

Abstract
The assembly of peptides and proteins into nanoscale amyloid fibrils via formation of intermolecular β-sheets not only plays an important role in the development of degenerative diseases but also represents a promising approach for the synthesis of functional nanomaterials. In many biological and technological settings, peptide assembly occurs in the presence of organic and inorganic interfaces with different physicochemical properties. In an attempt to dissect the relative contributions of the different molecular interactions governing amyloid assembly at interfaces, we here present a systematic study of the effects of terminal modifications on the adsorption and assembly of the human islet amyloid polypeptide fragment hIAPP(20–29) at organic self-assembled monolayers (SAMs) presenting different functional groups (cationic, anionic, polar, or hydrophobic). Using a selection of complementary in situ and ex situ analytical techniques, we find that even this well-defined and comparatively simple model system is governed by a rather complex interplay of electrostatic interactions, hydrophobic interactions, and hydrogen bonding, resulting in a plethora of observations and dependencies, some of which are rather counterintuitive. In particular, our results demonstrate that terminal modifications can have tremendous effects on peptide adsorption and assembly dynamics, as well as aggregate morphology and molecular structure. The effects exerted by the terminal modifications can furthermore be modulated in nontrivial ways by the physicochemical properties of the SAM surface. Therefore, terminal modifications are an important factor to consider when conducting and comparing peptide adsorption and aggregation studies and may represent an additional parameter for guiding the assembly of peptide-based nanomaterials.
1. Introduction
Molecular self-assembly plays an important role in biology and is nowadays increasingly utilized in bionanotechnology.1 A prominent example encountered in both disciplines is the assembly of proteins and peptides into nanoscale amyloid fibrils via the formation of intermolecular β-sheets. Although such fibrils may serve as nanoscale building blocks in the synthesis of functional materials and devices,2−5 amyloid assembly and fibrillization are also important steps in the development of so-called misfolding diseases such as Alzheimer’s disease, Parkinson’s disease, and type 2 diabetes mellitus (T2DM).6−8 Understanding and controlling amyloid assembly at a molecular level thus represent essential milestones along the route toward new functional materials and medical therapies.
Among the multitude of environmental parameters that are known to affect amyloid assembly kinetics and morphology, such as monomer concentration, temperature, pH, and ionic strength, the effects exerted by organic and inorganic interfaces with varying physicochemical properties on amyloidogenic peptides and proteins receive growing attention.9 This is to a large extent driven by the increasing number of medical applications that employ therapeutic and diagnostic nanoparticles that interact with numerous proteins and peptides in the body and may thereby induce or inhibit their assembly into toxic amyloid aggregates.10−13 On the other hand, amyloid fibrillization is increasingly explored with regard to applications in surface functionalization14,20 and the synthesis of hybrid materials that incorporate amyloid-like fibrils and other organic and inorganic materials.15−20 Consequently, amyloid assembly at interfaces has been the subject of numerous studies.21−51 These works as a whole paint a fairly complex picture of the adsorption and fibrillization of amyloidogenic peptides and proteins at interfaces, which appear to be governed by the interplay and competition of several inter- and intramolecular interactions, that is, hydrophobic interactions, electrostatic interactions, hydrogen bonding, and sometimes even covalent bonding, all of which may be significantly affected even by small variations in the properties of the surface35 or the molecule.21 As a result of this high degree of complexity, the adsorption and assembly behavior of a given peptide or protein at a given surface is almost impossible to predict and often counterintuitive.21
In an attempt to dissect the relative contributions of the different molecular interactions to amyloid assembly at interfaces, we here present a systematic study of the effects of terminal modifications on the adsorption and assembly of human islet amyloid polypeptide hIAPP(20–29) at chemically defined surfaces, that is, organic self-assembled monolayers (SAMs) presenting different functional groups. Human islet amyloid polypeptide (hIAPP) is a 37-residue hormone secreted by the insulin-producing β-cells in the pancreas, which may assemble into cytotoxic oligomeric and fibrillar amyloid structures.52 In the course of T2DM, these amyloid structures damage the membranes of the β-cells, resulting in long-term β-cell failure.53 The hIAPP fragment hIAPP(20–29) is able to form amyloid fibrils on its own, which are known to show a pronounced polymorphism, both in bulk solution54 and at interfaces.22 Its native sequence (Figure 1) features five hydrophobic amino acids but no charged residues. Under physiological conditions, its interaction with hydrophobic interfaces will thus involve only the hydrophobic amino acid residues, whereas electrostatic interactions with charged interfaces will be mediated by the charged termini only. By acetylation and amidation of the N and the C termini, respectively (see Figure 1), the relative contributions of attractive and repulsive electrostatic interactions can be controlled.
Figure 1.
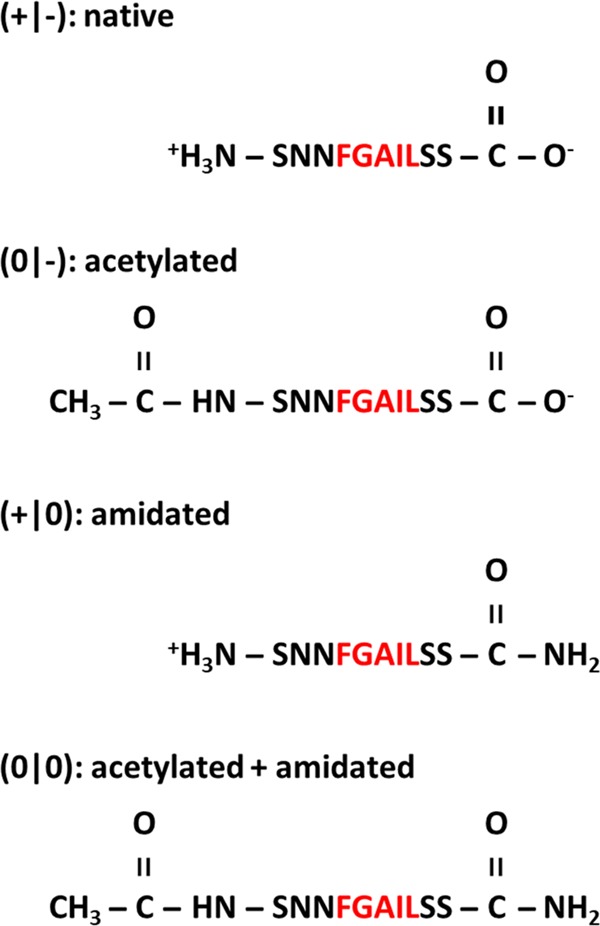
Structure of hIAPP(20–29) with various terminal modifications. Hydrophobic residues are indicated in red.
Several previous studies have addressed the effects of terminal modifications on amyloid assembly for various peptides and proteins.45,55−63 In general, terminal modifications were found to affect fibrillization propensity and kinetics, as well as the morphology, mechanical properties, and molecular structures of the assembled fibrils. For instance, Andreasen et al. investigated the assembly of hIAPP(20–29) with and without terminal modifications in bulk solution.55 Whereas the native fragment formed flat fibrils, C-terminal amidation induced the formation of twisted fibrils. N-terminal acetylation on the other hand completely inhibited fibrillization. This was also verified using infrared spectroscopy, which revealed a purely random coil conformation of the acetylated fragment, whereas the other peptides assumed conformations rich in β-sheets. Intriguingly, the acetylated and amidated peptide formed fibrils similar to those of the native peptide but at a much lower rate. Despite the significant interest in the effects of terminal modifications on amyloid fibrillization, only a few studies have so far evaluated its role in peptide adsorption and surface-mediated assembly. Karsai et al. investigated the epitaxial growth of Aβ(25–35) fibrils on mica surfaces.45 These fibrils were aligned along certain crystallographic directions as a result of the specific interaction of positive charges in the peptide with the negative charges of the mica surface. Acetylation of the N terminus thus resulted in reduced crystallographic alignment.
In this work, we studied the interaction of different hIAPP(20–29) fragments (see Figure 1) with NH3+-, COO–-, OH-, and CH3-terminated SAMs in situ using quartz crystal microbalance with dissipation monitoring (QCM-D), which allowed us to assess differences in adsorption kinetics and adsorbate film structure. Ex situ atomic force microscopy (AFM) and polarization-modulated infrared reflection absorption spectroscopy (PM-IRRAS) investigations provided us with information regarding the morphology and molecular structure of the amyloid assemblies formed at the different surfaces. The combination of these complementary, surface-sensitive techniques allows for a thorough investigation of molecular processes involved in peptide adsorption and surface-mediated assembly.21 Using this experimental approach, we find that even this well-defined and comparatively simple model system is governed by a rather complex interplay of electrostatic interactions, hydrophobic interactions, and hydrogen bonding with terminal modifications affecting peptide adsorption and assembly dynamics, aggregate morphology, and molecular structure. These termini-specific effects can furthermore be modulated in nontrivial ways by the physicochemical properties of the SAM surface.
2. Results and Discussion
2.1. Bulk Solution
To assess any intrinsic effects of the terminal modifications on hIAPP(20–29) assembly, we first investigated amyloid formation for all fragments in bulk solution using Thioflavin T (ThT) fluorescence spectroscopy to monitor β-sheet formation.64,65 As can be seen in Figure S1, no increase in ThT fluorescence intensity is observed for the four fragments over 24 h of incubation. This is at variance with the results obtained by Andreasen et al., who observed strong hIAPP(20–29) fibrillization over similar time scales with terminal modifications having a strong effect on aggregation propensity and kinetics.55 This discrepancy can be attributed to the different buffer conditions used and especially the significantly higher monomer concentrations employed in the study of Andreasen et al. Despite the absence of increased ThT fluorescence, the AFM images in Figures 2a and S2 show prefibrillar aggregates with some differences in aggregate morphology between the different fragments. For the fragments with amidated C termini (+|0) and (0|0), rather homogeneous particles with similar sizes are observed. Because of their rather homogeneous sizes and almost spherical shapes, we believe that these particle-like aggregates represent amyloid oligomers. For the acetylated fragment (0|-), smaller particle-like aggregates seem to dominate, whereas additional large, irregularly shaped aggregates can sometimes be observed as well. The aggregates of the native fragment (+|-) are not as homogeneous in size and shape and may thus include also amorphous aggregates. The molecular structure of the aggregates was assessed by PM-IRRAS, which allowed us to quantify their β-sheet contents (see the Supporting Information (SI)).21 In contrast to the ThT fluorescence data in Figure S1, which do not show any β-sheet-specific increase in fluorescence, PM-IRRAS reveals significant β-sheet contents for all fragments (Figures S10 and 2b). This discrepancy may be attributed to the limited sensitivity of the ThT assay, which may not detect all oligomeric species.66 The PM-IRRAS results in Figure 2b further reveal some significant differences between the fragments. Whereas the fragments with native C termini (+|-) and (0|-) have virtually identical β-sheet contents of ∼33%, the acetylated and amidated fragment (0|0) has a slightly higher content of 39%. For the amidated fragment with native N terminus (+|0), however, the total β-sheet content is decreased to about 24%. These results are again at variance with the report by Andreasen et al., who observed that acetylation completely abolished any hIAPP(20–29) aggregation and resulted only in random coil conformations.55 This can again be attributed to the different buffer conditions and especially the different ionic strengths. At the comparatively high ionic strength of the phosphate buffered saline (PBS) buffer used in our experiments, stronger screening of electrostatic interactions may result in a weaker influence of the terminal charges on peptide assembly, which thus may be less sensitive to terminal modification. Furthermore, the two fragments with amidated C termini (+|0) and (0|0) show comparatively high contents of antiparallel β-sheets around 10%, whereas the native (+|-) and acetylated (0|-) fragments feature only about 4 and 1% of antiparallel β-sheets, respectively. High content of antiparallel β-sheets may thus be related to the larger, homogeneous particle-like aggregates in Figure 2a, which are not as dominant for the native (+|-) and acetylated (0|-) fragments. Whereas previous NMR investigations have demonstrated the possibility of acetylated and amidated hIAPP(20–29) to form two molecularly different fibrillary species with either parallel or antiparallel β-sheets,67 our results indicate that this structural polymorphism may be modulated by terminal modifications of hIAPP(20–29). These investigations thus show that terminal modifications may result in pronounced differences in the molecular structures of prefibrillar oligomeric aggregates.
Figure 2.
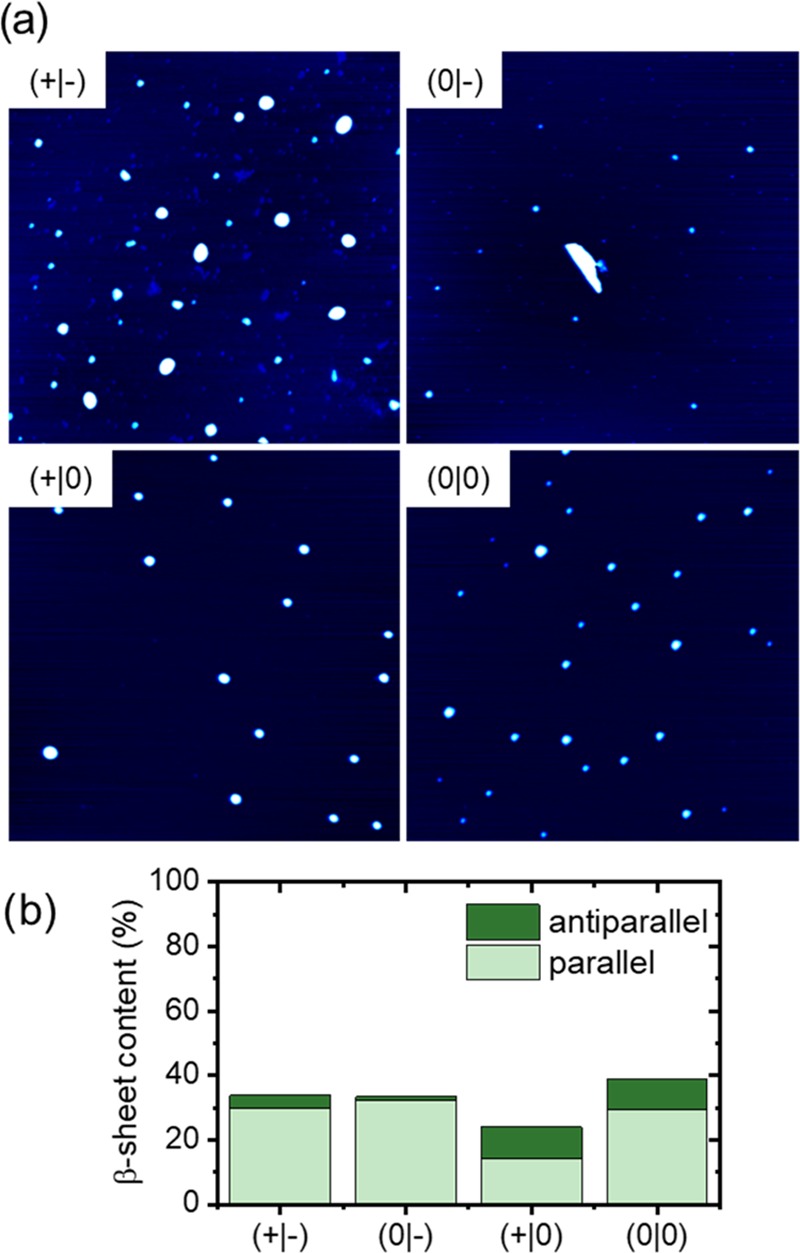
(a) AFM images (2 × 2 μm2) of the assembled hIAPP(20–29) aggregates recorded after 24 h incubation without ThT. The z-scales are 5 nm for all images. (b) β-Sheet contents (parallel and antiparallel) of the different hIAPP(20–29) fragments after assembly in bulk solution (without ThT) for 24 h as determined by PM-IRRAS.
2.2. Positively Charged NH2-Terminated SAM
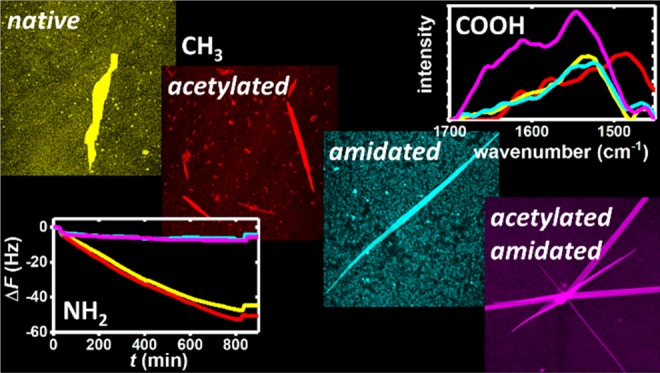
Under physiological conditions, that is, pH 7.5, the terminal groups of the NH2 SAM will be protonated and thus positively charged. Consequently, adsorption of hIAPP(20–29) should be governed by the interplay of electrostatic attraction and repulsion between the surface and the C and N termini of the peptide, respectively. This has been assessed in situ over the course of several hours using QCM-D. The shift of the resonance frequency ΔF shown in Figure 3 is a measure of the peptide mass adsorbed on the SAM-modified electrode surface.68 As can be seen in Figure 3, ΔF is decreasing for all fragments upon injection of the peptide-containing buffer at t = t0. However, for the native (+|-) and acetylated (0|-) fragments, adsorption proceeds for more than 12 h without any sign of saturation. The two amidated fragments (+|0) and (0|0) on the other hand quickly reach a plateau in which no significant adsorption is observed anymore. Flushing the QCM-D cells with peptide-free PBS buffer at t = t1 results in the desorption of reversibly adsorbed species. Similar amounts of desorption are observed for all fragments. The change in dissipation ΔD is also plotted in Figure 3 for all four fragments and mirrors the trends found in the ΔF plots. The fragments with native C termini (+|-) and (0|-) show a stronger increase in ΔD upon adsorption than the fragments with amidated C termini (+|0) and (0|0), indicating the formation of adsorbate films with higher viscoelasticity. Despite these pronounced differences in adsorption behavior, no significant variations can be deduced from the AFM images in Figure S7, which are dominated by the comparatively rough topography of the polycrystalline gold electrode surface.
Figure 3.

QCM-D results (ΔF, upper row; and ΔD, lower row) of the interaction of the different hIAPP fragments with the different SAMs. The arrows in the left column exemplarily indicate the times t0 and t1 of peptide injection and flushing with buffer, respectively.
These observations are in line with our hypothesis that hIAPP(20–29) adsorption at the positively charged NH2-terminated SAM is to a large extent governed by electrostatic interactions. The fragments with native, negatively charged C termini show the strongest frequency shifts, whereas the fragments with amidated C termini that lack any negative charge adsorb only weakly. Based on these observations, one could assume that the (+|-) and (0|-) monomers adsorb in an upright conformation with their C termini electrostatically bound to the NH3+ groups of the SAM and their N-terminal regions dangling from the surface. The weak adsorption of the amidated fragments (0|-) and (0|0) in the absence of any electrostatic attraction, however, is most likely the result of hydrogen bond formation between the NH3+ groups of the SAM and the peptides’ backbone and amino acid residues.
To assess the molecular structure of the adsorbate films, we have again turned to PM-IRRAS and first quantified the amount of adsorbed peptide on the NH2-terminated SAM after the QCM-D experiments by integrating the amide I intensities (see SI). The results are shown in Figure 4 and compared to the corresponding ΔF values obtained from the QCM-D measurements. Surprisingly, this comparison reveals a significant deviation from the overall trend observed in the QCM-D measurements. In fair agreement with the ΔF data, the native fragment (+|-) shows the highest amide I intensity and the two amidated fragments (+|0) and (0|0) show the lowest, almost identical amide I intensities. For the acetylated fragment (0|-), however, an integrated amide I intensity close to those of the amidated fragments is obtained, whereas the corresponding ΔF value is close to that of the native fragment. It is well known that the adsorbed mass measured by QCM-D includes not only the adsorbed peptide but also water molecules trapped in or hydrodynamically coupled to the adsorbate film. In fact, the contribution of coupled water can easily overcome that of the adsorbed peptide or protein.69,70 Therefore, the observed discrepancies may indicate different amounts of trapped or coupled water for the different fragment films.
Figure 4.

Integrated amide I intensities of the different hIAPP(20–29) adsorbate films at the different SAMs as determined by PM-IRRAS (see SI). The black squares give the corresponding −ΔF values (right axis) obtained from the QCM-D measurements (Tables 1–4).
To investigate this effect in more detail, we have calculated the loss factor −ΔD/ΔF at the end of the QCM-D experiment (see Table 1), which is a mass-independent measure of the dissipation.21,69−71 As can be seen from the corresponding values in Table 1, a decrease in the loss factor from 0.29 to 0.17 × 10–6 Hz–1 is observed upon acetylation of the native fragment. Acetylated hIAPP(20–29) monomers thus dissipate less energy than native ones, which indicates that native and acetylated monomers adopt different conformations upon adsorption. In particular, the high loss factor of the native fragment (+|-) indicates a rather loosely attached adsorbate film in which the monomers adopt an upright conformation and dangle from the surface. In contrast, the lower loss factor of the acetylated fragment (0|-) indicates a more firmly attached adsorbate film with the peptide monomers forming multiple contacts with the SAM surface, presumably via hydrogen bonding. The reason for this rather drastic change in conformation may be the reduction of electrostatic repulsion between the N terminus and the NH2-terminated SAM upon acetylation. Due to this conformation, the acetylated peptide film seems to trap large amounts of water. Although the amidated fragments (+|0) and (0|0) show a similar, albeit less pronounced reduction in loss factor upon acetylation, trapped water does not play such a significant role. This indicates significantly different adsorbate film structures.
Table 1. ΔF, ΔD, and −ΔD/ΔF Values of the Different hIAPP(20–29) Fragments Adsorbed on the Positively Charged NH2-Terminated SAMa.
| (+|-) | (0|-) | (+|0) | (0|0) | |
|---|---|---|---|---|
| ΔF (Hz) | –44.48 | –50.38 | –3.94 | –5.28 |
| ΔD (10–6) | 13.06 | 8.52 | 1.08 | 1.03 |
| –ΔD/ΔF (10–6 Hz–1) | 0.29 | 0.17 | 0.27 | 0.20 |
The values were obtained at the end of the QCM-D measurements, that is, after flushing with peptide-free buffer.
The β-sheet contents of the adsorbate films were again determined by deconvolution of the amide I bands (see SI), the results of which are given in Figure 5. As can be seen, the determined β-sheet content does not seem to correlate in any way with the adsorbed mass. Comparison with the β-sheet contents determined after assembly in bulk solution (black dots in Figure 5), however, reveals a rather different trend. With the exception of the acetylated fragment (0|-), which has an almost identical total β-sheet content in bulk and at the NH2 SAM, the β-sheet contents of all the other fragments increase significantly. The strongest increase of more than 30% is observed for the native fragment (+|-) and the smallest for the amidated (+|0) fragment (+14%). In addition, formation of antiparallel β-sheets seems to be reduced or fully suppressed at the NH2 SAM for all fragments. These results thus demonstrate that terminal modifications affect not only the adsorption of hIAPP(20–29) but also its propensity to form intermolecular β-sheets, as well as the structural arrangement of the corresponding β-strands. In particular, Figure 5 reveals that the positively charged NH2 SAM may promote or retard the formation of parallel and antiparallel β-sheets in hIAPP(20–29), depending on the terminal modifications.
Figure 5.

β-Sheet contents of the different hIAPP(20–29) adsorbate films at the different SAMs as determined by PM-IRRAS (see SI). The black dots give the corresponding total β-sheet contents (parallel + antiparallel) observed in bulk solution (Figure 2b).
2.3. Negatively Charged COOH-Terminated SAM
For the COOH-terminated SAM, whose terminal groups are deprotonated and thus negatively charged at physiological pH, we basically expected an inverted behavior compared to the positively charged NH2-terminated SAM. In particular, electrostatics-driven adsorption should result in strong adsorption of the fragments with native and thus positively charged N termini, whereas acetylation of the peptides should lead to only weak adsorption. Surprisingly, however, this assumption is not consistent with the results of the QCM-D measurements shown in Figure 3 and Table 2.
Table 2. ΔF, ΔD, and −ΔD/ΔF Values of the Different hIAPP(20–29) Fragments Adsorbed on the Negatively Charged COOH-Terminated SAMa.
| (+|-) | (0|-) | (+|0) | (0|0) | |
|---|---|---|---|---|
| ΔF (Hz) | –47.95 | –13.44 | –2.28 | –48.52 |
| ΔD (10–6) | 12.15 | 3.92 | 1.78 | 14.49 |
| –ΔD/ΔF (10–6 Hz–1) | 0.25 | 0.29 | 0.78 | 0.30 |
The values were obtained at the end of the QCM-D measurements, that is, after flushing with peptide-free buffer.
As can be seen in Figure 3, large frequency shifts of very similar magnitude are observed for the fragments (+|-) and (0|0) with zero net charge, whereas the positively charged fragment (+|0) shows almost no adsorption at all at the negatively charged SAM. Intermediate adsorption is observed for the negatively charged fragment (0|-) despite the presence of electrostatic repulsion. In our previous work, we have observed that such electrostatic repulsion can be overcome by hydrogen bond formation between peptide and SAM, which may lead to significant adsorption.21 Therefore, we assume that hydrogen bonding plays a dominant role also in the adsorption of the acetylated peptide (0|-) to the COOH-terminated SAM. Nevertheless, in the case of the native fragment (+|-), adsorption appears rather likely to involve the positively charged N-terminal region. This is in part because of the electrostatic repulsion between the terminal COO– groups of the SAM and the negatively charged C terminus, which should favor an upright conformation of the peptide similar to the case of the NH2-terminated SAM discussed above. Furthermore, it was previously observed that purely electrostatic adsorption of native hIAPP(20–29) at a negatively charged mica surface results first in the adsorption of monomers in an upright conformation that is stabilized by intermolecular β-sheets and finally in enhanced fibrillization.22 This is also observed in the AFM images shown in Figure S8, where few short protofibrils are visible for the native fragment (+|-).
The reduced yet significant adsorption of the acetylated fragment (0|-) is indicative of adsorption via hydrogen bonding. Considering the remaining negative charge at the C terminus of (0|-), it appears likely that this hydrogen bonding interaction involves mainly the N-terminal region, whereas the C-terminal region is to some extent dangling from the surface. This is consistent with the obtained loss factor (see Table 2), which is similar to the one obtained for the native fragment (+|-) and thus indicates a somewhat upright and dangling conformation. Removing also the negative charge of the C terminus via additional amidation should then result in reduced electrostatic repulsion between adsorbed peptide monomers and the SAM, as well as between neighboring monomers, and thus increase adsorption. This is observed for the uncharged fragment (0|0), which shows a similar frequency shift as the native one (+|-). However, the obtained loss factor is virtually identical to the one of the acetylated fragment (0|-) and thus indicates that the monomers still assume an upright conformation.
Even harder to explain is the low adsorption of the amidated fragment (+|0) (see Figure 3 and Table 2). Amidation of the negatively charged C terminus should result in decreased electrostatic repulsion both between neighboring monomers and between monomers and the negatively charged surface. Thus, we would expect to observe a stronger decrease in the resonance frequency compared to the native fragment, as well as a lower loss factor. However, this is obviously not happening here. On the contrary, as is evident from Figure 3 and Table 2, the amidated fragment (+|0) shows only very low adsorption at the COOH-terminated SAM and, even more astonishingly, a very high loss factor of 0.78 × 10–6 Hz–1.
To evaluate the role of bound water in the above QCM-D measurements, the obtained frequency shifts listed in Table 2 are again compared to the integral amide I intensities determined by PM-IRRAS in Figure 4. As in the case of the NH2 SAM, a significant discrepancy in the overall trend is observed. Whereas QCM-D yields similarly large frequency shifts for the native (+|-) and the acetylated and amidated (0|0) fragment, the native peptide actually exhibits the lowest amide I intensity. This is not too surprising, considering that the native fragment (+|-) is the only fragment that forms larger aggregates and even protofibrils at the COOH SAM (see Figure S8). It appears rather plausible that such large aggregates may trap larger amounts of water molecules in some buried cavities. Furthermore, the assembled protofibrils may to a certain extent dangle from the surface, resulting in an increased amount of hydrodynamically coupled water due to viscous drag.
The fact that both fragments (+|-) and (+|0) with native, positively charged N termini show similarly low adsorption on the negatively charged COOH-terminated SAM provides a clear indication that electrostatic interactions do not play a significant role in this particular system. At the same time, low monomer adsorption and high lateral mobility are generally considered a prerequisite for amyloid assembly at solid–liquid interfaces.21,22,24,72,73 This is evident from Figure S8, which shows large prefibrillar aggregates and a few protofibrils for native hIAPP(20–29). PM-IRRAS (Figure 5) reveals significantly increased β-sheet contents for all fragments compared to the assembly in bulk solution (Figure 2b). In contrast to the case of the NH2 SAM discussed above, the strongest, almost threefold increase in total β-sheet content is observed for the amidated fragment (+|0), followed by the native fragment (+|-), which shows a similar increase as for the NH2 SAM. Formation of antiparallel β-sheets is again largely suppressed for all fragments at the COOH SAM. These results indicate that the often observed effect that negatively charged interfaces promote hIAPP amyloid formation21,22,74−76 may depend on certain molecular features of the actual peptide in question.
2.4. Polar OH-Terminated SAM
The QCM-D results and AFM images obtained for the interaction of the different hIAPP(20–29) fragments with the polar OH-terminated SAM are shown in Figures 3 and S9, respectively. For the two acetylated fragments (0|-) and (0|0), similar large frequency shifts are observed, whereas the native fragment (+|-) shows a lower yet significant shift. The amidated fragment (+|0), however, does not show any irreversible adsorption. Similar trends are observed in the change of dissipation ΔD. No aggregate structures can be resolved on the rough gold electrode surfaces by AFM (Figure S9).
These results thus indicate that adsorption of the native fragment (+|-) involves the negatively charged C terminus, amidation of which suppresses adsorption. Acetylation of the positively charged N terminus on the other hand results in a change of the adsorption mode, which becomes considerably stronger and now involves the acetylated N terminus. This rather specific interaction with the acetylated N terminus is also reflected in the comparatively large loss factor of 0.29 obtained for the acetylated peptide (0|-) in Table 3, which is consistent with an upright monomer conformation. Additional amidation of the negatively charged C terminus results in further increased frequency shift and loss factor, which may be related to the reduced electrostatic repulsion between adsorbed (0|0) monomers or the fact that adsorption of the acetylated peptide (0|-) may involve both termini and thus cause a monomer conformation that is not dangling but rather looping from the surface. Most astonishing, however, is the observation that the native fragment (+|-) does not show any significant shift in dissipation, which results in a loss factor close to zero (see Table 3). This could indicate a very rigid adsorbate film, which, however, is at variance with the above interpretation of adsorption involving predominantly the native C terminus.
Table 3. ΔF, ΔD, and −ΔD/ΔF Values of the Different hIAPP(20–29) Fragments Adsorbed on the Negatively Charged OH-Terminated SAMa.
| (+|-) | (0|-) | (+|0) | (0|0) | |
|---|---|---|---|---|
| ΔF (Hz) | –10.77 | –26.81 | –0.04 | –32.05 |
| ΔD (10–6) | 0.33 | 7.80 | 11.84 | |
| –ΔD/ΔF (10–6 Hz–1) | 0.03 | 0.29 | 0.37 |
The values were obtained at the end of the QCM-D measurements, that is, after flushing with peptide-free buffer.
To shed some light on this issue, we again compare the observed frequency shifts to the integrated amide I intensities determined from PM-IRRA spectra in Figure 4. Here, two remarkable features can be observed. First, the integrated amide I intensity for the native peptide is close to zero. This might be attributed again to the dominant influence of bound and coupled water. In general, the wet mass of a given film of adsorbed proteins can exceed its dry mass by more than an order of magnitude.70,71,77 However, in this particular case, such a strong effect of coupled water is rather surprising, considering the loss factor that is virtually zero and thus indicates a very rigid adsorbate film that does not dissipate much energy. Similarly confusing is the observation that the adsorbate film of the amidated fragment (+|0) exhibits a significant amide I intensity despite QCM-D yielding a frequency shift of essentially zero. At this moment, we can only speculate that these discrepancies may result from a particular limitation of the PM-IRRAS technique. In PM-IRRAS, only bonds contribute to the recorded spectra that have a transition dipole moment with a component oriented perpendicular to the surface.78 Therefore, one could imagine that the monomers in a thin adsorbate film may adopt peculiar conformations upon drying that result in either an over- or an underestimation of the adsorbed mass determined by PM-IRRAS.
Nevertheless, we have attempted to determine the β-sheet contents also of these adsorbate films (see SI). As can be seen in Figure 5, the strongest deviation from assembly in the bulk solution is observed for the acetylated fragment (0|-), which shows a 30% increase in the total β-sheet content. Furthermore, the amidated fragment (+|0) is found to have a very low β-sheet content, <10%. This is most likely related to the almost absent adsorption of this peptide observed by QCM-D, which prevents assembly of the few adsorbed monomers. Furthermore, at the polar OH SAM, formation of antiparallel β-sheets seems completely suppressed in all fragments.
2.5. Hydrophobic CH3-Terminated SAM
The QCM-D results of hIAPP(20–29) adsorption to the hydrophobic CH3-terminated SAM are presented in Figure 3 and Table 4. The native (+|-), acetylated (0|-), and amidated (+|0) fragments show almost identical behaviors with intermediate frequency shifts around −10 Hz and only minor changes in dissipation. For the acetylated and amidated fragment (0|0), however, significantly higher ΔF values are observed throughout the experiment. Interestingly, the (0|0) fragment also shows the highest loss factor (see Table 4). Whereas adsorption of the other peptides results in very low loss factors between 0.04 and 0.11, indicating very rigid adsorbate films, the acetylated and amidated fragment (0|0) has a loss factor of 0.2. The reason for this large difference is revealed in the AFM images shown in Figure 6. All four fragments form large fibrillar structures at the CH3 SAM. However, whereas the native (+|-), acetylated (0|-), and amidated (+|0) fragments assemble into few, well-separated ribbonlike or straight fibrils, large clusters of crossing fibrils are observed for the acetylated and amidated peptide (0|0). Such clustering of fibrils may result in an increased loss factor.21
Table 4. ΔF, ΔD, and −ΔD/ΔF Values of the Different hIAPP(20–29) Fragments Adsorbed on the Negatively Charged CH3-Terminated SAMa.
| (+|-) | (0|-) | (+|0) | (0|0) | |
|---|---|---|---|---|
| ΔF (Hz) | –10.68 | –13.18 | –9.97 | –2.54 |
| ΔD (10–6) | 1.21 | 1.22 | 0.41 | 0.51 |
| –ΔD/ΔF (10–6 Hz–1) | 0.11 | 0.09 | 0.04 | 0.20 |
The values were obtained at the end of the QCM-D measurements, that is, after flushing with peptide-free buffer.
Figure 6.

AFM images (10 × 10 μm2) of the adsorbed fragments on the CH3 SAM recorded after the end of the QCM-D measurements. The ranges of the z-scales are given in the images.
The assembly of hIAPP(20–29) into mature fibrils at the hydrophobic CH3 SAM is a rather surprising observation. In our previous study, we have observed that fibrillization of native hIAPP(20–29) is delayed in contact with a hydrophobic hydrocarbon surface, whereas adsorption to a negatively charged mica surface resulted in strongly enhanced fibrillization compared to assembly in bulk solution.22 However, these experiments were performed at room temperature in water and not in PBS at 37 °C as the current experiments. Since amyloid assembly in general and aggregate polymorphism in particular are highly sensitive toward environmental conditions,79 these experimental differences may provide an explanation for these unexpected results.
Figure 4 shows the integrated amide I intensities of the adsorbed fragment films, together with the frequency shifts obtained at the end of the QCM-D experiments. Again, different trends are observed. Whereas QCM-D shows rather similar ΔF values for the (+|-), (0|-), and (+|0) fragments, PM-IRRAS yields very different amide I intensities, with the native (+|-) and amidated (+|0) fragments having the lowest and highest integrated intensity values, respectively. The acetylated fragments (0|-) and (0|0) have rather similar, intermediate amide I intensities. These discrepancies are most likely resulting from the formation of amyloid fibrils whose size, morphology, and density affect the amount of hydrodynamically coupled water.
To evaluate possible differences in the molecular structures of the formed hIAPP fibrils, the relative β-sheet contents have again been determined from deconvolution of the amide I bands (Figure 5). As in the above experiments at the OH-terminated SAM, contributions from antiparallel β-sheets are essentially absent for all four fragments. Interestingly, for the native fragment (+|-), the total β-sheet content shown in Figure 5 is almost identical to that obtained in the bulk experiments (parallel + antiparallel; see Figure 2b) despite the formation of amyloid fibrils. For the other three fragments, however, significant increases in β-sheet content of 20–30% are observed. This indicates that fibrillization of these fragment requires certain molecular conformations that are not present in the prefibrillar aggregates forming in bulk solution (see Figure 2).
According to the AFM images shown in Figure 6, all terminally modified fragments assemble into straight fibrils at the CH3 SAM, whereas the native fragment (+|-) forms sheetlike fibrils. Comparison with Figure 5 suggests that this fibrillary polymorphism is correlated with different β-sheet contents since all terminally modified fragments have significantly increased β-sheet contents between 50 and 60%, whereas the native fragment (+|-) has a β-sheet content similar to the bulk value of only about 35%.
The above observations of terminal modifications resulting in differences in adsorption, fibril morphology, and molecular structure are particularly remarkable considering that the interaction of hIAPP(20–29) with the hydrophobic CH3-terminated SAM should involve only the hydrophobic amino acid residues (see Figure 1) and thus be independent of the termini. However, it was shown for a variety of short peptides that terminal modifications may result in pronounced conformational changes in the monomers.80−82 Such conformational alterations could not only be responsible for the different aggregate morphologies and molecular structures observed in bulk solution (see Figure 2), but also result in different interactions with the CH3 SAM, for instance, due to different shielding of the hydrophobic amino acids from the environment.
3. Conclusions
In this work, we have investigated the effect of terminal modifications on the assembly of hIAPP(20–29) in bulk solution and at molecularly defined interfaces using a selection of complementary in situ and ex situ techniques. In bulk solution, fluorescence spectroscopy did not show any β-sheet-specific increase of ThT fluorescence. Nevertheless, AFM revealed the formation of prefibrillar particle-like aggregates with different sizes and morphologies. Whereas the amidated (+|0) and the acetylated and amidated (0|0) fragments were both found to form homogeneous particles of similar sizes, the acetylated (0|-) and especially the native (+|-) fragments form morphologically more diverse aggregates. These morphological differences translated into different contents of antiparallel β-sheets. In particular, the aggregates of the native (+|-) and acetylated (0|-) fragments showed only small contributions from antiparallel β-sheets (1–4%), whereas C-terminal amidation resulted in larger contents of around 10%. These results demonstrate that terminal modifications may significantly affect several aspects of peptide self-assembly, the results of which may not be obvious at first glance and require the application of several complementary analytical techniques to be fully revealed.
The terminal modifications were found to result also in sometimes pronouncedly different adsorption and assembly behaviors at different hydrophilic and hydrophobic surfaces. Also here, several complementary in situ and ex situ techniques were applied and revealed a fairly complex picture of the effect of the terminal modifications on the interaction with the different SAMs. Comparison of in situ QCM-D and ex situ PM-IRRAS data revealed significant differences in the trends of adsorbed mass with fragment species, indicating strong variations in the amount of trapped or hydrodynamically coupled water between fragments. However, these fragment-specific differences did not follow a general trend but varied from surface to surface, which makes them hard to rationalize. Also, the total β-sheet contents of the adsorbed hIAPP(20–29) films varied significantly from fragment to fragment in a surface-specific way. Most notably, for all terminally modified fragments in contact with the different SAMs, significantly reduced contents of antiparallel β-sheets were observed. This effect was most pronounced for the polar OH- and the hydrophobic CH3-terminated SAMs for which antiparallel β-sheet contents were essentially zero for all fragments.
Adsorption of hIAPP(20–29) at the positively charged NH2-terminated SAM seems to be governed to a large extent by electrostatic interactions, with fragment-specific contributions of hydrogen bond formation. Despite the apparent lack of any large aggregate structures at the NH2 SAM, PM-IRRAS revealed that this surface is able to promote β-sheet formation in a peptide-specific way.
In contrast to the positively charged NH2 SAM, electrostatic interactions seem to be less important for hIAPP(20–29) adsorption to the negatively charged COOH SAM. Most notably, we found that the often observed effect of negatively charged interfaces promoting hIAPP amyloid formation21,22,74−76 depends on the molecular structure of the peptide termini. Although increased β-sheet formation was observed for all fragments, the increase in β-sheet content varied drastically from fragment to fragment.
Our results obtained for the polar OH SAM are less conclusive. Here, the comparison between QCM-D and PM-IRRAS indicates a pronounced role of coupled water and the occurrence of peculiar monomer conformations in the adsorbate films upon drying that obscures the amide bonds from interrogation by PM-IRRAS. These issues seem to affect in particular the fragments with native N terminus, which prevents us from drawing any solid conclusions concerning this SAM.
For all hIAPP(20–29) fragments, contact with the hydrophobic CH3-terminated SAM resulted in low adsorption and the formation of amyloid fibrils several microns in length. However, significant differences in fibril morphology and density were observed and could be correlated with the β-sheet content. In particular, the native fragment assembled into ribbonlike fibrils with about 40% β-sheet content, whereas all terminally modified fragments formed straight fibrils with increased β-sheet contents of 50–60%. These observations are particularly noteworthy as the interaction between the hIAPP(20–29) fragments with the CH3 SAM should not involve the termini at all. Therefore, we assume that the observed differences in adsorbed mass, fibril morphology, and molecular structure are to a large extent the result of different monomer conformations with different accessibilities of the hydrophobic amino acid residues.
Our results demonstrate that terminal modifications can have tremendous effects on peptide adsorption and aggregation, even for comparatively simple model peptides such as hIAPP(20–29). The effects exerted by the terminal modifications can be further modulated in nontrivial ways by the physicochemical properties of the SAM surface. This also concerns cases in which adsorption is not mediated by the termini but by the amino acid residues only. Therefore, terminal modifications are an important factor to consider when conducting and comparing peptide adsorption and aggregation studies and may represent an additional parameter for guiding the assembly of peptide-based nanomaterials.
4. Experimental Section
4.1. Peptide Synthesis and Purification
Peptides were prepared using standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry with 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate as the coupling reagent on an automated solid-phase peptide synthesizer (ResPep SL, Intavis). For synthesizing peptide amide, Rink amide resin (TentaGel, Rapp Polymere, Germany) was used. For synthesizing peptide acid, Wang resin (TentaGel, Rapp Polymere, Germany) was used. Each amino acid was coupled twice at 5-fold excess followed by capping the nonreacted amino groups with acetic anhydride to achieve high-quality synthesis. For synthesizing acetylated peptide, an additional capping reaction was performed after the last Fmoc deprotection. The peptide was then cleaved from the resin with trifluoroacetic acid (TFA)/triisopropylsilane/water/dithiothreitol (90(v/v):5(v/v):2.5(v/v):2.5(m/v)) for 2 h. The product was precipitated and washed with ice-cold diethyl ether.
The precipitated peptide was solved in Milli-Q water/acetonitrile (1:1) and further purified using reverse-phase high-pressure liquid chromatography (HPLC) on semipreparative HPLC (Waters) equipped with a semipreparative column (PolymerX RP-1, Phenomenex). The mobile phase was represented by a gradient of 0.1% TFA in water (100–5%) and 0.1% TFA in acetonitrile (5–100%). The fraction containing the product was submitted to analytical reverse-phase ultrahigh-pressure liquid chromatography (UPLC Aquity with UV Detector) equipped with an analytical C18 column (ACQUITY UPLC BEH C18, bead size 1.7 μm, 50 × 2.1 mm2), which uses an anisocratic gradient and is directly connected to an electrospray ionization mass spectrometer (ACQUITY TQ Detector). Peptide structures can be confirmed on the basis of the molecular weight. Finally, the fraction containing the product was lyophilized and stored at −20 °C.
4.2. Sample Preparation
To prepare homogenous monomer solutions, one milligram of hIAPP(20–29) was carefully dissolved in 100 μL of 1,1,1,3,3,3-hexafluoro-2-propanol (≥99%, Sigma-Aldrich) for 1 h with occasional vortexing. The solution was then centrifuged at 15 000 rpm and 4 °C for 30 min, after which the top 80% of the total volume was removed and divided into 10 μL aliquots. Aliquots were frozen in liquid nitrogen and lyophilized for 5 h. The lyophilized hIAPP(20–29) aliquots were stored at −20 °C.
4.3. Peptide Assembly in Bulk Solution
Thioflavin T (ThT, Sigma-Aldrich) was freshly prepared before each experiment at 3 mg/mL in HPLC-grade water (ROTH). The solution was then filtered through a 0.2 μm filter (VWR) and ThT concentration calculated from its absorbance in water at 412 nm measured on an Implen Nanophotometer using a molar extinction coefficient of 36 000 M–1 cm–1.
Before the experiment, ∼2 μL of the freshly prepared ThT solution was mixed with phosphate buffered saline (PBS, 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, pH 7.5, VWR) to reach a final volume and ThT concentration of 495 μL and 20 μM, respectively. The frozen hIAPP(20–29) sample was allowed to reach room temperature and dissolved in 10 μL of dimethyl sulfoxide (DMSO, ≥99.7%, Sigma-Aldrich) by vortexing for 30 s. The dissolved peptide in DMSO was kept at room temperature for 10 min, after which 5 μL of the peptide–DMSO solution was added to the test tube containing 495 μL of PBS and ThT to yield a final concentration of 100 μM. After vortexing for 15 s, the sample was transferred into a clean fluorescence cuvette (Hellma), and a fluorescence emission spectrum was recorded using a JASCO FP-8200ST fluorescence spectrometer in the range from 445 to 700 nm (440 nm excitation) at a speed of 500 nm/min. Spectra were averaged over three individual measurements. The cuvette was then incubated for 24 h at 37 °C and remeasured at intervals of a few hours.
An identical sample but without ThT was incubated in a test tube along the cuvette. After 24 h, the sample was vortexed before depositing 100 μL on a freshly cleaved mica surface. After incubation for 15 min, the mica sample was washed with MQ water and dried with a stream of ultrapure air for AFM imaging. For PM-IRRAS, ∼140 μL of the hIAPP(20–29) solution was deposited on RCA1-cleaned gold-coated silicon wafers. To record representative PM-IRRA spectra that include all hIAPP(20–29) species present in bulk solution, the samples were left to dry and characterized without additional washing.
4.4. SAM Formation
1-Octadecanethiol, 11-mercaptoundecanoic acid, 11-mercapto-1-undecanol, and 11-amino-1-undecanethiol hydrochloride (Sigma-Aldrich) were each dissolved in ethanol to a final concentration of 1 mM. Gold-coated quartz crystal sensors (Filtech Inc.) were cleaned in RCA1 (1:1:5 35% H2O2, 25% NH3, water) for 5 min at 75 °C and immersed in the respective solutions for 24 h to form SAM-terminated surfaces. Immediately prior to the experiment, the SAM-coated sensors were rinsed with ethanol and dried with nitrogen. Successful SAM formation was verified using contact angle measurements and X-ray photoelectron spectroscopy.21
4.5. QCM-D
Right before the experiment, eight hIAPP(20–29) aliquots were slowly brought to room temperature and dissolved in 10 μL of DMSO each. After incubation at room temperature for 10 min, all eight DMSO–peptide aliquots were transferred into a single tube containing 7920 μL of PBS to reach a final concentration of 100 μM hIAPP(20–29). After vortexing for 15 s, the sample solution was injected into the QCM-D chamber.
QCM-D measurements were performed using a Q-Sense E4 (Biolin Scientific) at 37 °C in dynamic mode. After stabilization of the QCM-D system with peptide-free PBS, hIAPP(20–29)-containing buffer was injected into the cells at 10 μL/min. After 13.5 h, the QCM-D cells were flushed with peptide-free PBS for 120 min. Frequency and dissipation shifts were evaluated for the 7th overtone. All QCM-D measurements have been performed up to three times, and similar trends have been observed.
After the measurements, the quartz sensors were removed from the cells, washed with MQ water, and dried in a stream of ultrapure air for AFM imaging and PM-IRRAS.
4.6. AFM
Ex situ AFM imaging was performed in intermittent contact mode in air using JPK Nanowizard II and JPK Nanowizard III AFMs and HQ:NSC18/Al BS cantilevers from MikroMasch with a nominal force constant and tip radius of 2.8 N/m and 8 nm, respectively.
4.7. PM-IRRAS
The chemical analysis of the dried hIAPP(20–29) fragments on gold surfaces was performed by means of PM-IRRAS using a Bruker Vertex 70 (Bruker Optics). The samples were analyzed with a resolution of 4 cm–1 at an angle of 80°. A ZnSe Photo-Elastic-Modulator (PMA50, Bruker) was used to apply 50 kHz modulation to an aluminum wire grid to receive p-polarized light. The light, reflected by the sample, was focused by a ZnSe lens and collected with a liquid-nitrogen-cooled mercury cadmium telluride detector. The obtained original spectra were constrained to the relevant spectral range and background corrected between 1725 and 1500 cm–1 (amide I and amide II regions) for subsequent deconvolution. A second derivative analysis has been carried out to unveil the unresolved spectral contributions, mostly in the amide I peak region.83 Subsequently, spectra were fitted with Gaussian band profiles using OPUS 5.5 software (see SI for details of the deconvolution of the spectra). The integral areas corresponding to the amide I peak have been obtained by adding the contributions of the different components in the range of 1610–1700 cm–1.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b03028.
ThT fluorescence spectra; additional AFM images for assembly in bulk solution; different overtones recorded in the QCM-D measurements; AFM images of the different adsorbate films at NH2, COOH, and OH SAMs; deconvolution of PM-IRRA spectra (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mendes A. C.; Baran E. T.; Reis R. L.; Azevedo H. S. Self-assembly in nature: using the principles of nature to create complex nanobiomaterials. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2013, 5, 582–612. 10.1002/wnan.1238. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Mezzenga R. Amyloid Fibrils as Building Blocks for Natural and Artificial Functional Materials. Adv. Mater. 2016, 28, 6546–6561. 10.1002/adma.201505961. [DOI] [PubMed] [Google Scholar]
- Wei G.; Su Z.; Reynolds N. P.; Arosio P.; Hamley I. W.; Gazit E.; Mezzenga R. Self-assembling peptide and protein amyloids: from structure to tailored function in nanotechnology. Chem. Soc. Rev. 2017, 46, 4661–4708. 10.1039/C6CS00542J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humenik M.; Lang G.; Scheibel T. Silk nanofibril self-assembly versus electrospinning. Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol. 2018, 10, e1509 10.1002/wnan.1509. [DOI] [PubMed] [Google Scholar]
- Hauser C. A. E.; Maurer-Stroh S.; Martins I. C. Amyloid-based nanosensors and nanodevices. Chem. Soc. Rev. 2014, 43, 5326–5345. 10.1039/C4CS00082J. [DOI] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Ke P. C.; Sani M.-A.; Ding F.; Kakinen A.; Javed I.; Separovic F.; Davis T. P.; Mezzenga R. Implications of peptide assemblies in amyloid diseases. Chem. Soc. Rev. 2017, 46, 6492–6531. 10.1039/C7CS00372B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberg D.; Jucker M. The amyloid state of proteins in human diseases. Cell 2012, 148, 1188–1203. 10.1016/j.cell.2012.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefani M. Protein folding and misfolding on surfaces. Int. J. Mol. Sci. 2008, 9, 2515–2542. 10.3390/ijms9122515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parveen R.; Shamsi T. N.; Fatima S. Nanoparticles-protein interaction: Role in protein aggregation and clinical implications. Int. J. Biol. Macromol. 2017, 94, 386–395. 10.1016/j.ijbiomac.2016.10.024. [DOI] [PubMed] [Google Scholar]
- Zhang M.; Mao X.; Yu Y.; Wang C.-X.; Yang Y.-L.; Wang C. Nanomaterials for reducing amyloid cytotoxicity. Adv. Mater. 2013, 25, 3780–3801. 10.1002/adma.201301210. [DOI] [PubMed] [Google Scholar]
- Mahmoudi M.; Kalhor H. R.; Laurent S.; Lynch I. Protein fibrillation and nanoparticle interactions: opportunities and challenges. Nanoscale 2013, 5, 2570–2588. 10.1039/c3nr33193h. [DOI] [PubMed] [Google Scholar]
- John T.; Gladytz A.; Kubeil C.; Martin L. L.; Risselada H. J.; Abel B. Impact of nanoparticles on amyloid peptide and protein aggregation: a review with a focus on gold nanoparticles. Nanoscale 2018, 10, 20894–20913. 10.1039/C8NR04506B. [DOI] [PubMed] [Google Scholar]
- Li C.; Qin R.; Liu R.; Miao S.; Yang P. Functional amyloid materials at surfaces/interfaces. Biomater. Sci. 2018, 6, 462–472. 10.1039/C7BM01124E. [DOI] [PubMed] [Google Scholar]
- Li C.; Mezzenga R. The interplay between carbon nanomaterials and amyloid fibrils in bio-nanotechnology. Nanoscale 2013, 5, 6207–6218. 10.1039/c3nr01644g. [DOI] [PubMed] [Google Scholar]
- Li C.; Born A.-K.; Schweizer T.; Zenobi-Wong M.; Cerruti M.; Mezzenga R. Amyloid-hydroxyapatite bone biomimetic composites. Adv. Mater. 2014, 26, 3207–3212. 10.1002/adma.201306198. [DOI] [PubMed] [Google Scholar]
- Shen Y.; Nyström G.; Mezzenga R. Amyloid Fibrils form Hybrid Colloidal Gels and Aerogels with Dispersed CaCO3 Nanoparticles. Adv. Funct. Mater. 2017, 27, 1700897 10.1002/adfm.201700897. [DOI] [Google Scholar]
- Humenik M.; Mohrand M.; Scheibel T. Self-Assembly of Spider Silk-Fusion Proteins Comprising Enzymatic and Fluorescence Activity. Bioconjugate Chem. 2018, 29, 898–904. 10.1021/acs.bioconjchem.7b00759. [DOI] [PubMed] [Google Scholar]
- Humenik M.; Scheibel T. Nanomaterial building blocks based on spider silk-oligonucleotide conjugates. ACS Nano 2014, 8, 1342–1349. 10.1021/nn404916f. [DOI] [PubMed] [Google Scholar]
- Molina A.; Scheibel T.; Humenik M. Nanoscale patterning of surfaces via DNA directed spider silk assembly. Biomacromolecules 2018, 20, 347–352. 10.1021/acs.biomac.8b01333. [DOI] [PubMed] [Google Scholar]
- Hajiraissi R.; Hanke M.; Yang Y.; Duderija B.; Gonzalez Orive A.; Grundmeier G.; Keller A. Adsorption and Fibrillization of Islet Amyloid Polypeptide at Self-Assembled Monolayers Studied by QCM-D, AFM, and PM-IRRAS. Langmuir 2018, 34, 3517–3524. 10.1021/acs.langmuir.7b03626. [DOI] [PubMed] [Google Scholar]
- Hajiraissi R.; Giner I.; Grundmeier G.; Keller A. Self-Assembly, Dynamics, and Polymorphism of hIAPP(20–29) Aggregates at Solid-Liquid Interfaces. Langmuir 2017, 33, 372–381. 10.1021/acs.langmuir.6b03288. [DOI] [PubMed] [Google Scholar]
- Jeworrek C.; Hollmann O.; Steitz R.; Winter R.; Czeslik C. Interaction of IAPP and insulin with model interfaces studied using neutron reflectometry. Biophys. J. 2009, 96, 1115–1123. 10.1016/j.bpj.2008.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.-C.; Petersson E. J.; Fakhraai Z. Surface Effects Mediate Self-Assembly of Amyloid-β Peptides. ACS Nano 2014, 8, 10178–10186. 10.1021/nn5031669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shezad K.; Zhang K.; Hussain M.; Dong H.; He C.; Gong X.; Xie X.; Zhu J.; Shen L. Surface Roughness Modulates Diffusion and Fibrillation of Amyloid-β Peptide. Langmuir 2016, 32, 8238–8244. 10.1021/acs.langmuir.6b01756. [DOI] [PubMed] [Google Scholar]
- Yu Y.-P.; Zhang S.; Liu Q.; Li Y.-M.; Wang C.; Besenbacher F.; Dong M. 2D amyloid aggregation of human islet amyloid polypeptide at the solid–liquid interface. Soft Matter 2012, 8, 1616–1622. 10.1039/C1SM06599H. [DOI] [Google Scholar]
- Accardo A.; Shalabaeva V.; Di Cola E.; Burghammer M.; Krahne R.; Riekel C.; Dante S. Superhydrophobic Surfaces Boost Fibril Self-Assembly of Amyloid β Peptides. ACS Appl. Mater. Interfaces 2015, 7, 20875–20884. 10.1021/acsami.5b06219. [DOI] [PubMed] [Google Scholar]
- Kowalewski T.; Holtzman D. M. In situ atomic force microscopy study of Alzheimer’s -amyloid peptide on different substrates: New insights into mechanism of -sheet formation. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 3688–3693. 10.1073/pnas.96.7.3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losic D.; Martin L. L.; Aguilar M.-I.; Small D. H. Beta-amyloid fibril formation is promoted by step edges of highly oriented pyrolytic graphite. Biopolymers 2006, 84, 519–526. 10.1002/bip.20549. [DOI] [PubMed] [Google Scholar]
- Moores B.; Drolle E.; Attwood S. J.; Simons J.; Leonenko Z. Effect of surfaces on amyloid fibril formation. PLoS One 2011, 6, e25954 10.1371/journal.pone.0025954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H.; Fung S.-Y.; Pritzker M.; Chen P. Surface-assisted assembly of an ionic-complementary peptide: controllable growth of nanofibers. J. Am. Chem. Soc. 2007, 129, 12200–12210. 10.1021/ja073168u. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Shah N.; Zhao J.; Wang C.; Zhao C.; Liu L.; Li L.; Zhou F.; Zheng J. Structural, morphological, and kinetic studies of β-amyloid peptide aggregation on self-assembled monolayers. Phys. Chem. Chem. Phys. 2011, 13, 15200–15210. 10.1039/c1cp21156k. [DOI] [PubMed] [Google Scholar]
- Ragaliauskas T.; Mickevicius M.; Budvytyte R.; Niaura G.; Carbonnier B.; Valincius G. Adsorption of β-amyloid oligomers on octadecanethiol monolayers. J. Colloid Interface Sci. 2014, 425, 159–167. 10.1016/j.jcis.2014.03.042. [DOI] [PubMed] [Google Scholar]
- Hovgaard M. B.; Dong M.; Otzen D. E.; Besenbacher F. Quartz crystal microbalance studies of multilayer glucagon fibrillation at the solid-liquid interface. Biophys. J. 2007, 93, 2162–2169. 10.1529/biophysj.107.109686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A.; Fritzsche M.; Yu Y.-P.; Liu Q.; Li Y.-M.; Dong M.; Besenbacher F. Influence of hydrophobicity on the surface-catalyzed assembly of the islet amyloid polypeptide. ACS Nano 2011, 5, 2770–2778. 10.1021/nn1031998. [DOI] [PubMed] [Google Scholar]
- Giacomelli C. E.; Norde W. Influence of hydrophobic Teflon particles on the structure of amyloid beta-peptide. Biomacromolecules 2003, 4, 1719–1726. 10.1021/bm034151g. [DOI] [PubMed] [Google Scholar]
- Giacomelli C. E.; Norde W. Conformational changes of the amyloid beta-peptide (1–40) adsorbed on solid surfaces. Macromol. Biosci. 2005, 5, 401–407. 10.1002/mabi.200400189. [DOI] [PubMed] [Google Scholar]
- Lin Y.-C.; Li C.; Fakhraai Z. Kinetics of Surface-Mediated Fibrillization of Amyloid-β (12–28) Peptides. Langmuir 2018, 34, 4665–4672. 10.1021/acs.langmuir.7b02744. [DOI] [PubMed] [Google Scholar]
- Nayak A.; Dutta A. K.; Belfort G. Surface-enhanced nucleation of insulin amyloid fibrillation. Biochem. Biophys. Res. Commun. 2008, 369, 303–307. 10.1016/j.bbrc.2008.01.159. [DOI] [PubMed] [Google Scholar]
- Rabe M.; Soragni A.; Reynolds N. P.; Verdes D.; Liverani E.; Riek R.; Seeger S. On-surface aggregation of α-synuclein at nanomolar concentrations results in two distinct growth mechanisms. ACS Chem. Neurosci. 2013, 4, 408–417. 10.1021/cn3001312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sluzky V.; Tamada J. A.; Klibanov A. M.; Langer R. Kinetics of insulin aggregation in aqueous solutions upon agitation in the presence of hydrophobic surfaces. Proc. Natl. Acad. Sci. U.S.A. 1991, 88, 9377–9381. 10.1073/pnas.88.21.9377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Zhang Y.; Zhang F.; Pillai S.; Liu J.; Li R.; Dai B.; Li B.; Zhang Y. Hierarchical ordering of amyloid fibrils on the mica surface. Nanoscale 2013, 5, 4816–4822. 10.1039/c3nr00886j. [DOI] [PubMed] [Google Scholar]
- Kang S.-g.; Li H.; Huynh T.; Zhang F.; Xia Z.; Zhang Y.; Zhou R. Molecular mechanism of surface-assisted epitaxial self-assembly of amyloid-like peptides. ACS Nano 2012, 6, 9276–9282. 10.1021/nn303740j. [DOI] [PubMed] [Google Scholar]
- Dai B.; Kang S.-g.; Huynh T.; Lei H.; Castelli M.; Hu J.; Zhang Y.; Zhou R. Salts drive controllable multilayered upright assembly of amyloid-like peptides at mica/water interface. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 8543–8548. 10.1073/pnas.1220711110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karsai Á.; Grama L.; Murvai Ü.; Soós K.; Penke B.; Kellermayer M. S. Z. Potassium-dependent oriented growth of amyloid β25–35 fibrils on mica. Nanotechnology 2007, 18, 345102 10.1088/0957-4484/18/34/345102. [DOI] [Google Scholar]
- Goldsbury C.; Kistler J.; Aebi U.; Arvinte T.; Cooper G. J. Watching amyloid fibrils grow by time-lapse atomic force microscopy. J. Mol. Biol. 1999, 285, 33–39. 10.1006/jmbi.1998.2299. [DOI] [PubMed] [Google Scholar]
- Zhu M.; Souillac P. O.; Ionescu-Zanetti C.; Carter S. A.; Fink A. L. Surface-catalyzed amyloid fibril formation. J. Biol. Chem. 2002, 277, 50914–50922. 10.1074/jbc.M207225200. [DOI] [PubMed] [Google Scholar]
- Mao X.; Wang Y.; Liu L.; Niu L.; Yang Y.; Wang C. Molecular-level evidence of the surface-induced transformation of peptide structures revealed by scanning tunneling microscopy. Langmuir 2009, 25, 8849–8853. 10.1021/la901342r. [DOI] [PubMed] [Google Scholar]
- Rocha S.; Krastev R.; Thünemann A. F.; Pereira M. C.; Möhwald H.; Brezesinski G. Adsorption of amyloid beta-peptide at polymer surfaces: a neutron reflectivity study. ChemPhysChem 2005, 6, 2527–2534. 10.1002/cphc.200500158. [DOI] [PubMed] [Google Scholar]
- Smith M. I.; Sharp J. S.; Roberts C. J. Nucleation and growth of insulin fibrils in bulk solution and at hydrophobic polystyrene surfaces. Biophys. J. 2007, 93, 2143–2151. 10.1529/biophysj.107.105338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campioni S.; Carret G.; Jordens S.; Nicoud L.; Mezzenga R.; Riek R. The presence of an air-water interface affects formation and elongation of α-Synuclein fibrils. J. Am. Chem. Soc. 2014, 136, 2866–2875. 10.1021/ja412105t. [DOI] [PubMed] [Google Scholar]
- Cao P.; Abedini A.; Raleigh D. P. Aggregation of islet amyloid polypeptide: from physical chemistry to cell biology. Curr. Opin. Struct. Biol. 2012, 23, 82–89. 10.1016/j.sbi.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caillon L.; Hoffmann A. R. F.; Botz A.; Khemtemourian L. Molecular Structure, Membrane Interactions, and Toxicity of the Islet Amyloid Polypeptide in Type 2 Diabetes Mellitus. J. Diabetes Res. 2016, 2016, 5639875 10.1155/2016/5639875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Andreasen M.; Nielsen J. T.; Liu L.; Nielsen E. H.; Song J.; Ji G.; Sun F.; Skrydstrup T.; Besenbacher F.; et al. Coexistence of ribbon and helical fibrils originating from hIAPP(20–29) revealed by quantitative nanomechanical atomic force microscopy. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 2798–2803. 10.1073/pnas.1209955110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M.; Skeby K. K.; Zhang S.; Nielsen E. H.; Klausen L. H.; Frahm H.; Christiansen G.; Skrydstrup T.; Dong M.; Schiøtt B.; et al. The importance of being capped: Terminal capping of an amyloidogenic peptide affects fibrillation propensity and fibril morphology. Biochemistry 2014, 53, 6968–6980. 10.1021/bi500674u. [DOI] [PubMed] [Google Scholar]
- Forloni G.; Lucca E.; Angeretti N.; Della Torre P.; Salmona M. Amidation of β-Amyloid Peptide Strongly Reduced the Amyloidogenic Activity Without Alteration of the Neurotoxicity. J. Neurochem. 1997, 69, 2048–2054. 10.1046/j.1471-4159.1997.69052048.x. [DOI] [PubMed] [Google Scholar]
- Hernik-Magoń A.; Fedorczyk B.; Dec R.; Puławski W.; Misicka A.; Dzwolak W. Effects of terminal capping on the fibrillation of short (L-Glu)n peptides. Colloids Surf., B 2017, 159, 861–868. 10.1016/j.colsurfb.2017.08.061. [DOI] [PubMed] [Google Scholar]
- Kang L.; Janowska M. K.; Moriarty G. M.; Baum J. Mechanistic insight into the relationship between N-terminal acetylation of α-synuclein and fibril formation rates by NMR and fluorescence. PLoS One 2013, 8, e75018 10.1371/journal.pone.0075018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M.; Choi H.; Na S. Effects of End-Terminal Capping on Transthyretin (105–115) Amyloid Protofibrils Using Steered Molecular Dynamics. J. Nanomater. 2016, 2016, 1–10. 10.1155/2016/1863065. [DOI] [Google Scholar]
- Lee M.; Chang H. J.; Choi H.; Na S. Capping effects on polymorphic Aβ16-21 amyloids depend on their size: A molecular dynamics simulation study. Biophys. Chem. 2018, 232, 1–11. 10.1016/j.bpc.2017.09.003. [DOI] [PubMed] [Google Scholar]
- Ryan D. M.; Doran T. M.; Anderson S. B.; Nilsson B. L. Effect of C-terminal modification on the self-assembly and hydrogelation of fluorinated Fmoc-Phe derivatives. Langmuir 2011, 27, 4029–4039. 10.1021/la1048375. [DOI] [PubMed] [Google Scholar]
- Tao K.; Wang J.; Zhou P.; Wang C.; Xu H.; Zhao X.; Lu J. R. Self-assembly of short aβ(16–22) peptides: effect of terminal capping and the role of electrostatic interaction. Langmuir 2011, 27, 2723–2730. 10.1021/la1034273. [DOI] [PubMed] [Google Scholar]
- Yonemoto I. T.; Kroon G. J. A.; Dyson H. J.; Balch W. E.; Kelly J. W. Amylin proprotein processing generates progressively more amyloidogenic peptides that initially sample the helical state. Biochemistry 2008, 47, 9900–9910. 10.1021/bi800828u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue C.; Lin T. Y.; Chang D.; Guo Z. Thioflavin T as an amyloid dye: fibril quantification, optimal concentration and effect on aggregation. R. Soc. Open Sci. 2017, 4, 160696 10.1098/rsos.160696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freire S.; de Araujo M. H.; Al-Soufi W.; Novo M. Photophysical study of Thioflavin T as fluorescence marker of amyloid fibrils. Dyes Pigm. 2014, 110, 97–105. 10.1016/j.dyepig.2014.05.004. [DOI] [Google Scholar]
- Garai K.; Frieden C. Quantitative analysis of the time course of Aβ oligomerization and subsequent growth steps using tetramethylrhodamine-labeled Aβ. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 3321–3326. 10.1073/pnas.1222478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madine J.; Jack E.; Stockley P. G.; Radford S. E.; Serpell L. C.; Middleton D. A. Structural insights into the polymorphism of amyloid-like fibrils formed by region 20–29 of amylin revealed by solid-state NMR and X-ray fiber diffraction. J. Am. Chem. Soc. 2008, 130, 14990–15001. 10.1021/ja802483d. [DOI] [PubMed] [Google Scholar]
- Dixon M. C. Quartz crystal microbalance with dissipation monitoring: enabling real-time characterization of biological materials and their interactions. J. Biomol. Tech. 2008, 19, 151–158. [PMC free article] [PubMed] [Google Scholar]
- Hemmersam A. G.; Foss M.; Chevallier J.; Besenbacher F. Adsorption of fibrinogen on tantalum oxide, titanium oxide and gold studied by the QCM-D technique. Colloids Surf., B 2005, 43, 208–215. 10.1016/j.colsurfb.2005.04.007. [DOI] [PubMed] [Google Scholar]
- Höök F.; Vörös J.; Rodahl M.; Kurrat R.; Böni P.; Ramsden J. J.; Textor M.; Spencer N. D.; Tengvall P.; Gold J.; et al. A comparative study of protein adsorption on titanium oxide surfaces using in situ ellipsometry, optical waveguide lightmode spectroscopy, and quartz crystal microbalance/dissipation. Colloids Surf., B 2002, 24, 155–170. 10.1016/S0927-7765(01)00236-3. [DOI] [Google Scholar]
- Hemmersam A. G.; Rechendorff K.; Foss M.; Sutherland D. S.; Besenbacher F. Fibronectin adsorption on gold, Ti-, and Ta-oxide investigated by QCM-D and RSA modelling. J. Colloid Interface Sci. 2008, 320, 110–116. 10.1016/j.jcis.2007.11.047. [DOI] [PubMed] [Google Scholar]
- Lin Y.-C.; Repollet-Pedrosa M. H.; Ferrie J. J.; Petersson E. J.; Fakhraai Z. Potential Artifacts in Sample Preparation Methods Used for Imaging Amyloid Oligomers and Protofibrils due to Surface-Mediated Fibril Formation. J. Phys. Chem. B 2017, 121, 2534–2542. 10.1021/acs.jpcb.6b12560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L.; Adachi T.; Vanden Bout D.; Zhu X.-Y. A Mobile Precursor Determines Amyloid-β Peptide Fibril Formation at Interfaces. J. Am. Chem. Soc. 2012, 134, 14172–14178. 10.1021/ja305398f. [DOI] [PubMed] [Google Scholar]
- Jayasinghe S. A.; Langen R. Lipid membranes modulate the structure of islet amyloid polypeptide. Biochemistry 2005, 44, 12113–12119. 10.1021/bi050840w. [DOI] [PubMed] [Google Scholar]
- Knight J. D.; Miranker A. D. Phospholipid catalysis of diabetic amyloid assembly. J. Mol. Biol. 2004, 341, 1175–1187. 10.1016/j.jmb.2004.06.086. [DOI] [PubMed] [Google Scholar]
- Caillon L.; Lequin O.; Khemtémourian L. Evaluation of membrane models and their composition for islet amyloid polypeptide-membrane aggregation. Biochim. Biophys. Acta, Biomembr. 2013, 1828, 2091–2098. 10.1016/j.bbamem.2013.05.014. [DOI] [PubMed] [Google Scholar]
- Höök F.; Kasemo B.; Nylander T.; Fant C.; Sott K.; Elwing H. Variations in Coupled Water, Viscoelastic Properties, and Film Thickness of a Mefp-1 Protein Film during Adsorption and Cross-Linking: A Quartz Crystal Microbalance with Dissipation Monitoring, Ellipsometry, and Surface Plasmon Resonance Study. Anal. Chem. 2001, 73, 5796–5804. 10.1021/ac0106501. [DOI] [PubMed] [Google Scholar]
- Desroches M. J.; Chaudhary N.; Omanovic S. PM-IRRAS investigation of the interaction of serum albumin and fibrinogen with a biomedical-grade stainless steel 316LVM surface. Biomacromolecules 2007, 8, 2836–2844. 10.1021/bm070289d. [DOI] [PubMed] [Google Scholar]
- Pedersen J. S.; Andersen C. B.; Otzen D. E. Amyloid structure--one but not the same: the many levels of fibrillar polymorphism. FEBS J. 2010, 277, 4591–4601. 10.1111/j.1742-4658.2010.07888.x. [DOI] [PubMed] [Google Scholar]
- Kang T. S.; Vivekanandan S.; Jois S. D. S.; Kini R. M. Effect of C-terminal amidation on folding and disulfide-pairing of alpha-conotoxin ImI. Angew. Chem., Int. Ed. 2005, 44, 6333–6337. 10.1002/anie.200502300. [DOI] [PubMed] [Google Scholar]
- Katayama H.; Ohira T.; Aida K.; Nagasawa H. Significance of a carboxyl-terminal amide moiety in the folding and biological activity of crustacean hyperglycemic hormone. Peptides 2002, 23, 1537–1546. 10.1016/S0196-9781(02)00094-3. [DOI] [PubMed] [Google Scholar]
- Sforça M. L.; Oyama S.; Canduri F.; Lorenzi C. C. B.; Pertinhez T. A.; Konno K.; Souza B. M.; Palma M. S.; Ruggiero Neto J.; Azevedo W. F.; et al. How C-terminal carboxyamidation alters the biological activity of peptides from the venom of the eumenine solitary wasp. Biochemistry 2004, 43, 5608–5617. 10.1021/bi0360915. [DOI] [PubMed] [Google Scholar]
- Yang H.; Yang S.; Kong J.; Dong A.; Yu S. Obtaining information about protein secondary structures in aqueous solution using Fourier transform IR spectroscopy. Nat. Protoc. 2015, 10, 382–396. 10.1038/nprot.2015.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.