

Abstract
The concept of electrical protection of the ischemic myocardium is in constant evolution and has recently been supported by experimental and clinical studies. Historically, antiplatelet agents, angiotensin‐converting enzyme inhibitors, β‐blockers, and statins have been all proposed as drugs conferring anti‐ischemic cardioprotection. This was supported by the evidence consistently indicating that all these drugs were capable of reducing mortality and the risk of repeat myocardial infarction. The electrical plasticity paradigm is, however, a novel concept that depicts the benefits of improved sodium channel blockade with drugs such as ranolazine and cariporide. Although it has been hypothesized that the protective role of ranolazine depends on decreased fatty acid β‐oxidation affecting preconditioning, we speculate against such a hypothesis, because inhibition of β‐oxidation requires higher concentrations of the drug, above the therapeutic range. Rather, we discuss the key role of calcium overload reduction through inhibition of the late sodium current (INa). Mechanisms driving cardioprotection involve the block of a cascade of complex ionic exchanges that can result in intracellular acidosis, excess cytosolic calcium, myocardial cellular dysfunction, and eventually cell injury and death. In this review we discuss the studies that demonstrate how electrical plasticity through sodium channel blockers can promote cardioprotection against ischemia in coronary heart disease.
Introduction
The term cardioprotection indicates all mechanisms that contribute to the preservation of the heart from ischemic damage, reducing or even preventing it in various settings, including primary and secondary prevention of coronary heart disease, cardiac surgery, and thrombolysis in acute myocardial infarction.1 In the broad scope of treatments affording protection preventing myocardial damage or limiting its extension after initial damage has already ensued, mechanisms of cardioprotection can be classified as physiological adaptive and compensatory mechanisms, and therapeutic approaches. Acute adaptive mechanisms mainly work by increasing myocardial energy supply, whereas chronic mechanisms compensate for long‐lasting unfavorable influences, for example affecting the structural remodeling occurring after the damage.2 In this review we will discuss some of the classical therapeutic approaches yielding cardioprotection, mostly focusing on the novel possibility offered by membrane electrical remodeling through sodium channel blockers. This review will not discuss all of the numerous adaptive mechanisms operating in the heart, which have been covered by other recent reviews.3, 4, 5
Classical Therapeutic Approaches to Cardioprotection
Similar to the physiological adaptive and compensatory mechanisms, treatments for cardioprotection can be classified as acute or chronic.6 Acute interventions, such as thrombolysis and/or percutaneous coronary interventions in acute myocardial infarction, or prosthetic valve replacement in severe aortic stenosis, aim to prevent the loss of functional myocardium and to preserve ventricular function.7, 8 Artificial cardiac arrest during cardiac surgery, known also as cardioplegia, is a special form of cardioprotection that limits myocardial damage and reduces postoperative mortality.9 Coronary interventions in patients with stable coronary heart disease aim at relieving angina, reducing myocardial ischemia, or improving prognosis. Among medical therapies, antiplatelet drugs,10, 11, 12 angiotensin‐converting enzyme (ACE) inhibitors,13, 14 angiotensin II receptor blockers,15 and statins16, 17, 18 reduce mortality and morbidity in patients with chronic stable angina and preserved left ventricular (LV) function. Other therapies, such as nitrates,19, 20 calcium channel blockers, and β‐blockers have been shown to improve symptoms and exercise performance, but their effects on survival in patients with stable angina have not been proven. Nitrates, by releasing nitric oxide, when administered by intravenous, oral, or sublingual route, are very effective drugs in reducing angina. However, only a few small retrospective studies suggested improvement in prognosis and prevention of coronary events.20, 21, 22 Therefore, their chronic use, especially in asymptomatic patients without documentation of transient ischemia, is controversial. In theory, calcium channel blockers should have cardioprotective effects. However, studies of nifedipine, diltiazem, and verapamil did not demonstrate mortality benefit in patients with unstable angina and acute myocardial infarction. Similarly, studies of secondary prevention in the postinfarction period with these drugs also failed to reduce mortality, with the possible exception of a subgroup of patients without signs of heart failure. The addition of verapamil to trandolapril therapy reduced adverse coronary event rates by >60% in patients who developed congestive heart failure after myocardial infarction.23 On the other hand, unlike nitrates and calcium channel blockers, β‐blockers do improve prognosis.24 Indeed, in patients with stable angina and LV dysfunction following myocardial infarction, clinical trials have consistently indicated that ACE inhibitors and β‐blockers reduce mortality and the risk of repeat myocardial infarction. Therefore, these agents are recommended in such patients regardless of the presence of angina, along with aspirin and lipid‐lowering drugs.25
Role of Cardiac Sodium Currents in the Pathophysiology of Myocardial Injury From Ischemia
Myocardial ischemia is characterized by an imbalance between myocardial oxygen supply and oxygen demand, causing a drastic reduction of the energy necessary for the activity of ion pumps critical to the process of contraction‐relaxation of the cardiac myocyte. As a result, myocardial ischemia produces a cascade of complex ionic changes that reduce the intracellular pH (acidosis), and lead to an increase in intracellular sodium and calcium concentrations, cardiomyocyte dysfunction, and if the ischemia is severe and/or prolonged enough, to cell necrosis.26 In the initial phase of ischemia there is an increase of intracellular sodium, which quickly returns to preischemic levels during reperfusion if the duration of ischemia is short.27 Several mechanisms cause the increase in the intracellular sodium concentration in the ischemic cardiac myocyte. Ischemia (1) markedly increases the amplitude of the late sodium current (INa)28; (2) activates the sodium/hydrogen exchanger (NHE‐1) in response to intracellular acidosis, thus causing the increased entry of sodium27; and (3) decreases sodium extrusion from the cell as a result of an inhibition of the Na+/K+ ATPase pump.29 Ischemia also determines the opening of sarcolemmal adenosine triphosphate (ATP)‐dependent potassium channels (KATP). As a direct result of the increased intracellular sodium concentration, the concentration of intracellular calcium also increases by virtue of reduced extrusion and increased entry of calcium.30 In contrast, the opening of KATP channels leads to hyperpolarization, action potential shortening, a decrease in voltage‐dependent Ca2+ entry, reduction in contractility, decline in cellular metabolism, and eventually to a decrease in oxygen consumption. The ischemic changes in the intracellular sodium and calcium concentrations cause significant changes in electrophysiologic, metabolic, and contractile properties, which in turn may lead to irreversible damage ending with cardiomyocyte death (Figure 1).
Figure 1.
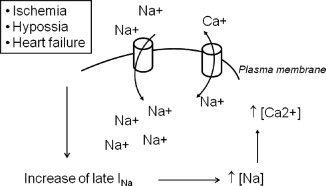
Pathophysiology of sodium‐induced calcium overload. In those situations in which late INa increases (ischemia, heart failure, hypoxia), the intracellular concentration of sodium increases, which in turn runs the Na+/Ca++ exchanger in reverse mode (ie, by sodium exit and calcium enter in the cardiac cell). The result is an overload of calcium that contributes to the appearance of electrical, mechanical, and metabolic alterations that are associated with cardiac ischemia. In the figure, cylinders represent the Na+/Ca++ exchanger in reverse mode.
Electrical Instability
Because the net current output of potassium during phase 2 of the action potential is small, an increase of late INa generates a depolarizing current that prolongs cardiac repolarization and extends the duration of the action potential and of the QT interval on the electrocardiogram.31 However, due to the nonhomogeneous distribution of the late INa in the ventricular wall, an increase in the late INa may extend much more than the duration of the action potential in epicardial and endocardial myocytes. This, in turn, causes increased dispersion of repolarization and electrical instability, leading to ventricular tachyarrhythmias.32 Furthermore, when phases 2 and 3 of the cardiac action potential are prolonged, calcium and sodium channels can reopen, with consequent increase of cation currents that may cause the development of early and post‐potential cardiac arrhythmias. Thus, the heterogeneity in the prolongation of ventricular action potential due to an increased late INa can generate the necessary electrical substrate for the occurrence of arrhythmias due to reentry and/or triggered activity (focal‐triggered arrhythmias).32
Contractile Dysfunction
The increased duration of the action potential along with the increased intracellular concentration of calcium during diastole gives rise to a tonic contraction that slows down LV diastolic relaxation (negative lusitropic effect), thus increasing LV end‐diastolic pressure and myocardial stiffness.33 This sustained contraction increases cardiac workload and oxygen demand, and compresses intramural, small, coronary vessels, in particular those in the subendocardial area, with a consequent reduction of coronary blood flow in the ischemic area during diastole.34
Metabolic Changes
A dysfunction at the mitochondrial level leads to decreased energy production and ATP synthesis, causing cell degeneration and death.35
Exogenous activation of KATP channels with the class of drugs known as KATP‐channel openers (eg, pinacidil) facilitates cardioprotection and improves outcome after acute impairment of myocardial circulation. At the same time, KATP‐channel openers are known to be associated with considerable side effects, including disturbances in cardiac rhythm such as ventricular fibrillation, inhibition of insulin production, and hypotension. Thus, elaboration of new KATP‐channel openers remains important. In contrast, a blockade of the late INa reduces the intracellular concentration of sodium, and indirectly of calcium, and may represent an effective pharmacological strategy to prevent cardiac arrhythmias and contractile dysfunction, and may have a protective effect in the ischemic myocardium. The best way to test this hypothesis is to use drugs that selectively block the late INa. Ranolazine is the first such drug that specifically inhibits the late INa. In this review we will focus on the studies that demonstrate how electrical plasticity through sodium channel blockers can promote cardioprotection against ischemia in coronary heart disease.
Therapeutic Potential of Sodium Channel Blockers
Sodium‐Hydrogen Exchange Inhibitors
Several experimental36, 37, 38, 39 and clinical studies40, 41, 42 have shown reduction of infarct size with the NHE inhibitor cariporide. Furthermore, cariporide is thought to play a critical role in vascular injury in diabetes.43, 44, 45 Cariporide decreases the influx of sodium into ischemic cells, which ultimately inhibits the exchange of sodium for calcium. It has been shown41 that 40 mg cariporide given intravenously over 10 minutes followed by a percutaneous coronary intervention in acute myocardial infarction patients decreased end‐systolic volume over 3 weeks (from 77 to 69 mL in the cariporide group vs from 80 to 97 mL in the placebo group; P = 0.048), increased the ejection fraction (from 44% to 50% in the cariporide group; P < 0.05 vs from 40% to 40% [unchanged] in the placebo group). There was also improvement in regional wall motion abnormalities and a reduction of myocardial infarct size, as well as of the area under the curve for the creatine kinase‐myocardial band release (P = 0.047). In the Guard During Ischemia Against Necrosis (GUARDIAN) trial,40 the effects of different doses of cariporide vs placebo in patients with various ischemic syndromes (unstable angina, non–ST‐elevation myocardial infarction, and high‐risk percutaneous or surgical revascularization) was investigated. The primary end point of all‐cause mortality or myocardial infarction at 36 days did not differ between groups. The highest dose (120 mg) was associated with a nonsignificant 10% reduction in the primary outcome. At 120 mg, the benefit was limited to patients undergoing coronary artery bypass grafting (CABG). Overall, in patients undergoing CABG, cariporide reduced the incidence of perioperative myocardial infarction, perhaps underscoring the importance of early therapy in an ischemic event. Cariporide may also exert protective effects on other cellular targets such as endothelial cells.46, 47, 48 Because the NHE‐1 isoform (with the water channel aquaporin‐1 [AQP1]) is also a membrane‐associated protein involved in the transmembrane transport of water driven by osmotic gradients, we have studied the involvement of NHE‐1 (through its blockage mediated by cariporide) and AQP1 in the hyperosmolarity‐mediated changes of endothelial function, in terms of decreased nitric oxide production and increased vascular cell adhesion molecule (VCAM)‐1 expression, in endothelial cells exposed to high glucose.48 We found that osmotic stress, at levels produced by diabetic hyperglycemia, increases VCAM‐1 expression and decreases endothelial nitric oxide synthase (eNOS) activity in cultured human endothelial cells.48 The inhibition of NHE‐1 and AQP1 prevented the hyperosmolarity‐induced enhancement of VCAM‐1 expression and the parallel reduction in eNOS activity and expression.48 Therefore, cariporide may contribute to protect endothelial cells from the glucose‐induced chain of events determined by hyperosmolarity. Correction of hyperosmolarity by targeting NHE‐1 through cariporide may thus represent a novel strategy to counteract the detrimental vascular consequences of diabetic hyperglycemia.
Eniporide, another inhibitor of the NHE, given to acute ST‐elevation myocardial infarction patients, did not yield differences in infarct size.49 There was actually a trend toward excess death and stroke in patients receiving eniporide. However, there was a significant reduction of the incidence of heart failure in patients reperfused late (<4 hours). In conclusion, the administration of the NHE‐1 inhibitor eniporide before reperfusion therapy, in patients with acute ST‐elevation myocardial infarction, did not limit infarct size or improve clinical outcome. Further research on NHE inhibitors is needed.
Inhibitors of the Late Sodium Current
Ranolazine (Ranexa; Gilead Sciences, Inc., Foster City, CA) ([(1)N‐(2,6‐dimethylphenyl)‐4(2‐hydroxy‐3‐(2‐metho xyphenoxy)‐propyl)‐1‐piperazine acetamide dihydrochloride]) is a racemic mixture that contains enantiomeric forms (S‐ranolazine and R‐ranolazine). In ventricular myocytes from dog and guinea pig hearts, ranolazine, at therapeutically relevant concentrations (up to 10 mmol/L), has been demonstrated to selectively inhibit the late INa (IC50 55 to 21 mmol/L) without affecting either the fast sodium current responsible for the upstroke of the action potential (IC50 value of 244 mmol/L for peak INa) or the NHE.50 The drug acts as an atrial‐selective sodium channel blocker, which predominantly inhibits early INa, late INa, IKr, and late ICa at a concentration within the therapeutic range (<10 μM). The drug is manufactured in a sustained‐release form that has a prolonged absorption phase with maximal plasma concentrations typically seen 4 to 6 hours after administration. Although it inhibits cardiac metabolism of fatty acids, this cannot explain the clinically relevant effects of the drug, because inhibition of β‐oxidation requires concentrations of the drug above the therapeutic range (>100 μM).35 Recent evidence suggests that ranolazine reduces calcium overload in the ischemic myocyte through inhibition of the late INa, which can block the cascade of complex ionic exchanges that can result in intracellular acidosis, excess cytosolic calcium, myocardial cellular dysfunction, and if sustained, cell injury and death.51 In isolated heart preparations of various animal species, ranolazine selectively inhibits late INa in a manner that is concentration, voltage, and frequency dependent.52, 53 This inhibition leads directly to a decrease in the intracellular concentration of sodium and indirectly to a reduction of calcium overload. At therapeutic concentrations, ranolazine has no effect on peak INa, which is the sodium current responsible for the action potential. Furthermore, ranolazine does not inhibit the calcium current flowing through the L‐type channels or modify the activity of the NHE‐1 exchanger.54 In accordance with these data, ranolazine does not depress the contractility of the atria or ventricles. All of these data confirm that the reduction of calcium overload caused by ranolazine is not due to the reduction of calcium entry through the L‐type calcium channels or to the inhibition of NHE‐1, but rather to an indirect effect mediated by reduction of the late INa.
In isolated, perfused, guinea pig hearts in which ischemia is induced by reducing flow, ranolazine exerts a cardioprotective effect with preservation of ATP cellular levels and a decrease in creatine kinase (CK) and lactate dehydrogenase (LDH), which are markers of cell death. These data suggest that the myocytes tolerate ischemia better when exposed to ranolazine.55 The effect of ranolazine on late INa is more pronounced in ischemic or failing myocytes in which the current is amplified. In a rat model of ischemia/reperfusion injury, the administration of ranolazine 10 minutes before the ligation of a coronary artery reduced the release of CK and LDH during the reperfusion period, suggesting that the drug was able to reduce cell damage.56 In rats subjected to coronary occlusion for 25 minutes and subsequent reperfusion for 2 hours, treatment with ranolazine reduced the release of troponin T and the size of the infarcted area.56 In a rabbit model of stunned myocardium (15 minutes of coronary occlusion followed by 3 hours of reperfusion), the animals pretreated with ranolazine 10 minutes before occlusion showed a higher ejection fraction and stroke volume.57 Moreover, ranolazine decreased the number of akinetic or dyskinetic segments in the left ventricle wall and infarct size.57 These data suggest that the inhibition of the late INa may represent a new therapeutic strategy for reducing myocardial stunning and increasing cardioprotection during ischemia and myocardial infarction.
Cardioprotection of Ranolazine in Clinical Trials
In controlled clinical trials carried out in patients with chronic stable angina and acute coronary syndrome, ranolazine, either alone or in combination with conventional antianginal drugs (diltiazem, amlodipine, and atenolol), has proven to be an effective and well‐tolerated antianginal drug. In patients with chronic angina and demand‐induced ischemia, ranolazine has the potential to partially disrupt the consequences of cell hypoxia during transient myocardial ischemia by reducing excess late sodium influx, thereby reducing calcium overload and ultimately reducing the concomitant increase in left ventricular wall tension. Reduction in diastolic left ventricular wall tension would decrease myocardial oxygen requirements in marginally ischemic myocytes and has the potential to reduce vascular compression, allowing more coronary blood flow to the affected area. Antianginal effects are observed at concentrations at which the heart rate, contractility, conduction velocity, and intracardiac blood pressure do not change. The antianginal effects of slow‐release ranolazine in patients with chronic stable angina have been evaluated in 3 studies: MARISA (Monotherapy Assessment of Ranolazine in Stable Angina), CARISA (Combination Assessment of Ranolazina in Stable Angina) and ERICA (Efficacy of Ranolazine in Chronic Angina). MARISA58 was a randomized, double‐blind, placebo‐controlled, crossover of 191 patients with effort angina present for at least 3 months responsive to β‐blockers, calcium antagonists, and/or nitrates. Patients were randomized to ranolazine 500, 1000, and 1500 mg twice daily or placebo. After a week of treatment, ranolazine significantly increased the total exercise duration, time to onset of angina, and time of appearance of ST depression >1 mm compared to placebo. Because with 1500 mg twice a day there was an increase of adverse events (decreased heart rate, blood pressure, and increased QTc), it was decided not to continue to use the dose of 1500 mg. CARISA59 was a randomized, double‐blind, placebo‐controlled, dose–response study achieved in 823 highly symptomatic patients (4 episodes of angina per week). Patients received placebo or ranolazine (750 or 1000 mg twice daily) for 12 weeks associated with classical antianginal therapy, which included atenolol, amlodipine, diltiazem, and nitrates as needed. Compared with placebo, ranolazine produced a significant and dose‐dependent increase of duration of exercise, increased time to onset of angina, and time of appearance of ST depression >1 mm. ERICA60 was a randomized, double‐blind, placebo‐controlled trial that compared the antianginal effects of ranolazine and placebo in 565 patients with more than 3 anginal attacks per week despite treatment with amlodipine 10 mg per day. Patients received an initial dose of ranolazine 500 mg twice daily or placebo for a week, followed by 6 weeks of treatment with 1000 mg twice a day or placebo, in addition to amlodipine. Ranolazine significantly decreased the number of angina attacks by 23%.
The anti‐ischemic effects of slow‐release ranolazine in patients with non–ST‐elevation acute coronary syndrome have been evaluated in MERLIN TIMI‐36 study (Metabolic Efficiency With Ranolazine for Less Ischemia in Non–ST‐Elevation Acute Coronary Syndromes),61 a randomized, double‐blind, placebo‐controlled trial that included 6560 patients. Within 48 hours from the onset of angina due to non–ST‐elevation myocardial infarction and/or unstable angina, in addition to standard therapy, patients were randomized 1:1 to intravenous ranolazine (200 mg bolus followed by infusion at a dose of 80 mg/h for a minimum of 12 hours and a maximum of 96 hours) or placebo. Slow‐release ranolazine (1000 mg twice daily) or placebo were then continued as long‐term treatment. In patients with renal failure, intravenous dose was reduced by 50%. The study demonstrated that the drug was ineffective in reducing the composite end point of cardiovascular death, myocardial infarction, or recurrent ischemia in patients with unstable angina or non–ST‐elevation myocardial infarction, but did reduce recurrent ischemia and improved exercise tolerance and frequency of angina without evidence of arrhythmias.
Adverse Events With Ranolazine Treatment
Even though ranolazine has potential benefits in patients with chronic stable angina and diastolic heart failure, dose‐related QTc interval prolongation secondary to IKr blockade remains a concern. This effect may cause ventricular arrhythmias especially in patients who are on other QTc prolonging drugs or patients with liver failure. Current evidence did not demonstrate increased sudden death in patients with acute coronary syndromes; however, there are very limited data on high doses (>1000 mg twice daily) of ranolazine use. In addition, ranolazine has side effects, including nausea, dizziness, and headache, which may render its prolonged use in some patients. Blockage of late Na channels in smooth muscle cells may potentially lead to constipation and urinary retention.
Future Research
The following studies can be done to prove the clinical utility of ranolazine:
Randomized clinical studies comparing ranolazine to long acting nitrate in patients with chronic stable angina on maximum tolerable dose of B‐blockers and calcium channel blockers: The effect of ranolazine in patients with refractory angina despite medical therapy and revascularization deserves investigation.
Patients with angina secondary to hypertrophic cardiomyopathy and aortic stenosis due to the anti‐ischemic effects in the absence of coronary vasodilatation: It has been demonstrated that ranolazine treatment improved symptoms in a young patient with hypertrophic cardiomyopathy.62 Similarly, it has been reported that an 88‐year‐old female with severe aortic stenosis had her angina episodes relieved subsequent to ranolazine 375 mg twice daily treatment.63 These reports suggest that ranolazine may be useful for the symptomatic treatment of patients with left ventricular hypertrophy who did not respond to traditional therapies.
Utility of ranolazine in the prevention of cardiac arrhythmias including atrial fibrillation: Ranolazine prolongs atrial refractoriness and inhibits after depolarizations and triggered activity. It has been demonstrated that ranolazine (1000 mg twice daily for 10–14 days) significantly reduced postoperative atrial fibrillation following coronary artery bypass surgery.64 This benefit was not associated with increased adverse events.
Clinical outcomes of QT prolongation induced by ranolazine: Ranolazine has direct effects on ion channels. This effect may be augmented by more effective energy production by cardiomyocytes, which may lead to ventricular arrhythmias and death. The clinical outcomes of QT prolongation by ranolazine should be investigated in large‐scale studies.
Utility of ranolazine in patients with left ventricular dyssynchrony: Ranolazine theoretically improves myocardial perfusion and reduces ischemia. This effect may be reflected as a reduction of dyssynchrony, especially in patients with coronary artery disease. It has been demonstrated that ranolazine improved systolic and diastolic dyssynchrony by gated single photon‐emission computed tomographic myocardial perfusion by an imaging study including 32 patients with reversible perfusion defects.65
Clinical outcomes of ranolazine treatment, especially in a subgroup of patients such as women or African Americans: Similar to each new drug, ranolazine treatment outcomes should be investigated thoroughly among all patient subgroups.
Conclusion
Selective sodium channel blockers may offer a safe and effective strategy for the management of stable angina. Preclinical and clinical studies have shown that ranolazine and cariporide inhibit sodium channels, reduce the overhead of calcium, and improve diastolic function, which translates clinically as an anti‐ischemic effect. These drugs exert these effects without any significant change in heart rate or cardiac contractility, blood pressure, and coronary flow. Ranolazine should be reserved for patients who have not achieved an adequate response with other antianginal drugs and should not be used as an alternative to β‐blockers. Large, randomized, controlled trials investigating the effects of these drugs on mortality and hospitalization rates in patients with coronary artery disease are required.
The authors have no funding, financial relationships, or conflicts of interest to disclose.
References
- 1. Bolli R, Becker L, Gross G, et al. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res. 2004;95:125–134. [DOI] [PubMed] [Google Scholar]
- 2. Sanz‐Rosa D, Garcia‐Prieto J, Ibanez B. The future: therapy of myocardial protection. Ann N Y Acad Sci. 2012;1254:90–98. [DOI] [PubMed] [Google Scholar]
- 3. Liu SQ, Tefft BJ, Zhang D, et al. Cardioprotective mechanisms activated in response to myocardial ischemia. Mol Cell Biomech. 2011;8:319–338. [PubMed] [Google Scholar]
- 4. Ravingerova T. Intrinsic defensive mechanisms in the heart: a potential novel approach to cardiac protection against ischemic injury. Gen Physiol Biophys. 2007;26:3–13. [PubMed] [Google Scholar]
- 5. Ong SG, Hausenloy DJ. Hypoxia‐inducible factor as a therapeutic target for cardioprotection. Pharmacol Ther. 2012;136:69–81. [DOI] [PubMed] [Google Scholar]
- 6. Michel D. Cardioprotection with drugs. 1: Coronary heart disease [in German]. Fortschr Med. 1990;108:40–44. [PubMed] [Google Scholar]
- 7. Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST‐elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol. 2004;44:E1–E211. [DOI] [PubMed] [Google Scholar]
- 8. Kloner RA, Schwartz Longacre L. State of the science of cardioprotection: challenges and opportunities—proceedings of the 2010 NHLBI Workshop on Cardioprotection. J Cardiovasc Pharmacol Ther. 2011;16:223–232. [DOI] [PubMed] [Google Scholar]
- 9. Chambers DJ, Fallouh HB. Cardioplegia and cardiac surgery: pharmacological arrest and cardioprotection during global ischemia and reperfusion. Pharmacol Ther. 2011;127:41–52. [DOI] [PubMed] [Google Scholar]
- 10. Juul‐Moller S, Edvardsson N, Jahnmatz B, et al. Double‐blind trial of aspirin in primary prevention of myocardial infarction in patients with stable chronic angina pectoris. The Swedish Angina Pectoris Aspirin Trial (SAPAT) Group. Lancet. 1992;340:1421–1425. [DOI] [PubMed] [Google Scholar]
- 11. CAPRIE Steering Committee . A randomised, blinded, trial of clopidogrel versus aspirin in patients at risk of ischaemic events (CAPRIE). CAPRIE Steering Committee. Lancet. 1996;348:1329–1339. [DOI] [PubMed] [Google Scholar]
- 12. Berger PB, Bhatt DL, Fuster V, et al. Bleeding complications with dual antiplatelet therapy among patients with stable vascular disease or risk factors for vascular disease: results from the Clopidogrel for High Atherothrombotic Risk and Ischemic Stabilization, Management, and Avoidance (CHARISMA) trial. Circulation. 2010;121:2575–2583. [DOI] [PubMed] [Google Scholar]
- 13. Yusuf S, Sleight P, Pogue J, et al. Effects of an angiotensin‐converting‐enzyme inhibitor, ramipril, on cardiovascular events in high‐risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. N Engl J Med. 2000;342:145–153. [DOI] [PubMed] [Google Scholar]
- 14. Fox KM. Efficacy of perindopril in reduction of cardiovascular events among patients with stable coronary artery disease: randomised, double‐blind, placebo‐controlled, multicentre trial (the EUROPA study). Lancet. 2003;362:782–788. [DOI] [PubMed] [Google Scholar]
- 15. Ruilope LM, Segura J, Zamorano JL. New clinical concepts after the ONTARGET trial. Expert Rev Cardiovasc Ther. 2011;9:685–689. [DOI] [PubMed] [Google Scholar]
- 16. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high‐risk individuals: a randomised placebo‐controlled trial. Lancet. 2002;360:7–22. [DOI] [PubMed] [Google Scholar]
- 17. Ford I, Murray H, Packard CJ, et al. Long‐term follow‐up of the West of Scotland Coronary Prevention Study. N Engl J Med. 2007;357:1477–1486. [DOI] [PubMed] [Google Scholar]
- 18. Ridker PM, Danielson E, Fonseca FA, et al. Rosuvastatin to prevent vascular events in men and women with elevated C‐reactive protein. N Engl J Med. 2008;359:2195–2207. [DOI] [PubMed] [Google Scholar]
- 19. Cohn JN. Vasodilator therapy of dilated cardiomyopathy. Postgrad Med J. 1986;62:599–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gori T, Fineschi M, Parker JD, et al. Current perspectives. Therapy with organic nitrates: newer ideas, more controversies. Ital Heart J. 2005;6:541–548. [PubMed] [Google Scholar]
- 21. Chaturvedi P. Isis‐4. Lancet. 1995;345:1374; author reply 1374–1375. [PubMed] [Google Scholar]
- 22. Kiraly C, Kiss A, Timar S, et al. Effects of long‐term transdermal nitrate treatment on left ventricular function in patients following myocardial infarction. Clin Cardiol. 2003;26:120–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hansen JF, Hagerup L, Sigurd B, et al. Cardiac event rates after acute myocardial infarction in patients treated with verapamil and trandolapril versus trandolapril alone. Danish Verapamil Infarction Trial (DAVIT) Study Group. Am J Cardiol. 1997;79:738–741. [DOI] [PubMed] [Google Scholar]
- 24. Go AS, Iribarren C, Chandra M, et al. Statin and beta‐blocker therapy and the initial presentation of coronary heart disease. Ann Intern Med. 2006;144:229–238. [DOI] [PubMed] [Google Scholar]
- 25. Freemantle N, Cleland J, Young P, et al. beta blockade after myocardial infarction: systematic review and meta regression analysis. BMJ. 1999;318:1730–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Silverman C, Stern M. Ionic basis of ischaemic cardiac injury: insights from cellular studies Cardiovasc Res 1994;28:581–597. [DOI] [PubMed] [Google Scholar]
- 27. Tani M, Neely JR. Role of intracellular Na + and Ca2+ overload and depressed recovery of ventricular function of reperfused ischemic rat hearts. Possible involvement of H + −Na + and Na + −Ca2+ exchange. Circ Res. 1989;65:1045–1056. [DOI] [PubMed] [Google Scholar]
- 28. Hammarstrom AK, Gage PW. Hypoxia and persistent sodium current. Eur Biophys. 2002;31:323–333. [DOI] [PubMed] [Google Scholar]
- 29. Xiao XH, Allen DG. Role of Na+/H + exchanger during ischemia and preconditioning in the isolated rat heart. Circ Res. 1999;85:723–730. [DOI] [PubMed] [Google Scholar]
- 30. Imahashi K, Kusuoka H, Hashimoto K, et al. Intracellular sodium accumulation during ischemia as the substrate for reperfusion injury. Circ Res. 1999;84:1401–1406. [DOI] [PubMed] [Google Scholar]
- 31. Wu L, Shryock JC, Song Y, et al. An increase in late sodium current potentiates the proarrhytymic activities of low‐risk QT‐prolonging drugs in female rabbit hearts. J Pharmacol Exp Ther. 2006;316:718–726. [DOI] [PubMed] [Google Scholar]
- 32. Noble D, Noble PJ. Late sodium current in the pathophysiology of cardiovascular disease: consequences of sodium‐calcium overload. Heart. 2006;92:(suppl 4):IV1–IV5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bers DM, Eisner DA, Valdivia HH. Sarcoplasmic reticulum Ca2+ and heart failure: roles of diastolic leak and Ca2+ transport. Circ Res. 2003;93:487–490. [DOI] [PubMed] [Google Scholar]
- 34. Ver Donck L, Borgers M, Verdonck F. Inhibition of sodium and calcium overload pathology in the myocardium: a new cytoprotective principle. Cardiovasc Res. 1993;27:349–399. [DOI] [PubMed] [Google Scholar]
- 35. Chaitman B. Efficacy and safety of a metabolic drug in chronic stable angina: review of evidence from clinical trials. J Cardiovasc Pharmacol Ther. 2004;9:S47–S64. [DOI] [PubMed] [Google Scholar]
- 36. Garcia‐Dorado D, Gonzalez MA, Barrabes JA, et al. Prevention of ischemic rigor contracture during coronary occlusion by inhibition of Na(+)‐H + exchange. Cardiovasc Res. 1997;35:80–89. [DOI] [PubMed] [Google Scholar]
- 37. Linz WJ, Busch AE. NHE‐1 inhibition: from protection during acute ischaemia/reperfusion to prevention/reversal of myocardial remodelling. Naunyn Schmiedebergs Arch Pharmacol. 2003;368:239–246. [DOI] [PubMed] [Google Scholar]
- 38. Karmazyn M, Sostaric JV, Gan XT. The myocardial Na+/H + exchanger: a potential therapeutic target for the prevention of myocardial ischaemic and reperfusion injury and attenuation of postinfarction heart failure. Drugs. 2001;61:375–389. [DOI] [PubMed] [Google Scholar]
- 39. Gumina RJ, Buerger E, Eickmeier C, et al. Inhibition of the Na(+)/H(+) exchanger confers greater cardioprotection against 90 minutes of myocardial ischemia than ischemic preconditioning in dogs. Circulation. 1999;100:2519–2526; discussion 2469–2572. [DOI] [PubMed] [Google Scholar]
- 40. Theroux P, Chaitman BR, Danchin N, et al. Inhibition of the sodium‐hydrogen exchanger with cariporide to prevent myocardial infarction in high‐risk ischemic situations. Main results of the GUARDIAN trial. Guard during ischemia against necrosis (GUARDIAN) Investigators. Circulation. 2000;102:3032–3038. [DOI] [PubMed] [Google Scholar]
- 41. Rupprecht HJ, vom Dahl J, Terres W, et al. Cardioprotective effects of the Na(+)/H(+) exchange inhibitor cariporide in patients with acute anterior myocardial infarction undergoing direct PTCA. Circulation. 2000;101:2902–2908. [DOI] [PubMed] [Google Scholar]
- 42. Mentzer RM Jr, Bartels C, Bolli R, et al. Sodium‐hydrogen exchange inhibition by cariporide to reduce the risk of ischemic cardiac events in patients undergoing coronary artery bypass grafting: results of the EXPEDITION study. Ann Thorac Surg. 2008;85:1261–1270. [DOI] [PubMed] [Google Scholar]
- 43. Anzawa R, Seki S, Nagoshi T, et al. The role of Na+/H + exchanger in Ca2+ overload and ischemic myocardial damage in hearts from type 2 diabetic db/db mice. Cardiovasc Diabetol. 2012;11:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang S, Peng Q, Zhang J, et al. Na+/H + exchanger is required for hyperglycaemia‐induced endothelial dysfunction via calcium‐dependent calpain. Cardiovasc Res. 2008;80:255–262. [DOI] [PubMed] [Google Scholar]
- 45. Vial G, Dubouchaud H, Couturier K, et al. Na+/H + exchange inhibition with cariporide prevents alterations of coronary endothelial function in streptozotocin‐induced diabetes. Mol Cell Biochem. 2008;310:93–102. [DOI] [PubMed] [Google Scholar]
- 46. Wang SX, Sun XY, Zhang XH, et al. Cariporide inhibits high glucose‐mediated adhesion of monocyte‐endothelial cell and expression of intercellular adhesion molecule‐1. Life Sci. 2006;79:1399–1404. [DOI] [PubMed] [Google Scholar]
- 47. Zerbini G, Roth T, Podesta F, et al. Activity and expression of the Na+/H + exchanger in human endothelial cells cultured in high glucose. Diabetologia. 1995;38:785–791. [DOI] [PubMed] [Google Scholar]
- 48. Madonna R, Montebello E, Lazzerini G, et al. NA+/H + exchanger 1‐ and aquaporin‐1‐dependent hyperosmolarity changes decrease nitric oxide production and induce VCAM‐1 expression in endothelial cells exposed to high glucose. Int J Immunopathol Pharmacol. 2010;23:755–765. [DOI] [PubMed] [Google Scholar]
- 49. Zeymer U, Suryapranata H, Monassier JP, et al. The Na(+)/H(+) exchange inhibitor eniporide as an adjunct to early reperfusion therapy for acute myocardial infarction. Results of the evaluation of the safety and cardioprotective effects of eniporide in acute myocardial infarction (ESCAMI) trial. J Am Coll Cardiol. 2001;38:1644–1650. [DOI] [PubMed] [Google Scholar]
- 50. Bonadei I, Vizzardi E, Quinzani F, et al. Effects of ranolazine on cardiovascular system. Recent Pat Cardiovasc Drug Discov. 2011;6:215–221. [DOI] [PubMed] [Google Scholar]
- 51. Shryock JC, Belardinelli L. Inhibition of late sodium current to reduce electrical and mechanical dysfunction of ischaemic myocardium. Br J Pharmacol. 2008;153:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Gralinski MR, Black SC, Kilgore KS, et al. Cardioprotective effects of ranolazine (RS‐43285) in the isolated perfused rabbit heart. Cardiovasc Res. 1994;28:1231–1237. [DOI] [PubMed] [Google Scholar]
- 53. McCormack JG, Barr RL, Wolff AA, et al. Ranolazine stimulates glucose oxidation in normoxic, ischemic, and reperfused ischemic rat hearts. Circulation. 1996;93:135–142. [DOI] [PubMed] [Google Scholar]
- 54. Antzelevitch C, Belardinelli L, Zygmunt AC, et al. Electrophysiological effects of ranolazine, a novel antianginal agent with antiarrhythmic properties. Circulation. 2004;110:904–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Clarke B, Spedding M, Patmore L, et al. Protective effects of ranolazine in guinea‐pig hearts during low‐flow ischaemia and their association with increases in active pyruvate dehydrogenase. Br J Pharmacol. 1993;109:748–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Allely MC, Brown CM, Kenny BA, et al. Modulation of alpha 1‐adrenoceptors in rat left ventricle by ischaemia and acyl carnitines: protection by ranolazine. J Cardiovasc Pharmacol. 1993;21:869–873. [DOI] [PubMed] [Google Scholar]
- 57. Hale SL, Leeka JA, Kloner RA. Improved left ventricular function and reduced necrosis after myocardial ischemia/reperfusion in rabbits treated with ranolazine, an inhibitor of the late sodium channel. J Pharmacol Exp Ther. 2006;318:418–423. [DOI] [PubMed] [Google Scholar]
- 58. Chaitman BR, Skettino SL, Parker JO, et al. Anti‐ischemic effects and long‐term survival during ranolazine monotherapy in patients with chronic severe angina. J Am Coll Cardiol. 2004;43:1375–1382. [DOI] [PubMed] [Google Scholar]
- 59. Chaitman BR, Pepine CJ, Parker JO, et al. Effects of ranolazine with atenolol, amlodipine, or diltiazem on exercise tolerance and angina frequency in patients with severe chronic angina: a randomized controlled trial. JAMA. 2004;291:309–316. [DOI] [PubMed] [Google Scholar]
- 60. Stone PH, Gratsiansky NA, Blokhin A, et al. Antianginal efficacy of ranolazine when added to treatment with amlodipine: the ERICA (Efficacy of Ranolazine in Chronic Angina) trial. J Am Coll Cardiol. 2006;48:566–575. [DOI] [PubMed] [Google Scholar]
- 61. Scirica BM, Morrow DA, Hod H, et al. Effect of ranolazine, an antianginal agent with novel electrophysiological properties, on the incidence of arrhythmias in patients with non ST‐segment elevation acute coronary syndrome: results from the Metabolic Efficiency With Ranolazine for Less Ischemia in Non ST‐Elevation Acute Coronary Syndrome Thrombolysis in Myocardial Infarction 36 (MERLIN‐TIMI 36) randomized controlled trial. Circulation. 2007;116:1647–1652. [DOI] [PubMed] [Google Scholar]
- 62. Tomberli B, Girolami F, Coppini R, et al. Management of refractory symptoms in hypertrophic cardiomyopathy with restrictive pathophysiology: novel perspectives for ranolazine [in Italian]. G Ital Cardiol (Rome). 2012;13:297–303. [DOI] [PubMed] [Google Scholar]
- 63. Athauda‐Arachchi P, Lang C. Metabolic antianginal agent ranolazine offers good symptom relief in a patient with inoperable severe aortic stenosis. Cardiovasc Ther. 2012;30:e210–e211. [DOI] [PubMed] [Google Scholar]
- 64. Miles RH, Passman R, Murdock DK. Comparison of effectiveness and safety of ranolazine versus amiodarone for preventing atrial fibrillation after coronary artery bypass grafting. Am J Cardiol. 2011;108:673–676. [DOI] [PubMed] [Google Scholar]
- 65. Venkataraman R, Chen J, Garcia EV, et al. Effect of ranolazine on left ventricular dyssynchrony in patients with coronary artery disease. Am J Cardiol. 2012;110:1440–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]