

Abstract
Objective
To analyze clinical phenotypes associated with KCNC1 variants other than the Progressive Myoclonus Epilepsy‐causing variant p.Arg320His, determine the electrophysiological functional impact of identified variants and explore genotype‐phenotype‐physiological correlations.
Methods
Ten cases with putative pathogenic variants in KCNC1 were studied. Variants had been identified via whole‐exome sequencing or gene panel testing. Clinical phenotypic data were analyzed. To determine functional impact of variants detected in the Kv3.1 channel encoded by KCNC1, Xenopus laevis oocyte expression system and automated two‐electrode voltage clamping were used.
Results
Six unrelated patients had a Developmental and Epileptic Encephalopathy and a recurrent de novo variant p.Ala421Val (c.1262C > T). Functional analysis of p.Ala421Val revealed loss of function through a significant reduction in whole‐cell current, but no dominant‐negative effect. Three patients had a contrasting phenotype of Developmental Encephalopathy without seizures and different KCNC1 variants, all of which caused loss of function with reduced whole‐cell currents. Evaluation of the variant p.Ala513Val (c.1538C > T) in the tenth case, suggested it was a variant of uncertain significance.
Interpretation
These are the first reported cases of Developmental and Epileptic Encephalopathy due to KCNC1 mutation. The spectrum of phenotypes associated with KCNC1 is now broadened to include not only a Progressive Myoclonus Epilepsy, but an infantile onset Developmental and Epileptic Encephalopathy, as well as Developmental Encephalopathy without seizures. Loss of function is a key feature, but definitive electrophysiological separation of these phenotypes has not yet emerged.
Keywords: Encephalopathy, epilepsy, KCNC1
Introduction
Humans have over 70 genes encoding for different potassium channel subunits. Pathogenic variants in many of these genes have been associated with a variety of neurological, cardiac, and other diseases. Voltage‐gated potassium channels are important in setting membrane resting potential, regulating firing patterns and modifying action potential duration and neurotransmitter release.1, 2 Mutations in genes encoding voltage‐gated potassium channels have been implicated in several conditions including spinocerebellar ataxia, paroxysmal movement disorders, and various epilepsies.3, 4, 5
KCNC1, which encodes the Kv3.1 subunit, was not known to be associated with human disease until the recent description of Myoclonus Epilepsy and Ataxia due to KCNC1 mutation (MEAK).6, 7 MEAK is a type of Progressive Myoclonus Epilepsy (PME) due to a specific recurrent heterozygous missense mutation, p.Arg320His. Kv3.1 expression is largely restricted to the central nervous system, with predominant expression in GABAergic interneurons.8 When analyzed in vitro, the PME causing mutation was shown to have a dominant‐negative loss of function effect.6 Subsequently, a truncating variant in KCNC1, p.Arg339X, was described in three related individuals with intellectual disability (ID) and dysmorphic features without epilepsy.9 This raises the possibility of a wider spectrum of phenotypes associated with pathogenic KCNC1 variants. We thus sought cases with other variants in KCNC1 and analyzed the clinical, electrographic, and radiological features.
Here, we describe an emerging genotype‐phenotype correlation with pathogenic variants in KCNC1. Analysis of ten patients with putative pathogenic variants in KCNC1 facilitated identification of nine patients with novel pathogenic KCNC1 variants. Six of these patients carry a recurrent KCNC1 variant and present with a developmental and epileptic encephalopathy (DEE).10 Three patients had a contrasting phenotype of developmental encephalopathy without seizures and different KCNC1 variants. The cellular electrophysiological impact of these variants is also presented.
Methods
Ascertainment
We studied 10 cases with putative pathogenic variants in KCNC1. Eight cases were referred to the University of Melbourne by direct personal correspondence given our previously published work. Two cases were identified employing the web‐based matching platform DECIPHER.11
Variant identification
Variants were identified via clinical whole‐exome sequencing (WES) or gene panel testing via the subjects’ home institution. The variant in one patient (patient 1) was identified through research WES testing as part of the Deciphering Developmental Disorders study.12 Consent for genetic testing was obtained through treating institutions using local protocols.
Functional analysis
Mutagenesis and RNA preparation
Mutations were introduced into human KCNC1 cDNA encoding transcript 1 (NM_001112741; Kv3.1b) or transcript 2 (NM_004976; Kv3.1a) cloned into a pCMV‐Entry vector that were obtained from OriGene Technologies using QuikChange Lightning Site‐Directed Mutagenesis Kit (Agilent Technologies) according to the manufacturer’s instructions. Insertion of mutation was confirmed and additional mutations excluded by Sanger sequencing. cRNA was prepared using the T7 mMessage mMachine kit from Ambion (Austin, TX).
Oocyte preparation and injection
Xenopus oocytes (Dumont stage V or VI) were surgically removed from Xenopus laevis and prepared as described previously.13 Oocytes were kept in ND96 solution and stored at 17°C. Equal volumes (50 nL) of cRNAs with the concentrations adjusted to 0.2–0.5 μg/μL were injected using Robooinject® (Multi Channel Systems) in the same batch of oocytes plated in 96 well‐plates. Recordings were performed in parallel two days after injection. Amplitudes of interest for all currents recorded on the same day were normalized to the mean value obtained for the KV3.1 wild‐type on that day, so that the normalized data from different experiments could be pooled.
Automated oocyte two‐microelectrode voltage clamp
Potassium currents in oocytes were recorded at room temperature (20–22°C) on Roboocyte2® (Multi Channel Systems) using prepulled and prepositioned intracellular glass microelectrodes with a resistance of 0.3–1 MΩ when filled with 0.5 mol/L KCl/ 1.5 mol/L KAc. Oocytes were held at − 90 mV and perfused with bath solution containing (in mmol/L) 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2 and 5 HEPES, pH 7.5. Currents were sampled at 5 kHz. For the analysis of channel activation, we used 0.5‐s long depolarizing steps applied at 10 mV increments, from − 60 mV to + 60 mV, followed by a step to − 90 mV for 0.5 sec to analyze tail currents.6
Data analysis
Electrophysiological recordings from the Roboocyte2® were extracted using Roboocyte2+ (Multi Channel Systems) and analyzed using AxoGraph (AxoGraph Scientific, Sydney, Australia). Plotting of graphs and statistical analysis was performed using GraphPad Prism 7 (GraphPad Software, La Jolla, CA). Maximum current amplitudes were compared at the end of a 0.5‐s test pulse to + 60 mV. The voltage‐dependence of channel activation was derived from tail current amplitudes recorded at − 90 mV. A Boltzmann function was fit to the current‐voltage relationships, I(V) = I max/(1 + exp[(V − V 0.5)/k]) + C, where I max is the maximum tail current amplitude at test potential V, V 0.5 the half‐maximal activation potential, k a slope factor reflecting characteristics of voltage‐dependent channel gating and C a constant. All data are shown as mean values ± SEM. Statistically significant differences were determined using one way ANOVA with Dunnett’s or Tukey’s multicomparisons correction or with non‐parametric Mann–Whitney test.
Results
KCNC1 variants associated with developmental and epileptic encephalopathy (DEE)
We identified six unrelated patients with DEE and a recurrent missense variant p.Ala421Val (c.1262C > T). All were confirmed de novo variants. The variant p.Ala421Val is absent from the gnomAD database and predicted as deleterious using in silico methods. The six patients were from the United Kingdom, Germany, France, and North America. Patient 1 had a family history of epilepsy on both maternal and paternal sides; the remaining five patients had no significant family history. All presented between birth and 10 months with predominantly generalized seizure types (Table 1); myoclonic seizures were reported in all but one case (patient 2).
Table 1.
Clinical features of patients with Developmental and Epileptic Encephalopathy and recurrent KCNC1 missense variant p.Ala421Val.
| Patient | 1 | 2 | 3 | 4 | 5 | 6 |
|---|---|---|---|---|---|---|
| Inheritance | De novo | De novo | De novo | De novo | De novo | De novo |
| Gender | F | M | F | M | F | M |
| Age at onset | Birth | 3 months | 10 months | 9 months | 9.5 months | 5 months |
| Seizure types |
Myoclonic Atypical absence GTCS |
Focal to bilateral tonic clonic Focal |
GTCS Focal Myoclonic |
Myoclonic Atypical absence Atonic GTCS |
Myoclonic |
Myoclonic GTCS Absence with myoclonus Focal to bilateral tonic clonic |
| Response to treatment | Refractory | Response to LMT and VPA |
Refractory then response to VPA Clobazam Stiripentol |
Refractory to medication, response to VNS | Response to LEV and LMT | Response to VPA |
| Age at walking | 2.5 years | 3 years | 2.5 years | 2 years | 2.5 years | 3 years |
| Intelligible speech | Yes | Yes | Yes | Yes | Yes | Yes |
| Cognitive Function | Severe ID | Moderate‐severe ID | Moderate‐severe ID | Moderate‐severe ID | Global developmental delayb | Moderate ID |
| Neurological Features | Ataxiaa | Ataxiaa, severe hypotonia | Nil | Ataxiaa, mild decreased power, hypotonia | Ataxiaa, mild athetosis, hypotonia | Unsteady gait, hypotonia |
| Co‐morbidities | Behavioural disturbance |
EsophagitisSleeping disturbance Failure to thrive Pectus excavatum |
Nil |
GERD, Chronic diarrhoea, Failure to thrive, Central sleep apnoea, Autism Functional B cell deficiency |
Nil |
Sleep disturbance Hypermobile joints Stereotypies |
| Clinical course | Refractory seizures age 16 | Monthly seizures at 3 years | Seizure control improved age 15 | <1 seizure every 3 months post VNS | Seizures every 2–3 months | Seizure control improved at age 9 |
Abbreviations: GERD, gastro‐esophageal reflux disease; ID, intellectual disability; GTCS, generalized tonic clonic seizures; LEV, levetiracetam; LMT, lamotrigine; VNS, vagal nerve stimulator; VPA, sodium valproate.
Ataxia, where noted, was mild and non‐progressive.
Child too young for grading of intellectual disability.
Developmental delay without regression was seen in all patients, however, they were able to walk and achieved intelligible speech. Associated neurological features included mild non‐progressive ataxia (n = 4), as well as hypotonia (n = 4). Three patients (3, 4, and 6) exhibited similar dysmorphic features including a large mouth, smooth philtrum, up‐slanting palpebral fissure and dental enlargement (Fig. 1). Three patients were noted to have a combination of respiratory co‐morbidities (central sleep apnoea, sleep disturbance) and musculoskeletal abnormalities (pectus excavatum, hypermobile joints). Two of these patients also had gastrointestinal co‐morbidities.
Figure 1.
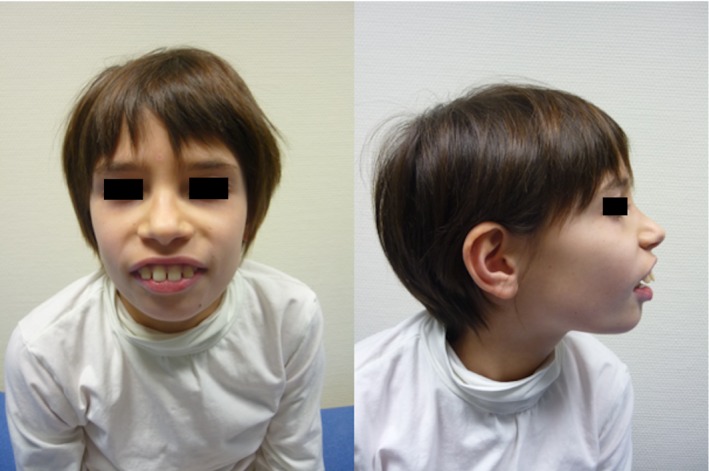
Facial features of patient 3 showing a large mouth, smooth philtrum, up‐slanting palpebral fissure, and dental enlargement.
All patients had abnormal EEGs. Five patients demonstrated slowing of background rhythms. Four patients demonstrated interictal generalized spike‐and‐wave discharges, and EEG in the other two patients captured electroclinical seizures with bi‐frontal onset. Myoclonus without electrographic correlate was noted in two patients, and normalization of electrographic abnormalities over a period of years was noted in two patients. MRI Brain imaging showed no specific abnormalities.
All patients had moderate‐severe intellectual disability. Long‐term seizure frequency and response to treatment was variable. Two patients achieved improved seizure control in late childhood (at 9 and 15 years of age, respectively) with combination antiepileptic therapy, and one patient had a good clinical response to vagal nerve stimulator insertion. The remaining three patients had ongoing refractory seizures despite multiple antiepileptic medications.
A single patient with DEE and KCNC1 missense variant p.Ala513Val (c.1538C > T) was also identified. The electro‐clinical phenotype was of Epilepsy of Infancy with Focal Migrating Seizures (EIFMS), and the patient died at 6 months of age from cardiorespiratory arrest. Recognized genetic causes of EIFMS were not detected on a 77 gene panel testing. The KCNC1 variant was confirmed de novo but it appears once in gnomAD, and thus was considered a variant of uncertain significance.
KCNC1 variants associated with developmental encephalopathy (DE) without seizures
Three patients, including a mother‐son pair, presented with a developmental encephalopathy without seizures (Table 2). The mother‐son pair with a truncating variant p.Gln492X (c.1474C > T) had mild‐moderate intellectual disability, no dysmorphic features and the son developed a nephroblastoma at four years of age. No other causative variants were identified on WES, The third case had missense variant p.Arg317His (c.950G > A), and presented with mild intellectual disability and autism. The patient had never been noted to have a seizure. A 23‐hour EEG video to investigate episodes of staring revealed rare irregular generalized spike wave discharges, with no clinical seizures. Brain MRI revealed cerebellar and posterior pontine atrophy. Trio WES also revealed a maternally inherited MAOA variant.
Table 2.
Clinical features of patients with KCNC1 variants and Developmental Encephalopathy without seizures.
| Patient | 1 | 2 | 3 |
|---|---|---|---|
| KCNC1 variant | p.Arg317His | p.Gln492X | p.Gln492X |
| Gender | M | F | M |
| Cognitive Function | Mild ID | Mild‐Moderate ID | Mild‐moderate ID |
| Seizures | No | No | No |
| Dysmorphic Features | Nil | Nil | Nil |
| Neurological Features | Autism | Nil | Nil |
| MRI | Cerebellar and posterior pontine atrophy | Not performed | Not performed |
| EEG | Rare generalized spike‐wave | Not performed | Not performed |
Functional analysis
To determine functional impact of variants detected in the Kv3.1 channel (Fig. 2A), encoded by KCNC1, we used Xenopus laevis oocyte expression system and automated two‐electrode voltage clamping. Current traces obtained from a set of depolarizing pulses from − 60 to + 60 mV (Fig. 2B) revealed a clear loss of function for all but the Ala513Val variant, being the variant observed once in gnomAD.
Figure 2.
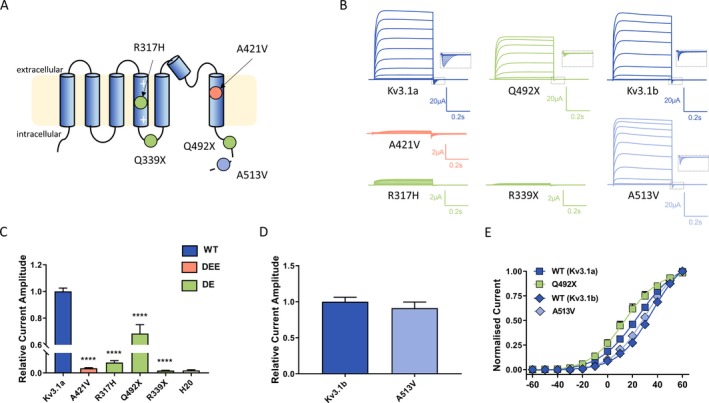
Functional expression of Kv3.1 variants in Xenopus laevis oocytes (A) Schematic of the Kv3.1 channel with putative positions of variants reported in this study (Orange ‐ Developmental and Epileptic Encephalopathy [DEE] variant; Green ‐ Developmental Encephalopathy variants [DE]; Light blue ‐ variant of uncertain significance). The dashed line shows the distal part of the C‐terminus that is present only in the longer transcript variant (Kv3.1b) indicating that the A513V variant is only found in this transcript. (B) Representative traces of whole‐cell currents recorded from Xenopus laevis oocytes injected with the same amount of cRNA encoding Kv3.1 wild‐type (Kv3.1a and Kv3.1b) and different variants during 0.5 sec voltage steps ranging from − 60 mV to + 60 mV. Insets show blown up tail currents, which were analyzed to generate conductance‐voltage relationships for WT and mutant channels shown in E. (C, D) Current amplitudes analyzed at the end of the voltage step to + 60 mV and normalized to the mean current amplitude of the corresponding WT recorded on the same day; Kv3.1a (n = 136), A421V (n = 62), R317H (n = 32), Q492X (n = 30), R339X (n = 48), and water (n = 35); Kv3.1b (n = 15), A513V (n = 17). ****P < 0.0001, using one‐way ANOVA with Dunnett’s multiple comparisons test (C). Mann–Whitney non‐parametric test (D) revealed P = 0.5. (E) Conductance‐voltage relationships for the WT transcripts and variants showing current amplitudes above the background level. V0.5 and k values were as follows: for Kv3.1a 23 ± 2 mV, 12.8 ± 0.6 (n = 37), for Kv3.1b 33.8 ± 1.1 mV, 12.80 ± 1.04 (n = 18), for Q492X 15 ± 3 mV, 12.7 ± 0.8 (n = 19), and for A513V 29 ± 2 mV, 11.6 ± 0.6 (n = 21); V0.5 Kv3.1a vs V0.5 Kv3.1b, ANOVA with Tukey’s multiple comparisons test.
Current amplitudes analyzed at the end of the current trace were obtained at maximum voltage (+60 mv) and normalized for all variants to the corresponding wild type (Kv3.1a for p.Ala421Val, p.Arg317His, p.Gln492X and p.Arg339X and Kv3.1b for p.Ala513Val) recorded on the same day, therefore providing the relative current amplitude (Fig. 2C and D). Voltage dependence of activation determined from tail current amplitudes recorded at − 90 mV for the variants showing current amplitudes above the background level (p.Gln492X and p.Ala513Val) showed no significant difference compared to the corresponding WT channels (Fig. 2E).
To determine potential dominant‐negative effect, we performed coexpression experiments, where both WT and mutant cRNA of the same concentration were injected at a 1:1 ratio. Two variants that showed very small or no currents, (Arg317His and Arg339X) caused a significant reduction compared to the WT current amplitude indicating a dominant‐negative effect (Fig. 3A and 3). For the coexpression of the Gln492X with the WT, the normalized current amplitudes were as follows: WT + H20 – 1.00 ± 0.03 (n = 111) and WT + Gln492X – 1.02 ± 0.12 (n = 21). In both cases, the injected amount of WT cRNA was the same, so that the resulting current amplitude of the coexpression should be the sum of the expressed WT + H2O (1.00 ± 0.03) and half of the amplitude recorded for the Gln492X alone (0.7 ± 0.1; Fig. 2C), that is, with a value of about 1.3. Since this predicted value is larger than the recorded one this might suggest the dominant‐negative effect of the Gln492X.
Figure 3.
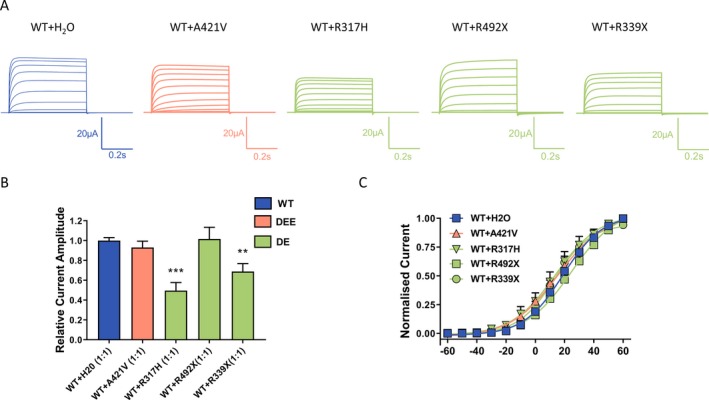
Dominant‐negative effect of Kv3.1 variants. (A) Representative current traces recorded from Xenopus laevis oocytes injected with the same amount of cRNA encoding Kv3.1a wild‐type with addition of either H2O or the same amount cRNA encoding Kv3.1 variants in a 1:1 ratio. (B) Current amplitudes analyzed at the end of the voltage step to + 60 mV and normalized to the mean current amplitude of WT + H2O recorded on the same day revealed a significant reduction for R317H and R339X but not for the A421V coexpression, ***P < 0.001, **P < 0.01 using one‐way ANOVA with Dunnett’s multiple comparisons test; WT + H2O (n = 111), WT + A421V (n = 62); WT + R317H (n = 11); WT + Q492X (n = 21); WT + R339X (n = 17). (C) Conductance‐voltage relationships of the WT and its coexpressions with A421V, R317H and R339X. V0.5 and slope factor (k) values were as follows: for WT + H2O 18.2 ± 1.4 mV, 12.4 ± 0.4 (n = 27), for WT + A421V 16.6 ± 1.8, 15.2 ± 0.4 (n = 36), for WT + R317H 21.1 ± 4.5 mV, 17.8 ± 1.9 (n = 8), for WT + Q492X 25 ± 3 mV, 13.9 ± 0.5 (n = 12), and for WT + R339X 15.8 ± 3.1 mV, 12.2 ± 1.1 (n = 15).
The conductance‐voltage relationships for Ala421Val + WT, Arg317His + WT, Gln492X + WT, and Arg339X + WT did not differ from the wild type alone (Fig. 3C). With no changes seen in the voltage dependence of activation, the observed reduction in current amplitudes may be explained by other mechanisms such as reduced number of mutant channels reaching the membrane or reduced open probability of these channels.
Discussion
We describe the phenotypic features of 10 patients with putative pathogenic variants in KCNC1, and also present the electrophysiological impact of these variants. These are summarized in Table 3 and compared to the previously described recurrent p.Arg320His variant that causes progressive myoclonus epilepsy.6,7
Table 3.
Phenotypes and electrophysiological changes associated with KCNC1 variants.
| Phenotype | Progressive myoclonus epilepsy | Developmental and epileptic encephalopathy | Developmental encephalopathy without seizures | ||
|---|---|---|---|---|---|
| KCNC1 variant | p.Arg320His | p.Ala421Val | p.Arg339X | p.Gln492X | p.Arg317His |
| Seizure types | Myoclonic tonic‐clonic (infrequent) | Myoclonic other generalized focal | Nil | Nil | Nil |
| Cognitive function | Normal with mild late decline in some | Moderate‐severe ID | Moderate ID | Mild‐moderate ID | Mild ID |
| Electrophysiological characteristics | |||||
| Whole‐cell current | Marked reduction | Marked reduction | Marked reduction | Moderate reduction | Marked reduction |
| Current‐voltage relationship | Gain of function | No change | No change | No change | No change |
| Dominant negative effect | Yes | No | Yes | Unknowna | Yes |
Not assessed as current with mutant was not markedly reduced (see Methods).
Six patients carry the recurrent KCNC1 missense variant p.Ala421Val and present with a DEE. These are the first reported cases of DEE due to KCNC1 mutation, and thus broaden the spectrum of phenotypes associated with KCNC1 missense variants to include infantile onset DEE. The Kv3 subfamily exhibits very fast activation and deactivation kinetics, enabling the generation of very high frequency action potentials.1 Kv3.1 expression is largely restricted to the central nervous system, predominantly in inhibitory GABAergic interneurons.8 An impact of KCNC1 variants on early development is supported by the fact that embryonic expression of Kv3.1 channels in animal models is well‐established,14, 15, 16 with gradual increases in transcript levels noted after birth in murine models, especially postnatal days 8–14.17 Embryonic expression when the fast spiking phenotype is not present suggests that these channels may also have other critical roles in early development, including in cell proliferation, migration, and regulation of neuronal and glial properties.17, 18
In this cohort of patients with DEE and KCNC1 variant p.Ala421Val, onset was within the first 10 months of life, with predominantly generalized seizure types seen. Myoclonus was a prominent feature in many patients but the pattern was not that of a PME. Seizures were generally refractory to treatment, persistent developmental delay without regression and moderate‐severe intellectual disability were seen. Mild non‐progressive ataxia was a common associated neurological feature. No consistent associated co‐morbidities were noted in this cohort, however musculoskeletal, respiratory, and gastrointestinal features were seen in several patients. Functional analysis of KCNC1 variant p.Ala421Val revealed loss of function through a significant reduction in whole‐cell current, but no dominant‐negative effect. This loss of function variant may cause disinhibition due to impaired firing of the fast‐spiking GABAergic interneurons, conceptually providing a plausible mechanism for the development of seizures.
One patient had a different missense KCNC1 variant p.Ala513Val, and had the more severe clinical phenotype of EIMFS. This variant appears once in gnomAD, and a different variant at the same amino acid location (p.Ala513Thr) appears three times. This, coupled with the fact that this variant did not show definitive in vitro changes, suggests that it is unlikely to be pathogenic even though it occurred de novo and other known causes of EIMFS (e.g., SCN2A and KCNT1 variants) were not detected. Although de novo mutation is the usual mechanism in DEEs, this highlights caution is necessary in attributing pathogenesis to a novel de novo variant in an otherwise persuasive clinico‐molecular context.
The other three patients with novel variants in KCNC1 presented with a different phenotype of developmental encephalopathy (DE) without seizures. These cases add to the published literature of three related patients from one family with a different KCNC1 truncation variant p.Arg339X, with DE but without epilepsy.9 Of note, the previously reported cases also had dysmorphic features (including prognathism, dysplastic ears, protruding tongue), which our patients did not.
Interestingly, quantification of mRNA in fibroblasts from a patient with Arg339X variant showed > 50% reduction of KCNC1 transcript compared to the control data, possibly due to nonsense mediated decay (NMD).9 This effect may explain why the observed dominant‐negative effect for this mutant was not more pronounced in our experiments. The pathogenicity of this variant may thus be a consequence of both reduced expression of KCNC1 and a damaging effect of the mutant protein that escaped NMD and interacted with the WT subunits. We were not able to obtain fibroblasts from Gln492X variant carriers. However, given that this variant produces a longer channel protein yielding potassium currents reaching about 70% of the WT, we do not expect the NMD effect would be the critical mechanism. Our functional analysis of the three different KCNC1 variants associated with this phenotype of DE without seizures reveals that again all variants cause a loss of function, all with reduced whole‐cell currents. Moreover, two of the three variants caused a dominant‐negative effect, which was not seen for the DEE‐causing variant p.Ala421Val. However, p.Gln492X showed a milder effect so that it is difficult to make conclusions about the electrophysiological features which distinguish these variants from the DEE‐causing variant.
Of note, the p.Arg317His variant resulted in functional changes similar to the PME‐causing p.Arg320His variant but the phenotype seen in this patient is a DE without epilepsy. The proximity of this variant to the PME causing one is noted, (Arg317 is the third arginine in the S4 segment of Kv3.1), and somewhat challenging to reconcile. However, we did observe that the loss of function and the dominant negative effect were not as prominent as seen for p.Arg320His. Moreover, no shift in the voltage‐dependence of activation was found for the Arg317His expressed with the WT. This is consistent with the findings reported for two S4 segment disease variants at the same position in the related Kv3.3. Both of these variants caused a dominant negative effect but only the alteration of the fourth arginine lead to a hyperpolarizing shift of the activation curve.19 The third and fourth arginine have been shown critical for the voltage sensor domain conformation. Analogous arginines in the crystal structure of the Kv1.2 channel are shown to face S1 and S2 helices, and establish salt bridge interactions with the acidic amino acids.20 Interestingly, removal of the positive charge within the S4 segment also affects protein turnover and surface expression of Kv3.3.21 We may thus propose that a similar mechanism may underlie the pathogenicity of Arg317His. This patient was also noted to have a maternally inherited MAOA variant. Mutation in MAOA has been implicated in DE, autism, and behavioral disturbances,22 with no reports of EEG abnormalities. The patient’s mother was unaffected.
A lack of clear distinguishing electrophysiological features between the DEE‐causing variants and those associated with DE without epilepsy may, at least in part, be a reflection of the fact that we have not examined the variants’ impact in the context of co‐expression of other Kv3 subunits. Kv3 subunits assemble either as homomers or heteromers to form voltage‐gated tetrameric potassium ion channels. In vitro, Kv3.1 can assemble with Kv3.3 and Kv3.4, and Kv3.1 and Kv3.2 co‐immunoprecipitate in vivo.23, 24 Kv3.1 and Kv3.3 have areas of overlapping expression, especially in the cerebellum, and a degree of functional redundancy is thought to explain why KCNC1 and KCNC3 knockout mice have relatively mild phenotypes, but double knockout mice exhibit myoclonus, tremor, and ataxia.25, 26, 27 Clearly there is significant interaction between subunits, and it is feasible that this may be relevant when considering an emerging spectrum of disease seen with different mutations in KCNC1.
The description of these cases expands the phenotypic spectrum seen with mutation in KCNC1 to now not only include a type of Progressive Myoclonus Epilepsy, but to also include an infantile onset DEE, as well as a DE without seizures. A genotype‐phenotype correlation is emerging with respect to pathogenic variants in KCNC1, whereby the recurrent missense mutation p.Arg320His causes MEAK, the recurrent missense variant p.Ala421Val gives rise to the infantile onset DEE, and truncation variants result in a developmental encephalopathy without seizures. At this stage, definitive electrophysiological functional correlations for these phenotypes have not emerged, which is a target for future investigation.
Author Contributions
J.C, S.M, S.P, and S.B were involved in conception and design of the study, acquisition and analysis of data and manuscript drafting and completion. All remaining authors were involved in acquisition and analysis of data and manuscript review.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgments
We thank Katherine Helbig for facilitating referral of a case and Karen Oliver for assistance. Supported by an NHMRC Program Grant (ID: 1091593) to Prof Berkovic and Petrou.
Cameron JM, Maljevic S, Nair U, et al. Encephalopathies with KCNC1 variants: genotype‐phenotype‐functional correlations. Ann Clin Transl Neurol. 2019;6:1263–1272. 10.1002/acn3.50822
Funding information
We thank Katherine Helbig for facilitating referral of a case and Karen Oliver for assistance. Supported by an NHMRC Program Grant (ID: 1091593) to Prof Berkovic and Petrou.
Funding Statement
This work was funded by National Health and Medical Research Council grant 1091593.
References
- 1. Rudy B, McBain CJ. Kv3 channels: voltage‐gated K+ channels designed for high‐frequency repetitive firing. Trends Neurosci 2001;24:517–526. [DOI] [PubMed] [Google Scholar]
- 2. Kaczmarek LK, Aldrich RW, Chandy KG, et al. International union of basic and clinical pharmacology. C. Nomenclature and properties of calcium‐activated and sodium‐activated potassium channels. Pharmacol Rev 2017;69:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang Y, Kaczmarek LK. Kv3.3 potassium channels and spinocerebellar ataxia. J Physiol 2016;594:4677–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kohling R, Wolfart J. Potassium channels in epilepsy. Cold Spring Harb Perspect Med 2016;6:a022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rajakulendran S, Schorge S, Kullmann DM, et al. Episodic ataxia type 1: a neuronal potassium channelopathy. Neurotherapeutics 2007;4:258–66. [DOI] [PubMed] [Google Scholar]
- 6. Muona M, Berkovic SF, Dibbens LM, et al. A recurrent de novo mutation in KCNC1 causes progressive myoclonus epilepsy. Nat Genet 2015;47:39–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Oliver KL, Franceschetti S, Milligan CJ, et al. Myoclonus epilepsy and ataxia due to KCNC1 mutation: analysis of 20 cases and K+ channel properties. Ann Neurol 2017;81:677–689. [DOI] [PubMed] [Google Scholar]
- 8. Gan L, When Kaczmarek LK. When, where, and how much? Expression of the Kv3.1 potassium channel in high‐frequency firing neurons. J Neurobiol 1998;37:69–79. [DOI] [PubMed] [Google Scholar]
- 9. Poirier K, Viot G, Lombardi L, et al. Loss of function of KCNC1 is associated with intellectual disability without seizures. Eur J Hum Genet 2017;25:560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Scheffer IE, Berkovic S, Capovilla G, et al. ILAE classification of the epilepsies: position paper of the ILAE commission for classification and terminology. Epilepsia 2017;58:512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Firth HV, Richards SM, Bevan AP, et al. DECIPHER: database of chromosomal imbalance and phenotype in humans using ensembl resources. Am J Hum Genet 2009;84:524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Firth HV, Wright CF, Deciphering Developmental Disorders Study . The deciphering developmental disorders (DDD) study. Dev Med Child Neurol 2011;53:702–703. [DOI] [PubMed] [Google Scholar]
- 13. Petrou S, Ugur M, Drummond RM, et al. P2X7 purinoceptor expression in Xenopus oocytes is not sufficient to produce a pore‐forming P2Z‐like phenotype. FEBS Lett 1997;411:339–345. [DOI] [PubMed] [Google Scholar]
- 14. Perney TM, Marshall J, Martin KA, et al. Expression of the mRNAs for the Kv3.1 potassium channel gene in the adult and developing rat brain. J Neurophysiol 1992;68:756–766. [DOI] [PubMed] [Google Scholar]
- 15. Feng JJ, Morest DK. Development of synapses and expression of a voltage‐gated potassium channel in chick embryonic auditory nuclei. Hear Res 2006;216–217:116–126. [DOI] [PubMed] [Google Scholar]
- 16. Kuenzel T, Wirth MJ, Luksch H, et al. Increase of Kv3.1b expression in avian auditory brainstem neurons correlates with synaptogenesis in vivo and in vitro. Brain Res 2009;1302:64–75. [DOI] [PubMed] [Google Scholar]
- 17. Boda E, Hoxha E, Pini A, et al. Brain expression of Kv3 subunits during development, adulthood and aging and in a murine model of Alzheimer's disease. J Mol Neurosci 2012;46:606–615. [DOI] [PubMed] [Google Scholar]
- 18. Yasuda T, Cuny H, Adams DJ. Kv3.1 channels stimulate adult neural precursor cell proliferation and neuronal differentiation. J Physiol 2013;591:2579–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Minassian NA, Lin MC, Papazian DM. Altered Kv3.3 channel gating in early‐onset spinocerebellar ataxia type 13. J Physiol 2012;590:1599–1614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Long SB, Campbell EB, Mackinnon R. Voltage sensor of Kv1. 2: structural basis of electromechanical coupling. Science 2005;309:903–908. [DOI] [PubMed] [Google Scholar]
- 21. Zhao J, Zhu J, Thornhill WB. Spinocerebellar ataxia‐13 Kv3.3 potassium channels: arginine‐to‐histidine mutations affect both functional and protein expression on the cell surface. Biochem J 2013;454:259–265. [DOI] [PubMed] [Google Scholar]
- 22. Piton A, Poquet H, Redin C, et al. 20 ans apres: a second mutation in MAOA identified by targeted high‐throughput sequencing in a family with altered behavior and cognition. Eur J Hum Genet 2014;22:776–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Baranauskas G, Tkatch T, Nagata K, et al. Kv3.4 subunits enhance the repolarizing efficiency of Kv3.1 channels in fast‐spiking neurons. Nat Neurosci 2003;6:258–266. [DOI] [PubMed] [Google Scholar]
- 24. Weiser M, Vega‐Saenz de Miera E, Kentros C, et al. Differential expression of Shaw‐related K+ channels in the rat central nervous system. J Neurosci 1994;14:949–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho CS, Grange RW, Joho RH. Pleiotropic effects of a disrupted K+ channel gene: reduced body weight, impaired motor skill and muscle contraction, but no seizures. Proc Natl Acad Sci USA 1997;94:1533–1538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Espinosa F, McMahon A, Chan E, et al. Alcohol hypersensitivity, increased locomotion, and spontaneous myoclonus in mice lacking the potassium channels Kv3.1 and Kv3.3. J Neurosci 2001;21:6657–6665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. McMahon A, Fowler SC, Perney TM, et al. Allele‐dependent changes of olivocerebellar circuit properties in the absence of the voltage‐gated potassium channels Kv3.1 and Kv3.3. Eur J Neurosci 2004;19:3317–3327. [DOI] [PubMed] [Google Scholar]