

Abstract
Objective
An epilepsy mouse model for Tuberous Sclerosis Complex (TSC) was developed and validated to investigate the mechanisms underlying epileptogenesis. Furthermore, the possible antiepileptogenic properties of commonly used antiepileptic drugs (AEDs) and new compounds were assessed.
Methods
Tsc1 deletion was induced in CAMK2A‐expressing neurons of adult mice. The antiepileptogenic properties of commonly used AEDs and inhibitors of the mTOR pathways were assessed by EEG recordings and by molecular read outs.
Results
Mice developed epilepsy in a narrow time window (10 ± 2 days) upon Tsc1 gene deletion. Seizure frequency but not duration increased over time. Seizures were lethal within 18 days, were unpredictable, and did not correlate to seizure onset, length or frequency, reminiscent of sudden unexpected death in epilepsy (SUDEP). Tsc1 gene deletion resulted in a strong activation of the mTORC1 pathway, and both epileptogenesis and lethality could be entirely prevented by RHEB1 gene deletion or rapamycin treatment. However, other inhibitors of the mTOR pathway such as AZD8055 and PF4708671 were ineffective. Except for ketogenic diet, none of commonly used AEDs showed an effect on mTORC1 activity. Vigabatrin and ketogenic diet treatment were able to significantly delay seizure onset. In contrast, survival was shortened by lamotrigine.
Interpretation
This novel Tsc1 mouse model is highly suitable to assess the efficacy of antiepileptic and ‐epileptogenic drugs to treat mTORC1‐dependent epilepsy. Additionally, it allows us to study the mechanisms underlying mTORC1‐mediated epileptogenesis and SUDEP. We found that early treatment with vigabatrin was not able to prevent epilepsy, but significantly delayed seizure onset.
Introduction
Tuberous Sclerosis Complex (TSC) is an autosomal dominant multisystem disorder caused by an inactivating mutation in either the TSC1 or TSC2 gene.1 The TSC1 and TSC2 genes encode for the proteins hamartin and tuberin respectively, forming a complex that inhibits the mammalian Target of Rapamycin Complex 1 (mTORC1) through Ras homolog enriched in brain (RHEB).
TSC is characterized by the formation of benign tumors, which affect multiple organ systems including the skin, heart, lungs, kidney, and brain.2 In the brain, lesions (referred to as tubers) are often linked to the presence of seizures within TSC patients, however, other factors are involved as well.3 Notably, studies in animal models show that acute deletion of the Tsc1 gene in adult animals is sufficient to induce epilepsy in the absence of discernable pathology.4
Up to 80–90% of the TSC patients develop severe epilepsy, causing a significant decrease in the quality of life and a high morbidity.5, 6 In the majority of TSC patients, epilepsy develops in the first years of life, and is associated with poor intellectual outcome and a developmental delay.7, 8, 9 Currently, vigabatrin is used as a first‐line treatment to suppress infantile spasms, in TSC.10 However, two‐thirds of the TSC patients do not reach sustained seizure remission, highlighting the need for better treatment.11, 12 Specific targeting of the mTOR pathway may in particular be a promising treatment method. A recent clinical study showed decreased seizure frequency in TSC patients treated with mTOR inhibitors13, 14 or with a ketogenic diet,15 which has also been shown to act on mTOR signaling. In addition, it might be profitable to start treatment well before seizures occur, thereby interfering with the process of epileptogenesis.16 Epileptogenesis is the molecular and cellular process that ultimately leads to the development of seizures or chronic epilepsy. However, it is largely unknown to what extend current antiepileptic drugs (AEDs) are effective in blocking (or delaying) mTOR‐driven epileptogenesis. A preclinical study would ideally utilize animal models in which epileptogenesis occurs in a well‐defined molecular and temporal fashion.
Previously, we generated an indicable Tsc1 mouse model in which Tsc1 was biallelic deleted throughout the entire body, resulting in a hyperactive mTORC1 pathway and spontaneous epilepsy4. However, seizure onset in this model was still quite variable and loss of Tsc1 in peripheral tissues induced early lethality, unrelated to epilepsy. Therefore, the first aim of this study was to develop and characterize a Tsc1 mouse model that showed mTORC1‐dependent epileptic seizures in a well‐defined time window, without the loss of Tsc1 in peripheral tissue. The second aim was to test the seizure‐suppressive and possible antiepileptogenic effects of commonly used AEDs as well as novel drugs, by electroencephalogram (EEG) recording and their effect on the mTOR pathway.
Material and Methods
Mice
Inducible Tsc1 knockout mice were generated by crossing transgenic mice carrying tamoxifen inducible CreERT2 under control of the Camk2a promoter (Tg(Camk2a‐cre/ERT2)2Gsc; MGI:3759305) in the C57BL/6J background17 with conditional biallelic floxed Tsc1 mutant mice (Tsc1tm1Djk; MGI:2656240).18 These latter mice were obtained in a mixed N2F5 background from Jackson Laboratories and after three crosses into C57BL/6J maintained by interbreeding. This resulted in Tsc1 f/f‐Tg(Camk2a‐CreERT2+) and Tsc1 f/f‐Tg(Camk2a‐CreERT2–) mice, hereafter named Tsc1‐Cre+ and Tsc1‐Cre– mice respectively. Tsc1‐Cre– mice were negative for Cre recombinase and used as control throughout all experiments. Inducible double knock out mice, in which both Tsc1 and Rheb1 were deleted upon tamoxifen injections, were created by crossing the Tsc1 f/f‐Tg(Camk2a‐CreERT2+) mice with a Rhebf/f mouse line (Rhebtm1Yelg; MGI:5752310) in the C57BL/6J background.19
Gene deletion was induced in male and female 12‐ to 20‐week‐old mice. Mice were housed in individual ventilated cages and kept on a 12‐h light/dark cycle, at 22 ± 2°C and had ad libitum access to food and water. Animal welfare was checked daily by animal caretakers and experimenters throughout the experiments. For each experiment, independent groups of mice were used. Only the mice used for EEG measurements were single housed. All cages contained basic bedding and nesting material. For the EEG experiments, groups of at least six mice were used and were sacrificed the latest on day 29 after gene deletion. Mice used for the western blot experiment were sacrificed on day 12 or 13 after gene deletion (indicated in the figure legend). All experiments were approved by the local animal experimentation center, in accordance with institutional animal care and use guidelines (license AVD1010020172684).
Immunohistochemistry and western blot analysis
Samples for immunohistochemstry and western blot analysis were prepared as described previously.4 To analyze the immunohistochemistry sections, a brightfield (CX41, Olympus, Japan) or a confocal microscope (Zeiss, LSM700) was used. Soma sizes were quantified in single‐section pictures with Zen (Zeiss, version 7.0). For western blot analysis, mice were sacrificed 2 h after AED administration. Lysates were prepared as described previously,4 and blots were imaged using a LI‐COR Odyssey Scanner system and quantified with Odyssey 3.0 software (LI‐COR Biosciences). Expression levels were corrected for actin loading control and normalized to control mice.
Antibodies
Primary antibodies against CAMK2A (Novus Biologicals NB100‐1983, 1:500 for immunohistochemistry (IHC)), Parvalbumin (SWANT, 1:9000 for IHC), NeuN ( 1:1000 IHC), TSC1 (CST #4906, 1:1000 for western blot), RHEB1 (CST #4935, 1:1000 for western blot), pS6S240/244 (CST #5634, 1:1000 for IHC, 1:2000 for western blot), S6 (CST #2217, 1:2000 for western blot), pAktSer473 (CST #9271 1:2000 for western blot) and Actin (Chemicon MAB1501R, 1:20.000 for western blot) were used.
PathScan Akt antibody array
Mice were euthanized at day 8 or 13 by decapitation under deep isoflurane anesthesia. Brains were dissected and isolated cortical and hippocampal tissues were homogenized in lysis buffer (0.02 mol/L Tris‐HCl (pH 7.5), 0.15 mol/L NaCl, 0.001 mol/L Na2EDTA, 0.001 mol/L EGTA, 1% Triton, 0.02 mol/L sodium pyrophosphate, 0.025M sodium fluoride, 0.001 mol/L β‐glycerophosphate, 0.001 mol/L Na3VO4, 1 μg/mL leupeptin, and protease inhibitor cocktail (P8340, Sigma‐Aldrich, 1:100). Samples were analyzed using the PathScan Akt Signaling Antibody Array Kit (CST#9700). With this kit, we assessed the phosphorylation of the following proteins: Akt (T308, S473), S6 (S235/236), AMPK‐a (T172), PRAS40 (T246), mTOR(S2481), GSK‐3a (S21), GSK‐3b (s9), p70S6K (T389, T421/S424), Bad (S112), RSK1 (S380), PTEN (S380), PDK1 (S241), Erk1/2 (T202/204), 4E‐BP1 (T37/46). LI‐COR Odyssey fluorescence imaging system was used to image the pad slide and Odyssey 3.0 software was used for quantitative analyses (LI‐COR Biosciences). The obtained signals were corrected for actin loading control and normalized to control mice.
EEG surgery and recordings
Mice were under anesthesia (2% isoflurane and 0.55% oxygen) and injected with 20 μL rimadyl (Zoetis, 5 mg/mL), an NSAID, to reduce pain and inflammation. Xilocaine (AstraZeneca, 100 mg/mL) was sprayed on the skull as a local anesthetic. In total, four EEG electrodes were placed in the skull bilateral on top of the motor cortex (Bregma AP: +1, ML: +1; AP: +1, ML: −1) and the somatosensory cortex (Bregma: AP: −1, ML −4; AP: −1, ML: +4). A reference electrode was placed on the cerebellar cortex. At the end of the surgery mice were injected with 50 μL temgesic (Reckitt Benckiser, 0.3 mg/mL). Mice recovered for a week from surgery before gene deletion was induced. For EEG recordings, mice were connected to a wireless EEG recorder (Neurologger, New behavior, TSE Systems) for 24 h per day. EEG measurements were analyzed using Spike2 software. A seizure was characterized by a rapid amplitude increase, high spike frequency, and a typical postictal phase. Seizure frequency for each mouse was averaged by day. The day on which a mouse died, was not included in the number of seizure measurements.
Drugs
Tamoxifen (concentration 20 mg/mL) was dissolved in sunflower oil and intraperitoneally injected for four consecutive days in all mice during the light phase of the day (100 mg/kg; day 0–3 see Fig. 2A), resulting in Tsc1 deletion in Cre+ mice. Mice received either vehicle (for baseline assessment, see below) or treatment. All AEDs were daily administered by intraperitoneal injections using hypodermic‐needle 25G × ypo mm (Sterican®/B‐Braun). Concentrations used for the AED treatments were based on effective concentrations described in literature for various animal models of epilepsy. Rapamycin (1, 3, 5 and 10 mg/kg; LC Laboratories), clobazam (30 mg/kg20, 21; Alliance healthcare), tiagabine (40 mg/kg22; Sigma‐Aldrich) and AZD8055 (15 mg/kg23; Adooq Bioscience, A10114) were dissolved in dimethyl sulfoxide (DMSO) (injection volume 1 μL/g). Vigabatrin (200 mg/kg24; Sanofi‐Aventis B.V.) was dissolved in 0.9% saline (injection volume 10 μL/g). Lamotrigine (25 mg/kg25; Alliance healthcare) and valproic acid (350 mg/kg26; Alliance healthcare) were27 dissolved in sterilized water (injection volume 10 μL/g). The S6 kinase inhibitor PF4708671 (75 mg/kg28; Sigma Aldrich, 1255517‐76‐0), was dissolved by adding a mixture of 10% Tween‐80 (Sigma Aldrich, 9005‐65‐6) to 40% DMSO. This mix was vortexed vigorously and sonicated for several minutes until the drug was dissolved. With saline, the mixture was adjusted to its final concentration and vortexed and/or sonicated till it was fully dissolved (injection volume 3 μL/g). The ketogenic diet with a nutrient distribution ratio of 4:1 of fat to protein and carbohydrates was given ad libitum (E15149‐30, Bio services ssniff EF R/M). Food consumption and body weight was monitored daily for both standard diet‐ and ketogenic diet‐fed mice. Ketone body levels were measured for both groups by loading 1.5 μL venous blood onto a ketone body testing strip (Freestyle Precision Xtra, Abbott Diabetes Care).
Figure 2.
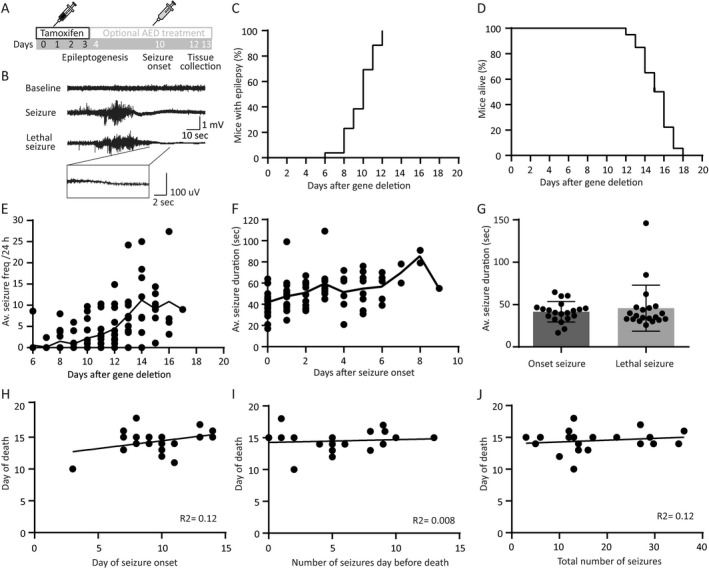
Tsc1‐Cre+ mice develop tonic–clonic seizures in a narrow time window without predictors for day of death. (A) Timeline of the experimental setup. All mice received four tamoxifen injections followed by optional AED treatment starting on day 4 after initiating gene deletion. Mice were sacrificed on day 12 or 13, 2 h after the last drug injection (B) Example traces of an EEG recording showing a baseline, a seizure, and a lethal seizure trace. All lethal seizures showed a typical flattening EEG trace visualized in the zoom in trace. (C) EEG recordings show that all Tsc1‐Cre+ mice develop epilepsy between 6 and 12 days after initiation of gene deletion. (D) Tsc1‐Cre+ mice died 12–18 days after gene deletion. (E) Average seizure frequency increases over time after gene deletion, whereas average seizure duration does not increase significantly over time (F). Plotted line is the average per day after gene deletion of all mice. (G) Seizure duration is not different between the day of onset and the day of death. (H) Correlation plot of day of death and day of seizure onset, showing no correlation. (I) Correlation plot of day of death and number of seizures the day before death, showing no correlation. (J) Correlation plot of day of death and total number seizures per mouse indicating no correlation. EEG recordings of animals were only taken into account if the indicated parameter could be unambiguously be assessed. This is the case for minimally 16 animals (day of seizure onset) and maximally 20 animals (average seizure duration per animal). The dots represent averages of individual mice.
Establishment of control cohort and replication study
Seizure characteristics and survival of untreated mice as presented in Figures 2 and 6, were calculated from combined data of all vehicle‐treated mice. To determine the robustness of this baseline (e.g., due to genetic drift, or differences with respect to surgery or analysis), we established again a baseline at the very end of all drug‐testing studies, performed by a different experimenter (n = 20) These results were completely overlapping (data not shown), indicating that the phenotype is very robust and can be assessed by different experimenters.
Figure 6.
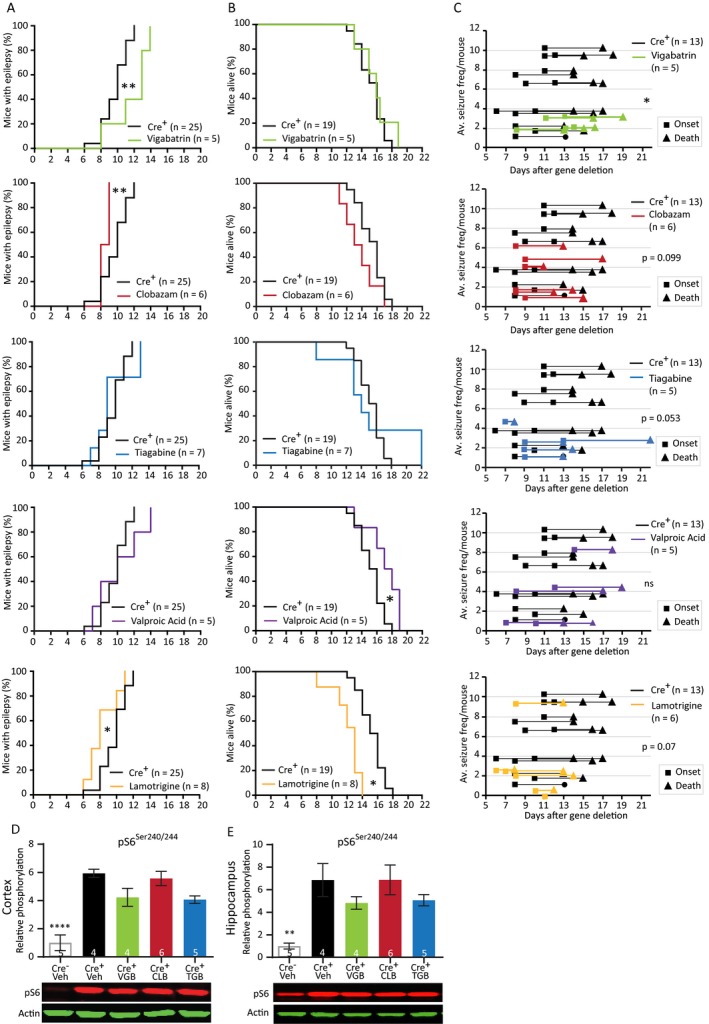
Preventive treatment with traditional AEDs reveal variable responses and do not affect the mTORC1 pathway. (A) Seizure onset of Tsc1‐Cre+ mice treated with the AEDs at 4 days after initiation of gene deletion, days before seizure onset. Tsc1‐Cre+ mice were treated with vigabatrin (200 mg/kg), clobazam (30 mg/kg), tiagabine (40 mg/kg), valproic acid (350 mg/kg), or lamotrigine (25 mg/kg). (B) Survival curves of the Tsc1‐Cre+ mice treated with the different AEDs. (C) Average seizure frequency of the different drug treatments in Tsc1‐Cre+ mice. Each line represents the average number of seizures per mouse with the squares indicating the day of onset and the triangles the day of death. (D and E) Cortical and hippocampal western blot data from a separate group of mice of the different treatment conditions of mice sacrificed on day 12, 2 h after the last drug injection. None of the used drugs are able to lower pS6Ser240/244. Behavior was analyzed with a Kapler–Meier Log Rank test. Seizure frequency was compared with an unpaired t‐test or a nonparametric Mann–Whitney test. Western blot data are presented as means with error bars representing SEM. Sample sizes are indicated in the figures. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test. Tsc1‐Cre+ mice were used as control group. *P < 0.05, **P < 0.01, ****P < 0.0001.
Analysis and statistics
Epilepsy onset, frequency, and survival were analyzed offline from the EEG recordings. For statistical analysis of the onset and survival, a Kaplan–Meier (Log Rank) analysis was performed. Correlation was tested with a Pearson correlation test. Seizure frequency per 24 h was calculated for each mouse, by averaging the total number of seizures by the total number of days the mouse was epileptic. The last day of seizures was excluded from this calculation. The average of all the mice within each drug condition was compared to the Tsc1‐Cre+ group.
Western blot data and Pathscan data were analyzed using a parametric unpaired t‐test or a one‐way ANOVA with a Dunnett’s post hoc test where the Tsc1‐Cre– mice or Tsc1‐Cre+ were used as controls (control group is specified in the figure legend). All statistics were done with GraphPad Prism (version 6, San Diego, CA). The results and further details of the performed statistical tests for the western blots, are presented in (Tables S1–S7). Error bars represent standard errors of the mean (SEM) and P‐values < 0.05 were considered as significant. Group sizes are indicated in each figure where n is the number of mice used.
Results
Tsc1‐Cre+ mice exhibit brain‐specific Tsc1 deletion and mTORC1 hyperactivation
We started the characterization of the Tsc1f/f‐Tg(Camk2a‐CreERT2) mouse model (for more details see the methods section), hereafter called Tsc1‐Cre+ or Tsc1‐Cre– mice, by examining whether the Tsc1 gene was effectively deleted upon four consecutive daily tamoxifen injections. A polymerase chain reaction (PCR) analysis showed Tsc1 gene deletion in the cortex and hippocampus of tamoxifen‐treated Tsc1‐Cre+ mice, with no discernable deletion outside the brain (e.g. in the liver) (Fig. 1A). We used the phosphorylation of S6 (pS6Ser240/244), a specific downstream target of mTORC1,18 as a mTORC1 activity read‐out to further characterize our mouse model. Increased pS6Ser240/244 levels were observed in DAB stainings of cortical and hippocampal slices from Tsc1‐Cre+ mice compared to Tsc1‐Cre– control mice (Fig. 1B). A strong co‐labeling of CAMK2A and pS6Ser240/244 was observed in Tsc1‐Cre+ mice, indicating that (nearly) all CAMK2A‐positive excitatory neurons in the cortex (Fig. 1C) and hippocampus (Fig. 1D) underwent Tsc1 gene deletion. In contrast, stainings with parvalbumin, a marker for parvalbumin‐positive inhibitory neurons, did not show any co‐labeling with pS6Ser240/244 (Fig. 1C and 1). Overall, there were no structural malformations observed. Furthermore, and in line with our previous published Tsc1 mouse model,4 we did not observe signs of astrogliosis. We did observe bigger pyramidal neuron soma’s in Tsc1‐Cre+ mice on day 12 after gene deletion (Tsc1‐Cre– M: 129.8 μm2, SEM: 1.95; Tsc1‐Cre+ M: 166.8 μm2, SEM: 2.75; T (288) = 11.16, P < 0.0001), which is a common observation after mTORC1 hyper activity.29, 30
Figure 1.
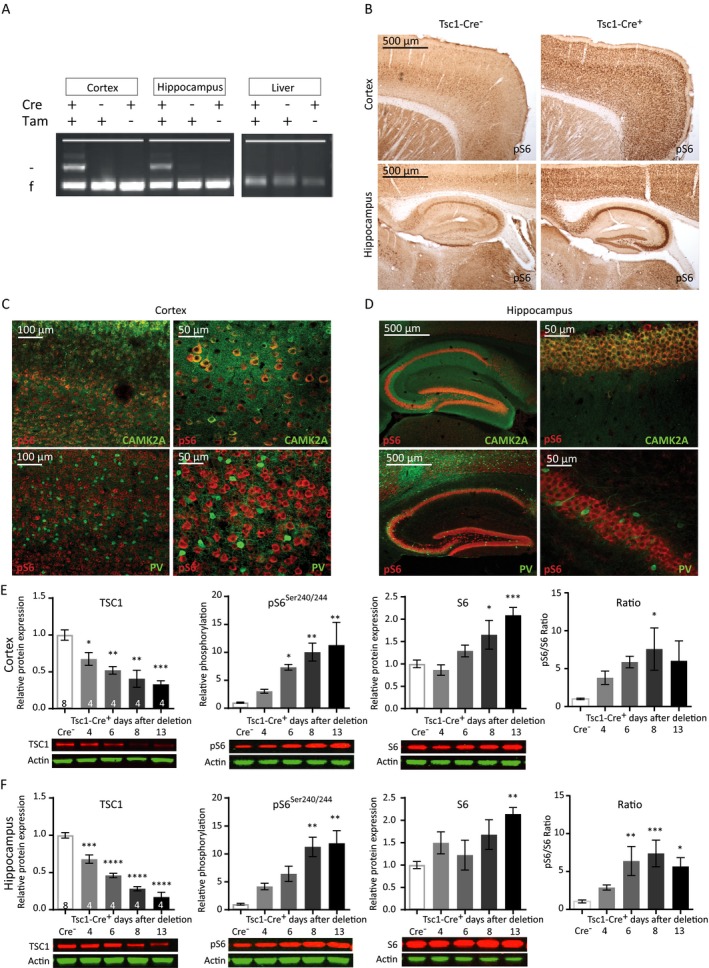
Cre‐mediated Tsc1 gene deletion in CAMK2A‐expressing neurons induces hyperactivation of the mTORC1 pathway. (A) Tsc1 PCR analysis of DNA isolated from the cortex, hippocampus, and liver after four tamoxifen (Tam) injections, inducing efficient and brain‐specific Tsc1 gene deletion of mice sacrificed at day 11. The higher band indicated with ‘–’ represents the knock‐out allele; the lower band ‘f’ represents the floxed (nondeleted) allele. (B) Immunohistochemistry (DAB staining) on sagittal sections of an adult Tsc1‐Cre+ and Tsc1‐Cre– mouse, 13 days after gene deletion stained for pS6Ser240/244. Increased levels of pS6Ser240/244 in tamoxifen injected Tsc1‐Cre+ mice are observed. (C and D) Fluorescent immunohistochemistry on coronal sections of Tsc1‐Cre+ for pS6Ser240/244 shows high colocalization with CAMK2A (green) but not with parvalbumin (PV; red), in the cortex and hippocampus. (E and F) Tsc1‐Cre+ mice show TSC1 reduction and increased levels of S6 and pS6Ser240/244 starting at day 6 after gene deletion in the cortex and hippocampus. Western blot data are presented as means with error bars representing SEM. Sample sizes are indicated in the figures with n the number of mice used. A one‐way ANOVA was used for statistical analysis with a Dunnett’s post hoc test. Tsc1‐Cre+ mice were used as control group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
To investigate how mTORC1 activity increased over time after Tsc1 gene deletion, we performed western blot analysis and investigated pS6Ser240/244 levels at different time points. Tsc1‐Cre+ and littermate control Tsc1‐Cre– mice, were sacrificed at day 4, 6, 8, or 13 after gene deletion (first tamoxifen injection on day 0). Western blot analysis confirmed significantly decreased TSC1 protein levels in cortical and hippocampal lysates already at day 4 after gene deletion (Fig. 1E and 1; Table S1). Furthermore, a gradual increase of pS6Ser240/244 was observed which reached statistical significance on day 6 in the cortex and day 8 in the hippocampus. pS6Ser240/244 levels reached a maximum on day 8 in both brain areas (Fig. 1E and 1; Table S1). In addition, an increase in S6 expression itself was observed, indicating that its translation is also controlled by the mTORC1 pathway. Taken together, this data show that we can successfully delete Tsc1 in CAMK2A‐expressing cells, resulting in cell‐specific activation of the mTORC1 pathway.
Acute loss of Tsc1 in adult excitatory forebrain neurons leads to spontaneous epilepsy and SUDEP
To examine whether Tsc1‐Cre+ mice develop epilepsy after Tsc1 deletion, we monitored brain activity continuously by using a wireless EEG device from the day of the first tamoxifen injection. Analysis of the EEG recordings (Fig. 2B), showed that all Tsc1‐Cre+ mice developed epilepsy in a narrow time window between day 8 and day 12 after gene deletion (median epilepsy onset is 10 days) (Fig. 2C). As shown in Figure 1, day 8 is the time point where TSC1 protein levels drop just below 50% and pS6Ser240/244 reaches its maximum. The Tsc1‐Cre+ mice suffered from tonic–clonic seizures, characterized by generalized limb clonus and loss of upright posture. During the postictal phase, mice showed immobilized behavior for several seconds. All Tsc1‐Cre+ mice died between 12 and 18 days (median 15 days; Fig. 2D) after a tonic–clonic seizure, characterized by abrupt flattening of the EEG correlated with immobilized behavior and at death (Fig. 2B arrow). Seizure frequency increased slightly over time with a striking variability (Fig. 2E). All seizures, including the lethal ones, lasted on average around 40 sec (Fig. 2F and G). Notably, death of the animal was entirely unpredictable, reminiscent of sudden unexpected death in epilepsy (SUDEP). Specifically, there was no correlation between the day of seizure onset and the day of death (Fig. 2H; (r = 0.34; n = 20; P = 0.12). Neither between seizure frequency and day of death (r = 0.09; n = 20; P = 0.69), nor the total number of seizures and the day of death (r = 0.16; n = 20; P = 0.49), and some animals even died at the very first seizure. Nevertheless, on average, seizure frequency increased over time with one seizure per 24 h at the day of epilepsy onset (median day 10), up to a median frequency to five seizures per 24 h preceding death (Fig. 2I and J).
In sum, biallelic Tsc1 gene deletion in adult mice results in epilepsy in a specific time window with increasing frequency over time and no change in seizure duration. Furthermore, none of the investigated parameters could predict the day of death, resembling SUDEP.
Tsc1 loss‐driven epileptogenesis is dependent on RHEB1 activation
To further confirm the mTORC1 dependency of the phenotypes observed in our mouse model, we crossed our Tsc1f/f‐Tg(CaMK2A‐CreERT2) mice with a Rheb1f/f mouse line.19 A conditional brain‐specific double homozygous mouse model was obtained, where both Tsc1 and Rheb1 could be deleted in CAMK2A‐positive neurons by tamoxifen injections (hereafter called Tsc1‐Rheb1‐Cre +). We have previously shown that deletion of Rheb1 from CAMK2A‐positive cells, significantly reduces mTORC1 activity (measured by the level of pS6), indicating that RHEB1 activation is the critical step for mTORC1 activation.19
In contrast to the Tsc1‐Cre+ mice, Tsc1‐Rheb1‐Cre + mice did not develop epilepsy, appeared healthy and showed normal survival until the last day of observation (n = 4, day 30 from the first day of tamoxifen injection (data not shown). To confirm the deletion of Rheb1 and Tsc1 genes and to assess the effect on the mTORC1 pathway, we performed western blot analysis on cortex and hippocampus on day 12 after the first tamoxifen injection. We observed significantly reduced levels of the RHEB1 and TSC1 protein, confirming the successful gene deletion (Fig. 3A and 3; Table S2). In addition, the level of pS6Ser240/244 in Tsc1‐Rheb1‐Cre + mice was significantly lower than Tsc1‐Cre+ mice and was not different from Tsc1‐Cre– mice or the rapamycin (10 mg/kg)‐treated Tsc1‐Cre+ mice. These results confirm that the phenotypes observed in our Tsc1‐Cre+ mice are specifically dependent on RHEB1‐mediated mTORC1 activation.
Figure 3.
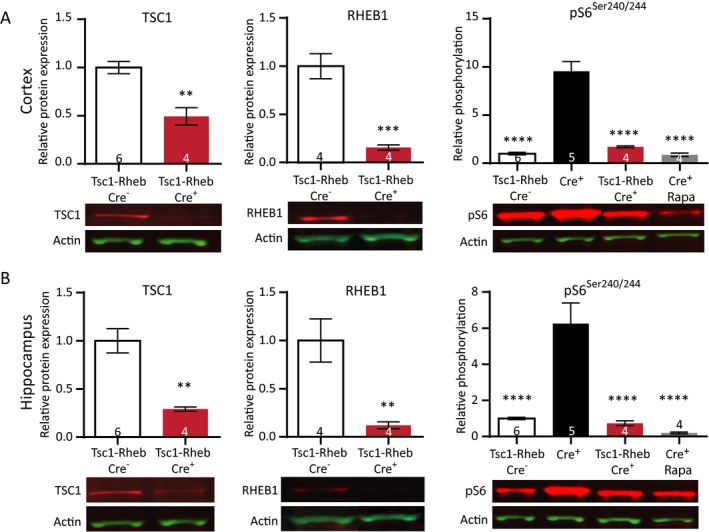
Simultaneous deletion of Tsc1 and Rheb1 prevents mTORC1 epileptogenesis and mTORC1 activation. (A) Cortical protein levels of both TSC1 and RHEB1 are significantly lower in Tsc1‐Rheb1‐Cre+ mice compared to Tsc1‐Rheb1‐Cre– mice. Levels of pS6Ser240/244 in Tsc1‐Rheb1‐Cre+ mice are comparable to Tsc1‐Rheb1‐Cre– mice and rapamycin‐treated (10 mg/kg) Tsc1‐Cre+ mice. (B) Within the same mice, hippocampal samples show significantly lower TSC1 and RHEB1 levels. Levels of pS6Ser240/244 in Tsc1‐Rheb1‐Cre+ mice are comparable to Tsc1‐Rheb1‐Cre– mice and rapamycin‐treated (10 mg/kg) Tsc1‐Cre+ mice. All mice were sacrificed on day 12. Western blot data is presented as means with error bars representing SEM. Sample sizes are indicated in the figures with n the number of mice used. An unpaired t‐test was performed to compare protein levels of TSC1 and RHEB1. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test for pS6Ser240/244 levels. Tsc1‐RHEB‐Cre– mice were used as control group. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Epileptogenesis driven by the loss of Tsc1 shows a dose‐dependent response to mTORC1 inhibitor rapamycin
Previous studies showed that rapamycin treatment effectively rescues and/or prevents seizures in TSC mouse models.4, 31, 32, 33, 34 To test the efficacy of rapamycin in our model, we treated mice with different doses of rapamycin, starting 4 days after initiation of gene deletion. The effect on epilepsy onset, epilepsy frequency, and animal survival was analyzed. Of the mice treated with 1 mg/kg rapamycin, 80% developed epilepsy. A dose of 3 mg/kg resulted in epilepsy in 60% of the mice and 5 mg/kg showed epilepsy in 20% of the mice. Treatment with 1 mg/kg rapamycin had no effect on the onset and on survival, while treatment with a dose of 3 or 5 mg/kg significantly delayed epilepsy onset and significantly extended survival (Fig. 4A and 4; Table S3). Furthermore, seizure frequency was reduced in a dose‐dependent manner (Fig. 4C; Table S3). Mice treated with 10 mg/kg rapamycin, did not develop epilepsy and survived throughout the duration of the experiment (data not shown). These results show the antiepileptogenic effects of rapamycin.
Figure 4.
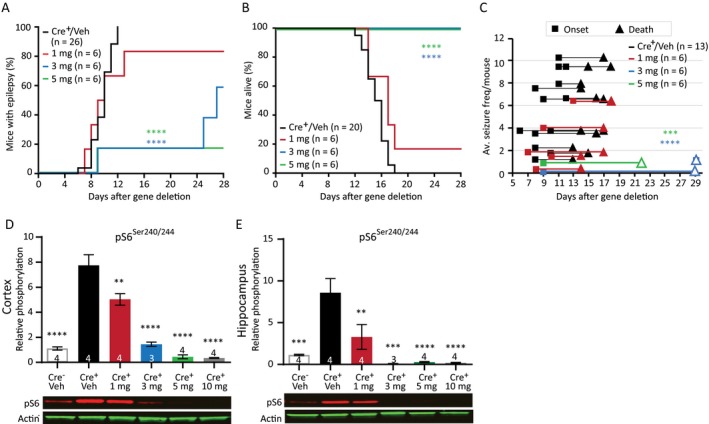
Rapamycin improves the epileptic phenotype in Tsc1‐Cre+ mice in a dose‐dependent manner. (A) A delayed seizure onset is observed in mice treated with 3 mg/kg and 5 mg/kg rapamycin compared to Tsc1‐Cre+ vehicle (Veh) control mice and mice treated with 1 mg/kg rapamycin. (B) All mice treated with 3 mg/kg or 5 mg/kg rapamycin stayed alive as long the treatment lasted. (C) Rapamycin‐treated mice show on average lower seizure frequencies. Each line represents the average number of seizures per mouse with the squares indicating the day of onset and the triangles the day of death. The open triangle indicates mice that were sacrificed by the experimenter. (D and E) Western blot data of cortical and hippocampal tissue from a separate group of mice, reveal significant lower pS6Ser240/244 levels in Tsc1‐Cre+ mice after treatment with rapamycin of 1 mg/kg or higher. Mice were sacrificed on day 13, 2 h after the last rapamycin injection. Seizure onset and survival were analyzed with a Kapler–Meier Log Rank test. Seizure frequency was analyzed with a nonparametric Kruskal–Wallis test. Western blot data is presented as means with error bars representing SEM. Sample sizes are indicated in the figures with n the number of mice used. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test. Tsc1‐Cre+ mice were used as control group. **P < 0.01, ***P < 0.001, ****P < 0.0001.
To assess whether the dose‐dependent effect of rapamycin on epilepsy in Tsc1‐Cre+ mice was reflected by the level of mTORC1 activity, we measured pS6Ser240/244 protein levels in Tsc1‐Cre+ mice treated with either vehicle or different concentrations of rapamycin. As expected, pS6Ser240/244 levels increased significantly in vehicle‐treated mice. Interestingly, treatment with rapamycin 1 mg/kg of rapamycin lowered pS6Ser240/244 in hippocampus and cortex, but this level was still well above Tsc1‐Cre– control mice. In contrast, doses of 3 mg/kg rapamycin or higher seemed to be more effective as both reduced pS6Ser240/244 levels near (or even below) control levels (Fig. 4D and 4; Table S3).
Taken together, these results show that epilepsy severity as measured by onset time, seizure frequency, and animal survival in this TSC mouse model, is directly related to the degree of neuronal mTORC1 activity.
Molecular analysis indicates changes in 4E‐BP1/2, RSK1 and AMPK activity
To study which proteins besides pS6 are dysregulated during epileptogenesis in Tsc1‐Cre+ mice, we performed a limited analysis of phosphorylated proteins in the TSC‐mTORC1 pathway (Fig. 5A). By using an antibody array, we assessed the phosphorylation of the following proteins: Akt (T308, S473), S6 (S235/236), AMPK‐a (T172), PRAS40 (T246), mTOR(S2481), GSK‐3a (S21), GSK‐3b (s9), p70S6K (T389, T421/S424), BAD (S112), RSK1 (S380), PTEN (S380), PDK1 (S241), ERK1/2 (T202/204), and 4E‐BP1 (T37/46). Because the vast majority of Tsc1‐Cre+ mice developed epilepsy around day 8 and onward, we were in particularly interested in the proteins that would show increased phosphorylation at this time point. Furthermore, day 13 was examined as well, to assess the effect of epilepsy on phosphorylation of the aforementioned proteins. Lastly, we wanted to know if treatment with rapamycin could minimize the phosphorylation of these proteins, comparable to control levels. To that end, mice were divided into four groups: (I) a control group of Tsc1‐Cre– mice, (II) Tsc1‐Cre+ mice treated with vehicle and sacrificed on day 8 or (III) on day 13, and (IV) Tsc1‐Cre+ mice treated with rapamycin (5 mg/kg) sacrificed on day 13. Out of the tested proteins, only pS6Ser235/236, p4E‐BP1Thr37/46, pRSK1Ser380, and pAMPKThr172 showed a significant upregulation in both cortex and hippocampus, and responded to rapamycin treatment. Of these four proteins, only pS6Ser235/236 (cortex and hippocampus) and pRSK1Ser380 (only cortex) were significantly elevated around epilepsy onset (day 8) (Fig. 5B and C; Table S4). p4E‐BP1Thr37/46, and pAMPKThr172 were only significant increased at day 13. Although the four proteins all responded to rapamycin treatment in the cortex, this was not the case for the hippocampus. Levels of p4E‐BP1Thr37/46, pRSK1Ser380, and pAMPKThr172 remained significantly increased, despite the high dose of rapamycin treatment used (Fig. 5C; Table S4).
Figure 5.
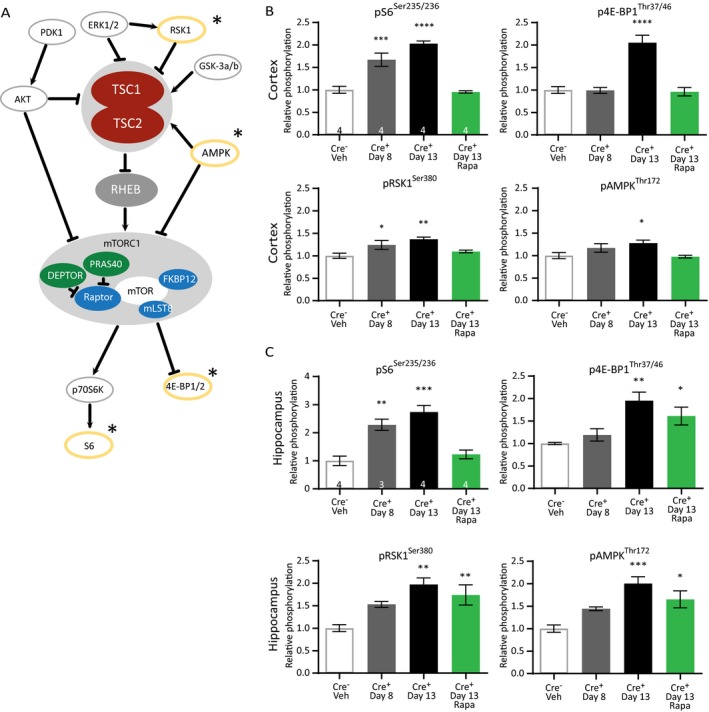
TSC1‐mediated epilepsy induces changes in the mTORC1‐pathway that are only partially reversed by rapamycin. (A) Proteins within the mTORC1 pathway that were selected for the pathscan phosphorylation analyses are depicted. The significantly changed proteins are circled in yellow and indicated with a star. (B) Pathscan results of the cortical samples where pS6Ser235/236 and pRSK1Ser380 are already significantly higher on day 8. (C) Pathscan results of hippocampal tissue of Tsc1‐Cre+ mice sacrificed on day 8, day 13, and day 13 after treatment with 5 mg/kg of rapamycin that started on day 4 after initiating gene deletion. pS6Ser235/236 levels are significantly higher on day 8 and rescued by rapamycin treatment. Pathscan data are presented as means and error bars represent SEM. Sample sizes are indicated in the figures with n the number of mice used. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test. Tsc1‐Cre– mice were used as control group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Pretreatment with AEDs does not prevent epileptogenesis
A clinical relevant advantage of our mouse model is the ability to investigate if treatment with AEDs before seizure onset could be antiepileptogenic by preventing or delaying the onset of epilepsy, as well as preventing the molecular changes that are induced upon Tsc1 gene deletion, in a similar way as rapamycin does. If a drug does not have such an effect, the seizure suppressive properties can further be investigated by determining the effect on seizure frequency as well as survival time.
We tested three main GABAergic drugs, increasing neuronal inhibition: vigabatrin (200 mg/kg), clobazam (30 mg/kg), and tiagabine (40 mg/kg). Furthermore, two drugs were tested that mainly lower neuronal excitation: valproic acid (350 mg/kg) and lamotrigine (25 mg/kg).35 Vigabatrin and Valproic acid are commonly used to treat infantile spasms including those in TSC‐infants.12, 36 Tsc1‐Cre+ mice received daily drug injections starting on day 4 after gene deletion. At this time point, TSC1 protein levels are still above 50% and it is well before the mice show a first seizure. Vehicle‐treated Tsc1‐Cre+ mice were used as control. Despite initiating treatment well before seizure onset, all mice developed epilepsy, and none of the drugs was able to prevent seizure‐related lethality (Fig. 6A and B, Table S5). Importantly, all drugs reduced the average seizure frequency, but due to the high variability combined with relative small group sizes, this effect did not always reach statistical significance (Fig. 6C).
Vigabatrin had the largest effect on seizure frequency and was the only drug that was able to significantly delay seizure onset and lower seizure frequency. Valproic acid did not delay seizure onset, but significantly improved survival. In contrast, lamotrigine significantly worsened it by showing an earlier onset and lethality. Clobazam also showed a significant earlier onset of seizures and a tendency toward decreased survival.
Given that vigabatrin was more effective than comparable drugs that increase neuronal inhibition (clobazam and tiagabine), we tested the effect of these drugs on mTORC1 signaling. Treated mice were sacrificed on day 12 and phosphorylation of pS6Ser240/244 in cortical and hippocampal tissues was assessed by western blots analysis (Fig. 6D and 6). pS6Ser240/244 levels remained high in all treatments, indicating that none of the drugs had an effect on mTORC1 pathway activity (Fig. 6D and 6; Table S5).
Ketogenic diet delays seizure onset and extends survival, but does not lower seizure frequency
It has been shown that ketogenic diet can be beneficial for treating TSC‐related epilepsy.15, 37 To test the efficacy of the ketogenic diet in our mouse model, Tsc1‐Cre+ mice were first fasted for 18 h and were placed on a ketogenic diet 6 weeks before Tsc1 gene deletion. Within these 6 weeks, the mice were fully used to the diet having a stabilized calorie intake and a ketone body‐based metabolism.38 Treatment was continued until the end of the experiment. Ketone bodies were measured on day 13 just before decapitation, and were significantly higher in mice on the ketogenic diet than in mice fed with standard diet. This indicates that ketosis had replaced a glucose‐based metabolism in these mice (Table S6). Although the ketogenic diet was not able to significantly lower seizure frequency (Fig. 7C), epilepsy onset showed a small but significant delay and mice on a ketogenic diet lived significantly longer than mice on a standard diet (Fig. 7A and 7; Table S6), Moreover, pS6Ser240/244 was significantly lower in mice treated with the ketogenic diet, but this effect was only observed in the cortex. Besides, the phosphorylation levels in the ketogenic diet‐fed mice were still higher than Tsc1‐Cre– control animals (Fig. 7E and 7; Table S6).
Figure 7.
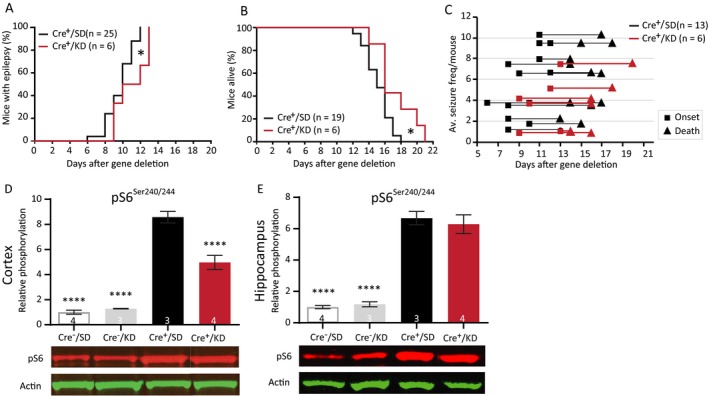
Ketogenic diet reduces cortical mTORC1 activity, delays epilepsy onset, and improves survival. (A) Seizure onset curves of Tsc1‐Cre+ mice treated with the ketogenic diet (Cre+/KD) or standard diet (Cre+/SD). The KD‐fed mice were on the diet for 6 weeks before gene deletion. Seizure onset is significantly delayed in Cre+/KD mice. (B) Survival curve of the Cre+/KD and Cre+/SD mice. (C) Each line represents the average number of seizures per mouse with the squares indicating the day of onset and the triangles the day of death. (E and F) Western blot analysis of hippocampal and cortical tissue of an independent group of mice sacrificed on day 13. Significantly lower levels of cortical pS6Ser240/244) in the Cre+/KD mice are found. Seizure onset and survival were analyzed with a Kapler–Meier Log Rank test. Seizure frequency was compared with an unpaired t‐test. Western blot data is presented as means with error bars representing SEM. Sample sizes are indicated in the figures with n the number of mice used. Blots shown are representative but in some cases the order of the samples was adjusted to allow alignment with the corresponding bar. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test. Cre+/SD mice were used as control group. *P < 0.05, ****P < 0.0001.
AZD8055 and PF4708671 do not improve the epilepsy phenotype
We tested the effects of two novel compounds, AZD8055 and PF4708671 in our Tsc1 mouse model. The first drug, AZD8055 is an ATP‐competitive dual mTOR inhibitor inhibiting both mTORC1 and mTORC2.23 The second drug, PF4708671 is a specific S6K1 inhibitor, a downstream target of mTORC1.39 Because rapamycin could successfully prevent and treat epilepsy by inhibiting mTORC1 activity and reducing S6 phosphorylation, we hypothesized that AZD8055 would be equally or even more effective in treating or preventing epilepsy in Tsc1‐Cre+ mice. If S6K has a key role in epileptogenesis, the S6K inhibitor PF4708671 might also be able to inhibit epileptogenesis in this Tsc1 mouse model. To study the possible antiepileptic effects of these compounds, mice were treated daily with either AZD8055 (15 mg/kg) or PF4708671 (75 mg/kg) starting 4 days after initiation of gene deletion, and seizures were monitored by EEG. Surprisingly, both drugs did not show an effect on epilepsy onset or survival (Fig. 8A and 8). Although not significant, the drugs seem to lower average seizure frequency per mouse (Fig. 8C and 8; Table S7).
Figure 8.
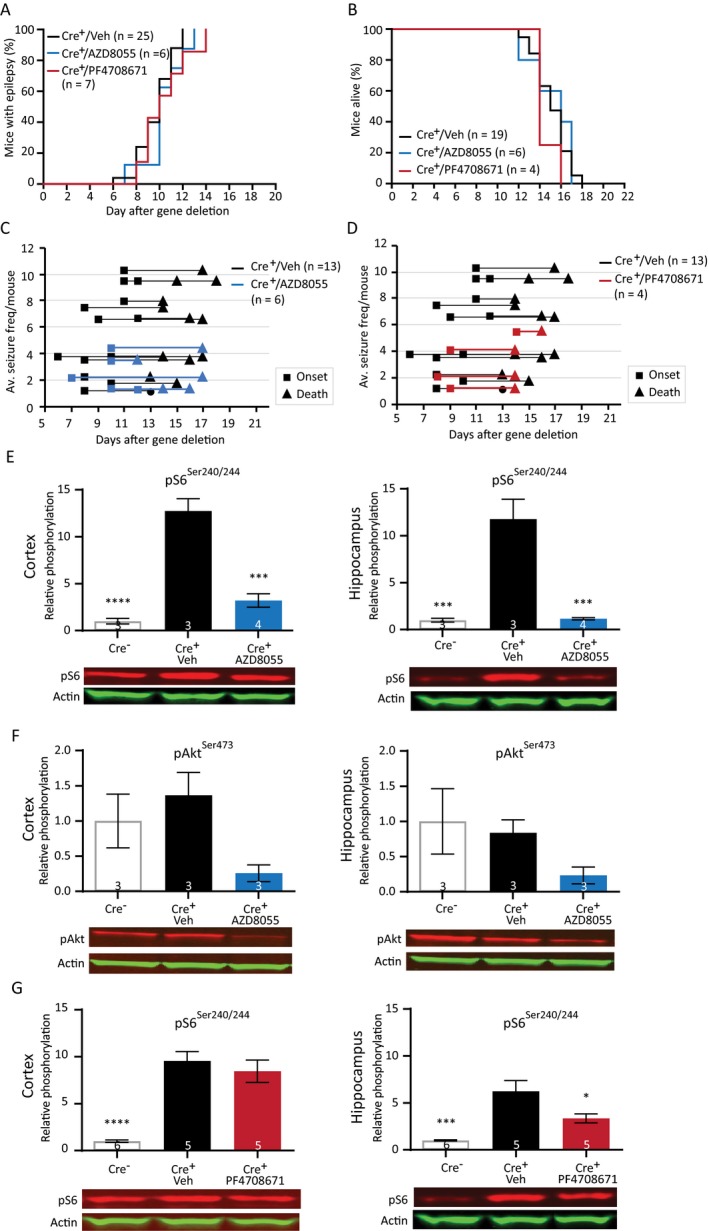
Preventive treatment with AZD8055 and PF4708671 does not improve the epileptic phenotype despite reduced levels of pS6Ser240/244. (A) Seizure onset graph of Tsc1‐Cre+ mice treated with vehicle, AZD8055 (15 mg/kg), or PF4708671 (75 mg/kg) started 4 days after initiation of gene deletion. (B) Survival curves of the Tsc1‐Cre+ mice treated with AZD8055 and PF4708671. (C and D) Average seizure frequency is not different between the treated mice and control mice. Each line represents the average number of seizures per mouse with the squares indicating the day of onset and the triangles the day of death. (E and F) Western blot of cortical and hippocampal tissue of an independent group of mice treated with AZD8055 sacrificed on day 12, 2 h after the last drug injection. Levels of pS6Ser240/244 are significantly lower in the cortex and hippocampus of the AZD8055‐treated mice. Although statistically not different, lower levels of pAkt are observed in AZD8055‐treated mice. (G) Mice treated with PF4708671 show significant lower pS6Ser240/244 levels in the hippocampus. Seizure onset and survival were analyzed with a Kapler–Meier Log Rank test. Average frequency was compared with a one‐way ANOVA and Dunnett’s post hoc test. Western blot data is presented as means with error bars representing SEM. Sample sizes are indicated in the figures with n the number of mice used. Blots shown are representative but in some cases the order of the samples was adjusted to allow alignment with the corresponding bar. A one‐way ANOVA was used for statistical analysis with a Dunnett’s test as post hoc test. Tsc1‐Cre + mice were used as control group. *P < 0.05, ***P < 0.001, ****P < 0.0001.
To establish that both drugs crossed the blood–brain barrier and lowered mTORC1/2 or S6K activity, we performed a western blot analysis. Mice were sacrificed 12 days after gene deletion and pS6Ser240/244 levels were assessed in hippocampal and cortical tissue of Tsc1‐Cre– mice, vehicle‐treated Tsc1‐Cre + mice, AZD8055‐treated mice, and PF4708671‐treated mice. In addition, protein levels of pAkt were used as a read‐out of the mTORC2 pathway of the AZD8055‐treated mice. Despite having no effect on epilepsy, pS6Ser240/244 levels were reduced to Tsc1‐Cre– control levels in the hippocampus and cortex of AZD8055‐treated Tsc1‐Cre+ mice, indicating a strong inhibition of the mTORC1 pathway. Although statistically not significant, levels of pAkt(473) were also much lower in the AZD8055‐treated mice (Fig. 8E and 8; Table S7). Brain tissue of PF4708671‐treated mice revealed slightly lower levels of pS6Ser240/244 in the hippocampus, but levels did not turn back to the levels observed in Tsc1‐Cre‐ mice. Moreover, no effect of PF4708671 was observed in the cortex (Fig. 8G and 8; Table S7).
Taken together, treatment with AZD8055 normalized mTORC1 and mTORC2 activity while it did not improve the epileptic phenotype. Treatment with PF4708671 failed to normalize mTORC1 signaling as well as the epilepsy phenotype.
Discussion
Poor responsiveness to AEDs is a major problem for patients with epilepsy in general, but in particular for TSC patients of which the majority do not respond to drug therapy.11, 12, 40 Rather than just suppress seizures, treatment should ideally interfere with the process of epileptogenesis.
Previous studies showed that Tsc1 deletion in different cell types can result in epilepsy, for example, local Tsc1 deletion in CAMK2A‐expressing neurons,41, 42, 43 or Tsc1 deletion specifically in glial cells.34 Although these models are of great value, they are not very suitable to study the process of TSC/mTOR‐associated epileptogenesis as seizure onset is unpredictable and poorly controlled. To get more insight into the process of mTORC1‐driven epileptogenesis, we previously developed a model where the Tsc1 gene was conditionally deleted in all cells. The somatic deletion of Tsc1 in all cells, including the peripheral tissue, resulted in severe epilepsy. However, these mice also suffered from other health issues resulting in lethality unrelated to epilepsy, limiting their usefulness.4 Therefore, we developed and characterized a new Tsc1 mouse model. In this model, the Tsc1 gene is acutely deleted and limited to CAMK2A‐expressing cells, which in the forebrain is restricted to excitatory neurons. This model showed seizure onset in a narrow time window (10 ± 2 days) upon gene deletion.
Characterizing the epileptic seizures in our inducible mouse model, we observed tonic–clonic seizures with generalized limb clonus and loss of upright posture, similar to previously reported TSC mouse models.18, 34 Average seizure duration remained quite constant, whereas average seizure frequency increased over time. Despite the highly distinct time window in which seizures started, seizure frequency varied greatly between animals as well as within animals.
Reminiscent of SUDEP, none of the investigated parameters (seizure onset, frequency, and duration) could predict the day of death. In fact, all mice died after a seizure and some even after the very first seizure. Moreover, the lethal seizure was also not longer than the very first seizure. Importantly, SUDEP is one of the leading causes of seizure‐related death and contributes substantial to the cause of death in patients who die from complications of TSC.44, 45 Besides, SUDEP cases were described in 10% of the families who carried mutations in genes of the GATOR1 complex, a regulator of the mTOR pathway.46 Characteristics of SUDEP are generalized tonic–clonic seizures and abrupt flattening of EEG patterns, where death is probably related to brainstem spreading depolarization.45, 47, 48, 49, 50, 51 This acute flattening of the EEG is also observed in our recordings and is likely responsible for the lethality, given that the gene deletion is brain specific. Lamotrigine significantly shortened survival in our mice, which is noteworthy as it has been suggested that lamotrigine could facilitate SUDEP.45, 48, 52
At the molecular level, we observed a strong increase in mTORC1 activation after Tsc1 deletion, as measured by the phosphorylation of S6, a downstream target of mTORC1. Seizures and associated lethality were fully prevented by genetic (RHEB1 deletion) or pharmacological (rapamycin) blockade of the mTORC1 pathway. In line with other studies, rapamycin prevented epilepsy in a dose‐dependent manner in our TSC1 mouse model.4, 31, 32, 33, 34 To identify future treatment targets, we further assessed the phosphorylation of proteins that are related/linked to the mTORC1 pathway. We observed a significant upregulation of pS6Ser240/244, p4E‐BP1Thr37/46, pRSK1Ser380, and pAMPKThr172 in both the cortex and hippocampus. Rapamycin treatment at 5 mg/kg reduced the phosphorylation of all these proteins in the cortex, although p4E‐BP1Thr37/46, pRSK1Ser380, and pAMPKThr172 remained significantly increased in the hippocampus at this dose. Notably, p4E‐BP1Thr37/46 and pAMPKThr172, were only significantly increased at day 13. Although rapamycin treatment at 5 mg/kg reduced seizures, it did not reduce p4E‐BP1Thr37/46 and pAMPKThr172 levels in the hippocampus. Taken together, these results suggest that these proteins are not key players in the mechanisms underlying epilepsy. Relating these findings to literature, it appears that 4E‐BP plays distinct roles in TSC‐driven cortical migration during development and in TSC‐driven epilepsy.53 Upstream of mTORC1, AMPK1 can phosphorylate TSC2 and Raptor, leading to mTORC1 inhibition. AMPK1 is activated by phosphorylation of the Threonine 172, which is mediated by CAMK2B and Serine/threonine kinase 11 (STK11; also known as Liver Kinase B1 (LKB1)).54, 55 This supports the notion that increased pAMPKThr172 phosphorylation at day 13 is a secondary response, induced either by the epilepsy itself, or by a feedback mechanism in reaction to the high mTORC1 levels.
Rapamycin showed strong antiepileptogenic properties in our mice: at higher doses, seizures were absent, lethality was not observed, and molecular changes were absent. Although rapamycin treatment of TSC patients has been shown to be effective in some cases treating epilepsy,13, 14, 56 the clinical effects are not as striking as the results seen in our mice. Since treatment with rapamycin in patients is typically initiated after a long period of epilepsy, other mechanisms may have been altered and unresponsive to rapamycin treatment. This might explain the observed variable responses in patients. In addition, our (acute) inducible Tsc1 mouse model does not model the epilepsy that is generated by aberrant neuronal migration (tuber formation), which is observed in many patients. Nevertheless, our mouse model does have important clinical value, showing that loss of a functional TSC complex in excitatory neurons is sufficient to induce epilepsy, even in the absence of developmental changes.
Out of the tested AEDs, vigabatrin showed the largest improvements on the epileptic phenotype in the TSC mice. Vigabatrin is known as a first‐line treatment in infantile spasm including TSC patients.10 It was the only AED within this study that was able to significantly delay seizure onset. Vigabatrin did not affect mTORC1 signaling, hence it is not truly antiepileptogenic and the basis of beneficial effect of vigabatrin remains to be identified. Interestingly, valproic acid did not delay seizure onset, but had a small but significant effect on survival. This drug enhances the GABAergic system, but also acts on different classes of voltage gated ion channels and on the NMDA receptors.57 The broad working mechanism of this AED makes it difficult to determine what mechanism contributes to the increased survival.
Another treatment that can be quite effective for TSC patients, is the ketogenic diet15, 37 as well as the related low glycemic index treatment.58 We confirmed the efficacy of the ketogenic diet by showing delayed seizure onset and extended survival in ketogenic diet‐treated TSC mice. The precise mechanism of the ketogenic diet is still under investigation, but there could be a mechanistic link between ketogenic diet and mTOR, through amino acid metabolism, which acts upon mTOR signaling.59, 60 In line with these findings, we observed lower pS6 levels in our mice, although levels remained significantly higher than controls.
To identify novel treatments and get more insight in the downstream targets of mTORC1, we tested two drugs that affect mTORC1 signaling: AZD8055 and PF4708671. The first drug, a dual mTOR inhibitor AZD8055 was previously used to investigate its effect on glioma development. Treatment with AZD could successfully slow down the growth of tumors and gliomas by inhibiting the mTOR complex.23, 53 Furthermore, the fact that another dual mTOR inhibitor (PQR620) was effectively able to improve epilepsy in a mouse model of chronic epilepsy,61 suggests that this dual mTOR inhibitor might have a beneficial effect. Comparable to rapamycin treatment, AZD8055 treatment resulted in a strong reduction of pS6Ser240/244. Furthermore, it reduced the protein levels of pAkt(473). In contrast, the reduction in seizure frequency was rather modest and no effect on seizure onset and survival was observed. A possible explanation for this negative finding could be attributed to the half‐life of AZD8055, which is 2–3 h in blood plasma62. However, detailed analysis of the EEG data did not reveal a difference in total number of seizures in the 2 h after drug injection compared to any of the other 2‐h windows within 24 h following injection. A second explanation could be that mTORC2 activation is neuroprotective instead61, 63, 64 and hence, a decrease in mTORC2 reduces the effectiveness of inhibiting mTORC1. The second specific drug that we tested, PF4708671, is a cell‐permeable inhibitor of p70 ribosomal S6 kinase (S6K1 isoform). It was previously shown that mTORC1 activity in a Tsc1 heterozygous knock out mouse, could be successfully reduced by PF4708671 treatment.28 We observed a modest reduction of pS6Ser240/244 in hippocampus, but not in the cortex, and no effect on epilepsy. Possibly, at the concentration used, the drug was not able to counteract the strong mTORC1 increase seen in our mice. Further investigation into the downstream mTORC1 targets remains necessary to identify novel treatments.
In conclusion, we present a novel TSC‐mTORC1‐dependent epilepsy model with high construct and face validity. We demonstrate the translational value of this model for drug testing by confirming clinical observations that vigabatrin, ketogenic diet, and rapamycin are among the most effective treatments for TSC‐associated epilepsy. We prove that these therapies (vigabatrin, ketogenic diet, and rapamycin) are most efficient in our mouse model affecting seizure onset, survival, and seizure frequency. Moreover, the model can be used to study the mechanisms underlying SUDEP as well as the mechanisms underlying epileptogenesis. The fact that vigabatrin improves all the epilepsy parameters in our mouse model suggests this treatment can indeed be suitable to treat TSC patients and delay/prevent epilepsy as seen in some clinical trials.16.
Author Contributions
L.M.C.K. and S.G. equally contributed to the manuscript, data acquisition, and data analysis. M.P.O., I.W., N.H.R.M.K., and A.O. contributed to acquisition and analysis of the data. Y.E. and J.S. designed the study and Y.E., S.G., and L.M.C.K. drafted the manuscript and figures.
Conflict of Interest
Nothing to report.
Supporting information
Table S1. Statistics figure 1 – “Cre mediated Tsc1 gene deletion in CAMK2A expressing neurons induces hyper activation of the mTORC1 pathway”.
Table S2. Statistics figure 3 – “Simultaneous deletion of Tsc1 and Rheb1 prevents mTORC1 epileptogenesis and mTORC1 activation”.
Table S3. Statistics figure 4 – “Rapamycin improves the epileptic phenotype in Tsc1‐Cre + mice in a dose dependent manner”.
Table S4. Statistics figure 5 – “TSC1‐mediated epilepsy induces changes in the mTORC1‐pathway that are only partially reversed by rapamycin”.
Table S5. Statistics figure 6 – “Preventive treatment with traditional AEDs reveal variable responses and do not affect the mTORC1 pathway”.
Table S6. Statistics figure 7 – “Ketogenic diet reduces cortical mTORC1 activity, delays epilepsy onset and improves survival”.
Table S7. Statistics figure 8 – “Preventive treatment with AZD8055 and PF4708671 does not improve the epileptic phenotype despite reduced levels of pS6Ser240/244”.
Acknowledgment
L. M. C .K. was supported by the Dutch Epilepsy Foundation, project number 16‐09. J. S, was funded by a fellowship from the Tuberous Sclerosis Association (TSA), United Kingdom (2014‐F02). We thank Minetta Elgersma‐Hooisma and Mehrnoush Aghadavoud Jolfaei for managing the mouse colony and genotyping. We thank Lucas Verwegen, Aniek Dillesse, Sandra van Iwaarden, and Catia A. Pinho Silva for their technical contribution, and Marie Claire de Wit, Diana C. Rotaru, Mark Nellist, and Fried Zwartkruis for fruitful discussions. We thank Geeske M. van Woerden for supervising students and her input on the data.
Funding information
L. M. C .K. was supported by the Dutch Epilepsy Foundation, project number 16‐09. J. S, was funded by a fellowship from the Tuberous Sclerosis Association (TSA), United Kingdom (2014‐F02).
Funding Statement
This work was funded by Dutch Epilepsy Foundation grant 16-09; Tuberous Sclerosis Association grant 2014-F02.
References
- 1. Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet 2008;372:657–668. [DOI] [PubMed] [Google Scholar]
- 2. Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med 2006;355:1345–1356. [DOI] [PubMed] [Google Scholar]
- 3. Curatolo P, Moavero R, van Scheppingen J, Aronica E. mTOR dysregulation and tuberous sclerosis‐related epilepsy. Expert Rev Neurother 2018;18:185–201. [DOI] [PubMed] [Google Scholar]
- 4. Abs E, Goorden SMI, Schreiber J, et al. TORC1‐dependent epilepsy caused by acute biallelic Tsc1 deletion in adult mice. Ann Neurol 2013;74:569–579. [DOI] [PubMed] [Google Scholar]
- 5. de Vries PJ. Targeted treatments for cognitive and neurodevelopmental disorders in tuberous sclerosis complex. Neurotherapeutics 2010;7:275–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Orlova KA, Crino PB. The tuberous sclerosis complex. Ann NY Acad Sci 2010;1184:87–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chu‐Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia 2010;51:1236–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. van Eeghen AM, Pulsifer MB, Merker VL, et al. Understanding relationships between autism, intelligence, and epilepsy: a cross‐disorder approach. Dev Med Child Neurol 2013;55:146–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Overwater IE, Verhaar BJH, Lingsma HF, et al. Interdependence of clinical factors predicting cognition in children with tuberous sclerosis complex. J Neurol 2017;264:161–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Curatolo P, Jóźwiak S, Nabbout R; TSC Consensus Meeting for SEGA and Epilepsy Management . Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol 2012;16:582–586. [DOI] [PubMed] [Google Scholar]
- 11. Cusmai R, Moavero R, Bombardieri R, et al. Long‐term neurological outcome in children with early‐onset epilepsy associated with tuberous sclerosis. Epilepsy Behav 2011;22:735–739. [DOI] [PubMed] [Google Scholar]
- 12. Overwater IE, Bindels‐de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia 2015;56:1239–1245. [DOI] [PubMed] [Google Scholar]
- 13. French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment‐resistant focal‐onset seizures associated with tuberous sclerosis (EXIST‐3): a phase 3, randomised, double‐blind, placebo‐controlled study. Elsevier, 2016. [DOI] [PubMed] [Google Scholar]
- 14. Overwater IE, Rietman AB, Bindels‐de Heus K, et al. Sirolimus for epilepsy in children with tuberous sclerosis complex: a randomized controlled trial. Neurology 2016;87:1011–1018. [DOI] [PubMed] [Google Scholar]
- 15. Kossoff EH, Thiele EA, Pfeifer HH, et al. Tuberous sclerosis complex and the ketogenic diet. Epilepsia 2005;46:1684–1686. [DOI] [PubMed] [Google Scholar]
- 16. Jóźwiak S, Kotulska K, Domańska‐Pakieła D, et al. Antiepileptic treatment before the onset of seizures reduces epilepsy severity and risk of mental retardation in infants with tuberous sclerosis complex. Eur J Paediatr Neurol 2011;15:424–431. [DOI] [PubMed] [Google Scholar]
- 17. Erdmann G, Schütz G, Berger S. Inducible gene inactivation in neurons of the adult mouse forebrain. BMC Neurosci 2007;8:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Meikle L, Talos DM, Onda H, et al. A mouse model of tuberous sclerosis: neuronal loss of Tsc1 causes dysplastic and ectopic neurons, reduced myelination, seizure activity, and limited survival. J Neurosci 2007;27:5546–5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Goorden SMI, Abs E, Bruinsma CF, et al. Intact neuronal function in Rheb1 mutant mice: implications for TORC1‐based treatments. Hum Mol Genet 2015;24:3390–3398. [DOI] [PubMed] [Google Scholar]
- 20. Jensen HS, Nichol K, Lee D, Ebert B. Clobazam and its active metabolite N‐desmethylclobazam display significantly greater affinities for α2‐ versus α1‐GABAA–receptor complexes. PLoS ONE 2014;9:e88456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hawkins NA, Zachwieja NJ, Miller AR, et al. Fine mapping of a dravet syndrome modifier locus on mouse chromosome 5 and candidate gene analysis by RNA‐Seq. PLOS Genet 2016;12:e1006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Morimoto K, Sato H, Yamamoto Y, et al. Antiepileptic effects of tiagabine, a selective GABA uptake inhibitor, in the rat kindling model of temporal lobe epilepsy. Epilepsia 1997;38:966–974. [DOI] [PubMed] [Google Scholar]
- 23. Chresta CM, Davies BR, Hickson I, et al. AZD8055 is a potent, selective, and orally bioavailable ATP‐competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res 2010;70:288–298. [DOI] [PubMed] [Google Scholar]
- 24. Zhang B, McDaniel SS, Rensing NR, Wong M. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS ONE 2013;8:e57445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Omrani A, van der Vaart T, Mientjes E, et al. HCN channels are a novel therapeutic target for cognitive dysfunction in Neurofibromatosis type 1. Mol Psychiatry 2015;20:1311–1321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Luszczki JJ, Trojnar MK, Ratnaraj N, et al. Interactions of stiripentol with clobazam and valproate in the mouse maximal electroshock‐induced seizure model. Epilepsy Res 2010;90:188–198. [DOI] [PubMed] [Google Scholar]
- 27. Younts TJ, Monday HR, Dudok B, et al. Presynaptic protein synthesis is required for long‐term plasticity of GABA release. Neuron 2016;92:479–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bartley CM, O’Keefe RA, Bordey A. FMRP S499 is phosphorylated independent of mTORC1‐S6K1 activity. PLoS ONE 2014;9:e96956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meikle L, Pollizzi K, Egnor A, et al. Neurobiology of disease response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved. Survival and Function 2008;28:5422–5432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tavazoie SF, Alvarez VA, Ridenour DA, et al. Regulation of neuronal morphology and function by the tumor suppressors Tsc1 and Tsc2. Nat Neurosci 2005;8:1727–1734. [DOI] [PubMed] [Google Scholar]
- 31. Goto J, Talos DM, Klein P, et al. Regulable neural progenitor‐specific Tsc1 loss yields giant cells with organellar dysfunction in a model of tuberous sclerosis complex. Proc Natl Acad Sci USA 2011;108:E1070–E1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Magri L, Cambiaghi M, Cominelli M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex‐associated lesions. Cell Stem Cell 2011;9:447–462. [DOI] [PubMed] [Google Scholar]
- 33. Rensing N, Han L, Wong M. Intermittent dosing of rapamycin maintains antiepileptogenic effects in a mouse model of tuberous sclerosis complex. Epilepsia 2015;56:1088–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zeng L‐H, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol 2008;63:444–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Greenhill SD, Jones RSG. Diverse antiepileptic drugs increase the ratio of background synaptic inhibition to excitation and decrease neuronal excitability in neurones of the rat entorhinal cortex in vitro. NSC 2010;167:456–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Curatolo P, Moavero R. mTOR inhibitors in tuberous sclerosis complex. Curr Neuropharmacol 2012;10:404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Park S, Lee EJ, Eom S, et al. Ketogenic diet for the management of epilepsy associated with tuberous sclerosis complex in children. J Epilepsy Res 2017;7:45–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kennedy AR, Pissios P, Otu H, et al. A high‐fat, ketogenic diet induces a unique metabolic state in mice. Am J Physiol Metab 2007;292:E1724–E1739. [DOI] [PubMed] [Google Scholar]
- 39. Pearce LR, Alton GR, Richter DT, et al. Characterization of PF‐4708671, a novel and highly specific inhibitor of p70 ribosomal S6 kinase (S6K1). Biochem J 2010;431:245–255. [DOI] [PubMed] [Google Scholar]
- 40. Thiele EA. Managing epilepsy in tuberous sclerosis complex. J Child Neurol 2004;19:680–686. [DOI] [PubMed] [Google Scholar]
- 41. Bateup HS, Johnson CA, Denefrio CL, et al. Excitatory/inhibitory synaptic imbalance leads to hippocampal hyperexcitability in mouse models of tuberous sclerosis. Neuron 2013;78:510–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Benthall KN, Ong SL, Bateup HS. Corticostriatal transmission is selectively enhanced in striatonigral neurons with postnatal loss of Tsc1. Cell Rep 2018;23:3197–3208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. McMahon JJ, Yu W, Yang J, et al. Seizure‐dependent mTOR activation in 5‐HT neurons promotes autism‐like behaviors in mice. Neurobiol Dis 2014;73C:296–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amin S, Lux A, Calder N, et al. Causes of mortality in individuals with tuberous sclerosis complex. Dev Med Child Neurol 2017;59:612–617. [DOI] [PubMed] [Google Scholar]
- 45. Devinsky O, Hesdorffer DC, Thurman DJ, et al. Sudden unexpected death in epilepsy: epidemiology, mechanisms, and prevention. Lancet Neurol 2016;15:1075–1088. [DOI] [PubMed] [Google Scholar]
- 46. Baldassari S, Picard F, Verbeek NE, et al. The landscape of epilepsy‐related GATOR1 variants. Genet Med 2018;1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Aiba I, Noebels JL. Spreading depolarization in the brainstem mediates sudden cardiorespiratory arrest in mouse SUDEP models. Sci Transl Med 2015;7:282ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aurlien D, Gjerstad L, Taubøll E. The role of antiepileptic drugs in sudden unexpected death in epilepsy. Seizure 2016;43:56–60. [DOI] [PubMed] [Google Scholar]
- 49. McLean BN, Wimalaratna S. Sudden death in epilepsy recorded in ambulatory EEG. J. Neurol. Neurosurg. Psychiatry 2007;78:1395–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ribierre T, Deleuze C, Bacq A, et al. Second‐hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia‐associated epilepsy. J Clin Invest 2018;128:2452–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sperling MR. Sudden unexplained death in epilepsy. Epilepsy Curr 2001;1:21–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Hesdorffer DC, Tomson T. Sudden unexpected death in epilepsy. CNS Drugs 2013;27:113–119. [DOI] [PubMed] [Google Scholar]
- 53. Lin F, de Gooijer MC, Hanekamp D, et al. PI3K‐mTOR pathway inhibition exhibits efficacy against high‐grade glioma in clinically relevant mouse models. Clin Cancer Res 2017;23:1286–1298. [DOI] [PubMed] [Google Scholar]
- 54. Hawley SA, Davison M, Woods A, et al. Characterization of the AMP‐activated protein kinase kinase from rat liver and identification of threonine 172 as the major site at which it phosphorylates AMP‐activated protein kinase. J Biol Chem 1996;271:27879–27887. [DOI] [PubMed] [Google Scholar]
- 55. Xiao B, Sanders MJ, Carmena D, et al. Structural basis of AMPK regulation by small molecule activators. Nat Commun 2013;4:3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Citraro R, Leo A, Constanti A, et al. MTOR pathway inhibition as a new therapeutic strategy in epilepsy and epileptogenesis. Pharmacol Res 2016;107:333–343. [DOI] [PubMed] [Google Scholar]
- 57. Tomson T, Battino D, Perucca E. Valproic acid after five decades of use in epilepsy: time to reconsider the indications of a time‐honoured drug. Lancet Neurol 2016;15:210–218. [DOI] [PubMed] [Google Scholar]
- 58. Larson AM, Pfeifer HH, Thiele EA. Low glycemic index treatment for epilepsy in tuberous sclerosis complex. Epilepsy Res 2012;99:180–182. [DOI] [PubMed] [Google Scholar]
- 59. McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett 2011;497:231–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yudkoff M, Daikhin Y, Melø TM, et al. The ketogenic diet and brain metabolism of amino acids: relationship to the anticonvulsant effect. Annu Rev Nutr 2007;27:415–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Brandt C, Hillmann P, Noack A, et al. The novel, catalytic mTORC1/2 inhibitor PQR620 and the PI3K/mTORC1/2 inhibitor PQR530 effectively cross the blood‐brain barrier and increase seizure threshold in a mouse model of chronic epilepsy. Neuropharmacology 2018;140:107–120. [DOI] [PubMed] [Google Scholar]
- 62. Rosborough BR, Raïch‐Regué D, Liu Q, et al. Adenosine triphosphate‐competitive mTOR inhibitors: a new class of immunosuppressive agents that inhibit allograft rejection. Am J Transplant 2014;14:2173–2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Talos DM, Jacobs LM, Gourmaud S, et al. Mechanistic target of rapamycin complex 1 and 2 in human temporal lobe epilepsy. Ann Neurol 2018;83:311–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Lopes MW, Soares FMS, de Mello N, et al. Time‐dependent modulation of mitogen activated protein kinases and AKT in rat hippocampus and cortex in the pilocarpine model of epilepsy. Neurochem Res 2012;37:1868–1878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Statistics figure 1 – “Cre mediated Tsc1 gene deletion in CAMK2A expressing neurons induces hyper activation of the mTORC1 pathway”.
Table S2. Statistics figure 3 – “Simultaneous deletion of Tsc1 and Rheb1 prevents mTORC1 epileptogenesis and mTORC1 activation”.
Table S3. Statistics figure 4 – “Rapamycin improves the epileptic phenotype in Tsc1‐Cre + mice in a dose dependent manner”.
Table S4. Statistics figure 5 – “TSC1‐mediated epilepsy induces changes in the mTORC1‐pathway that are only partially reversed by rapamycin”.
Table S5. Statistics figure 6 – “Preventive treatment with traditional AEDs reveal variable responses and do not affect the mTORC1 pathway”.
Table S6. Statistics figure 7 – “Ketogenic diet reduces cortical mTORC1 activity, delays epilepsy onset and improves survival”.
Table S7. Statistics figure 8 – “Preventive treatment with AZD8055 and PF4708671 does not improve the epileptic phenotype despite reduced levels of pS6Ser240/244”.