

Abstract
Objective
ANO5‐related myopathy is an important cause of limb‐girdle muscular dystrophy (LGMD) and hyperCKemia. The main descriptions have emerged from European cohorts, and the burden of the disease worldwide is unclear. We provide a detailed characterization of a large Brazilian cohort of ANO5 patients.
Methods
A national cross‐sectional study was conducted to describe clinical, histopathological, radiological, and molecular features of patients carrying recessive variants in ANO5. Correlation of clinical and genetic characteristics with different phenotypes was studied.
Results
Thirty‐seven patients from 34 nonrelated families with recessive mutations of ANO5 were identified. The most common phenotype was LGMD, observed in 25 (67.5%) patients, followed by pseudometabolic presentation in 7 (18.9%) patients, isolated asymptomatic hyperCKemia in 4 (10.8%) patients, and distal myopathy in a single patient. Nine patients presented axial involvement, including one patient with isolated axial weakness. The most affected muscles according to MRI were the semimembranosus and gastrocnemius, but paraspinal and abdominal muscles, when studied, were involved in most patients. Fourteen variants in ANO5 were identified, and the c.191dupA was present in 19 (56%) families. Sex, years of disease, and the presence of loss‐of‐function variants were not associated with specific phenotypes.
Interpretation
We present the largest series of anoctaminopathy outside Europe. The most common European founder mutation c.191dupA was very frequent in our population. Gender, disease duration, and genotype did not determine the phenotype.
Introduction
ANO5 encodes a putative calcium‐activated chloride channel, known as anoctamin 5, which plays a role in muscle cell membrane fusion and repair.1, 2 Recessive mutations in ANO5 cause limb‐girdle muscular dystrophy type 2L (LGMD2L), Miyoshi‐like distal myopathy (MMD3) and serum creatine kinase (CK) increase with variable exercise intolerance.3, 4, 5, 6 Dominant variants have been associated with the skeletal disorder gnathodiaphyseal dysplasia (GDD).7
The prevalence of ANO5 pathogenic variants and their phenotypes are variable in different populations and may be underestimated. The frequency of LGMD2L, the most common presentation of anoctaminopathy, in LGMD cohorts has been reported from 2% in Italian population and 7% in US population to around 25% in UK and Finland populations.4, 8, 9, 10, 11 Additionally, many other patients have been identified with non‐LGMD phenotypes, and up to 50% of patients harboring ANO5 mutations may have a distal myopathy or an isolated hyperCKemia as initial presentation.6, 11 A common mutation (c.191dupA), for which is suggested to be the result of a founder effect on Northern Europeans,4 and another variant (c.2272C > T), especially common in the Finnish population,9 are considered the most frequent mutations in patients with anoctaminopathy, but many other variants have been described.11
Studies describing the clinical and genetic patterns of anoctaminopathies from patients outside Europe are scarce, except for new small reports of families from China, Japan, and Saudi Arabia.12, 13, 14 Recently, with the advent of next‐generation sequence (NGS), patients with ANO5 mutations have been detected in Brazil. Thus, the objective of the present study is to provide a detailed characterization of a large Brazilian cohort of patients with ANO5 mutations from several reference centers around the country.
Patients and Methods
A national collaborative cross‐sectional study was conducted to describe the clinical, histopathological, radiological, and molecular features of consecutive patients with recessive pathogenic variants in ANO5 from 12 neuromuscular centers in Brazil, from January 2016 to November 2018. In each neuromuscular center, patients were selected for genetic panel addressed to recessive LGMD genes if they had a non‐congenital unexplained myopathy and/or hyperCKemia with no other obvious diagnosis.
The study was approved by the institutional ethic committees of the local centers. Patients or their legal representatives were invited to participate in the study, and informed consent was obtained for genetic studies and muscle imaging records according to the institutional ethical committee and regulating rules.
Clinical evaluation
All patients underwent detailed clinical examination, and medical chart reviews were collected by neuromuscular specialists in each center using a standardized protocol. Muscle strength was measured by manual muscle testing according to the Medical Research Council. Comprehensive records of ancillary exams including nerve conduction studies (NCS)/needle electromyography (EMG), maximal serum CK levels, pulmonary function tests, electrocardiograms, and echocardiograms were obtained.
Patients were classified into four different clinical phenotypes, based on the initial symptoms and presentation, and assessed retrospectively: LGMD, MMD3, and pseudometabolic and asymptomatic hyperCKemia. The LGMD phenotype was defined as progressive weakness and atrophy in a limb‐girdle pattern. The MMD3 phenotype was considered if patients presented initial and predominantly distal weakness and calf wasting. The pseudometabolic phenotype was defined by persistently elevated serum CK levels associated with exercise intolerance and/or myalgia without significant limb‐girdle weakness in the current neurological examination. Asymptomatic hyperCKemia was considered when the patient had no other complaint or weakness beyond high serum CK levels. For all phenotypes, the age of onset was considered the age that the first symptoms were perceived or the first abnormal CK measurement was observed.
Histological analysis
Muscle biopsies were reviewed by two authors (AMSS and EZ). All biopsies had been performed using an open technique in the biceps brachii or deltoid muscles. The samples were stained with hematoxylin and eosin, modified Gömöri trichrome and histochemical staining, such as reduced nicotinamide adenine dinucleotide tetrazolium reductase (NADH‐TR), cytochrome C oxidase (COX), succinate dehydrogenase (SDH), and ATPase in three different pH values (9.4, 4.6, and 4.3).
The following histological aspects were analyzed: changes in fiber size and fiber typing, amount of connective tissue, the presence of necrotic and regenerating fibers, inflammatory infiltrations, nuclear internalization, intracytoplasmic oxidative changes and neurogenic abnormalities. Based on these characteristics, the samples were classified into four different histologic patterns: (1) normal; (2) unspecific myopathic changes (mild oxidative defects, fiber size variation, mild increase in connective tissue); (3) myopathic with necrotic fibers (myopathic changes associated with necrosis as the main histological finding); and (4) dystrophic pattern (myopathic changes together with intense connective and fatty infiltration).
Muscle imaging analysis
Muscle magnetic resonance imaging (MRI) was performed in 18 patients, based on local protocols, although a common characteristic was acquisition of coronal and axial slices, 6‐mm‐thick, using a conventional T1‐weighted spin echo sequence. Whole‐body MRI study was available for 7 patients, and for the other 11 patients, the examination was limited to the lower limbs. The STIR sequence was available for 12 patients.
Two experienced radiologists (PVPH and LUA) and a neurologist (AMSS) evaluated all exams to reach a consensus. Muscles were systematically explored using stacks of axial and coronal slices encompassing the entire volume. Muscle fatty degeneration was assessed according to the distribution of abnormal muscle signal intensity on T1‐weighted sequences. The grade of involvement was ranked according to a modified 5‐point scale: 0, normal; 1, mild; 2, moderate (<50% of fatty replacement); 3, severe (>50% of fatty replacement); and 4, extreme or absent muscle (end‐stage involvement).15, 16
DNA analysis
DNA was extracted from peripheral blood and analyzed by a commercial targeted NGS panel that included the most common genes related to LGMD2 (CAPN3, DYSF, SGCA, SGCB, SGCD, SGCE, FKRP, ANO5, GAA). ANO5 sequences were searched for using the National Center for Biotechnology Information (NCBI) protein database, and variants were described based on the NM_213599.2 transcript. Sequence variations were compared to data available in the Human Gene Mutation Database (HGMD®) and ClinVar. Variants were classified according to 2015 American College of Medical Genetics and Genomics (ACMG) criteria.17 PolyPhen‐2, SIFT, CADD, M‐CAP, and Mutation Taster were used for in silico analysis.18, 19, 20, 21, 22 Phylogenetic conservation was estimated with Genomic Evolutionary Rate Profiling (GERP++), and allele frequencies were searched on the gnomAD and 1000 Genomes browsers.23, 24, 25
Statistical analysis
To evaluate the association of demographic and genetic characteristics with different phenotypes, patients were classified into two main groups (LGMD and non‐LGMD) and compared using the Student’s t‐test for quantitative variables and the chi‐squared test or the Fisher exact test for qualitative variables. Multivariate analysis was proceeded using the stepwise regression model with backward elimination. Correlation between MRI score and the duration of disease was analyzed by the Spearman’s rank correlation (ρ), and the Mann–Whitney U test was used to compare the MRI score between LGMD and non‐LGMD phenotypes. The software IBM SPSS Statistics V.1.0.0‐3239 (IBM, Armonk, New York, USA) was used, and two‐sided P < 0.05 was considered statistically significant.
Results
Thirty‐seven patients from 34 non‐related Brazilian families with recessive mutations of ANO5 were identified. The clinical findings of each patient are summarized in Table 1. Table 2 shows the most common clinical features and the results of NCS/EMG and muscle biopsy. The median of age at onset was 38 years, ranging from 4 to 67 years, and a male predominance was noticed (72.9%). Most patients (86.5%) were White, but there were one Brazilian African (#23) and four patients (#4, #30, #31 and #32) defined as mixed race (White and Brazilian African). The serum CK levels were elevated in all patients.
Table 1.
Clinical and genetic findings of the ANO5 patients
| Patients | Onset | CK (U/L) | Pattern of muscle involvement | Mutations | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| # | Age/sex | Age at onset | Initial symptoms | Atrophy | Physical examination summary | Functional status | Phenotype | Other features | Allele 1 | Allele 2 | |
| 1.1 | 18/M | 12 | Myalgia, exercise intolerance | 5665 | None | Normal | Ambulant | Pseudometabolic | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys | |
| 1.2 | 18/F | 18 | Asymptomatic hyperCKemia | 4008 | None | Normal | Ambulant | HyperCKemia | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys |
| 2 | 43/M | 37 | Mild lower limbs weakness | 4512 | None | Proximal and distal weakness (MRC 4) | Ambulant | LGMD | Dysphagia, calf hypertrophy | c.692G > T, p.Gly231Val | c.1359C > G, p.(Tyr453*) |
| 3 | 48/M | 42 | Mild lower limbs weakness | 3897 | None | Proximal lower limb weakness (MRC 4) | Ambulant | LGMD | Dysphagia | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys |
| 4 | 12/M | 11 | Asymptomatic hyperCKemia | 8928 | None | Normal | Ambulant | HyperCKemia | c.172C > T, p.Arg58Trp | c.172C > T, p.Arg58Trp | |
| 5 | 62/M | 54 | Mild lower limbs weakness | 7533 | Biceps and quadriceps (moderate) | Proximal and distal weakness (MRC 4) | Ambulant | LGMD | Asymmetry | c.692G > T, p.Gly231Val | c.1359C > G, p.(Tyr453*) |
| 6 | 40/M | 20 | Lower limbs weakness with myalgia | 5450 | Deltoids, biceps and quadriceps (moderate) | Neck (MRC 4), proximal (MRC 3) and distal (MRC 4) weakness | Assisted walking | LGMD | Asymmetry | c.692G > T, p.Gly231Val | c.692G > T, p.Gly231Val |
| 7 | 55/M | 35 | Exercise intolerance | 4806 | Biceps, triceps and quadriceps (severe) | Proximal and distal weakness (MRC 4) | Ambulant | LGMD | Asymmetry | c.1898 + 1G>A, p.(?) | c.2012A > G, p.Tyr671Cys |
| 8 | 48/M | 34 | Exercise intolerance | 4467 | Pectoralis (mild) | Proximal lower limb weakness (MRC 4) | Ambulant | LGMD | Dysphagia, calf hypertrophy | c.172C > T, p.Arg58Trp | c.172C > T, p.Arg58Trp |
| 9 | 68/F | 58 | Myalgia, exercise intolerance | 2051 | Gastrocnemius (mild) | Trunk (MRC 3) weakness | Ambulant | Pseudometabolic | Restrictive respiratory involvement (FVC 75%) | c.692G > T, p.Gly231Val | c.692G > T, p.Gly231Val |
| 10 | 66/M | 56 | Mild lower limb weakness | 882 | Quadriceps (severe) | Proximal lower limb weakness (MRC 4) | Ambulant | LGMD | c.368C > T, p.Ser123Leu | c.368C > T, p.Ser123Leu | |
| 11 | 66/M | 58 | Lower limb weakness with myalgia | 1802 | Biceps, triceps (moderate), quadriceps and gastrocnemius (severe) | Neck (MRC4) and proximal (MRC 3) weakness | Wheelchair bounded | LGMD | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys | |
| 12.1 | 52/M | 40 | Mild lower limb weakness | 10679 | None | Biceps and quadriceps weakness (MRC 4) | Ambulant | LGMD | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.2012A > G, p.Tyr671Cys |
| 12.2 | 50/F | 44 | Myalgia, exercise intolerance | 1138 | None | Normal | Ambulant | Pseudometabolic | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.2012A > G, p.Tyr671Cys |
| 13 | 66/F | 52 | Lower limb weakness with myalgia | 2179 | Quadriceps (severe) | Neck and proximal weakness (MRC 4) | Ambulant | LGMD | Asymmetry, calf hypertrophy | c.1359C > G, p.(Tyr453*) | c.689A > G, p.Asp230Gly |
| 14 | 63/M | 40 | Myalgia | 913 | None | Normal | Ambulant | Pseudometabolic | c.692G > T, p.Gly231Val | c.692G > T, p.Gly231Val | |
| 15 | 37/M | 28 | Progressive lower limbs weakness | 12640 | Quadriceps (mild) and gastrocnemius (severe) | Proximal (MRC 4) and distal weakness (MRC 3) | Ambulant | LGMD | Asymmetry, mild restrictive respiratory involvement (FVC 74%) | c.1210 C > T, p.(Arg404*) | c.1210 C > T, p.(Arg404*) |
| 16 | 53/F | 48 | Myalgia, exercise intolerance | 1850 | Gastrocnemius (severe) | Neck (MRC 3) and lower limb weakness (MRC 4) | Assisted walking | LGMD | Severe restrictive respiratory involvement (FVC 27%) with dyspnoea | c.1210 C > T, p.(Arg404*) | c.1210 C > T, p.(Arg404*) |
| 17 | 61/F | 49 | Myalgia, exercise intolerance | 7500 | Deltoids, biceps, gastrocnemius (moderate), quadriceps (severe) | Trunk and proximal weakness (MRC 4) | Ambulant | LGMD | Asymmetry | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys |
| 18 | 19/M | 14 | Lower limb weakness with myalgia | 2800 | Glutei (mild) | Proximal weakness (MRC 4) | Ambulant | LGMD | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.1295C > G, p.Ala432Gly |
| 19 | 45/M | 38 | Progressive lower limbs weakness | 8550 | Biceps and quadriceps (mild) | proximal weakness (MRC 4) | Ambulant | LGMD | c.191dupA, p.Asn64Lys | c.692G > T, p.Gly231Val | |
| 20 | 72/M | 67 | Progressive lower limbs weakness | 20000 | Deltoids, biceps, gastrocnemius and quadriceps (moderate) | Trunk (MRC 3), proximal and distal (MRC 4) weakness | Assisted walking | LGMD | Asymmetry, hypertrophic cardiomyopathy | c.191dupA, p.Asn64Lys | c.2498T > A, p.Met833Lys |
| 21 | 44/M | 37 | Lower limb weakness with myalgia | 1198 | Quadriceps and gastrocnemius (severe) | Trunk and proximal (MRC 4) weakness | Ambulant | LGMD | c.692G > T, p.Gly231Val | c.2498T > A, p.Met833Lys | |
| 22 | 54/F | 42 | Progressive proximal weakness | 505 | Biceps (mild) and quadriceps (moderate) | Proximal and distal weakness (MRC 4) | Ambulant | LGMD | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.2272C > T, p.Arg758Cys |
| 23 | 25/M | 25 | Rhabdomyolysis | 47562 | None | Normal | Ambulant | Pseudometabolic | Calf hypertrophy | c.191dupA, p.Asn64Lys | c.2272C > T, p.Arg758Cys |
| 24 | 40/M | 32 | Asymptomatic hyperCKemia | 12000 | None | Normal | Ambulant | HyperCKemia | c.191dupA, p.Asn64Lys | c.692G > T, p.Gly231Val | |
| 25 | 36/M | 28 | Lower limb weakness with myalgia | 950 | Deltoids, biceps, glutei and quadriceps (moderate) | Proximal weakness (MRC 4) | Ambulant | LGMD | Calf hypertrophy | 191dupA, p.Asn64Lys | c.2311_2312delCA, p.Gln771Alafs |
| 26 | 53/M | 50 | Rapid onset proximal weakness | 32105 | Deltoids, biceps, gastrocnemius (moderate), quadriceps (severe) | Trunk (MRC 3) and proximal (MRC 4) weakness | Assisted walking | LGMD | Asymmetry | 191dupA, p.Asn64Lys | c.2498T > A, p.Met833Lys |
| 27 | 27/F | 22 | Exercise intolerance | 2223 | Deltoids, biceps, quadriceps and gastrocnemius (mild) | Proximal weakness (MRC 4) | Assisted walking | LGMD | c.2272C > T, p.Arg758Cys | c.1295C > G, p.Ala432Gly | |
| 28 | 42/M | 40 | Lower limb weakness with myalgia | 3600 | Quadriceps and gastrocnemius (moderate) | Proximal and distal weakness (MRC 4) | Ambulant | LGMD | Asymmetry | 191dupA, p.Asn64Lys | c.2272C > T, p.Arg758Cys |
| 29.1 | 46/M | 39 | Lower limb weakness with myalgia | 20540 | Quadriceps (moderate) | Proximal lower limb weakness (MRC 4) | Ambulant | LGMD | Asymmetry | 191dupA, p.Asn64Lys | c.2498T > A, p.Met833Lys |
| 29.2 | 49/M | 30 | Myalgia and hyperCKemia | 3789 | Gastrocnemius (mild) | Normal | Ambulant | Pseudometabolic | 191dupA, p.Asn64Lys | c.2498T > A, p.Met833Lys | |
| 30 | 50/M | 40 | Progressive lower limbs weakness | 2200 | Quadriceps and gastrocnemius (moderate) | Quadriceps weakness (MRC 4) | Ambulant | LGMD | Asymmetry, mild restrictive respiratory involvement (FVC 75%), cardiomyopathy (FE 48%) | c.172C > T, p.Arg58Trp | c.172C > T, p.Arg58Trp |
| 31 | 70/F | 61 | Progressive lower limbs weakness | 2764 | None | Proximal lower limb weakness (MRC 4) | Ambulant | LGMD | Asymmetry | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys |
| 32 | 42/M | 26 | Distal lower limb weakness | 6182 | Quadriceps (moderate) and gastrocnemius (moderate) | Distal lower limb weakness (MRC 3) | Ambulant | MMD3 | Asymmetry | c.191dupA, p.Asn64Lys | c.191dupA, p.Asn64Lys |
| 33 | 43/M | 34 | Asymptomatic hyperCKemia | 3275 | None | Normal | Ambulant | Hyperckemia | c.191dupA, p.Asn64Lys | c.2190G > T, p.(Trp730Cys) | |
| 34 | 12/F | 4 | Myalgia, exercise intolerance | 3000 | None | Normal | Ambulant | Pseudometabolic | c.191dupA, p.Asn64Lys | c.1359C > G, p.(Tyr453*) | |
MRC, Medical Research Council; LGMD, limb‐girdle muscular dystrophy; MMD3, Miyoshi‐like distal myopathy. Novel variants are represented in bold.
Table 2.
Frequency of the most common clinical signs and symptoms and electrophysiological and histological findings in ANO5 patients
| N | % | |
|---|---|---|
| Clinical characteristics (N = 37) | ||
| Male | 27/37 | 72.9 |
| Proximal weakness | 22/37 | 59.4 |
| Myalgia | 21/37 | 56.7 |
| Quadriceps atrophy | 19/37 | 51.3 |
| Asymmetry of muscle weakness or atrophy | 14/37 | 37.8 |
| Calf atrophy | 13/37 | 35.1 |
| Calf hypertrophy | 10/37 | 27.0 |
| Axial weakness (neck or trunk weakness) | 9/37 | 24.3 |
| Distal weakness (associated or not with proximal weakness) | 9/37 | 24.3 |
| Respiratory involvement | 3/37 | 8.1 |
| Cardiomyopathy | 2/37 | 5.4 |
| EMG (N = 34) | ||
| Myopathic without PW/fib1 | 14/34 | 41.1 |
| Myopathic with PW/fib | 12/34 | 35.2 |
| Normal | 8/34 | 23.5 |
| Muscle biopsy (N = 28) | ||
| Myopathic without specific pathology | 11/28 | 39.2 |
| Dystrophic pattern | 7/28 | 25.0 |
| Myopathic with necrosis | 7/28 | 25.0 |
| Normal | 2/28 | 7.1 |
PW/Fib – positive waves and fibrillation.
Clinical characteristics and phenotypes
The most common phenotype was LGMD, which was observed in 25 (67.5%) patients. Pseudometabolic myopathy was present in seven (18.9%) patients, isolated asymptomatic hyperCKemia was present in four (10.8%) patients and MMD3 was present in a single patient (#32). Nine patients presented axial weakness, including one patient (#9) who presented isolated trunk weakness at examination. Calf involvement was common but variable: 13 patients presented calf atrophy, and another 10 had calf hypertrophy. Most patients (56.7%) reported myalgia at the initial presentation, and dysphagia was present in four patients (Tables 1 and 2).
Most of the patients had a mild disease: 31 patients were independently ambulant, 5 walked with assistance, and only 1 was wheelchair dependent at clinical evaluation. Cardiorespiratory manifestations were uncommon. In three patients without respiratory complaints, we detected mild restrictive pattern, and another patient had clinical manifestation of severe respiratory compromise (27% of predicted forced vital capacity), requiring nocturnal noninvasive ventilation. Two patients presented asymptomatic cardiomyopathy (Tables 1 and 2).
Electrophysiological and histologic findings
NCS/EMG study was performed in 34 patients, and a myogenic pattern was observed in 26 of them, of which 12 presented membrane instability (positive waves and fibrillations) (Table 2). Muscle biopsy was performed in 28 patients. Eleven samples showed unspecific myopathic findings; seven presented myopathic changes with a moderate‐to‐severe amount of necrotic fibers; seven samples presented a dystrophic pattern; and two samples were normal (Table 2, Fig. 1).
Figure 1.
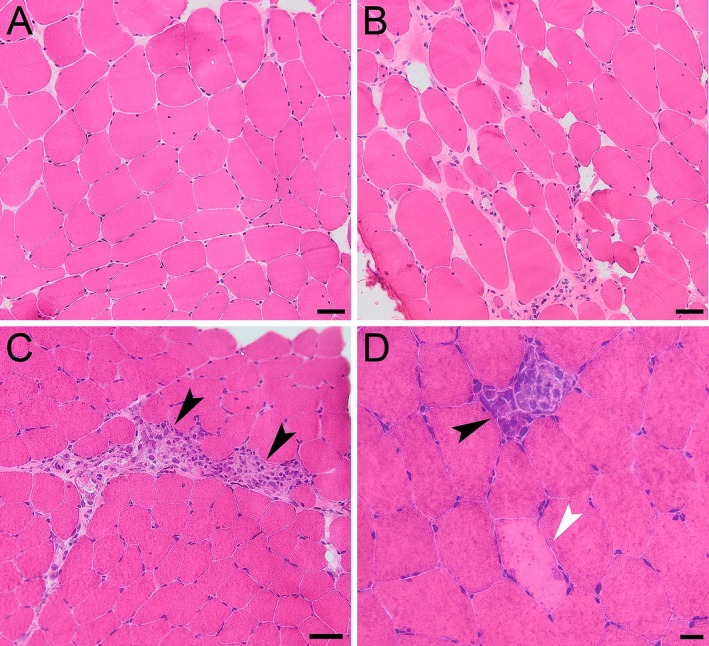
Muscle biopsy of patients with anoctaminopathy. Unspecific myopathic findings in patient #1.1 characterized by mild fiber size variability, increase in endomysial connective tissue, and increase in nuclei internalization (A) (H&E). More severe myopathic features (dystrophic aspect) in patient #2 (B) (H&E). Preserved muscle architecture with the presence of grouped necrotic fibers (C) (arrow heads) in patient #4; sparse necrotic fibers (D) in patient #26 (degeneration fiber with macrophagy indicated by the black arrowhead and without macrophagy indicated by the white arrowhead) (H&E). Bar = 50 µm (A, B and C); Bar = 20 µm (D).
Muscle MRI findings
In general, the most commonly affected muscles in the thighs were the semimembranosus (83%), biceps femoris long head (77%), and adductors (77%), whereas the vastus lateralis, medialis and intermedius, rectus femoris, biceps femoris short head, semitendinous, sartorius, and gracilis were variably less compromised (Figs. 2, and 3A and B). In the calves, the most affected muscles were the gastrocnemius (especially the medial head, in 81%) and the soleus (75%), whereas the tibialis and fibular muscles were usually spared (Figs. 2, and 3C and D). The iliopsoas muscles were spared in all patients (Fig. 2). In upper limbs imaging (available in seven patients), the most affected muscles were the biceps brachii, triceps, and deltoids (Fig. 2). Among 12 patients with available images, there was moderate to severe involvement of spinal muscles in 8 of them, and 7 patients had some involvement of abdominal musculature, ranging from mild to severe (Figs. 2 and 3E). Eight patients (#7, #9, #13, #15, #29.1, #29.2, #30, and #32) presented areas of STIR hyperintensity in the thigh and leg muscles (Fig. 3H).
Figure 2.
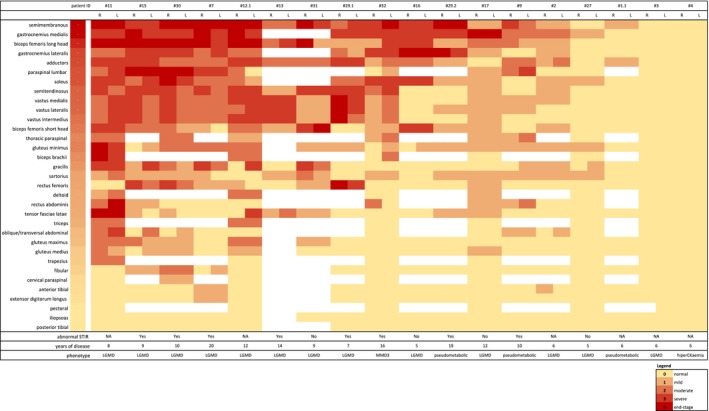
The heatmap for muscle involvement in MRI, using the 5‐point scale for each muscle (right and left) of each patient. Muscles and patients were ordered in grade of severity based on the mean score of the muscle and patient. The score of a muscle in a patient is indicated by the color of the square. White squares mean that data are not available. R, right; L, left; LGMD, limb‐girdle muscular dystrophy; MMD3, Miyoshi‐like distal myopathy; NA, not available
Figure 3.
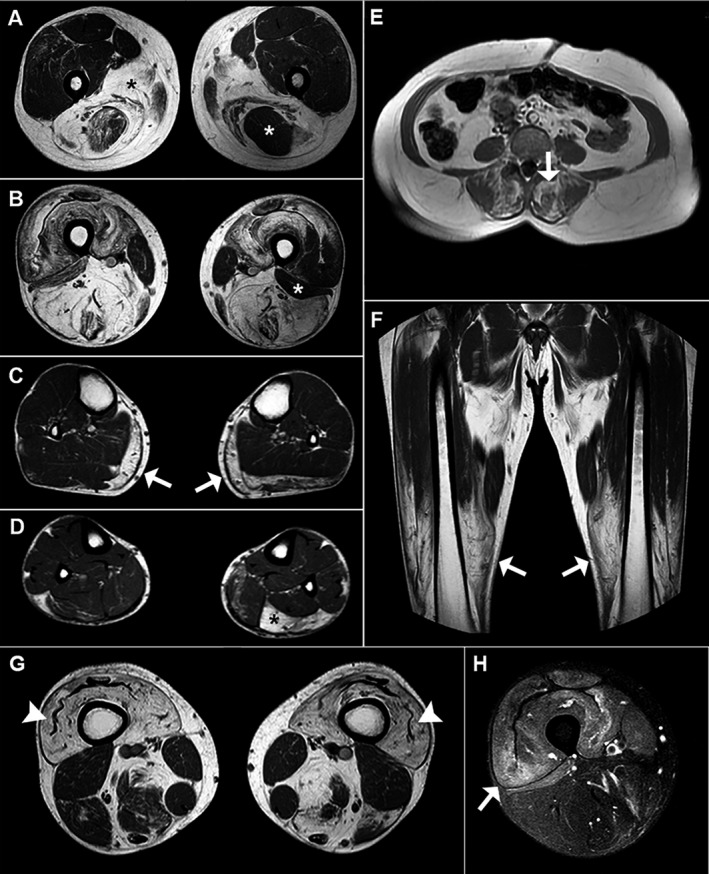
Typical MRI findings of ANO5‐related myopathy. (A and B) Axial T1‐weighted images of the thighs reveal striking involvement of the posterior compartment (mainly the semimembranosus and the adductors) in patient #7; black asterisks in A show bilateral fatty degeneration of the adductors, and the white asterisk in A shows asymmetrical involvement of the semitendinosus. White asterisks in B show asymmetrical sparing of the short head of the left biceps femoris. (C and D) Axial T1‐weighted images of the legs show bilateral fatty infiltration of the medial head of the gastrocnemius (white arrows in C) and asymmetrical involvement of the soleus (black asterisk in D) in patient #7. (E) Axial T1‐weighted image of the lumbar spine of patient #9 reveals severe fatty degeneration of the posterior paravertebral muscles (white arrow). (F) Coronal T1‐weighted image of the thighs in patient #15 shows a craniocaudal gradient of involvement (the distal portion of the quadriceps muscles is more affected than the proximal portion). (G and H) Axial T1‐weighted image of the thighs of patient #29.1 shows severe fatty infiltration of the quadriceps and the “undulating fascia” sign (white arrowhead) in G, and areas of hyperintensity indicate muscle edema (white head) in H (axial STIR‐weighted image)
Most patients had MRI evidence of definite asymmetric muscle involvement: 12 patients had at least two muscles with different stages of involvement, comparing the right side to the left side (Fig. 2). Furthermore, there were different levels of severity and patterns of muscle involvement, seen in MRI, even among siblings (patients #29.1 and #29.2). Disease duration and phenotype had some influence in these imaging differences: the 5‐point MRI score mean was slightly higher in the 13 LGMD patients than in the other 4 non‐LGMD patients (1.16 ± 0.67 vs. 0.29 ± 0.43, P‐value 0.05), and there was a moderate correlation between MRI score severity and disease duration (ρ 0.55, P‐value 0.027).
Additionally, a gradient of involvement in the thighs with a greater fat replacement in the distal portions of the quadriceps more than in the proximal ones was noticed in eight patients (#7, #11, #12.1, #15, #29.1, #29.2, #30, and #32), including in the patient with MMD3 phenotype, in addition to a sign named “undulating fascia” (Fig. 3F and G) defined by the presence of a wavy fascia between the severe atrophic and fat‐infiltrated vastus intermedius and vastus lateralis muscles.26
Genetic analysis
In the present cohort, 14 variants in ANO5 were detected (9 missense, 2 nonsense, 2 frameshift, and 1 splice site) (Table 1). The most common variants were c.191dupA, detected in 19 families (6 in homozygosis), and c.692G > T, identified in 8 families (3 in homozygosis) (Table 1). Five novel variants, one pathogenic nonsense variant, one likely pathogenic missense variant, and three missense variants currently classified as of unknown significance were found: c.1359C > G (p.Tyr453*) in four families (in compound heterozygosis); c.2012A > G (p.Tyr671Cys) in three families (in compound heterozygosis); c.689A > G (p.Asp230Gly) in one family (in compound heterozygosis); c.368C > T (p.Ser123Leu) in one family (in homozygosis); and c.2190G > T (p.Trp730Cys) in one family (in compound heterozygosis). All these variants have very low frequency in different databases and were damaging in prediction tools, but the missense c.689A > G and c.2190G > T mutations had divergent results according to prediction algorithms. The other missense variant (c.2012A > G) was found in three non‐related families and was defined as likely pathogenic (Table 3).
Table 3.
Novel variants in ANO5
| Nucleotide change | AA change | Mutation type | AF (gnomAD) | AF (1000 genomes) | SIFT | Poly Phen2 | Mutation taster | CADD | MCAP | GERP++ | N families | ClinVar | ACMG criteria 1 | Classification |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| c.1359C > G | p.Tyr4531 | nonsense | 0.000004 | 0 | NA | NA | 1 | 25.1 | NA | −3,91 | 3 | No | PVS1, PS4, PM2 | pathogenic |
| c.2012A > G | p.Tyr671Cys | missense | 0.000008 | 0 | 0.03 | 0.992 | 0.99 | 26.7 | 0.086 | 5.71 | 3 | Yes (1 VUS) | PS4, PM2, PP3 | likely pathogenic |
| c.689A > G | p.Asp230Gly | missense | 0.00011 | 0.0004 | 0.21 | 0.006 | 0.94 | 21.6 | 0.055 | 5.77 | 1 | Yes (2 VUS) | PM1, PM2 | VUS |
| c.368C > T | p.Ser123Leu | missense | 0.00002 | 0 | 0.03 | 0.997 | 0.81 | 29.2 | 0.060 | 5.94 | 1 | No | PM2, PP3 | VUS |
| c.2190G > T | p.Trp730Cys | missense | 0 | 0 | 0 | 0.980 | 0.99 | 34 | 0.272 | 5.57 | 1 | No | PM2,PP3 | VUS |
AA, amino acid; AF, allele frequency; NA not available; VUS, variant of unknown significance. For GERP++ data is shown as mean or raw value.
American College of Medical Genetics and Genomics criteria, Richards et al, 2015.
Comparison of LGMD and non‐LGMD patients
Patients were classified into two main groups (LGMD and non‐LGMD) and were compared regarding age, gender, disease duration, and the presence of loss‐of‐function (LoF) variants (frameshift, nonsense, and splice site) (Table 4). We found that sex, years of disease, and the presence of one or two LoF variants were not associated with a specific phenotype. However, the non‐LGMD patients were associated with an earlier mean age of disease onset than the LGMD patients (27.7 vs. 41.3, P 0.017) (Table 4).
Table 4.
Comparison of LGMD and non‐LGMD patients
| Characteristics | LGMD (N = 25) | Non‐LGMD (N = 12) | P‐value | |
|---|---|---|---|---|
| Univariate | Multivariate | |||
| Male, n (%) | 18 (72) | 9 (75) | 0.847 | 0.913 |
| Age at onset, mean (SD) | 41.3 (13.1) | 27.7 (14.9) | 0.008 | 0.017 |
| Disease duration, mean (SD) | 8.9 (4.6) | 8.8 (7.3) | 0.949 | 0.746 |
| Homozygous loss‐of‐function variants, n (%) | 7 (28) | 4 (33) | 1.0 | NA |
| Presence of at least 1 loss‐of‐function variant, n (%) | 19 (76) | 9 (75) | 1.0 | NA |
| Presence of homozygous c.191dupA, n (%) | 4 (16) | 3 (25) | 0.659 | NA |
NA, not applicable.
Discussion
We present herein a Brazilian cohort of ANO5 patients that is the largest detailed series of anoctaminopathy outside Europe. Although some phenotypic heterogeneity has occurred, similar characteristics were observed in most of the patients, such as onset after 30–40 years of age, male predominance, proximal weakness, calf involvement, myalgia, high‐serum CK levels, frequent necrotic fibers on muscle biopsy, and involvement of the posterior compartment muscles in lower limbs on MRI. Axial involvement, detected by physical examination and muscle MRI, was also frequent. Additionally, the common variants reported in Europe (c.191dupA, c.2272C > T and c.692G > T)9, 11 were frequent in our population, representing 91% of all variants in our cohort.
ANO5‐related myopathies have a wide clinical presentation. Two‐third of our patients had the LGMD phenotype, a third had pseudometabolic presentation or asymptomatic hyperCKemia and only one patient had MMD3 presentation. The pseudometabolic presentation and isolated hyperCKemia have been demonstrated in about half of patients in a French cohort.6 Another large multicentric European and North American cohort with 51 ANO5 patients found 25% of patients with hyperCKemia,11 which was similarly observed in our cohort. The MMD3 phenotype has been described in previous ANO5 cohorts, varying from 10% to 25%,6, 11 and we found less than expected MMD3 in our population, probably due to a selection bias since patients selected to perform the genetic panel were usually those with LGMD phenotype or with hyperCKemia. As observed in previous studies,27, 28 we noticed a third of patients with axial weakness in the physical examination and paravertebral involvement in the MRI study. Interestingly, one of our patients (#9) had an isolated axial weakness, with no clinical limb‐girdle involvement, although the lower limbs MRI pattern was typical for anoctaminopathy; this suggests ANO5 as a possible etiology in subjects with axial myopathies and that ANO5 must be included in the workup of these patients.
Most patients presented a mild disease when compared to other common LGMD type 2, such as calpainopathy, dysferlinopathy, and sarcoglycanopathy. Only one patient was wheelchair bound, and another five had assisted walking. Cardiopulmonary involvement was not common. No significant cardiac disease was observed in this cohort, and only one patient had significant pulmonary involvement with dyspnea and noninvasive ventilation need, suggesting that these are not major features of the condition. Other studies have shown a low incidence (less than 10%) of cardiac failure in ANO5 patients, although an increased risk of ventricular arrhythmia has been reported.29, 30
We found that patients with the LGMD phenotype were older than non‐LGMD patients and had a later mean age of onset compared with non‐LGMD patients, even with both groups having a similar disease duration. This suggests that the milder phenotypes are most commonly seen in younger individuals. A similar difference in the age of onset between these phenotypes was reported in a French cohort, in which non‐LGMD patients were almost 15 years younger than LGMD patients and remained oligosymptomatic over a follow‐up of 5 years, suggesting a different progression compared to the LGMD phenotype.6 However, the same study showed that around 30% of patients with LGMD reported exercise intolerance initially, comparable with our cohort, where 20% of LGMD patients presented exercise intolerance at onset (Table1). This difference in age at onset between LGMD and non‐LGMD phenotypes detected in our study may just reflect different time points in the natural history of the disease; therefore, prospective cohort studies are needed to better clarify whether patients with asymptomatic hyperCKemia and the pseudometabolic phenotype will develop LGMD.
Male predominance, a well‐recognized feature of ANO5 myopathy, was also noticed in our population.4 No correlation between sex and severity was observed in our cohort because males and females showed similar proportions of LGMD presentation. Some authors have claimed that females usually have a milder presentation, and a selection bias could explain their low frequency in cohorts 6, 9, 11 (i.e., women may be less likely to be tested for CK or taken seriously for symptoms of myalgia/exercise tolerance without weakness and therefore are under‐represented in the non‐LGMD group).
Most patients of our cohort carried the common mutation c.191dupA – 6 families (18%) presenting with homozygosity and 13 families (38%) presenting with compound heterozygosity. This variant has been previously suggested as a founder mutation from Northern Europe, based on high frequency and polymorphism studies in this population, and was suggested as being spread worldwide.4 Additionally, another frequent variant reported in a large European cohort (c.692G > T)11 was found in eight (24%) families. The common Finish mutation (c.2272C > T) was detected in four (12%) families in our cohort and has been demonstrated to be from central European origin.9 The Brazilian population has a high genetic admixture, and previous studies have shown that European background represents more than 60% of the ancestry proportion, mostly from Portuguese and Spanish origins (due to colonization during the 16th and 17th centuries), followed by Africans (21%) and Amerindians (17%).31, 32 Such genetic background may explain that 31 of 34 (91%) of our families had at least one of the three common variants described in Europe. Interestingly, the incidence of ANO5 myopathy seems to be very low in non‐European origin populations, and the few cases reported in Asian patients did not include these frequent variants.12, 13, 14, 33, 34 These findings may corroborate the founder effect of the c.191dupA and c.2272C > T from the European population. Additionally, we investigated the impact of truncating variants (mainly represented by the c.191dupA) in determining phenotype, but no genotype‐phenotype correlation was detected in our cohort, in consonance with previous studies.6, 9, 28, 35
Muscle imaging studies confirmed a rather homogeneous pattern of muscle involvement on MRI, regardless of the clinical phenotype, with a predominant involvement of muscles from the posterior compartment of the thighs and calves, asymmetric involvement and an intrafamilial severity variability, as previously reported.36 STIR abnormalities were relatively frequent in our cohort, occurring in eight patients. Additionally, an interesting finding observed in some of our patients was the presence of a gradient of involvement in the thighs, characterized by a greater involvement in the more distal portions of the thighs, regardless the phenotype. Such a finding, associated with the presence of the “undulating fascia” signal (Fig. 3G), a distal quadriceps atrophy consequence, may cause diagnostic confusion with sporadic inclusion body myositis (sIBM),26 especially because sIBM also occurs in individuals over 40 years of age and has a male predominance.37 However, sIBM presents a distal involvement in upper limbs, while in our patients, there was a predominant involvement of proximal muscles of the upper and lower limbs, and the typical degenerative histological changes observed in sIBM muscles are not described in patients with ANO5 mutations.
Regarding the only patient presenting the MMD3 phenotype, the MRI showed a predominant and asymmetrical leg involvement along with moderate fatty infiltration in the distal portion of the thighs. In this case, other muscle groups were also mildly affected, such as thoracic paraspinal and left biceps brachii. These findings show that even in individuals with predominant distal phenotype, the involvement depicted by MRI is widespread, with a similar muscle involvement distribution, STIR abnormalities and the distal‐to‐proximal gradient shown in LGMD patients.
Most of our patients presented a myopathic pattern on muscle biopsy, ranging from normal to clearly dystrophic. A finding noticed in several cases was the presence of necrotic fibers, without inflammatory infiltrations. Although muscle biopsy did not show evident pathology of inflammation, in four patients (#2, #3, #13, and #26), the initial diagnosis was inflammatory/necrotizing myopathy based on the finding of proximal weakness, increased CK serum levels and the predominant presence of necrotic fibers on biopsy (data not shown). However, no satisfactory response was obtained with immunosuppressive therapy in these cases. The presence of STIR changes and instability signs on EMG are other data that can cause misdiagnosis in the direction of inflammatory diseases.
This study was conducted based on a national collaboration from different neuromuscular centers in Brazil, and a wide range of clinical and molecular characteristics was evaluated in detail, which shows the feasibility and importance of collaborative studies in a developing country. New contributions evolved in the knowledge of ANO5‐related myopathy from a non‐European population. However, this study has some limitations. First, our study was observational and not prospective, so data about the disease progression and its clinical predictors were not conclusive. Second, patients were selected based on a genetic panel usually used for those with LGMD phenotype or with hyperCKemia, which might explain the low frequency of MMD3 phenotype. Third, we included three patients with novel variants classified as VUS, and we did not have segregation analysis available for them, although they had typical ANO5 phenotype and carried a pathogenic mutation in the other allele, except one (#10) in whom variant was in homozygosity.
In conclusion, we found that ANO5‐related myopathy is present in the Brazilian population with similar characteristics to previous reported cohorts, including the very well‐described phenotypes, the homogeneous MRI pattern of muscle involvement, the high frequency of the European founder mutation c.191dupA, and the low incidence of cardiopulmonary complications. Some patients may be misdiagnosed with inflammatory myopathies due to EMG signs of membrane instability, STIR abnormalities and necrotic fibers on muscle biopsy. Furthermore, the description of predominant axial weakness expands the ANO5 phenotype.
Author Contributions
AMSS, PBW, JAMS, MCFJ, and EZ conceived and designed the study. All authors contributed directly to the acquisition of data. AMSS, PBW, JAMS, and EZ organized and interpreted the data. AMSS, ARCN, PVSS, PBW, JAMS, MCFJ, and EZ drafted the manuscript. All authors contributed to the critical revision of the manuscript for important scientific or intellectual content. AMSS and EZ had full access to all the data in the study and had responsibility for the integrity and accuracy of the data and performed statistical analysis. All authors read and approved the final manuscript before submission. The corresponding author EZ had final responsibility for the decision to submit for publication.
Conflict of Interest
Nothing to declare.
Acknowledgments
The authors are grateful to patients and their families for participation in this study and professionals who also attended these individuals, but were not directly involved in this research project.
Funding information
None.
References
- 1. Hartzell HC, Yu K, Xiao Q, et al. Anoctamin/TMEM16 family members are Ca2+‐activated Cl– channels. J Physiol 2009;587(Pt 10):2127–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Griffin DA, Johnson RW, Whitlock JM, et al. Defective membrane fusion and repair in anoctamin5‐deficient muscular dystrophy. Hum Mol Genet 2016;25:1900–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bolduc V, Marlow G, Boycott KM, et al. Recessive mutations in the putative calcium‐activated chloride channel Anoctamin 5 cause proximal LGMD2L and distal MMD3 muscular dystrophies. Am J Hum Genet 2010;86:213–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hicks D, Sarkozy A, Muelas N, et al. A founder mutation in Anoctamin 5 is a major cause of limb‐girdle muscular dystrophy. Brain 2011;134:171–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Schessl J, Kress W, Schoser B. Novel ANO5 mutations causing hyper‐CK‐emia, limb girdle muscular dystrophy and miyoshi type muscular dystrophy. Muscle Nerve 2012;45:740–2. [DOI] [PubMed] [Google Scholar]
- 6. Papadopoulos C, Laforêt P, Nectoux J, et al. Hyperckemia and myalgia are common presentations of anoctamin‐5‐related myopathy in French patients. Muscle Nerve 2017;56:1096–100. [DOI] [PubMed] [Google Scholar]
- 7. Tsutsumi S, Kamata N, Vokes TJ, et al. The novel gene encoding a putative transmembrane protein is mutated in gnathodiaphyseal dysplasia (GDD). Am J Hum Genet 2004;74:1255–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Magri F, Del Bo R, D'Angelo MG, et al. Frequency and characterisation of anoctamin 5 mutations in a cohort of Italian limb‐girdle muscular dystrophy patients. Neuromuscul Disord 2012;22:934–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Penttilä S, Palmio J, et al. E Eight new mutations and the expanding phenotype variability in muscular dystrophy caused by ANO5. Neurology 2012;78:897–903. [DOI] [PubMed] [Google Scholar]
- 10. Nallamilli BRR, Chakravorty S, Kesari A, et al. Genetic landscape and novel disease mechanisms from a large LGMD cohort of 4656 patients. Ann Clin Transl Neurol 2018;5:1574–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sarkozy A, Hicks D, Hudson J, et al. ANO5 gene analysis in a large cohort of patients with anoctaminopathy: confirmation of male prevalence and high occurrence of the common exon 5 gene mutation. Hum Mutat 2013;34:1111–8. [DOI] [PubMed] [Google Scholar]
- 12. Kadoya M, Ogata K, Suzuki M, et al. A Japanese male with a novel ANO5 mutation with minimal muscle weakness and muscle pain till his late fifties. Neuromuscul Disord 2017;27:477–80. [DOI] [PubMed] [Google Scholar]
- 13. Hu B, Xiong L, Zhou Y, et al. First familial limb‐girdle muscular dystrophy 2L in China: Clinical, imaging, pathological, and genetic features. Medicine (Baltimore). 2018;97:e12506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bohlega S, Monies DM, Abulaban AA, et al. Clinical and genetic features of anoctaminopathy in Saudi Arabia. Neurosciences (Riyadh). 2015;20:173–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fischer D, Kley RA, Strach K, et al. Distinct muscle imaging patterns in myofibrillar myopathies. Neurology 2008;71:758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Mercuri E, Cini C, Counsell S, et al. Muscle MRI findings in a three‐generation family affected by Bethlem myopathy. Eur J Paediatr Neurol 2002;6:309–14. [DOI] [PubMed] [Google Scholar]
- 17. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Adzhubei IA, Schmidt S, Peshkin L, et al. A method and server for predicting damaging missense mutations. Nat Methods 2010;7:248–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 2001;11:863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Rentzsch P, Witten D, Cooper GM, et al. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res 2019;47(D1):D886–D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Jagadeesh KA, Wenger AM, Berger MJ, et al. M‐CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat Genet 2016;48:1581–6. [DOI] [PubMed] [Google Scholar]
- 22. Schwarz JM, Cooper DN, Schuelke M, et al. MutationTaster2: mutation prediction for the deep‐sequencing age. Nat Methods 2014;11:361–2. [DOI] [PubMed] [Google Scholar]
- 23. Cooper GM, Stone EA, Asimenos G, et al. Distribution and intensity of constraint in mammalian genomic sequence. Genome Res 2005;15:901–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. gnomAD, Genome Aggregation Database, http://gnomad.broadinstitute.org. 2018. (Accessed December 2018).
- 25. Browser Genomes, https://www.ncbi.nlm.nih.gov/variation/tools/1000genomes. 2018. (Accessed December 2018).
- 26. Guimaraes JB, Zanoteli E, Link TM, et al. Sporadic inclusion body myositis: MRI findings and correlation with clinical and functional parameters. Am J Roentgenol 2017;209:1340–1347. [DOI] [PubMed] [Google Scholar]
- 27. van der Kooi AJ, Ten Dam L, Frankhuizen WS, et al. ANO5 mutations in the Dutch limb girdle muscular dystrophy population. Neuromuscul Disord 2013;23:456–60. [DOI] [PubMed] [Google Scholar]
- 28. Liewluck T, Winder TL, Dimberg EL, et al. ANO5‐muscular dystrophy: clinical, pathological and molecular findings. Eur J Neurol 2013;20:1383–9. [DOI] [PubMed] [Google Scholar]
- 29. Wahbi K, Béhin A, Bécane HM, et al. Dilated cardiomyopathy in patients with mutations in anoctamin 5. Int J Cardiol 2013;168:76–9. [DOI] [PubMed] [Google Scholar]
- 30. Witting N, Duno M, Petri H, et al. Anoctamin 5 muscular dystrophy in Denmark: prevalence, genotypes, phenotypes, cardiac findings, and muscle protein expression. J Neurol 2013;260:2084–93. [DOI] [PubMed] [Google Scholar]
- 31. Guerreiro‐Junior G, Bisso‐Machado R, Marrero A, et al. Genetic signatures of parental contribution in black and white populations in Brazil. Genet Mol Biol 2009;32:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moura RR, Coelho, et al. Meta‐analysis of Brazilian genetic admixture and comparison with other Latin America Countries. Am J Hum Biol 27:674–80. [DOI] [PubMed] [Google Scholar]
- 33. Lahoria R, Winder TL, Lui J, et al. Novel ANO5 homozygous microdeletion causing myalgia and unprovoked rhabdomyolysis in an Arabic man. Muscle Nerve 2014;50:610–3. [DOI] [PubMed] [Google Scholar]
- 34. Little AA, McKeever PE, Gruis KL. Novel mutations in the Anoctamin 5 gene (ANO5) associated with limb‐girdle muscular dystrophy 2L. Muscle Nerve 2013;47:287–91. [DOI] [PubMed] [Google Scholar]
- 35. Savarese M, Di Fruscio G, Tasca G, et al. Next generation sequencing on patients with LGMD and nonspecific myopathies: findings associated with ANO5 mutations. Neuromuscul Disord 2015;25:533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sarkozy A, Deschauer M, Carlier RY, et al. Muscle MRI findings in limb girdle muscular dystrophy type 2L. Neuromuscul Disord 2012;22(Suppl 2):S122–9. [DOI] [PubMed] [Google Scholar]
- 37. de Camargo LV, de Carvalho MS, Shinjo SK, et al. Clinical, histological, and immunohistochemical findings in inclusion body myositis. Biomed Res Int 2018;2018:5069042. [DOI] [PMC free article] [PubMed] [Google Scholar]