

Abstract
Background:
The potential of iron to generate reactive oxygen species has motivated a long‐standing interest in whether excess iron is causally linked to atherosclerotic heart disease. Circulating catalytic iron (“free” iron) is that which is not bound to transferrin or ferritin and is available to generate reactive oxygen species that may have deleterious vascular effects.
Hypothesis:
We hypothesized that increased levels of catalytic iron would be associated with increased cardiovascular events.
Methods:
We investigated the association of catalytic iron with clinical outcomes in 1701 patients with unstable angina, non–ST‐segment elevation myocardial infarction (MI), or ST‐segment elevation MI who were followed for a median of 10 months. All endpoints were adjudicated by a blinded Clinical End Points Committee.
Results:
The median catalytic iron level was significantly higher in those who died, 0.45 µmol/L (0.37, 0.57), compared with survivors, 0.37µmol/L (0.31, 0.46; P = 0.016). Catalytic iron was associated with a stepwise increased risk of death, with the highest quartile at an almost 4‐fold risk compared with baseline (hazard ratio: 3.94, P = 0.035), which persisted after adjustment for age, diabetes, prior MI, prior congestive heart failure, ST‐segment deviation, creatinine clearance, B‐type natriuretic peptide, smoking, and Killip class (adjusted hazard ratio: 3.97, P = 0.036). There was no association between catalytic iron and risk of MI, recurrent ischemia, heart failure, or bleeding.
Conclusions:
Increasing catalytic iron levels were associated with increased all‐cause mortality. Although our findings suggest that catalytic iron is not likely to add to available tools as a routine biomarker for risk stratification of recurrent ischemic events, its association with mortality is intriguing and leaves open the question of whether cardiovascular therapeutics aimed at catalytic iron may be useful.
The TIMI Study Group has received research grant support from the Muljibhai Patel Society for Research in Nephro‐Urology. There are no other financial relationships, or conflicts of interest relevant to this manuscript to disclose.
Introduction
The potential of iron to generate reactive oxygen species (ROS) has motivated a long‐standing interest in whether excess iron is causally linked to atherosclerotic heart disease. Ferritin, total iron‐binding capacity, transferrin saturation, serum iron, and dietary intake are established indirect measures of total body iron reserves but may not reliably assess its deleterious potential.1 Circulating catalytic iron (“free” iron) is that which is not bound to transferrin or ferritin and is available to generate ROS that may have deleterious vascular effects.
Results of small studies have demonstrated higher catalytic iron levels in patients with stable coronary artery disease2 and suspected acute coronary syndrome (ACS) compared with healthy individuals.3 Moreover, in the latter study of 127 patients with acute myocardial infarction (MI) and 51 patients with suspected ACS without MI, catalytic iron concentration was associated with the risk of recurrent coronary ischemic events and all‐cause mortality at 30 days. These studies were limited by small size and a constrained ability to assess confounding clinical characteristics. Therefore, we investigated the association of catalytic iron with clinical outcomes in a large, well‐characterized population across the spectrum of ACS.
Methods
Study Population
The design and results of the Oral Glycoprotein IIb/IIIa Inhibition with Orbofiban in Patients with Unstable Coronary Syndromes (OPUS)‐Thrombolysis in Myocardial Infarction (TIMI) 16 trial have been reported.4 Briefly, OPUS‐TIMI 16 was a randomized, multicenter trial comparing an oral platelet glycoprotein IIb/IIIa receptor inhibitor, orbofiban, with placebo in 10288 patients with ACS. Patients with acute MI or high‐ risk unstable angina (UA) were included if they could be enrolled within 72 hours from the onset of symptoms. Eligible patients received aspirin and were randomized 1:1:1 to receive 50 mg of orbofiban twice daily; 50 mg of orbofiban twice daily for 1 month, followed by 30 mg of orbofiban twice daily; or placebo. The median duration of follow‐up was 10 months. A biomarker substudy was conducted within the arm assigned to 50 mg twice daily.5 In the present study, catalytic iron was measured in 1701 patients within the biomarker cohort who had a baseline plasma specimen available for analysis.
Blood Sampling and Biochemical Analyses
At the time of enrollment, blood specimens were collected in citrate‐treated tubes and centrifuged for ≥12 minutes to isolate plasma. The plasma component was frozen within 60 minutes and shipped on dry ice to the TIMI Biomarker Core Laboratory (Boston, MA), where samples were stored at −70°C or colder.
Catalytic Iron:
The bleomycin‐detectable iron (BDI) assay is based on the fact that bleomycin, in the presence of ferric iron and a suitable reducing agent, binds to and degrades DNA, with the formation of a product that reacts with thiobarbituric acid to form a chromogen.6 The quantity of chromogen formed (ie, the intensity of the color) is measured in a spectrophotometer (DU 800; Beckman Coulter, Brea, CA) at 532 nm against a blank sample. The assay measures the ferric iron complexes that can catalyze free‐radical reactions in biological systems. Catalytic iron is reported in µmol/L.
Samples were sent frozen for blinded analysis by the BDI assay to the Muljibhai Patel Society for Research in Nephro‐Urology, Nadiad, India. This study employed a modification of the original assay,7 one that is currently used for renal and cardiovascular‐disease research. All reactions were carried out in polypropylene tubes to avoid iron contamination. All soluble reagents except bleomycin were treated with chelex (Bio‐Rad Laboratories, Inc., Hercules, CA) at 300 mg for 10 mL solution, to remove any iron contamination. At a serum BDI concentration of 0.15–1.0 µmol/L, the intra‐assay coefficient of variation (CV) was 1.22% and the interassay CV was 3.42%.
Other Biomarkers:
Plasma specimens were previously thawed and analyzed for B‐type natriuretic peptide (BNP) and troponin I (TnI). The cutpoints for BNP and TnI were 80 pg/mL5 and 1.5 ng/mL8, respectively, based on our prior work. Creatine kinase‐MB (CK‐MB) testing was performed locally at each participating hospital and recorded in the case‐record form. Estimated creatinine clearance (CrCl) was calculated using the Cockcroft‐Gault formula.
Endpoints
Clinical endpoints for this analysis included all‐cause mortality, nonfatal MI, and a composite of major cardiovascular events (all‐cause mortality, nonfatal MI, ischemia leading to urgent revascularization, recurrent ischemia requiring rehospitalization, and stroke) as defined for the OPUS‐TIMI 16 trial.4 All components of the composite were adjudicated by the study Clinical Events Committee blinded to study treatment and investigational biomarkers. The endpoint of new or worsened heart failure or cardiogenic shock was collected as reported by the local investigator and recorded on the case‐record form. Severe bleeding was defined as an intracranial hemorrhage or bleeding associated with severe hemodynamic compromise; major bleeding was that associated with >15% absolute reduction in hematocrit or requiring a blood transfusion.
Statistical Analysis
The distribution of catalytic iron was summarized using the median (interquartile range [IQR]). Patients were divided into quartiles on the basis of their catalytic iron concentration at the time of enrollment. Baseline characteristics were compared among quartiles with the use of linear regression for continuous variables and log‐linear analysis for categorical variables. The correlation between catalytic iron levels and other continuous baseline variables was assessed with the Spearman correlation coefficient. Catalytic iron concentrations were compared between those with and without a clinical endpoint using the Wilcoxon rank sum test.
Cox regression analysis was used to evaluate the association between the quartile of catalytic iron and clinical outcomes at 10 months. In multivariable analyses, we adjusted for the effects of age, diabetes mellitus (DM), prior MI, prior congestive heart failure (CHF), ST‐segment deviation >1 mm, estimated CrCl, BNP, current smoking, and Killip class.
Results
Catalytic iron was assessed in 1701 patients with either an ST‐segment elevation MI (STEMI; 566, 33%), non–ST‐segment elevation MI (NSTEMI; 407, 24%), or UA (728, 43%). The mean time from the onset of ischemic symptoms to enrollment was 41 ± 20 hours. The catalytic iron level ranged from 0.14 to 84.5 µmol/L, with a median of 0.38 µmol/L and 25th‐percentile and 75th‐percentile values of 0.17 and 12.7 µmol/L, respectively. Catalytic iron levels varied slightly by index event, with median (25th, 75th percentile values) of 0.39 (0.20, 2.02), 0.38 (0.18, 2.90), and 0.37 (0.18, 12.3) for STEMI, NSTEMI, and UA, respectively (P = 0.036).
Association With Baseline Clinical Variables
Higher levels of catalytic iron were associated with current smoking, higher heart rate, and higher BNP (P < 0.05) and were inversely associated with systolic blood pressure (P < 0.05; table 1). Catalytic iron levels correlated only very weakly with clinical characteristics of the presentation, including time from onset of chest pain in the total cohort (ρ = 0.08, P < 0.01) and in the STEMI subgroup (ρ = 0.13, P < 0.01). Catalytic iron levels did not vary significantly based on the time interval after percutaneous coronary intervention in the subset of patients who underwent revascularization prior to randomization. Catalytic iron was not significantly correlated with sex, history of DM, duration of ischemic discomfort at rest, age, or body mass index (age and body mass index measured as continuous variables; P = not significant).
Table 1.
Baseline Characteristics by Catalytic Iron Quartile
| Characteristic | Total | Biomarker Quartile | P Value for Trend Across Quartiles | |||
|---|---|---|---|---|---|---|
| 1 (0.14–0.31) | 2 (0.32–0.38) | 3 (0.39–0.47) | 4 (0.48–84.5) | |||
| Demographics | ||||||
| Age, y | 1696 | 60.0 (52.0, 68.0) | 61.0 (53.0, 69.0) | 61.0 (53.0, 71.0) | 58.0 (51.0, 68.0) | 0.37 |
| M, % | 1245 | 344 (75.4) | 302 (68.8) | 286 (73.5) | 313 (75.1) | 0.75 |
| Race, % white | 1597 | 434 (95.2) | 406 (92.5) | 364 (93.6) | 393 (94.2) | 0.71 |
| BMI, kg/m2 | 1646 | 27.4 (24.8, 30.4) | 27.6 (24.9, 30.3) | 27.3 (24.7, 30.1) | 27.3 (25.0, 30.3) | 0.93 |
| Past medical history, n (%) | ||||||
| Hypertension | 704 | 197 (43.4) | 186 (42.4) | 150 (38.6) | 171 (41.1) | 0.32 |
| Current smoker | 622 | 147 (32.3) | 159 (36.4) | 155 (39.9) | 161 (38.7) | 0.027 |
| DM | 349 | 94 (20.7) | 76 (17.3) | 79 (20.3) | 100 (24.0) | 0.15 |
| Hypercholesterolemia | 478 | 138 (30.4) | 118 (26.9) | 102 (26.3) | 120 (28.8) | 0.57 |
| Prior MI | 470 | 136 (29.9) | 123 (28.0) | 103 (26.5) | 108 (25.9) | 0.16 |
| Prior PCI | 182 | 59 (12.9) | 43 (9.8) | 34 (8.7) | 46 (11.0) | 0.30 |
| Prior CABG | 184 | 52 (11.4) | 53 (12.1) | 32 (8.2) | 47 (11.3) | 0.54 |
| PAD | 113 | 29 (6.4) | 33 (7.5) | 27 (7.0) | 24 (5.8) | 0.67 |
| CHF | 79 | 19 (4.2) | 23 (5.2) | 17 (4.4) | 20 (4.8) | 0.81 |
| Index hospitalization | ||||||
| ACS‐type STEMI | 566 | 130 (28.5) | 153 (34.9) | 128 (32.9) | 155 (37.2) | 0.12 |
| NSTEMI | 407 | 112 (24.6) | 99 (22.6) | 103 (26.5) | 93 (22.3) | |
| UA | 728 | 214 (46.9) | 187 (42.6) | 158 (40.6) | 169 (40.5) | |
| Onset of chest pain to randomization (h) | 1681 | 36.9 (23.3, 52.6) | 41.1 (25.3, 57.1) | 41.7 (25.5, 56.7) | 43.1 (25.6, 58.7) | 0.002 |
| SBP, mm Hg | 1699 | 130 (115, 150) | 128 (113, 140) | 125 (114, 140) | 126 (110, 140) | 0.023 |
| HR, bpm | 1697 | 70 (62, 80) | 72 (62, 80) | 72 (63, 80) | 72 (62, 83) | 0.027 |
| ST‐segment deviation >1 mm | 796 | 203 (44.5) | 210 (47.8) | 196 (50.4) | 187 (44.8) | 0.73 |
| Killip class, II–V | 1676 | 35 (7.8) | 32 (7.4) | 37 (9.6) | 30 (7.3) | 0.90 |
| CrCl, mL/min | 1429 | 101.0 (79.9, 125.7) | 100.8 (76.7, 127.8) | 95.7 (78.2, 126.3) | 103.8 (79.3, 134.5) | 0.55 |
| Other biomarkers | ||||||
| BNP, pg/mL | 1701 | 74.8 (42.5, 132.2) | 78.7 (43.5, 138.8) | 90.3 (50.6, 153.6) | 86.6 (45.5, 139.3) | 0.099 |
| BNP ≥80 pg/mL | 867 | 216 (47.4) | 213 (48.5) | 220 (56.6) | 218 (52.3) | 0.035 |
| TnI ≥1.5 ng/mL | 1692 | 123 (27.1) | 120 (27.5) | 115 (29.8) | 124 (29.8) | 0.29 |
| Peak CK‐MB, max/ULN | 1163 | 3.40 (1.05, 9.50) | 3.46 (1.16, 10.80) | 3.70 (1.00, 11.63) | 3.06 (1.00, 12.02) | 0.84 |
| Hemoglobin, g/dL | 1583 | 13.9 (13.1, 14.9) | 13.8 (12.8, 14.6) | 13.6 (12.6, 14.5) | 13.7 (12.7, 14.7) | 0.004 |
Abbreviations: ACS, acute coronary syndrome; BMI, body mass index; BNP, B‐type natriuretic peptide; CABG, coronary artery bypass grafting; CHF, congestive heart failure; CK‐MB, creatine kinase MB; CrCl, creatinine clearance; DM, diabetes mellitus; HR, heart rate; IQR, interquartile range; M, male; max, maximum; MI, myocardial infarction; NSTEMI, non–ST‐segment elevation myocardial infarction; PAD, peripheral arterial disease; PCI, percutaneous coronary intervention; SBP, systolic blood pressure; STEMI, ST‐segment elevation myocardial infarction; TnI, troponin I; UA, unstable angina; ULN, upper limit of normal.
Continuous variables are represented by median (IQR). Categorical variables are represented by n (column %).
Catalytic iron levels were only weakly correlated with hemoglobin (ρ = −0.07, P < 0.01) and white blood cell count (ρ = 0.06, P = 0.02). There were no significant associations between catalytic iron with other biomarkers, including BNP (ρ = 0.04, P = 0.10), TnI (ρ = 0.05, P = 0.05), CrCl (ρ = 0.01, P = 0.60), or the ratio of peak CK‐MB/upper limit of normal (ρ = −0.01, P = 0.84).
Clinical Outcomes
The median baseline concentration of catalytic iron was significantly higher in those who died compared with survivors: 0.45 vs 0.37 µmol/L, respectively (P = 0.016). All‐cause mortality increased in a significant graded manner across quartiles of rising catalytic iron with 10‐month rates of 0.7%–3.2% (P = 0.027; Table 2). After adjusting for age, history of DM, history of prior MI, history of CHF, ST‐segment deviation, CrCl, BNP, current smoking, and Killip class, a stepwise increase in risk of death persisted with increasing quartiles of catalytic iron (Table 3). The highest quartile of catalytic iron demonstrated a 3.97‐fold increase in risk compared with the lowest quartile (P = 0.036; Figure 1).
Table 2.
Kaplan‐Meier Event Rates at 10 Months of Follow‐Up
| Quartile 1 (0.14–0.31) N = 456 | Quartile 2 (0.32–0.38) N = 439 | Quartile 3 (0.39–0.47) N = 389 | Quartile 4 (0.48–84.5) N = 417 | P Value | |
|---|---|---|---|---|---|
| All‐cause mortality | 0.7% | 1.9% | 2.5% | 3.2% | 0.027 |
| MI | 2.3% | 1.9% | 3.2% | 1.9% | 0.63 |
| Ischemia leading to urgent revascularization | 2.6% | 3.2% | 2.0% | 2.2% | 0.56 |
| Ischemia requiring rehospitalization | 2.3% | 2.7% | 3.8% | 3.0% | 0.75 |
| Stroke | 0.0% | 0.7% | 0.3% | 0.6% | 0.41 |
| Composite | 6.9% | 8.8% | 10.1% | 8.5% | 0.77 |
| CHF | 1.3% | 2.3% | 3.8% | 2.7% | 0.28 |
Abbreviations: CHF, congestive heart failure; MI, myocardial infarction.
Kaplan‐Meier event rates include only the endpoints for which time/date data were available.
Table 3.
Unadjusted and Multivariate Adjusted HRs for 10‐Month Clinical Endpoints
| HR (95% CI) | ||
|---|---|---|
| Unadjusted | Multivariate Adjusted | |
| All‐cause death (n = 25) | ||
| Quartiles | ||
| 1 | 1.00 | 1.00 |
| 2 | 2.22 (0.55–8.86) | 1.51 (0.33–6.83) |
| 3 | 2.16 (0.52–9.06) | 1.86 (0.44–7.87) |
| 4 | 3.94 (1.10–14.13)a | 3.97 (1.09–14.41)a |
| MI (n = 31) | ||
| Quartiles | ||
| 1 | 1.00 | 1.00 |
| 2 | 0.75 (0.29–1.97) | 0.67 (0.24–1.85) |
| 3 | 0.86 (0.33–2.27) | 0.74 (0.27–2.04) |
| 4 | 0.75 (0.29–1.98) | 0.71 (0.27–1.88) |
| Primary composite (n = 117) | ||
| Quartiles | ||
| 1 | 1.00 | 1.00 |
| 2 | 1.20 (0.73–1.98) | 1.10 (0.66–1.85) |
| 3 | 1.12 (0.68–1.90) | 0.99 (0.57–1.70) |
| 4 | 1.11 (0.66–1.84) | 0.98 (0.57–1.66) |
| CHF (n = 27) | ||
| Quartiles | ||
| 1 | 1.00 | 1.00 |
| 2 | 1.29 (0.43–3.85) | 1.34 (0.44–4.08) |
| 3 | 1.27 (0.41–3.95) | 0.76 (0.22–2.58) |
| 4 | 1.58 (0.56–4.44) | 1.73 (0.60–4.97) |
Abbreviations: BNP, B‐type natriuretic peptide; CHF, congestive heart failure; CI, confidence interval; CrCl, creatinine clearance; HR, hazard ratio; MI, myocardial infarction. Multivariate adjustment of risk included the following variables: age, diabetes, prior MI, prior CHF, ST deviation (>1 mm), CrCl, BNP (>80 pg/mL), current smoking, and Killip class (I vs II–IV).
The interval increased HR for the fourth quartile of catalytic iron was statistically significant for both the unadjusted (P = 0.035) and multivariate adjusted models (P = 0.036).
Figure 1.
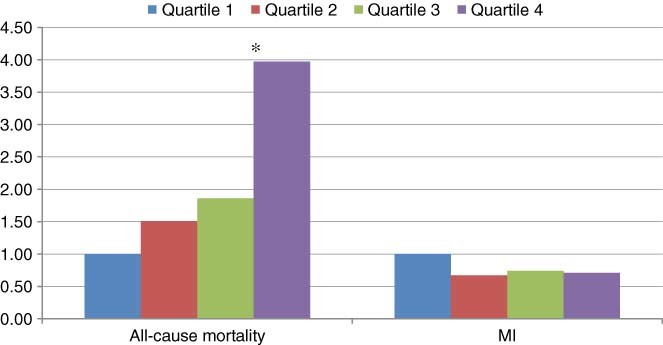
Risk for all‐cause mortality and MI. Adjusted for age, history of DM, prior MI, prior CHF, ST‐segment deviation (>1 mm), CrCl, NT‐pro‐BNP (>80 pg/mL), current smoking, and Killip class (I vs II–IV). ∗︁ = The interval increased HR for all‐cause mortality in the fourth quartile of catalytic iron was statistically significant (P = 0.036). Abbreviations: CHF, congestive heart failure; CrCl, creatinine clearance; CV, cardiovascular; DM, diabetes mellitus; HR, hazard ratio; NT‐pro‐B‐type natriuretic peptide, N terminal pro‐B‐type natriuretic peptide; MI, myocardial infarction.
In contrast, individual ischemic endpoints, MI, ischemia requiring revascularization, ischemia requiring hospitalization, stroke, as well as their composite, did not differ significantly according to baseline catalytic iron levels. Adjustment for other clinical characteristics did not reveal any risk relationship with catalytic iron after accounting for potential negative confounders (Table 3).
To assess other potential contributors to the higher mortality risk with increased catalytic iron, we examined the relationship between catalytic iron and bleeding and heart failure events. We found that neither CHF (Table 2) nor bleeding (P trend for major bleeding = 0.12 and major/severe bleeding = 0.17) were associated with catalytic iron levels. The site‐reported etiology of the 25 deaths in the catalytic‐iron cohort is summarized in Table 4. Notably, 21 of 25 deaths were classified as cardiac in nature. More than half of the deaths in the highest quartile were observed >3 months after the index event (Figure 2).
Table 4.
Etiology of Death in Patients With Baseline Catalytic Iron
| Cause of Death | Catalytic Iron Quartiles | |||
|---|---|---|---|---|
| 1 (0.14‐0.31) (n = 456) | 2 (0.32‐0.38) (n = 459) | 3 (0.39‐0.47) (n = 389) | 4 (0.48‐84.5) (n = 417) | |
| Acute MI | 1 | 1 | 3 | |
| Mechanical: CHF/pulmonary edema/cardiogenic shock/rupture | 1 | 2 | 2 | 5 |
| Arrhythmia | 1 | 1 | 1 | 1 |
| Stroke | ||||
| Ischemic | 1 | |||
| Hemorrhagic | 1 | |||
| Bleeding (not ICH) | 1 | |||
| Unknown | 1 | 1 | 1 | |
| Total mortality | 3 | 6 | 5 | 11 |
Abbreviations: CHF, congestive heart failure; ICH, intracerebral hemorrhage; MI, myocardial infarction.
Arrhythmia is defined as witnessed arrhythmia and presumed sudden cardiac death; mechanical complications are defined as heart failure, cardiogenic shock, or cardiac rupture.
Figure 2.
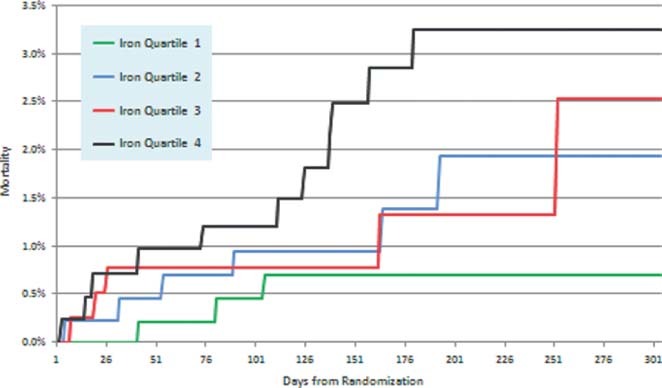
Kaplan‐Meier curves for mortality by quartile of catalytic iron at baseline.
Discussion
In this large, well‐characterized cohort, we found that catalytic iron measured after ACS was associated with all‐cause mortality independently of traditional risk indicators. We did not observe a significant association between catalytic iron and ischemic endpoints or heart failure. Our findings suggest that catalytic iron is not likely to add to currently available tools as a routine clinical biomarker for risk stratification of recurrent ischemic events. However, the association with mortality is intriguing and leaves open the question of whether cardiovascular therapeutics aimed at catalytic (“free”) iron may be useful.
Iron in Cardiovascular Disease
The association of catalytic iron with atherosclerotic disease and events, and particularly its causal potential in this disease, has been of interest for more than 30 years.9 In the absence of a transition metal like iron, ROS, such as superoxide radicals and hydrogen peroxide, have a relatively low cellular toxicity. However, in the presence of ferric iron,10 the generation of hydroxyl radicals is catalyzed, and along with other toxic compounds generated by the Fenton reaction, potentially contributes to lipid peroxidation, leading to atherogenesis and cellular oxidative damage during periods of ischemia and reperfusion.11 The pathologic contribution of circulating catalytic iron vs that which is produced (or localized) within tissues (eg, atherosclerotic plaque) is uncertain.
Catalytic iron, as measured by the BDI assay, is that component of iron stores that can participate in the Fenton reaction and potentially increase the production of oxygen radicals. In the circulation, this represents iron bound to low‐molecular‐mass chelating agents such as citrate or loosely bound to certain proteins such as albumin.6 Increased levels of circulating catalytic iron may result from release of intracellular stores such as lysosomes or microsomes during periods of tissue injury,12, 13 dissociation from circulating proteins such as transferrin or ferritin (by acidemia or free radicals), and in states of transferrin saturation.14, 15 The variation in circulating levels of catalytic iron during healthy and diseased states is not well established.
Experimental evidence that increased serum and tissue catalytic iron is associated with worsened cellular injury is derived primarily from various ischemia/reperfusion animal models evaluating tissue homogenates including myocardium10, 16 and kidney.17 In a rat model, Voogd et al demonstrated significant increases in the low‐molecular‐weight‐iron content of cardiac tissue after ischemia; similar results were found in a kidney model of ischemia. Although acute ischemia results in a spike in circulating and tissue catalytic iron, this does not appear to be solely a marker, but also a causal agent of worsened injury. Improved recovery of myocardial function after ischemia has been demonstrated with iron chelation (eg, deferoxamine) in animal models10, 16 and in one human study, providing indirect support for the toxicity of catalytic iron. Paraskevaidis et al suggested deferoxamine infusion was able to reduce myocardial stunning following elective coronary artery bypass grafting and improve long‐term ejection fraction. Circulating catalytic iron was not assessed in that study and no tissue was available to examine the effect on intracellular catalytic iron, so the mechanism by which the benefit was conferred can only be inferred.18
Evidence to support the hypothesis that iron overload broadly contributes to atherosclerosis is mixed.19, 20 More evidence exists for the deleterious role of catalytic iron once it is within arterial plaque. Evidence exists for a direct relationship between intraplaque BDI and lipid peroxidation products.21 Smith et al demonstrated the presence of BDI in homogenized atherosclerotic plaques from cadavers and some evidence for the ability of this iron to induce lipid peroxidation and hydroxyl‐radical production.22 Experimental data suggest catalytic iron is an important contributor to increased oxidation of low‐density lipoprotein and has been shown in plaques to be localized near ceroid accumulations. Erythrophagocytosis by circulating macrophages with subsequent exocytosis of iron along with intraplaque hematomas resulting from plaque fissures or hemorrhages of the vasa vasorum are the most likely sources of catalytic iron outside of acute insults (eg, ischemia). Enhanced expression of endothelial ferritin and heme oxygenase‐1 have been observed and are to thought to be protective adaptations to iron‐induced toxicity and lipid peroxidation.23, 24
Implications of the Results
Together, these preclinical data have pointed to free iron as a potential participant in cardiovascular disease and a potential therapeutic target. Our results add to previous smaller studies to support a relationship between catalytic iron and mortality in patients with ACS. Notably, in contrast to one prior smaller study, we did not observe an association between catalytic iron and recurrent ischemic events. Previous studies in a rat ischemia/reperfusion model demonstrated that global cardiac ischemia for prolonged periods led to increased levels of circulating iron, which rapidly returned to preischemic levels within 1 minute.25 It is possible that because of a less‐severe ischemic insult (eg, single‐vessel territory and perhaps with transient reperfusion) and later collection of sample after ischemia onset, an elevation in circulating catalytic iron was no longer detectable. The origin of the circulating catalytic iron and mechanism by which it may have contributed to plaque formation, index ACS, or mortality during follow‐up cannot be determined from this study. As mentioned, circulating catalytic iron may not correlate with intraplaque catalytic iron, and thus not identify those subjects at higher risk of recurrent ischemic events. Nevertheless, the finding that catalytic iron measured on average at 40 hours after onset of an ischemic syndrome is not associated with recurrent ischemic events suggests catalytic iron should not be used as a predictor of recurrent ischemic events in clinical practice.
With respect to the association with mortality in this study, it was interesting that most of the deaths were related to ischemic or mechanical complications, and therefore we can speculate that catalytic iron may be a marker or a contributor to a higher case fatality rate of ischemic injury, even though it was not a predictor of recurrent ischemia per se. This pattern of risk relationships is consistent with other investigational biomarkers that we have studied in ACS.26, 27
Study Limitations
Mechanistic insights into the causal role catalytic iron may play in atherosclerotic events and mortality are limited from this study; this would have been aided by earlier and more frequent assessments. In addition, the entire cohort received orbofiban 50 mg twice daily; and thus, although it is unlikely, it is possible that this may have altered the association between catalytic iron and recurrent ischemic events and all‐cause mortality.
Conclusion
We found that catalytic iron measured after presenting with an ACS was associated with all‐cause mortality, independently of traditional risk indicators. Our findings in this large, well‐characterized population suggest that although catalytic iron is not likely to add to currently available tools as a routine clinical biomarker for risk stratification of recurrent ischemic events, its association with mortality is intriguing and leaves open the question of whether cardiovascular therapeutics aimed at catalytic iron may be useful.
References
- 1. Danesh J, Appleby P. Coronary heart disease and iron status: meta‐analyses of prospective studies. Circulation. 1999;99:852–854. [DOI] [PubMed] [Google Scholar]
- 2. Rajapurkar MM, Shah SV, Lele SS, et al. Association of catalytic iron with cardiovascular disease. Am J Cardiol. 2012;109: 438–442. [DOI] [PubMed] [Google Scholar]
- 3. Lele S, Shah S, McCullough PA, et al. Serum catalytic iron as a novel biomarker of vascular injury in acute coronary syndromes. EuroIntervention. 2009;5:336–342. [DOI] [PubMed] [Google Scholar]
- 4. Cannon CP, McCabe CH, Wilcox RG, et al. Oral glycoprotein IIb/IIIa inhibition with orbofiban in patients with unstable coronary syndromes (OPUS‐TIMI 16) trial. Circulation. 2000;102:149–156. [DOI] [PubMed] [Google Scholar]
- 5. De Lemos JA, Morrow DA, Bentley JH, et al. The prognostic value of B‐type natriuretic peptide in patients with acute coronary syndromes. N Engl J Med. 2001;345:1014–1021. [DOI] [PubMed] [Google Scholar]
- 6. Evans PJ, Halliwell B. Measurement of iron and copper in biological systems: bleomycin and copper‐phenanthroline assays. Methods Enzymol. 1994;233:82–92. [DOI] [PubMed] [Google Scholar]
- 7. Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview. Methods Enzymol. 1990;186:1–85. [DOI] [PubMed] [Google Scholar]
- 8. O'Donoghue M, de Lemos JA, Morrow DA, et al. Prognostic utility of heart‐type fatty acid binding protein in patients with acute coronary syndromes. Circulation. 2006;114:550–557. [DOI] [PubMed] [Google Scholar]
- 9. Sullivan JL. Iron and the sex difference in heart disease risk. Lancet. 1981;1:1293–1294. [DOI] [PubMed] [Google Scholar]
- 10. Williams RE, Zweier JL, Flaherty JT. Treatment with deferoxamine during ischemia improves functional and metabolic recovery and reduces reperfusion‐induced oxygen radical generation in rabbit hearts. Circulation. 1991;83:1006–1014. [DOI] [PubMed] [Google Scholar]
- 11. De Valk B, Marx JJ. Iron, atherosclerosis, and ischemic heart disease. Arch Intern Med. 1999;159:1542–1548. [DOI] [PubMed] [Google Scholar]
- 12. Baliga R, Zhang Z, Baliga M, et al. Evidence for cytochrome P‐450 as a source of catalytic iron in myoglobinuric acute renal failure. Kidney Int. 1996;49:362–369. [DOI] [PubMed] [Google Scholar]
- 13. Paller MS, Jacob HS. Cytochrome P‐450 mediates tissue‐damaging hydroxyl radical formation during reoxygenation of the kidney. Proc Natl Acad Sci U S A. 1994;91: 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pepper JR, Mumby S, Gutteridge JM. Transient iron overload with bleomycin‐detectable iron present during cardiopulmonary bypass surgery. Free Radic Res. 1994;21:53–58. [DOI] [PubMed] [Google Scholar]
- 15. Sengoelge G, Rainer V, Kletzmayr J, et al. Dose‐dependent effect of parenteral iron therapy on bleomycin‐detectable iron in immune apheresis patients. Kidney Int. 2004;66:295–302. [DOI] [PubMed] [Google Scholar]
- 16. Voogd A, Sluiter W, van Eijk HG, et al. Low‐molecular‐weight iron and the oxygen paradox in isolated rat hearts. J Clin Invest. 1992;90:2050–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Baliga R, Ueda N, Shah SV. Increase in bleomycin‐detectable iron in ischaemia/reperfusion injury to rat kidneys. Biochem J. 1993;291(part 3):901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Paraskevaidis IA, Iliodromitis EK, Vlahakos D, et al. Deferoxamine infusion during coronary artery bypass grafting ameliorates lipid peroxidation and protects the myocardium against reperfusion injury: immediate and long‐term significance. Eur Heart J. 2005;26:263–270. [DOI] [PubMed] [Google Scholar]
- 19. Kirk EA, Heinecke JW, LeBoeuf RC. Iron overload diminishes atherosclerosis in apoE‐deficient mice. J Clin Invest. 2001;107: 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004;24: 949–954. [DOI] [PubMed] [Google Scholar]
- 21. Berger TM, Polidori MC, Dabbagh A, et al. Antioxidant activity of vitamin C in iron‐overloaded human plasma. J Biol Chem. 1997;272: 15656–15660. [DOI] [PubMed] [Google Scholar]
- 22. Smith C, Mitchinson MJ, Aruoma OI, et al. Stimulation of lipid peroxidation and hydroxyl‐radical generation by the contents of human atherosclerotic lesions. Biochem J. 1992;286(part 3):901–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nagy E, Eaton JW, Jeney V, et al. Red cells, hemoglobin, heme, iron, and atherogenesis. Arterioscler Thromb Vasc Biol. 2010;30:1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan XM, Anders WL, Olsson AG, et al. Iron in human atheroma and LDL oxidation by macrophages following erythrophagocytosis. Atherosclerosis. 1996;124:61–73. [DOI] [PubMed] [Google Scholar]
- 25. Chevion M, Jiang Y, Har‐El R, et al. Copper and iron are mobilized following myocardial ischemia: Possible predictive criteria for tissue injury. Proc Natl Acad Sci U S A. 1993;90: 1102–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Shimpo M, Morrow DA, Weinberg EO, et al. Serum levels of the interleukin‐1 receptor family member ST2 predict mortality and clinical outcome in acute myocardial infarction. Circulation. 2004;109:2186–2190. [DOI] [PubMed] [Google Scholar]
- 27. Scirica BM, Morrow DA, Cannon CP, et al. Clinical application of C‐reactive protein across the spectrum of acute coronary syndromes. Clin Chem. 2007;53:1800–1807. [DOI] [PubMed] [Google Scholar]