

Abstract
Objective
To determine the key inflammatory pathways that are activated in the peripheral and CNS compartments at the mild cognitive impairment (MCI) stage of Alzheimer’s disease (AD).
Methods
A cross‐sectional study of patients with clinical and biomarker characteristics consistent with MCI‐AD in a discovery cohort, with replication in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. Inflammatory analytes were measured in the CSF and plasma with the same validated multiplex analyte platform in both cohorts and correlated with AD biomarkers (CSF Aβ42, total tau (t‐tau), phosphorylated tau (p‐tau) to identify key inflammatory pathway activations. The pathways were additionally validated by evaluating genes related to all analytes in coexpression networks of brain tissue transcriptome from an autopsy confirmed AD cohort to interrogate if the same pathway activations were conserved in the brain tissue gene modules.
Results
Analytes of the tumor necrosis factor (TNF) signaling pathway (KEGG ID:4668) in the CSF and plasma best correlated with CSF t‐tau and p‐tau levels, and analytes of the complement and coagulation pathway (KEGG ID:4610) best correlated with CSF Aβ42 levels. The top inflammatory signaling pathways of significance were conserved in the peripheral and the CNS compartments. They were also confirmed to be enriched in AD brain transcriptome gene clusters.
Interpretation
A cell‐protective rather than a proinflammatory analyte profile predominates in the CSF in relation to neurodegeneration markers among MCI‐AD patients. Analytes from the TNF signaling and the complement and coagulation pathways are relevant in evaluating disease severity at the MCI stage of AD.
Introduction
Alzheimer’s disease (AD) is characterized by a progressive decline in cognitive function and is the most common cause of dementia worldwide. Even as classical AD neuropathology is described as amyloid plaques and neurofibrillary tangles in the brain, it is recognized that a complex cascade of events involving additional molecular players likely play a crucial role in the disease onset and progression.
Accumulating evidence implicates inflammatory changes in AD.1, 2, 3 The role of systemic inflammation and occurrence of amyloid plaque‐dependent inflammation have been well documented in both human autopsy specimens and animal models.4, 5 Altered expression of multiple cytokines and chemokines in AD patients compared to controls have been reported.3, 6, 7, 8, 9, 10 Alterations in systemic inflammation, including plasma levels of tumor necrosis factor (TNF)‐α‐related cytokines have been linked to worsened cognition in AD.3, 10, 11 In addition, genome‐wide association studies have demonstrated that polymorphisms in several inflammatory genes are associated with modestly increased risk for AD.12, 13 A role for inflammation and immune mechanisms in AD therefore appears highly likely.14, 15
In spite of recognition of the role for inflammation in the pathophysiology of AD, there are some key outstanding questions. (1) Which key inflammatory pathways are activated in clinical AD? Among most prior studies of inflammatory changes in AD patients, small number of inflammatory analytes were measured, typically 10 or fewer.6, 10, 11, 16, 17, 18 To help identify inflammatory pathways or networks of inflammatory analytes pertinent to AD and to evaluate if they are helpful or harmful to cognition, it is essential to develop approaches that evaluate multiple analytes concomitantly and interrogate their biological significance when expressed together, (2) Prior studies have predominantly focused on the peripheral compartment and it remains unclear how the inflammatory changes in the CNS relates to changes in the peripheral compartment for a large set of inflammatory molecules. This knowledge is critical to a mechanistic understanding of the pathophysiological changes related to inflammation in the peripheral and CNS compartments simultaneously and to potentially evaluate the utility of monitoring these inflammatory changes in AD for clinical outcomes.
We therefore took a systematic approach to answer these two questions among mild cognitive impairment (MCI)‐AD clinical patients. After evaluation in our discovery cohort we validated the results among MCI patients in the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort. We also determined if the genes related to soluble inflammatory analytes evaluated in our discovery cohort have also been transcribed in the brain tissue, using transcriptomic data from autopsied AD brains. Using functional network enrichment analysis we determined the biological pathways most likely related to the inflammatory analytes and genes of significance and its potential convergence across multiple datasets of brain, CSF, and plasma in the presence of markers of AD pathology.
Materials and Methods
Discovery cohort
A cross‐sectional discovery study of 48 MCI‐AD patients was conducted. All patient consent was obtained according to the Declaration of Helsinki and the study was approved by the Cleveland Clinic Institutional Review Board. The patients were recruited from a specialized memory clinic. The diagnosis of MCI‐AD was confirmed by the presence of CSF Aβ42 and p‐tau levels consistent with a diagnosis of AD and consensus evaluation of two neurologists and a neuropsychologist by published criteria.19 A commercially available test from Athena Diagnostics, ADmark® Alzheimer's evaluation Innotest sandwich Enzyme Linked Immunosorbant Assay (ELISA) kits was used for measuring CSF levels of Aβ42, t‐tau, and p‐tau levels. The subjects met cut offs CSF Aβ42 ≤ 530pg/mL, CSF p‐tau ≥ 60pg/mL which are consistent with Amyloid positive status on the Amyvid TM PET at our center. An additional marker of neuronal degeneration in the CSF, neuron‐specific enolase (NSE), was measured in the rules‐based‐medicine (RBM) multiplex analyte platform described below. NSE is often highly elevated in diseases which result in relative rapid neuronal destruction (hours/days) and levels are therefore supportive of a more recent neuronal loss in relation to analytes measured. APOE status was determined by blood samples (10 ng per subject) dispensed into 96 well plates for TaqMan allelic discrimination detection of single‐nucleotide polymorphisms that discriminate the APOE alleles (rs429358, rs7412) (Life Technologies). PCR reactions were carried out using a 9700 Gene Amp PCR system (Applied Biosystems, CA) and an end‐point read in a 7500 Real‐Time PCR system (Applied Biosystems, CA). Table 1 provides data on discovery cohort demographics and Figure 1 provides a methodological overview of the study.
Table 1.
Demographics and medical history of Discovery and ADNI cohorts.
| Factor | Discovery cohort (N = 48) | ADNI cohort (N = 43) | P‐value | ||
|---|---|---|---|---|---|
| n | Statistics | n | Statistics | ||
| Age at enrollment | 48 | 68.1 ± 7.3 | 43 | 76.1 ± 6.5 | <0.001a |
| Gender | 48 | 43 | 0.11c | ||
| Male | 28 (58.3) | 32 (74.4) | |||
| Female | 20 (41.7) | 11 (25.6) | |||
| Years of education | 48 | 16.0 [12.5, 18.0] | 43 | 16.0 [15.0, 18.0] | 0.12b |
| APOEε4 positive | 48 | 37 (77.1) | 43 | 21 (48.8) | 0.005c |
| MMSE | 48 | 24.8 ± 3.1 | 43 | 26.6 ± 1.6 | <0.001a |
| CDR‐SB | 48 | 2.2 ± 1.3 | 43 | 1.5 ± 0.79 | 0.003a |
| DRS | 47 | 125.6 ± 10.3 | NA | ||
| CSF Aβ42, pg/mL | 48 | 305.9 [216.1, 367.1] | 43 | 144.0 [135.0, 167.0] | <0.001b |
| CSF t‐tau, pg/mL | 48 | 454.3 [335.1, 771.] | 43 | 91.0 [64.0, 132.0] | <0.001b |
| CSF p‐tau, pg/mL | 48 | 79.6 [59.3, 104.6] | 43 | 33.0 [21.0, 42.0] | <0.001b |
Statistics presented as Mean ± SD, Median [P25, P75] or N (column %).
P‐values: a = ANOVA, b = Kruskal–Wallis test, c = Pearson's chi‐square test.
MMSE, Mini‐mental state exam; CDR‐SB, Clinical dementia rating scale‐sum of boxes.
DRS, Dementia rating scale. P values in bold meet statistical significance <0.05.
Figure 1.
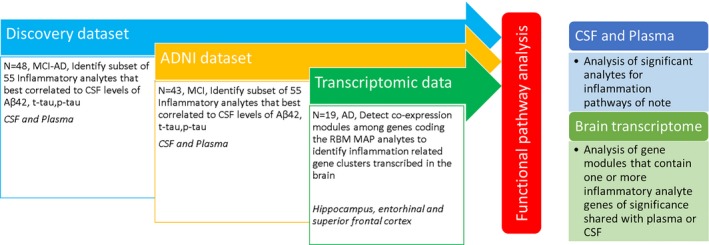
Methodological overview.
Characterizing clinical and environmental factors
Age, sex, education, medical comorbidities family history, smoking, and other addictions and medication use, including nonsteroidal anti‐inflammatory drugs (NSAIDs) were characterized. Confounding treatment with steroids, trauma, and surgical intervention and infectious disease in the last 6 months, in addition to uncontrolled diabetes and vascular disease as part of clinical evaluation were screened. Additional preexisting proinflammatory disorders (e.g., systemic lupus erythematosus) were recorded and levels of C‐reactive protein (CRP) and sedimentation rate related to systemic inflammation was quantified in the blood. Table S1.
Neuropsychological tests
Mini‐Mental State Examination (MMSE),20 WMS‐IV Logical Memory 1 and 2,21 Clinical Dementia Rating scale (CDR‐SB and Global),22 and Dementia Rating Scale (DRS)23 tests were conducted to characterize the degree of their baseline cognitive and functional deficits.
Inflammatory biomarkers
CSF and plasma were collected and analyzed by an independent laboratory via the validated RBM Multi‐Analyte Profile (MAP) platform from Myriad Genetics (Salt Lake City, UT). Samples were evaluated for levels of 86 analytes using a custom MAP: HumanMAP® v2.0 + IL1 and 16 using a Luminex platform. Validation has been performed as defined by the Clinical and Laboratory Standards Institute and is therefore replicable across multiple runs. CSF and plasma samples were collected contemporaneously. They were both frozen within 15 min after collection and processed (at −70°C in dry ice) and maintained at a freezing temperature (−80°C at a maximum nonfrost‐free type refrigerator) continuously. The samples were shipped frozen, in a Styrofoam container with sufficient dry ice to maintain temperature less than −70°C for at least 48 h. Samples therefore underwent a single freeze thaw cycle prior to analyses. Each analyte measure had to pass a threshold of measurement and quality control criteria set by Myriad Genetics.
The consistency of the data was further evaluated by internal and external validation. There were two technical replicates of CSF and plasma samples chosen randomly from three patients to evaluate consistency of data. Expected high degree of correlation within individual subjects analyte profiles between markers of positive acute phase response (CRP, haptoglobin, ferritin, α2 macroglobulin, fibrinogen) was further confirmed and the results from the discovery cohort were next evaluated in ADNI.
ADNI validation cohort
ADNI is a longitudinal multicenter study designed to develop clinical, imaging, genetic, and biochemical biomarkers for the early detection and tracking of AD. ADNI was launched by the National Institute of Aging and is a multicenter project with additional support from private pharmaceutical companies, and nonprofit organizations. ADNI 1 eligibility criteria are described in the ADNI 1 protocol http://adni.loni.usc.edu/methods/documents/. Briefly, participants are 55–90 years of age, had an informant able to provide an independent evaluation of functioning, and spoke either English or Spanish. Participants had completed at least 6 years of education (or had a work history sufficient to exclude mental retardation). General inclusion/exclusion criteria for MCI subjects are as follows: MMSE scores between 24 and 30 (inclusive; exceptions made on a case by case basis by neurologists at the center of follow up), a memory complaint, objective memory loss measured by education adjusted scores on Wechsler Memory Scale Logical Memory II, a CDR of 0.5, absence of significant levels of impairment in other cognitive domains, essentially preserved activities of daily living, and an absence of dementia.
The demographics at baseline among the subset of all 43 ADNI MCI participants who had CSF and plasma multiplex data were used in the validation analysis are shown in Table 1. These subjects were noted to have CSF Aβ42, t‐tau, and p‐tau levels supportive of underlying AD pathology.24 Details on the Luminex method used for ADNI AD biomarkers measurement are detailed elsewhere.25 CSF samples were measured for levels of 159 analytes using the RBM DiscoveryMAP® v.1.0 panel. The RBM HumanMAP® v.2.0 used in the discovery cohort is a subset of the RBM DiscoveryMAP® v.1.0 and uses a Luminex platform with the same quality control and thresholding process used in ADNI dataset and are comparable. We analyzed data from ADNI's well‐characterized subset of MCI participants who had curated CSF and plasma multiplex data, the details of which have been published previously.26 The data from CSF QC multiplex data used in this analysis is the cleaned, quality controlled data according to methodology described in the statistical analysis of Biomarkers Consortium data primer.27
Bioinformatics and statistical analysis
Ontology analysis and network analysis to identify inflammatory analytes
Gene/protein annotations and gene ontologies were downloaded from the Gene Ontology (GO)28 and processed into tabular files containing GO terms annotated to genes and the gene ontology tree. The RBM analyte names were mapped to HUGO Gene Nomenclature Committee gene symbols by manual curation. All analytes had a one to one correspondence with coding genes with the exceptions of ferritin (heavy chain coding gene FTH1), and fibrinogen (alpha chain coding gene FGA). The full ontology tree rooted at the inflammatory response node (GO:0006954) was constructed and all genes labeled by this term were extracted (n = 458). The analytes identified in the proprietary RBM InflammationMAP® v1.0 (n = 45 analytes) and the inflammatory response genes from GO analysis were combined (n = 483) and used in a network analysis to identify additional analytes from the RBM HumanMAP® v2.0 that are related to inflammation. The Crosstalker algorithm was run using the 483 seeds to rank the RBM HumanMAP® by their proximity to the seeds.29, 30 The analysis was run using three reference networks (STRINGv10.5, BioGRIDv3.4.163, and Human Protein Reference Database, all latest versions), and repeated three times on each network since the algorithm is nondeterministic.21, 31 For a target analyte to be deemed significant it had to pass the significance threshold in all three runs for the same reference network where significance was set at P < 0.2 (z> 0.85). This identified an additional eight analytes from the RBM HumanMAP® that were combined with the RBM InflammationMAP® (n = 45 analytes) to form our final comprehensive list of 53 candidate inflammatory analytes. The list of inflammatory analytes analyzed in the study is provided in Table 2.
Table 2.
List of inflammatory analytes analyzed.
| RBM Name | Gene | RBM Name | Gene |
|---|---|---|---|
| 1. Alpha‐1‐Antitrypsin | SERPINA1 | 27. Interleukin‐12 Subunit p40 | IL12B |
| 2. Alpha‐2‐Macroglobulin | A2M | 28. Interleukin‐12 Subunit p70 | IL12P70 |
| 3. Apolipoprotein A‐I | APOA1 | 29. Interleukin‐15 | IL15 |
| 4. Beta‐2‐Microglobulin | B2M | 30. Interleukin‐17 | IL17A |
| 5. Brain‐Derived Neurotrophic Factor | BDNF | 31. Interleukin‐18 | IL18 |
| 6. Complement C3 | C3 | 32. Interleukin‐8 | CXCL8 |
| 7. C‐Reactive Protein | CRP | 33. Interleukin‐23 | IL23A |
| 8. Eotaxin‐1 | CCL11 | 34. Macrophage Inflammatory Protein‐1 alpha | CCL3 |
| 9. Fibrinogen | FGA | 35. Macrophage Inflammatory Protein‐1 beta | CCL4 |
| 10. Factor VII | F7 | 36. Matrix Metalloproteinase‐3 | MMP3 |
| 11. Ferritin | FTH1 | 37. Matrix Metalloproteinase‐9 | MMP9 |
| 12. Granulocyte‐Macrophage Colony‐Stimulating Factor | CSF2 | 38. Monocyte Chemotactic Protein 1 | CCL2 |
| 13. Granulocyte Colony‐Stimulating Factor | CSF3 | 39. Matrix Metalloproteinase‐2 | MMP2 |
| 14. Haptoglobin | HP | 40. Myeloperoxidase | MPO |
| 15. Intercellular Adhesion Molecule 1 | ICAM1 | 41. Neuron‐Specific Enolase | ENO2 |
| 16. Interferon gamma | IFNG | 42. Plasminogen Activator Inhibitor 1 | SERPINE1 |
| 17. Interleukin‐1 alpha | IL1A | 43. Serotransferrin | TF |
| 18. Interleukin‐1 beta | IL1B | 44. Stem Cell Factor | SCF |
| 19. Interleukin‐1 receptor antagonist | IL1RN | 45. T‐Cell‐Specific Protein RANTES | CCL5 |
| 20. Interleukin‐2 | IL2 | 46. Tissue Inhibitor of Metalloproteinases 1 | TIMP1 |
| 21. Interleukin‐3 | IL3 | 47. Tumor Necrosis Factor alpha | TNF |
| 22. Interleukin‐4 | IL4 | 48. Tumor Necrosis Factor beta | LTA |
| 23. Interleukin‐5 | IL5 | 49. Tumor Necrosis Factor Receptor 2 | TNFRSF1B |
| 24. Interleukin‐6 | IL6 | 50. Vascular Cell Adhesion Molecule‐1 | VCAM1 |
| 25. Interleukin‐7 | IL7 | 51. Vascular Endothelial Growth Factor | VEGFA |
| 26. Interleukin‐10 | IL10 | 52. Vitamin D‐Binding Protein | GC |
| 53. von Willebrand Factor | VWF |
Statistical analysis
The CSF and plasma analytes of interest identified above were log transformed, which limits the impact of extreme measures. Only those with at least 50% response rate of analytes above the limit of detection were included for further analysis Figure S1. The absolute values of the inflammatory markers and AD biomarkers were compared. Normality of biomarkers was evaluated using graphical methods and the Shapiro–Wilk test and a log (base 2) transformation allowed Pearson correlations to be fit. Along with estimates of correlation, 95% confidence intervals and P‐values with and without false discovery rate (FDR) adjustment were calculated. If we assume 53 analytes (corresponding to a Bonferroni adjusted significance level of 0.009), with 48 CSF samples and 46 plasma samples used in each analysis, there would be 80% power to detect correlations above 0.5 as statistically significant. Analysis was performed unadjusted and then adjusting for age, sex, baseline MMSE, and APOEε4 status (present vs. absent). Agreement on relative ordering of response of analytes between the discovery and ADNI data was evaluated using Kendall’s W coefficient of concordance statistic. 95% confidence intervals for kappa statistics and P‐values testing whether more agreement than expected by chance were calculated for both measures. Analysis was performed using SAS software (version 9.4) and an overall significance level of 0.05 was assumed for all tests.
Subgroup searching for analyte synergistic relationships
In order to evaluate analyte levels that show higher correlation when considered together (synergistic relationship) rather than individual analyte correlation by univariate analysis alone, we performed an exhaustive search to find analyte subgroups whose aggregate levels maximally correlated with AD CSF biomarkers. The full panel of analytes was filtered to contain only those which were detected in at least 50% of the data. All subgroups of analytes were enumerated up to maximum group size of six analytes. Aggregate activity of each subgroup was computed by first normalizing each analyte by subtracting the mean and dividing by the standard deviation, and then summing normalized analyte levels for each patient.32, 33 The Pearson correlation coefficient was computed between the aggregate activity of each analyte group and all response markers (e.g., t‐tau levels). Only the highest scoring analyte subgroup was reported for each response marker in CSF and plasma, respectively. Significance P‐values for findings were estimated by two tests using an approach that has been previously described.32 Hypothesis 1(H1) tested how likely we were to see greater or equal correlation with random analyte subgroups by sampling 10,000 random analyte subgroups (of the same size as each actual best scoring analyte subgroup) from among all analytes that met the 50% detection threshold (i.e., not just the inflammatory analytes) and computing the correlation values. P‐values were estimated as the fraction of random subgroups that achieved greater or equal correlation with the response marker as the actual analyte subgroup. Hypothesis 2(H2) tested how likely we were to randomly observe greater or equal correlation between the aggregate activity of an analyte subgroup and a response marker by permuting the values of each response marker 10,000 times and computing the correlation values to the aggregate analyte levels. P‐values were estimated as the proportion of randomized responses with equal or greater correlation with aggregate analyte levels than the actual response.
Functional pathway analysis on analytes of interest
The analytes of significance identified in the univariate statistical analysis and confirmed in the analyte subgroup search above in relation to CSF neurodegeneration markers were entered into STRING: functional protein association networks for pathway enrichment analysis.34 The top pathway for each analysis with the largest gene count and the lowest P value following FDR correction is reported.
Weighted gene coexpression network analysis (WGCNA) in autopsy AD brains
The R package WGCNA35 was used to construct coexpression networks as in prior studies36 using Alzheimer's Disease Dataset: GSE 48350 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE48350). Here, postmortem brain tissue had been collected from ADRC brain banks, data from all 19 AD brains in the dataset with hippocampal, entorhinal cortex, and superior frontal cortex transcriptomic data were used for the current analysis. The GSE48350 expression data were reduced to probe sets corresponding to the 84 RBM full panel genes. Four HumanMAP® RBM analytes were not represented by any probe sets (IL12, IgE, Chorionic Gonadotropin Subunit Beta 3, and CA‐19‐9). The remaining 80 RBM analytes were represented by 198 probe sets. The modules with one or more inflammatory analyte genes of significance shared with plasma or CSF were characterized using GO Elite to control the network‐wide false discovery rate, with all enriched pathways comprising at least 10 genes at Z > 2 and FDR < 0.01.37 The gene modules were characterized for their functional enrichment of biological pathways using STRING34 to evaluate their functional role.
Results
Subject demographics of the discovery and ADNI cohorts are in Table 1. Summary statistics of the 53 inflammatory analytes evaluated in CSF and plasma are provided in Table S2.
Discovery cohort
Univariate analysis CSF
In the unadjusted analyses, after applying the FDR correction, TNFR2, SCF, Ferritin, and α2macroglobulin were positively correlated with CSF t‐tau, p‐tau, and NSE. After adjusting for covariates (age, sex, baseline MMSE, APOEε4 status), the same associations were significant. In addition, MMP2, MMP3, β2 microglobulin, VCAM1, and VEGF were positively correlated with CSF t‐tau, and p‐tau but not NSE. After adjustment for the same covariates in addition to the above five analytes, CCL2 (MCP1) and vWF were also found to be significantly correlated with CSF t‐tau and p‐tau. (Table 3). There were no significant correlations of the CSF inflammatory analytes to CSF Aβ42.
Table 3.
Significant associations between CSF inflammatory analytes and CSF neurodegenerative markers (adjusted for age, sex, APOEε4, and MMSE) in discovery cohort.
| Factor 1 | Factor 2 | N | Pearson r (95% CI) | Raw P‐value | False discovery rate (FDR) P‐value |
|---|---|---|---|---|---|
| Aβ42 | VDBP | 47 | 0.37 (0.08, 0.60) | 0.014 | 0.16 |
| Total Tau | TNFR2 | 48 | 0.73 (0.55, 0.84) | <0.001 | <0.001 |
| SCF | 48 | 0.66 (0.44, 0.80) | <0.001 | <0.001 | |
| B2M | 47 | 0.62 (0.38, 0.77) | <0.001 | <0.001 | |
| Ferritin | 48 | 0.58 (0.33, 0.74) | <0.001 | <0.001 | |
| A2M | 48 | 0.50 (0.24, 0.69) | <0.001 | 0.002 | |
| MMP3 | 48 | 0.49 (0.22, 0.69) | <0.001 | 0.002 | |
| VCAM1 | 48 | 0.46 (0.18, 0.66) | 0.002 | 0.012 | |
| VEGF | 48 | 0.42 (01.4, 0.64) | 0.004 | 0.012 | |
| CCL2 (MCP1) | 48 | 0.38 (0.09, 0.61) | 0.009 | 0.023 | |
| vWF | 48 | 0.34 (0.05, 0.58) | 0.022 | 0.047 | |
| PhosphoTau | TNFR2 | 48 | 0.72 (0.54, 0.84) | <0.001 | <0.001 |
| SCF | 48 | 0.71 (0.52, 0.83) | <0.001 | <0.001 | |
| B2M | 47 | 0.60 (0.36, 0.76) | <0.001 | <0.001 | |
| Ferritin | 48 | 0.59 (0.35, 0.75) | <0.001 | <0.001 | |
| A2M | 48 | 0.57 (0.32, 0.74) | <0.001 | 0.002 | |
| VCAM1 | 48 | 0.48 (0.21, 0.68) | <0.001 | 0.003 | |
| MMP3 | 48 | 0.46 (0.18, 0.66) | 0.002 | 0.006 | |
| VEGF | 48 | 0.43 (0.14, 0.64) | 0.003 | 0.009 | |
| vWF | 48 | 0.41 (0.12, 0.63) | 0.005 | 0.013 | |
| CCL2 (MCP1) | 48 | 0.35 (0.05, 0.58) | 0.020 | 0.045 | |
| NSE | Ferritin | 48 | 0.75 (0.58, 0.85) | <0.001 | <0.001 |
| SCF | 48 | 0.71 (0.51, 0.83) | <0.001 | <0.001 | |
| TNFR2 | 48 | 0.68 (0.47, 0.81) | <0.001 | <0.001 | |
| CCL4 (M1P1β) | 48 | 0.48 (0.21, 0.68) | <0.001 | 0.004 | |
| vWF | 48 | 0.48 (0.21, 0.68) | <0.001 | 0.005 | |
| A2M | 48 | 0.46 (0.18, 0.66) | 0.002 | 0.008 | |
| MMP2 | 47 | 0.39 (0.10, 0.62) | 0.009 | 0.036 | |
| VCAM1 | 48 | 0.36 (0.06, 0.59) | 0.017 | 0.045 | |
| MMP3 | 48 | 0.31 (0.01, 0.55) | 0.043 | 0.10 | |
| AAT | 47 | −0.36 (−0.60, −0.07) | 0.016 | 0.045 |
FDR P values in bold meet statistical significance < 0.05.
Univariate analysis plasma
In unadjusted analysis after applying the FDR correction, only α2‐macroglobulin was positively correlated with CSF t‐tau and p‐tau measures and MMP9 was positively correlated with CSF NSE. After adjustment for covariates, the associations between α2‐macroglobulin and CSF p‐tau and between MMP9 and CSF NSE remained significant (Table 4). There were no significant correlations of the plasma inflammatory analytes to CSF Aβ42.
Table 4.
Significant associations between Plasma inflammatory analytes and CSF neurodegenerative markers (adjusted for age, sex, APOEε4, and MMSE) in discovery cohort.
| Factor 1 | Factor 2 | N | Pearson r (95% CI) | Unadjusted P‐value | False discovery rate (FDR) P‐value |
|---|---|---|---|---|---|
| ABeta42 | CCL4 | 46 | 0.39 (0.09, 0.61) | 0.011 | 0.35 |
| AAT | 46 | 0.31 (0.01, 0.56) | 0.042 | 0.41 | |
| Total Tau | A2M | 46 | 0.38 (0.08, 0.61) | 0.013 | 0.20 |
| ICAM1 | 45 | −0.40 (−0.63, −0.10) | 0.009 | 0.20 | |
| PhosphoTau | A2M | 46 | 0.49 (0.22, 0.69) | <0.001 | 0.023 |
| ICAM1 | 45 | −0.36 (−0.60, −0.05) | 0.020 | 0.31 | |
| VEGF | 45 | −0.43 (−0.65, −0.13) | 0.005 | 0.10 | |
| NSE | MMP9 | 45 | 0.57 (0.31, 0.74) | <0.001 | 0.002 |
| Haptoglobin | 46 | −0.39 (−0.62, −0.09) | 0.010 | 0.16 |
FDR P values in bold meet statistical significance < 0.05.
Secondary analyses
None of the CSF and plasma inflammatory analytes had a significant degree of correlation with any of the cognitive measures after applying the FDR correction. No difference was noted in the analytes of significance when adjusted for NSAID exposure. Adjusting for individual CSF/plasma albumin ratio noted no decrease in the number of analytes of significance on the Pearson correlations corrected for FDR. While, complement C3 and vWF were the additional analytes from the unadjusted correlations with CSF t‐tau now meeting FDR correction.
When comparing univariate correlations between plasma and CSF inflammatory analytes and CSF AD biomarkers in the discovery cohort, the correlation values to AD biomarkers for plasma inflammatory analytes were lower than those for CSF (Tables 3 and 4 and Fig. S2). On Synergistic group analysis A2M, CCL2, ENO2, MMP3, and TNFRSF1B shared significance between plasma and CSF t‐tau, whereas CCL4, HP, and SERPINA1 shared synergistic correlations with Aβ42 (Table 5).
Table 5.
Analyte subgroup analysis with most significant inflammatory analytes in discovery cohort.
| CSF factor (+correlation, −correlation) | r | H1 P‐value | H2 P‐value | Analyte subgroup | Shared with plasma | Shared with ADNI dataset |
|---|---|---|---|---|---|---|
| Aβ42+ | 0.69 | <0.0001 | <0.0001 | SERPINA1,CRP,HP,CCL4,ENO2,CCL5,VEGFA,GC,VWF | CCL4,HP, SERPINA1 | |
| Aβ42− | −0.039 | 0.074 | 0.40 | SERPINE1 | ||
| t‐tau+ | 0.917 | <0.0001 | <0.0001 | A2M,CRP,F7,FTH1,MMP3,CCL2,ENO2,TNFRSF1B,VWF | A2M,CCL2,ENO2,MMP3,TNFRSF1B | FTH1,MMP3 |
| t‐tau − | −0.148 | 0.018 | 0.14 | SERPINA1,CRP | CRP | |
| p‐tau+ | 0.963 | <0.0001 | <0.0001 | A2M,F7,FTH1,MMP3,CCL2,ENO2,TNFRSF1B,VWF | A2M,CCL2,ENO2,MMP3 | FTH1,MMP3 |
| PTAU− | −0.137 | 0.069 | 0.17 | SERPINA1 |
| Plasma factor (+correlation, −correlation) | r | H1 P‐value | H2 P‐value | Analyte subgroup | Shared with CSF | Shared with ADNI dataset |
|---|---|---|---|---|---|---|
| Aβ42+ | 0.745 | <0.0001 | <0.0001 | SERPINA1,BDNF,F7,FTH1,HP,CCL4,MMP3 | CCL4,HP, SERPINA1 | FTH1,HP |
| Aβ42− | −0.795 | <0.0001 | <0.0001 | C3,IL1RN,IL18,MMP2,MMP9,ENO2,CCL5,GC | MMP2,MMP9 | |
| t‐tau+ | 0.64 | <0.0001 | 0.0011 | A2M,B2M,IL12B,MMP3,CCL2,ENO2,TNFRSF1B,GC | A2M,CCL2,ENO2,MMP3,TNFRSF1B | |
| t‐tau − | −0.735 | <0.0001 | <0.0001 | B2M,CCL11,ICAM1,MMP9,SERPINE1,KITLG,CCL5,VCAM1,VEGFA,GC | B2M,CCL11,MMP9,SERPINE1 | |
| p‐tau+ | 0.685 | <0.0001 | 0.0003 | A2M,B2M,IL12B,CCL4,MMP2, MMP3,CCL2,ENO2,TIMP1,GC | A2M,CCL2,ENO2,MMP3 | CCL4,MMP2 |
| p‐tau − | −0.71 | <0.0001 | <0.0001 | B2M,CCL11,ICAM1,MMP9,SERPINE1,KITLG,CCL5,VCAM1,VEGFA,GC |
H1 and H2 P values in bold meet statistical significance < 0.05.
Comparing inflammatory analytes in ADNI versus the discovery cohort
On univariate comparisons none of the inflammatory analyte correlations with CSF neurodegeneration markers within the ADNI dataset met P < 0.05 FDR correction threshold. However, most inflammatory analytes shared similar direction and similar relative ordering/ranking of correlation distances between ADNI and discovery data (Fig. 2). Comparing ADNI and the discovery cohorts, the ordering of the correlations between CSF inflammatory analytes and neurodegeneration markers was significantly higher than chance in relation to CSF t‐tau and p‐tau(mean diff in correlation: 0.32, W statistic 0.92, P = 0.013 for t‐tau). On average, the correlations between the cohorts were 0.3 or higher in the discovery cohort. The correlation distances between the two cohorts were lower for measures in relation CSF Aβ42 (mean diff in correlation: 0.11, W statistic 0.64, P = 0.19 for Aβ42) and plasma inflammatory analytes (t‐tau, Aβ42: mean diff in correlation: 0.01, 0.03, Wstatistic 0.55, 0.43, P = 0.33, 0.68) (Table S3, Fig. S3).
Figure 2.
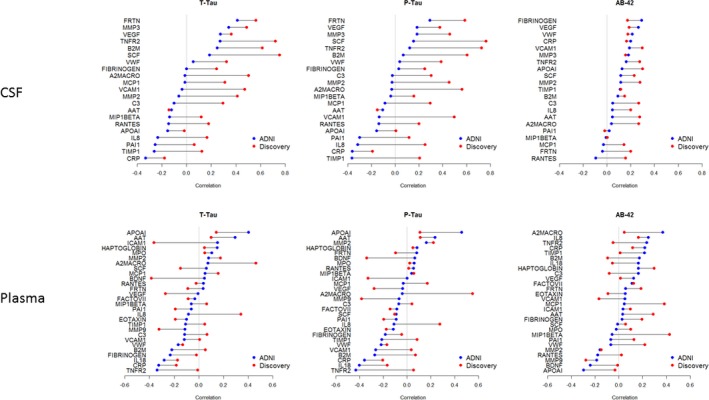
Cluster plot noting similar correlation ordering among the inflammatory analytes in ADNI and discovery cohorts, with shared ordering of analytes more notable in CSF than plasma measures.(Colored points: Pearson correlation coefficients of discovery and ADNI analytes, Connector lines: difference in correlation values for each analyte compared between the two datasets).
Analyte subgroup analysis for synergestic relationships in discovery and ADNI cohorts
On evaluating analyte levels that show higher correlation when considered together rather than the individual component analytes (synergistic analyte analysis), the results replicated the significant results of univariate Pearson correlations above. In addition it identified groups of analytes that did not meet statistical significance when considered alone. The analyte subgroups that best correlated with t‐tau, p‐tau, and NSE had analytes cross represented between them and differed from the analyte subgroup that best correlated with Aβ42 in both the CSF and plasma (Table 5). The synergistic analytes in the discovery cohort that correlated with t‐tau that were noted in both plasma and CSF included α‐2 Macroglobulin, CCL2, NSE(ENO2), MMP3, and TNFR2 (TNFRS1B). Haptoglobin and α‐1‐antitrypsin (SERPINA1), while CCL4 was best correlated with Aβ42 in both plasma and CSF synergistic analyte analysis. On comparing ADNI and discovery data subanalyte analysis, they again shared common analyte groups meeting statistical significance that is detailed in Table 5 and Table S4.
Functional pathway analysis
When CSF and plasma analytes that positively correlated with t‐tau and p‐tau measures from univariate and subanayte analyses were entered into STRING,34 the top hit among the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways38 in both CSF (gene count 4, P < 0.0001) and plasma (gene count = 3, P = 0.0021) was the TNF signaling pathway (KEGG entry: hsa04668). In relation to Aβ42 levels both in the CSF (gene count 3, P = 0.0012) and plasma (gene count 3, P = 0.0022), the complement and coagulation cascade (KEGG entry: hsa04610) was noted consistently among the top KEGG pathway hits (Table S5).
AD brain transcriptomic analysis
Using autopsy AD brain whole‐genome transcriptomic data, three coexpression modules (M1‐M3 see Fig. 3 for module details) were identified. The modules represent network gene clusters that share highly similar expression patterns in the three AD brain regions evaluated. Functional analyses by STRING showed that the transciptomic gene modules again included key inflammatory pathways identified in the CSF and plasma analytes that correlated with AD biomarkers (cytokine–cytokine receptor interaction, complement and coagulation cascade, and TNF signaling pathway) (Fig. S3). The modules share genes with significant analytes from the synergic group analysis (Module 1: A2M, SCF, MMP2, F7, FTH1, CRP, and AAT. Module 2: TNFRSF1B, B2M, TIMP1, ICAM1, Module 3: did not enrich to a KEGG cluster).
Figure 3.
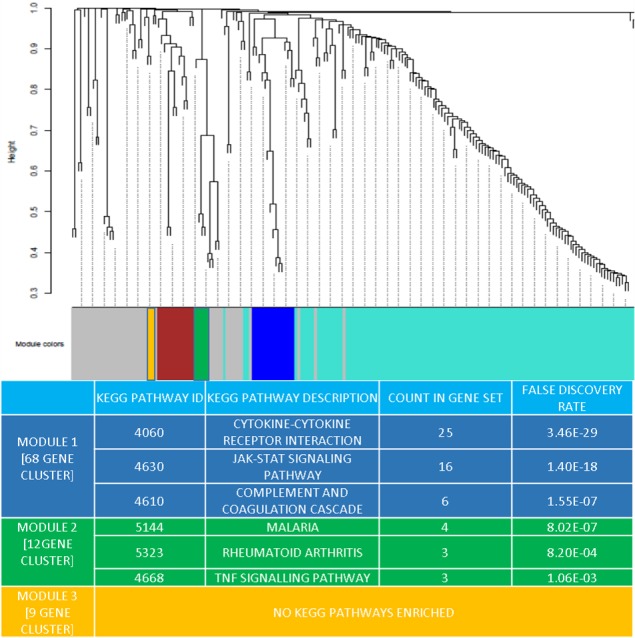
Network analysis dendrogram showing modules based on the coexpression topological overlap of genes related to inflammatory analytes and brain transcriptome. Color bars give information on module membership in they hold one or more analytes of significance identified in CSF and plasma. The table provides the related enrichment for KEGG pathways within these specific inflammation‐related gene clusters of interest and false discovery rate.
Discussion
This study clearly demonstrates activation of key shared inflammatory pathways across the CSF, plasma and brain tissue in human subjects with AD. In our discovery dataset, the inflammatory analytes that best correlated with levels of t‐tau, p‐tau, and NSE levels were in TNF signaling pathway and those that best correlated to Aβ42 were in the complement and coagulation pathway. These pathway activations were also noted to be enriched in AD brain tissue following a transcriptomic gene coexpression analysis.
Replication of CSF and plasma results in ADNI
The AD biomarkers shared similar relationships to significant inflammatory analytes in both the cohorts (Fig. 2). The discovery cohort with a larger range of AD biomarker values had consistently larger correlation coefficients than ADNI. The median and range of CSF Aβ42 and t‐tau, p‐tau were different between ADNI and discovery cohorts (even though both met MCI‐AD criteria) likely due to different analyte platforms for AD biomarkers. This likely impacts the correlation strengths between biomarkers and inflammatory analytes. However, these differences in absolute correlations did not notably impact the relative ordering/ranking of analyte correlations between the cohorts.
The relative ordering of the CSF inflammatory analyte correlations with CSF t‐tau and t‐tau was significantly high between the ADNI and discovery cohorts, with slightly lower correspondence for Aβ42 and plasma analytes. Furthermore, in the synergistic subgroup analysis after FDR correction, among the analytes with at least 50% of values above the limit of detection in the discovery panel, 30% were replicated in ADNI CSF. The functional analysis of synergistic analytes in ADNI also independently pointed to shared inflammatory pathway activations as in the discovery cohort.
The differences in analyte correlation strengths between the discovery and the ADNI cohorts could be related to number of potential factors, (1) given the well‐known challenges in standardization of AD biomarker quantification and these measurements being undertaken by necessity across different laboratories (Table 1) (2) Differences in patient recruitment characteristics: a memory clinic sample of MCI subjects with notable cognitive concerns in the discovery cohort, versus a longitudinal MCI cohort in ADNI with slightly lower CDR‐SB and higher MMSE (Table 1). This could be related to an earlier stage of MCI‐AD or potential biases against atypical forms of AD in ADNI (e.g., frontal variant, logopenic aphasia) c) Fewer shared correlation trends in the plasma than the CSF between the two cohorts (Fig. 2), also supports the possibility that these differences could be related to concomitant environmental exposures and/or medical comorbidities that were different between the cohorts, that could potentially have had a stronger effect on the plasma analyte levels than CSF analytes.
Brain transcriptome replication
Our use of AD brain transcriptome to evaluate the enrichment of inflammatory pathway activation provides additional level of confidence in the key inflammation‐related pathways identified. The correspondence in key inflammatory analyte genes, and inflammatory pathways between brain, CSF, and plasma (A2M, SCF, MMP2, F7, FTH1, CRP, AAT, TNFRSF1B, B2M, TIMP1, ICAM1) following these results also provides a starting point for further experimental investigation of targeted inflammatory analytes in the CSF/ plasma knowing that a high degree of conserved biology exists with the brain.
Key analytes of note
Tumor necrosis factor receptor 2 (TNFR2)
Among the TNF signaling pathway analytes, TNFR2 had a consistently high correlation value to CSF t‐tau, p‐tau, and NSE. TNFR2 is expressed primarily in immune and endothelial cells. Signaling through TNFR2 activates inflammatory and prosurvival signaling pathways through subsequent activation of cellular inhibitor of apoptosis (cIAPs) and the NF‐κB pathway.38 Interestingly, prior reports note a negative correlation between CSF levels of proinflammatory TNFα and t‐tau.39 Unlike TNFR2, it is notable that TNFα and related proinflammatory cytokines IL‐6 and IL‐1β did not meet the limit of detection in greater than 50% of subjects in both the cohorts used in the analysis. Taken together these suggest a cell protective role for the TNFR2 mediated pathway in MCI‐AD. Additional analytes that correlated to the neurodegeneration markers are known to be part of the TNF signaling pathways mediating downstream functions including leukocyte recruitment across the blood brain barrier (CCL2), remodeling of extra cellular matrix (MMP3), cell adhesion (VCAM1), and vascular effects (VGEF).38, 40
Stem cell factor (SCF)
SCF levels also correlated highly to CSF t‐tau and p‐tau. It is a hematopoeitic growth factor that is critically involved in regulation of blood cell production and mobilization of bone marrow stem cells and cell migration.41 SCF is highly overexpressed by neurons at sites of brain injury. It has been noted to mediate chemoattractant activity for neural stem/progenitor cell migration and thought to play a repair role in models of stroke.42 In the context of AD it has been reported to be low in the CSF and plasma of AD patients43 and therapeutic effects of systemic SCF administration in mice models have shown promise in reducing amyloid.44
The analytes related to the Complement and Coagulation pathways that were strongly related to CSF Aβ42 include α‐1‐antitrypsin, plasminogen activator inhibitor (PAI) Type 1, and von Willebrand factor in the CSF and α‐1‐antitrypsin, complement factor 3 and coagulation factor VII in the plasma. The main consequences of the activation of this pathway are the opsonization of pathogens, the recruitment of inflammatory and immunocompetent cells, and the direct killing of pathogens.38 The association between levels of these analytes and Aβ burden has been noted in multiple previous studies.45, 46, 47, 48 Among the other inflammatory analytes of significance in our results, the relationships between AD diagnosis and CCL2,11 MMP2,49 and VGEF50 have been previously reported, whereas VCAM1 has been reported in relation to both AD and vascular cognitive impairment.51
Even as TNF‐α, IL‐1β, IL‐6 and, IL‐12 did not meet threshold for analysis, among proinflammatory cytokines that met analysis threshold, CSF and plasma CRP had negative correlations with CSF t‐tau in both cohorts, whereas IL‐18 in the plasma had negative correlations with CSF t‐tau in both cohorts (Fig. 2). The direction of correlation in the above two analytes were consistent between the ADNI and discovery cohorts. In the plasma, the lack of some classical proinflammatory analytes as reported in previous AD meta‐analysis, is notable.18 No prior studies have evaluated inflammatory analytes measured both in CSF and plasma in relation to CSF AD biomarker levels, using Luminex technology to enable direct comparisons. Additionally as some of the prior studies lacked data on clinical variables that could impact proinflammation markers including comorbidities and concomitant medications (evaluated in this study), that could also account for difference in results.
Another inference from our CSF data is that, at the MCI stage of AD, TNF pathway cytokines that have been noted in AD animal and in vitro models in relation to amyloid,52 were here best correlated not to CSF Aβ42 levels but to CSF t‐tau, p‐tau, and NSE pointing to potential TNF pathway activation in relation to beta amyloid deposition starting earlier in preclinical AD53 that warrants investigation.
Strengths and limitations
There are several differences between the current study design and previous reports. First, even though some prior reports had a larger number of subjects than this study, they often lacked characterization by AD biomarkers, and often focused on the dementia stage of AD, both of which could potentially add confounders to the results. Furthermore, none of these previous studies had a validation cohort to evaluate replicability of results on the same analysis platform. Additionally the methods used in prior analyses were based on in‐house methodologies, mostly ELISA, and not from a clinically validated shared resource that others could replicate their results against, limiting the generalizability. Second, there are concomitant CSF and plasma measurements to evaluate both peripheral and CNS‐related inflammation changes in this report. Third, this study has multiple internal and external validity checks to account for quality of data and measurements. Fourth, we were able go beyond single analyte associations to meaningfully assess multiple analytes and narrow our focus to key activated biological pathways. Fifth, we could assess the relevance of functional pathways identified in CSF and plasma across two different cohorts and for their activation in the AD brain tissue as well and validate these results.
Despite these strengths both Type I and II errors have to be considered in this study where we do not confirm some plasma analyte results reported previously, while other analytes (e.g., YKL‐40) were not analyzed in the RBM MAP. Our results pass a stringent multiple comparisons cut‐off but it is possible that with weaker enrichment patterns other analytes of significance may become more salient with increased sample sizes. Lack of neuropathologic confirmation of diagnosis also limits our understanding of the role for mixed pathology.
Conclusion
We report key inflammation‐related pathway activations related to the TNF signaling pathway and complement and coagulation cascade were conserved in plasma, CSF and brain tissue in symptomatic AD. A cell protective rather than the proinflammatory analyte profile predominates in the CSF at the MCI‐stage of AD in relation to neurodegeneration markers. Exploring their modulation toward therapeutic outcomes could be of clinical interest.
Author Contributions
1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
J.A.P.: 1) A, B, C 2) A, B 3) A
B.T.L.: 1) A 2) C 3) B
S.M.: 2) A, B 3) B
J.B.: 2) A, B 3) B
L.M.B.: 1) B, 2) C, 3) B
S.M.R.: 1) B, 2) C, 3) B
M.C.: 1) B, 2) C, 3) B
J.B.L.: 1) B, 2) C, 3) B
Conflict of Interest
Full Financial Disclosures of all Authors for the Past Year:
Jagan A Pillai has obtained funding for research from the National Institutes of Health, Alzheimer’s Association and Keep Memory Alive foundation. Bruce T Lamb has received honoraria or consulting fees from Eli Lilly, Amgen and Eisai and research funding from the National Institutes of Health, US Department of Defense, the Alzheimer’s Association and the BrightFocus Foundation. Stephen M. Rao has received honoraria, royalties or consulting fees from: Biogen, Genzyme, Novartis, American Psychological Association, International Neuropsychological Society and research funding from the National Institutes of Health, US Department of Defense, National Multiple Sclerosis Society, CHDI Foundation, Biogen, and Novartis. James B. Leverenz has received consulting fees from Axovant, GE Healthcare, Navidea Biopharmaceuticals, Takeda, and Grant support from Alzheimer’s Association, Alzheimer’s Drug Discovery Foundation, Biogen, Genzyme/Sanofi, Lundbeck, Michael J Fox Foundation, National Institute of Health. Sean Maxwell, James Bena, Lynn Bekris, Mark Chance declared no conflict of interest.
Supporting information
Figure S1. Plot noting analytes that met lower limit of detection in at least 50% of subjects.
Figure S2. Cluster plot noting correlation coefficients of inflammatory analytes in plasma and CSF with AD biomarkers CSF t‐tau, p‐tau, and Aβ42 in the discovery cohort.
Figure S3. Scatter plot of individual analyte correlations between ADNI and discovery cohort.
Table S1. Additional clinical and environmental characteristics of the discovery cohort.
Table S2. Summary statistics of analyte markers in plasma and CSF.
Table S3. Agreement statistics for correlations between inflammatory analytes and AD biomarkers between ADNI and discovery cohorts.
Table S4. Analyte subgroup analysis with most significant inflammatory analytes in the ADNI cohort.
Table S5. Significant KEGG pathways enriched in synergetic analyte analysis in discovery cohort.
Acknowledgment
We thank the patients and families who took part in the discovery cohort at Cleveland Clinic Lou Ruvo Center for Brain Health.
Data collection and sharing for this project was funded by the Alzheimer's Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904) and DOD ADNI (Department of Defense award number W81XWH‐12‐2‐0012). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol‐Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann‐La Roche Ltd and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. The Canadian Institutes of Health Research is providing funds to support ADNI clinical sites in Canada. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Pillai JA, Maxwell S, Bena J, et al; for the Alzheimer’s Disease Neuroimaging Initiative . Key inflammatory pathway activations in the MCI stage of Alzheimer’s disease. Ann Clin Transl Neurol. 2019;6:1248–1262. 10.1002/acn3.50827
Funding information
Funded by 2014‐NIRG‐305310 Alzheimer’s Association, NIA K23AG055685‐01, NIA R01AG022304‐12, the Jane and Lee Seidman fund and Keep Memory Alive scholar grant.
Funding Statement
This work was funded by National Institute on Aging grants K23AG055685-01 , R01AG022304-12, and U01 AG024904; Jane and Lee Seidman fund grant ; Alzheimer's Association grant 2014-NIRG-305310 ; Department of Defense grant W81XWH-12-2-0012; National Institute of Biomedical Imaging and Bioengineering grant ; AbbVie grant ; BioClinica grant ; Biogen grant ; Bristol‐Myers Squibb Company grant ; Eisai grant ; Elan grant ; Eli Lilly and Company grant ; F. Hoffmann‐La Roche Ltd grant ; Genentech grant ; Fujirebio grant ; GE Healthcare grant ; Johnson & Johnson grant ; Merck grant ; Meso Scale Diagnostics grant ; Novartis grant ; Pharmaceuticals grant ; Pfizer grant ; Takeda Pharmaceutical Company grant ; Health Research grant ; National Institutes of Health grant ; Northern California Institute for Research and Education grant ; University of Southern California grant .
References
- 1. Wyss‐Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med 2006;12:1005–1015. [DOI] [PubMed] [Google Scholar]
- 2. Holtzman DM, Mandelkow E, Selkoe DJ. Alzheimer disease in 2020. Cold Spring Harb Perspect Med 2012;1:a011585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bettcher BM, Kramer JH. Longitudinal inflammation, cognitive decline, and Alzheimer’s disease: a mini‐review. Clin Pharmacol Ther 2014;96:464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Perry VH, Cunningham C, Holmes C. Systemic infections and inflammation affect chronic neurodegeneration. Nat Rev Immunol 2007;7:161–167. [DOI] [PubMed] [Google Scholar]
- 5. Holmes C, Cunningham C, Zotova E, et al. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009;73:768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Song F, Poljak A, Smythe GA, Sachdev P. Plasma biomarkers for mild cognitive impairment and Alzheimer's disease. Brain Res Rev 2009;61:69–80. [DOI] [PubMed] [Google Scholar]
- 7. Lucin KM, Wyss‐Coray T. Immune activation in brain aging and neurodegeneration: too much or too little? Neuron 2009;15:110–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Craig‐Schapiro R, Perrin RJ, Roe CM, et al. YKL‐40: a novel prognostic fluid biomarker for preclinical Alzheimer's disease. Biol Psychiatry 2010;15:903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Réaux‐Le Goazigo A, Van Steenwinckel J, Rostène W et al. Current status of chemokines in the adult CNS. Prog Neurogibol 2013;104:67–92. [DOI] [PubMed] [Google Scholar]
- 10. Hye A, Riddoch‐Contreras J, Baird AL, et al. Plasma proteins predict conversion to dementia from prodromal disease. Alzheimer’s Dement 2014;10:799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Westin K, Buchhave P, Nielsen H, et al. CCL2 is associated with a faster rate of cognitive decline during early stages of Alzheimer's disease. PLoS ONE 2012;7:e30525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tosto G, Reitz C. Genome‐wide association studies in Alzheimer's disease: a review. Curr Neurol Neurosci Rep 2013;13:381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wyss‐Coray T, Rogers J. Inflammation in Alzheimer disease‐a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med 2012;2:a006346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Heneka MT, Carson MJ, El Khoury J, et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol 2015;14:388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Swardfager W, Lanctôt K, Rothenburg L, et al. A meta‐analysis of cytokines in Alzheimer's disease. Biol Psychiatry 2010;68:930–941. [DOI] [PubMed] [Google Scholar]
- 17. Saleem M, Herrmann N, Swardfager W, et al. Inflammatory markers in mild cognitive impairment: a meta‐analysis. J Alzheimers Dis 2015;47:669–679. [DOI] [PubMed] [Google Scholar]
- 18. Lai K, Liu CS, Rau A, et al. Peripheral inflammatory markers in Alzheimer's disease: a systematic review and meta‐analysis of 175 studies. J Neurol Neurosurg Psychiatry 2017;88:876–882. [DOI] [PubMed] [Google Scholar]
- 19. Albert MS, DeKosky ST, Dickson D, et al. The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging‐Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s Dement 2011;7:270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Folstein MF, Folstein SE, McHugh PR. "Mini‐mental state". A practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res 1975;12:189–198. [DOI] [PubMed] [Google Scholar]
- 21. Wechsler D. Wechsler memory scale—revised. San Antonio, TX: The Psychological Corporation, 1987. [Google Scholar]
- 22. Morris JC. The Clinical Dementia Rating (CDR): current version and scoring rules. Neurology 1993;43:2412–2414. [DOI] [PubMed] [Google Scholar]
- 23. Mattis S. Dementia rating scale: professional manual. Odessa, FL: Psychological Assessment Resources, 1988. [Google Scholar]
- 24. Shaw LM, Vanderstichele H, Knapik‐Czajka M, et al. Alzheimer's disease neuroimaging initiative. Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 2009;65:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Olsson A, Vanderstichele H, Andreasen N, et al. Simultaneous measurement of beta‐amyloid(1–42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clin Chem 2005;51:336–345. [DOI] [PubMed] [Google Scholar]
- 26. Khan W, Aguilar C, Kiddle SJ, et al. A subset of cerebrospinal fluid proteins from a multi‐analyte panel associated with brain atrophy, disease classification and prediction in Alzheimer's disease. PLoS ONE 2015;10:e0134368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Biomarkers Consortium ADNI CSF Targeted Proteomics Project ‐ Data Primer Version 28 Dec 2011. Downloaded from "http://adni.loni.usc.edu/methods/" on December 1st 2018.
- 28. The Gene Ontology Consortium . Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res 2017;45:D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nibbe RK, Koyutürk M, Chance MR. An integrative ‐omics approach to identifyfunctional sub‐networks in human colorectal cancer. PLoS Comput Biol 2010;6:e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Maxwell S, Chance MR, Koyutürk M. Linearity of network proximity measures: implications for set‐based queries and significance testing. Bioinformatics 2017;33:1354–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Chatraryamontri A, Oughtred R, Boucher L, et al. The BioGRID interaction database: 2017 update. Nucleic Acids Res 2017;45:D369–D379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nibbe RK, Markowitz S, Myeroff L, et al. Discovery and scoring of protein interaction subnetworks discriminative of late stage human colon cancer. Mol Cell Proteomics 2009;8:827–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chuang HY, Lee E, Liu YT, et al. Network‐based classification of breast cancer metastasis. Mol Syst Biol 2007;3:140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res 2017;45:D362–D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Langfelder P, Zhang B, Horvath S. Defining clusters from a hierarchical cluster tree: the Dynamic Tree Cut package for R. Bioinformatics 2008;24:719–720. [DOI] [PubMed] [Google Scholar]
- 36. Parikshak NN, Luo R, Zhang A, et al. Integrative functional genomic analyses implicate specific molecular pathways and circuits in autism. Cell 2013;155:1008–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zambon AC, Gaj S, Ho I, et al. GO‐Elite: a flexible solution for pathway and ontology over‐representation. Bioinformatics 2012;28:2209–2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kanehisa M, Sato Y, Kawashima M, et al. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res 2016;44:D457–D462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor‐alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol 1999;19:223–230. [DOI] [PubMed] [Google Scholar]
- 40. Lancet D, Safran M, Olender T, et al.GeneCards tools for combinatorial annotation and dissemination of human genome information GIACS Conference on Data in Complex Systems April, 2008. www.genecards.org.
- 41. Sanchez‐Ramos J, Song S, Cao C, Arendash G. The potential of hematopoietic growth factors for treatment of Alzheimer's disease: a mini‐review. BMC Neurosci 2008;9(Suppl 2):S3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhao LR, Singhal S, Duan WM, et al. Brain repair by hematopoietic growth factors in a rat model of stroke. Stroke 2007;38:2584–2591. [DOI] [PubMed] [Google Scholar]
- 43. Laske C, Stellos K, Stransky E, et al. Decreased plasma and cerebrospinal fluid levels of stem cell factor in patients with early Alzheimer's disease. J Alzheimers Dis 2008;15:451–460. [DOI] [PubMed] [Google Scholar]
- 44. Li B, Gonzalez‐Toledo ME, Piao CS, et al. Stem cell factor and granulocyte colony‐stimulating factor reduce β‐amyloid deposits in the brains of APP/PS1 transgenic mice. Alzheimers Res Ther 2011;3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nilsson LN, Bales KR, DiCarlo G, et al. Alpha‐1‐antichymotrypsin promotes beta‐sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer's disease. J Neurosci 2001;21:1444–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Akhter H, Huang WT, van Groen T, et al. A small molecule inhibitor of plasminogen activator inhibitor‐1 reduces brain amyloid‐β Load and improves memory in an animal model of Alzheimer's disease. J Alzheimers Dis 2018;64:447–457. [DOI] [PubMed] [Google Scholar]
- 47. Shi Q, Chowdhury S, Ma R, et al. Complement C3 deficiency protects against neurodegeneration in aged plaque‐rich APP/PS1 mice. Sci Transl Med 2017;9:eaaf6295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cortes‐Canteli M, Zamolodchikov D, Ahn HJ, et al. Fibrinogen and altered hemostasis in Alzheimer's disease. J Alzheimers Dis 2012;32:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Duits FH, Hernandez‐Guillamon M, Montaner J, et al. Matrix metalloproteinases in Alzheimer's disease and concurrent cerebral microbleeds. J Alzheimers Dis 2015;48:711–720. [DOI] [PubMed] [Google Scholar]
- 50. Hohman TJ, Bell SP, Jefferson AL. Alzheimer’s disease neuroimaging initiative. The role of vascular endothelial growth factor in neurodegeneration and cognitive decline: exploring interactions with biomarkers of Alzheimer disease. JAMA Neurol 2015;72:520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zuliani G, Cavalieri M, Galvani M, et al. Markers of endothelial dysfunction in older subjects with late onset Alzheimer's disease or vascular dementia. J Neurol Sci 2008;15:164–170. [DOI] [PubMed] [Google Scholar]
- 52. McAlpine FE, Tansey MG. Neuroinflammation and tumor necrosis factor signaling in the pathophysiology of Alzheimer’s disease. J Inflamm Res 2008;1:29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sutphen CL, Jasielec MS, Shah AR, et al. Longitudinal cerebrospinal fluid biomarker changes in preclinical Alzheimer disease during middle age. JAMA Neurol 2015;72:1029–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Plot noting analytes that met lower limit of detection in at least 50% of subjects.
Figure S2. Cluster plot noting correlation coefficients of inflammatory analytes in plasma and CSF with AD biomarkers CSF t‐tau, p‐tau, and Aβ42 in the discovery cohort.
Figure S3. Scatter plot of individual analyte correlations between ADNI and discovery cohort.
Table S1. Additional clinical and environmental characteristics of the discovery cohort.
Table S2. Summary statistics of analyte markers in plasma and CSF.
Table S3. Agreement statistics for correlations between inflammatory analytes and AD biomarkers between ADNI and discovery cohorts.
Table S4. Analyte subgroup analysis with most significant inflammatory analytes in the ADNI cohort.
Table S5. Significant KEGG pathways enriched in synergetic analyte analysis in discovery cohort.