in the presence of the catalyst without UV irradiation (UAPF 2), and (iii) in the absence of the catalyst with UV irradiation (UAPF 3).
Another important aspect to consider is that the mobility of LDH in the HB catalyst. To hinder the LDH diffusion inside the pores of ACB, different drying methods were applied for HB. The closing down of pores was achieved via air-drying HB, thus LDH diffusing inside pores was completely arrested which could occur under grafting reaction conditions. In addition, it made the HB hard and mechanically stable. The drying experiments were also carried out under a freeze-drying condition, which is mostly used to obtain high surface area because of pores not closing down. Nevertheless, later, the HBs became soft and fragile. When they used in catalytic coupling reaction between EA and cellulose on PFs, they broke down and became powder, which made their separation/regeneration difficult from the reaction mixture (Figure S1). Hence, they have not proven to be ideal separable heterogeneous catalysts. Therefore, drying methods had a huge influence on LDH mobility viz-a-viz pores in HB and stability of HB (Figure 5f,i).
Acrylated PFs
The characterization results for the APF obtained from ATR–FTIR, time of flight–secondary ion mass spectroscopy (TOF-SIMS), SEM–EDX, and thermogravimetric analysis (TGA)/differential thermal analysis (DTA) are discussed here to highlight the gained acrylate groups on the cellulose backbone and its homogenous distribution.
The ATR–FTIR measurements were performed to understand the bonding interaction between EA and PFs (Figure 8). The new functional group formed during this reaction could be either (cellulose–CH2–O)···C=O–CH=CH2 (acrylate) or (cellulose–CH2–O)···CH2–CH2–COO–C2H5 (an alkyl ether group). The IR spectra of the pulps (UAPF 1), (UAPF 2), and (UAPF 3) showed the peaks similar to that of the reference PFs without any new peak, indicating that no grafting happened or was initiated without the catalyst and the UV light source. When the pulp was treated with a definite amount of catalyst with UV radiation for 15 min, there appeared a peak for the carbonyl group around 1700 cm–1, confirming that grafting has taken place (APF 1).37 In order to increase the grafting efficiency, the irradiation time was increased to 60 min. As the duration of UV irradiation increased, the intensity of the carbonyl group also increased sharply (APF 2). The TOF-SIMS studies revealed the fragmentation pattern of APF 2, and the predominant peak (m/z = 55) was found to be the acroloyl radical, (CH2=CH–C*=O), which could have been the potential intermediate in the formation of cellulose acrylate. The visual images showed plain fibers in the PF sample, whereas islets of acrylate groups on fibers as bright spots in the APF 2 sample (Figure 9). Thus, the bonding emerged could be due to the radical-mediated esterification reaction between the acryloyl CH2=CH–C*=O group and the cellulosic −CH2O* group as schematized in Figure 7.
Figure 8.
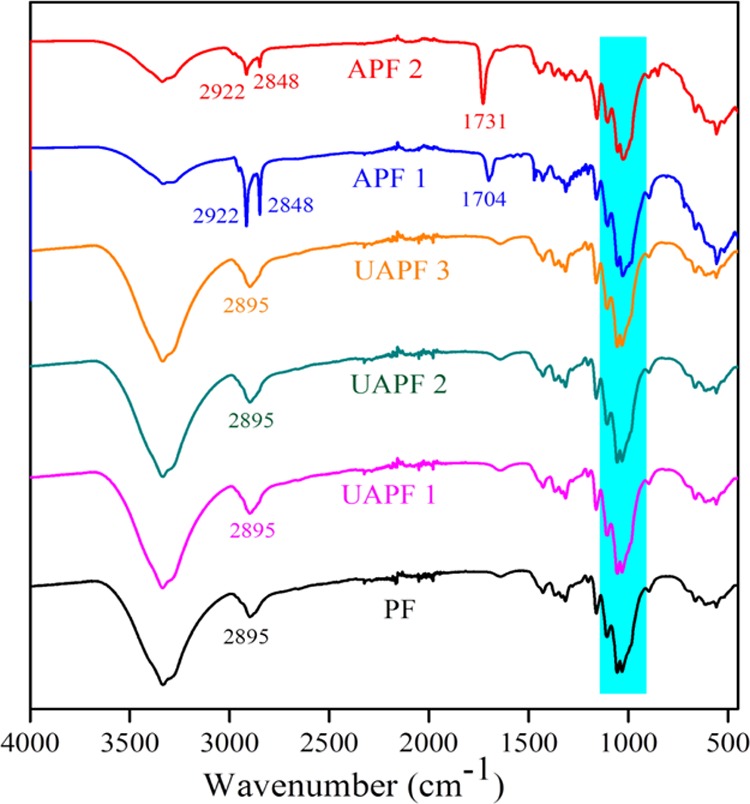
FTIR spectra of the modified fibers.
Figure 9.

TOF-SIMS image of the APF 2.
The morphology of kraft pulp found from SEM images revealed the complex weblike fibrous network in reference fibers (PF) (Figure 10a) and modified fibers (Figure 10c,e). The low-magnification images (Figure 10c,e) have also shown slight fibrillation on the surfaces of PFs caused by sulfuric acid/EA environment. The high-magnification images depicted that the wrinkled surface of the reference fibers (Figure 10b) have been smoothened and became a shine-coated surface (Figure 10d,f) after treatment with EA. The coating difference was seen clearly with 1 h treated fiber (APF 2) than the 15 min reacted fiber (APF 1) with respect to PF. This proved that APF 2 had excessive functionalization by the acrylate group. Thus, the morphology gave indication about EA functionalization/plasticization on fibers. EDX analysis (Figure 11) was carried out on the spent catalyst and EA-modified fibers (APF 2) to make sure that the HB catalyst did not undergo any chemical reaction and there was no leaching of metals. If there is any chemical reaction between LDH and PFs, there should be a presence of Ni and Fe in APF 2. The analysis showed that the APF 2 did not have any metal ions present in it. In other words, it was clear that the LDH in HBs acted only as a catalyst and does not interfere with the final product. This ensures that the HB catalyst will not have any bonding with the feedstock fibers in large-scale applications; therefore, the final materials will be metal-free, safe, and environmentally friendly.
Figure 10.

SEM spectra of the modified fibers (a,b) PF, (c,d) APF 1, and (e,f) APF 2.
Figure 11.

EDX spectra of the catalyst and modified fibers. The spectra reveal the absence of any metal ions (Ni and Fe, here) in the final modified fibers. In other words, it is clear that the LDH acts only as a catalyst and does not interfere with the final product, which will not affect the applications.
The EA-grafted pulp (APF) was further analyzed for their thermal stability using TGAs in order to evaluate the effect of the grafting on their thermal behavior. The TGA (Figure 12) showed a different weight loss profile for reference PFs and APF 2. The reference pulp lost its weight in two instances sequentially (i) at 350 °C corresponding to depolymerization and the destruction of intramolecular and intermolecular hydrogen bonding and (ii) at 480 °C due to the complete decomposition of cellulose. APF 2 was stable until 250 °C and beyond this temperature, the weight loss occurred in three consecutive instances: (i) 250–300 °C due to intramolecular and intermolecular hydrogen bonding (minus one −OH group), (ii) 400 °C corresponding to ester group removal, and (iii) 480 °C due to incineration of the cellulose backbone in pulp. There was no weight loss observed at 350 °C, which confirms the absence of the EA homopolymer.38,39 The DTA curves suggested that the weight loss temperatures obtained in TGA measurements was exactly fitting in, where the temperature difference was steep high in DTA curves. Thus, both the thermal profiles were in good agreement and confirmed the decomposition temperatures.
Figure 12.

TGA and DTA analysis of the reference (PF) and APF 2.
Hydrophobicity/Water Repellency Behavior of APF
The water contact-angle (CA) measurements were carried out in order to evaluate the extent of cellulose acrylate formation in PFs (Figure 13). The more the acrylate formation, the better was the water CA. The better the CA, the more suitable it will be for water-proof/oil adhesive applications. The reference (PF), dissolving pulp (ACel Enocell) was highly hydrophilic in nature because of inherent −OH groups in cellulose; hence, water droplets wetted the pulp sheet in a fraction of second and there was no CA measured. The APFs obtained via control experiments got wetted swiftly and showed very poor CA (19–20°), whereas UV-irradiated/HB-catalyzed esterification reactions showed slight increase in CA (26°) for APF 1, and much improved CA (90°) for APF 2. Thus, APF 2 avoided wetting by water droplets. This showed that the APF 2 was largely functionalized by acrylate groups and highly hydrophobic. Therefore, APF 2 material can be employed in hydrophobic (water repellency) or lipophilic (oil affinity) applications. Pulp, paper, packaging, thin film, and textile industries are those that can become potential exploiters of this biocomposite material if their technical specification requires high water repellency and oil affinity. They would benefit from this material as it employs a nonlaborious synthesis technique, kraft PFs and recyclable eco-friendly catalyst material, which reduces cost of solvents, radical initiators, and energy consumption. The final material can be easily made into hand paper sheet for facile characterization and transport or even pressed into deep three-dimensional shapes for printed materials applications. Thus, it could encourage the forest product industries to develop new materials and support circular economy.
Figure 13.
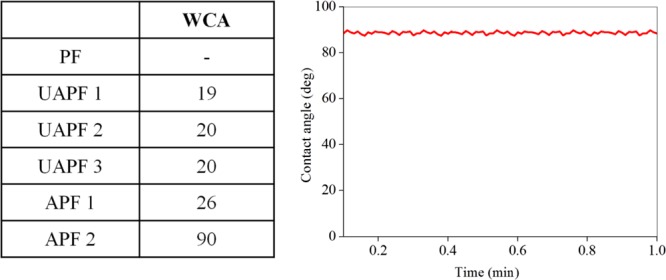
Water CA of the reference (PF), control (UAPF) and APF. The water CA of APF 2 remains constant for at least 1 min.
Conclusions
The NiFe LDH catalyst particles were made to grow in situ and immobilized over the surface of anionic CBs (HB) as a novel attempt. The HBs were applied as heterogeneous catalysts, which made their separation from the reaction mixture feasible and easy. This way, the catalyst can be washed and reused multiple times, that made our finding cost-effective. Hydrophilic pulp was converted into hydrophobic pulp photocatalytically in kraft PFs. The photon-harvesting HB catalyst facilitated the creation of radical formation in the cellulose backbone and EA, for the esterification reaction between cellulose and EA. This successfully brought novel functionality (acrylate group) covalently anchored to the cellulose chain (APF) under aqueous conditions and it is reported for the first time. This tailor-made catalyst system was stable chemically, thermally, and photonically. When the duration of UV irradiation was increased, the pulp exhibited higher grafting efficiency and the APF 2 hand sheets showed better hydrophobicity compared to our previous report. Thus, grafting acrylate functionality on the cellulose backbone brought new properties to the PFs for advanced utilization. APF can be used in water-repellent packaging of goods, food, and groceries (meat, fish, vegetables, cooked food). The strategic value of the material could be that it will have the potential to replace the existing nonrenewable petrochemical-based thermoformable plastic composites with its unique features such as low cost, hazardless, recyclability, ease of making, converting harmful EA into nonharmful material and thus fixing the EA waste problem. These features will have very high impact on cellulose economy and clean environment. In academy, this study addresses photocatalytic way of functionalizing inactive −OH groups of cellulose to form ester, chemical bonding between cellulose and hydrophobic moiety/plasticizer and its stability. This investigation can also be disseminated to graft other co-materials like acrylonitrile and acrylic acid on cellulose covalently. Hence, the economic potential and environmental protection can be widened.
Materials
Kraft pulp (ACel Enocell) was obtained from VTT Technical Research Centre of Finland. Microcrystalline cellulose (Avicel PH 101), sodium dihydrogen phosphate (NaH2PO4), sodium hypochlorite (NaClO), sodium chlorite (NaClO2), TEMPO, EA were procured from Sigma-Aldrich. Nickel(II) nitrate hexahydrate Ni(NO3)2·6H2O, iron(III) nitrate nonahydrate Fe(NO3)3·9H2O, sodium carbonate Na2CO3, sodium hydroxide (NaOH) 98%, urea (CO(NH2)2), and nitric acid (HNO3) were procured from Merck and VWR. All the reagents in the synthesis were used as received and Milli-Q water was used in all the experiments.
Experimental Methods
Catalyst Preparation
Preparation of Anionic CBs
Microcrystalline cellulose was dissolved in 7% NaOH–12% urea–water at −10 °C aiming the final concentration of cellulose 5%. The cellulose solution was further carefully dropped in a beaker containing 2 M HNO3 at 25 °C at systematic time intervals and allowed them to grow into a spherical shape followed by aging to become hard. The formed beads were kept standing still for a couple of hours and then washed thoroughly in running tap water for 24 h to ensure complete neutralization of the CBs. The beads (CB) were then subjected to TEMPO oxidation using a mixture of TEMPO/NaClO2/NaClO for 5 h at 40 °C. These oxidized beads were washed thoroughly and stored in water for further use. These beads are denoted as ACBs.
Synthesis of an Anionic CB-Supported LDH Catalyst
The NiFe LDH material was prepared via the co-precipitation method in the presence of ACBs. To do that, the as-prepared beads were suspended in solution containing nickel(II) nitrate hexahydrate Ni(NO3)2·6H2O and iron(III) nitrate nonahydrate Fe(NO3)3·9H2O overnight, where the Ni/Fe ratio was 3:1. Further, this mixture was treated with sodium carbonate–sodium hydroxide solution for 24 h at pH 9, to form NiFe LDH and immobilize on the beads in situ. The whole reaction was performed at room temperature and air atmosphere. Thus, NiFe LDH was synthesized in the presence of anionic CBs and subsequently, anchored on these beads uniformly as first of its kind for catalysis purpose. The ACB-supported LDH material was filtered and washed repeatedly until neutral pH was reached to remove any mobile LDH particles. The LDH on ACB were air-dried in one portion and freeze-dried in another portion for further comparison. The LDH–ACB beads are denoted as HBs.
Synthesis of Unsupported LDH
The NiFe LDH material was also prepared in the absence of catalyst support (anionic CBs) via the abovementioned co-precipitation method by exactly following the other preparation parameters.
Catalyst Testing
Topochemical Grafting on PFs
PFs (500 mg) were dispersed in 100 mL of deionized water. Further, 50 mg of the HB catalyst was added to this mixture followed by the addition of a small quantity of scavenger reagent, methyl viologen. The mixture was stirred well and then 5 mg of plasticizing/hydrophobic organic moiety, EA, was added. The whole mixture was continued to stir under nonirradiation conditions and UV light conditions for 15 min, to induce topochemical functionalization of pulp fibers. The product material was pressed into sheet and dried at 21 °C temperature in a protective room with 53% of relative humidity. Similar experiments were performed without adding catalyst beads in the mixture to study the difference in grafting on PFs.
Characterization
The synthesized catalyst materials and photocatalytically produced APF products were analyzed systematically using the range of spectroscopic, microscopic, and thermal analytical techniques.
XRD Spectroscopy
As such, the beads were macroparticle, and it was not possible to subject them for the PXRD technique. Therefore, the beads (pristine and hybrid) were pressed into pellets with a force of 1 ton using a laboratory press prior to diffraction studies. Then, the pelleted beads were investigated for PXRD using a Siemens D501 diffractometer with copper Kα radiation (λ = 0.15415 nm). Patterns were recorded in the 2θ range of 5–70° in steps of 0.04° with a counting time per step of 8 s. The analyses performed were triplicated to correctly check the reproducibility of the diffraction from the crushed beads.
ATR–FTIR and Raman Spectroscopy
ATR–FTIR spectra were measured in the range 4000–400 cm–1 on a Thermo Scientific, Nicolet iS50 FTIR, Madison, WI, USA, instrument with a Raman module. It was equipped with a diamond crystal and a pressure gauge. The beads and APF product material were gently and carefully placed on the diamond crystal and the IR spectra were recorded. A total 64 scans were recorded for each sample and corrected by the OMNIC spectral suite software that provided ambient background. ATR correction was done for 45° incident angle with 1 refraction index, assuming a 1.50 refractive index for all samples. Raman measurements were performed for ACBs using a diode laser as source radiation (P = 0.5 W, λ = 1064 nm) and a gold plate as a sample holder in order to strengthen the signal. An indium gallium nitride detector with CaF2 splitter was used. The total number of scans for each sample was 1024 with the resolution 8.00 cm–1.
Time of Flight–Secondary Ion Mass Spectrometry
Secondary-ion mass spectra in TOF mode were obtained for the APFs using a physical electronics TOF-SIMS TRIFT II spectrometer. The spectrometer operates with a primary ion beam of the 69Ga+ liquid metal ion source. A raster size of 200 × 200 μm with the resolution of 256 × 256 pixels was scanned, and at least five different spots were analyzed. The surface distribution of acrylate groups was obtained with the best spatial resolution using the ion gun at 25 kV. The spectra were acquired for 10 min ensuring static conditions. The electron flood was used to compensate the charge with respect to the ion gun.
Solid-State CP MAS 13C NMR
CP MAS 13C NMR spectra were obtained with a Bruker AVANCE-III HD 400 MHz spectrometer. The powdered samples of CB, ACB, and HB were spun at a 12 kHz spin rate in a Bruker 1H broadband double-resonance 4 mm CP MAS probe. The proton 90° high-power pulse was 2.9 μs with contact time 2 ms. The recovery delay time was set to 2 s and 30 000 scans were accumulated.
Scanning Electron Microscopy–Energy-Dispersive X-ray Spectroscopy
A Leo Gemini 1530 field-emission scanning electron microscope with an in-lens detector (LEO Electron Microscopy Ltd., Oberkochen, Germany) was utilized for morphological characterization of the CB, HB, and APF. The samples were coated on carbon in a Temcarb TB500 sputter coater (Emscope Laboratories, Ashford, UK). The optimum accelerating voltage was 2.70 kV. EDX spectra were also obtained using the same equipment in energy-dispersive X-ray spectra mode.
ThermoGravimetric Analysis and Differential Thermal Analysis
The thermal behavior of the samples was studied with a TA Instruments SDT Q600 simultaneous TGA/DSC apparatus between 25 and 600 °C in a flowing air (flow rate: 100 cm3 min–1). A heating rate of 10 °C min–1 and sample weight of ca. 10 mg was used. The sample pan was aluminum oxide and the reference material was an empty aluminum oxide pan.
Water CA Measurements
The hydrophobicity/water repellency of all the pulp handsheets was assessed by CA measurements. These were performed with an optical contact angle meter, CAM 200 (KSV Instruments Ltd., Finland), using deionized water. A water droplet size of 1.6 ± 0.2 μL was placed on a 1 × 10 cm sample hand sheet strip. Then, the CA values were collected at 200 ms intervals in the beginning and subsequently at 1 and 2 s intervals until either the water droplet absorbed or no changes during wetting occurred. The results were analyzed and interpreted with Attention Theta software (Biolin Scientific, Sweden) based on the Young–Laplace function for iterative CA calculation.
Acknowledgments
L.S.S. and P.F. appreciate funding from “Advanced Cellulose to Novel Cellulose Products (ACel)” project and “Magnus Ehrnrooth Foundation” to conduct this research at Åbo Akademi University. L.K. acknowledges the Turku Collegium for Science & Medicine (TCSM) and University of Turku for the funding aid and hosting his collaborative postdoctoral research. The authors thank Ida Mattsson MSc (Tech), Laboratory of Organic Chemistry, Åbo Akademi University, Finland for conducting NMR analysis.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03061.
Visualization in understanding the difference between homogeneous and heterogeneous catalysis conditions (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Tajeddin B.; Thakur V. K.. Lignocellulosic Polymer Composites: Processing, Characterization, and Properties, Cellulose Based Polymers for Packaging Applications; Scrivener Publishing LLC, 2014; Chapter 21. [Google Scholar]
- Emblem A.; Emblem H.. Packaging Technology, Fundamentals, Materials and Processes Book, 2012, ISBN 978-1-84569-665-8. [Google Scholar]
- Sirviö J. A.; Liimatainen H.; Niinimäki J.; Hormi O. Sustainable packaging materials based on wood cellulose. RSC Adv. 2013, 3, 16590–16596. 10.1039/c3ra43264e. [DOI] [Google Scholar]
- He M.; Xu M.; Zhang L. Controllable Stearic Acid Crystal Induced High Hydrophobicity on Cellulose Film Surface. ACS Appl. Mater. Interfaces 2013, 5, 585–591. 10.1021/am3026536. [DOI] [PubMed] [Google Scholar]
- Sobhana S. S. L.; Zhang X.; Kesavan L.; Liias P.; Fardim P. Layered double hydroxide interfaced stearic acid - Cellulose fibres: A new class of super-hydrophobic hybrid materials. Colloids Surf., A 2017, 522, 416–424. 10.1016/j.colsurfa.2017.03.025. [DOI] [Google Scholar]
- Huang X.; Wang A.; Xu X.; Liu H.; Shang S. Enhancement of Hydrophobic Properties of Cellulose Fibers via Grafting with Polymeric Epoxidized Soybean Oil. ACS Sustainable Chem. Eng. 2016, 5, 1619–1627. 10.1021/acssuschemeng.6b02359. [DOI] [Google Scholar]
- Ohara T.; Sato T.; Shimizu N.; Schwind G. P. H.; Weiberg O.; Marten K.; Greim H.. “Acrylic Acid and Derivatives” in Ullmann’s Encyclopedia of Industrial Chemistry; Wiley-VCH: Weinheim, 2003. [Google Scholar]
- Miyata S. The syntheses of hydrotalcite-like compounds and their structures and physico-chemical properties I: the systems, Mg2+-A13+-NO3-,Mg2+-A13+-C1-,Mg2+-A13+-ClO4-, Ni2+- A13+-C1- and Zn2+-A13+-C1-. Clays Clay Miner. 1975, 23, 369–375. 10.1346/ccmn.1975.0230508. [DOI] [Google Scholar]
- Liji Sobhana S. S.; Kesavan L.; Fardim P. Topochemical Engineering of Cellulose-based Functional Materials. Langmuir 2018, 34, 9857–9878. 10.1021/acs.langmuir.7b04379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhana L.; Sarakha M.; Prevot V.; Fardim P. Layered double hydroxides decorated with Au-Pd nanoparticles to photodegradate Orange II from water. Appl. Clay Sci. 2016, 134, 120–127. 10.1016/j.clay.2016.06.019. [DOI] [Google Scholar]
- Miyata S.; Kumura T. Synthesis of new hydrotalcite-like compounds and their physico-chemical properties. Chem. Lett. 1973, 2, 843–848. 10.1246/cl.1973.843. [DOI] [Google Scholar]
- Ramanathan G.; S. S L. S.; Fardim P.; Sivagnanam U. T. Fabrication of 3D dual-layered nanofibrous graft loaded with layered double hydroxides and their effects in osteoblastic behavior for bone tissue engineering. Process Biochem. 2018, 64, 255–259. 10.1016/j.procbio.2017.09.025. [DOI] [Google Scholar]
- Géraud E.; Prévot V.; Leroux F. Synthesis and characterization of macroporous MgAl LDH using polystyrene spheres as template. J. Phys. Chem. Solids 2006, 67, 903–908. 10.1016/j.jpcs.2006.01.002. [DOI] [Google Scholar]
- Kloprogge J. T.; Frost L. In Layered Double Hydroxides: Present and Future; Rives V., Eds.; Nova Science Publishers, Inc.: New York, 2001. [Google Scholar]
- Silva C. G.; Bouizi Y.; Fornés V.; García H. Layered double hydroxides as highly efficient photocatalysts for visible light oxygen generation from water. J. Am. Chem. Soc. 2009, 131, 13833–13839. 10.1021/ja905467v. [DOI] [PubMed] [Google Scholar]
- Evans D. G.; Duan X. Preparation of layered double hydroxides and their applications as additives in polymers, as precursors to magnetic materials and in biology and medicine. Chem. Commun. 2006, 5, 485–496. 10.1039/b510313b. [DOI] [PubMed] [Google Scholar]
- Mohapatra L.; Parida K. A review on the recent progress, challenges and perspective of layered double hydroxides as promising photocatalysts. J. Mater. Chem. A 2016, 4, 10744–10766. 10.1039/c6ta01668e. [DOI] [Google Scholar]
- Gong M.; Li Y.; Wang H.; Liang Y.; Wu J. Z.; Zhou J.; Wang J.; Regier T.; Wei F.; Dai H. An Advanced Ni-Fe Layered Double Hydroxide Electrocatalyst for Water Oxidation. J. Am. Chem. Soc. 2013, 135, 8452–8455. 10.1021/ja4027715. [DOI] [PubMed] [Google Scholar]
- Nayak S.; Mohapatra L.; Parida K. Visible light-driven novel g-C3N4/NiFe-LDH composite photocatalyst with enhanced photocatalytic activity towards water oxidation and reduction reaction. J. Mater. Chem. A 2015, 3, 18622–18635. 10.1039/c5ta05002b. [DOI] [Google Scholar]
- Zhou H.; Song Y.; Liu Y.; Li H.; Li W.; Chang Z. Fabrication of CdS/Ni Fe LDH heterostructure for improved photocatalytic hydrogen evolution from aqueous methanol solution. Int. J. Hydrogen Energy 2018, 43, 14328–14336. 10.1016/j.ijhydene.2018.05.172. [DOI] [Google Scholar]
- Gao X.; Pan X.; Long X.; Yi Z. Room-Temperature Synthesis FeNiCo Layered Double Hydroxide as an Excellent Electrochemical Water Oxidation Catalyst. J. Electrochem. Soc. 2017, 164, H755–H759. 10.1149/2.0561706jes. [DOI] [Google Scholar]
- Trygg J.; Fardim P.; Gericke M.; Mäkilä E.; Salonen J. Physicochemical design of the morphology and ultrastructure of cellulose beads. Carbohydr. Polym. 2013, 93, 291–299. 10.1016/j.carbpol.2012.03.085. [DOI] [PubMed] [Google Scholar]
- Gericke M.; Trygg J.; Fardim P. Functional Cellulose Beads: Preparation, Characterization, and Applications. Chem. Rev. 2013, 113, 4812–4836. 10.1021/cr300242j. [DOI] [PubMed] [Google Scholar]
- Sobhana S. S. L.; Bogati D. R.; Reza M.; Gustafsson J.; Fardim P. Cellulose biotemplates for layered double hydroxides networks. Microporous Mesoporous Mater. 2016, 225, 66–73. 10.1016/j.micromeso.2015.12.009. [DOI] [Google Scholar]
- Ogiwara T.; Katsumura A.; Sugimura K.; Teramoto Y.; Nishio Y. Calcium Phosphate Mineralization in Cellulose Derivative/Poly(acrylic acid) Composites Having a Chiral Nematic Mesomorphic Structure. Biomacromolecules 2015, 16, 3959–3969. 10.1021/acs.biomac.5b01295. [DOI] [PubMed] [Google Scholar]
- Gupta K. C.; Sahoo S. Graft Copolymerization of Acrylonitrile and Ethyl Methacrylate Comonomers on Cellulose Using Ceric Ions. Biomacromolecules 2001, 2, 239–247. 10.1021/bm000102h. [DOI] [PubMed] [Google Scholar]
- Gupta K. C.; Sahoo S.; Khandekar K. Graft Copolymerization of Ethyl Acrylate onto Cellulose Using Ceric Ammonium Nitrate as Initiator in Aqueous Medium. Biomacromolecules 2002, 3, 1087–1094. 10.1021/bm020060s. [DOI] [PubMed] [Google Scholar]
- Trygg J.; Yildir E.; Kolakovic R.; Sandler N.; Fardim P. Solid-State Properties and Controlled Release of Ranitidine Hydrochloride from Tailored Oxidised Cellulose Beads. Macromol. Mater. Eng. 2014, 300, 210–217. 10.1002/mame.201400175. [DOI] [Google Scholar]
- Trygg J.; Yildir E.; Kolakovic R.; Sandler N.; Fardim P. Anionic cellulose beads for drug encapsulation and release. Cellulose 2014, 21, 1945–1955. 10.1007/s10570-014-0253-z. [DOI] [Google Scholar]
- Kim I. S.; Kim J. P.; Kwak S. Y.; Ko Y. S.; Kwon Y. K. Novel regenerated cellulosic material prepared by an environmentally-friendly process. Polymer 2006, 47, 1333. 10.1016/j.polymer.2005.12.070. [DOI] [Google Scholar]
- Sobhana S. S. L.; Mehedi R.; Malmivirta M.; Paturi P.; Lastusaari M.; Dîrtu M. M.; Garcia Y.; Fardim P. Heteronuclear nanoparticles supported hydrotalcites containing Ni(II) and Fe(III) stable photocatalysts for Orange II degradation. Appl. Clay Sci. 2016, 132–133, 641–649. 10.1016/j.clay.2016.08.016. [DOI] [Google Scholar]
- Wang Y.; Wu P.; Li Y.; Zhu N.; Dang Z. Structural and spectroscopic study of tripeptide/layered double hydroxide hybrids. J. Colloid Interface Sci. 2013, 394, 564–572. 10.1016/j.jcis.2012.11.031. [DOI] [PubMed] [Google Scholar]
- Toraishi T.; Nagasaki S.; Tanaka S. Adsorption behavior of IO3 by CO32–- and NO3–- hydrotalcite. Appl. Clay Sci. 2002, 22, 17–23. 10.1016/s0169-1317(02)00108-4. [DOI] [Google Scholar]
- Koilraj P.; Srinivasan K. ZnAl Layered Double Hydroxides as Potential Molybdate Sorbents and Valorize the Exchanged Sorbent for Catalytic Wet Peroxide Oxidation of Phenol. Ind. Eng. Chem. Res. 2013, 52, 7373–7381. 10.1021/ie302782c. [DOI] [Google Scholar]
- Lee S.; Shin G.-O.; Park K.; Chang P.-S.; Kim Y.-S. Determination of odor release in hydrocolloid model systems containing original or carboxylated cellulose at different pH values using static headspace gas chromatographic (SHS-GC) analysis. Sensors 2013, 13, 2818–2829. 10.3390/s130302818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong K. H.; Liu N.; Sun G. UV-induced graft polymerization of acrylamide on cellulose by using immobilized benzophenone as a photo-initiator. Eur. Polym. J. 2009, 45, 2443–2449. 10.1016/j.eurpolymj.2009.04.026. [DOI] [Google Scholar]
- Qian Y.-q.; Han N.; Bo Y.-w.; Tan L.-l.; Zhang L.-f.; Zhang X.-x. Homogeneous synthesis of cellulose acrylate- g -poly ( n -alkyl acrylate) solid-solid phase change materials via free radical polymerization. Carbohydr. Polym. 2018, 193, 129–136. 10.1016/j.carbpol.2018.03.057. [DOI] [PubMed] [Google Scholar]
- Zhang C.; Xu T.; Bao Z.; Chen L. Synthesis and characterization of fluorinated polyacrylate latex emulsified with novel surfactants. Des. Monomers Polym. 2016, 20, 118–124. 10.1080/15685551.2016.1231040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Guerrero M.; López-Beceiro J.; Sánchez-Jiménez P. E.; Pascual-Cosp J. Comparison of thermal behavior of natural and hot-washed sisal fibers based on their main components: cellulose, xylan and lignin. TG-FTIR analysis of volatile products. Thermochim. Acta 2014, 581, 70–86. 10.1016/j.tca.2014.02.013. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.