

Abstract
Objective
To analyze the microRNA profile in serum of patients with Adult Onset Pompe disease (AOPD).
Methods
We analyzed the expression of 185 microRNAs in serum of 15 AOPD patients and five controls using microRNA PCR Panels. The expression levels of microRNAs that were deregulated were further studied in 35 AOPD patients and 10 controls using Real‐Time PCR. Additionally, the skeletal muscle expression of microRNAs which showed significant increase levels in serum samples was also studied. Correlations between microRNA serum levels and muscle function test, spirometry, and quantitative muscle MRI were performed (these data correspond to the study NCT01914536 at ClinicalTrials.gov).
Results
We identified 14 microRNAs that showed different expression levels in serum samples of AOPD patients compared to controls. We validated these results in a larger cohort of patients and we found increased levels of three microRNAs, the so called dystromirs: miR‐1‐3p, miR‐133a‐3p, and miR‐206. These microRNAs are involved in muscle regeneration and the expression of these was increased in patients' muscle biopsies. Significant correlations between microRNA levels and muscle function test were found.
Interpretation
Serum expression levels of dystromirs may represent additional biomarkers for the follow‐up of AOPD patients.
Introduction
Pompe disease is an autosomal recessive disorder produced by mutations in the GAA gene that encodes for the lysosomal enzyme acid alpha‐glucosidase.1 This enzyme is essential for the degradation of glycogen to glucose in lysosomes.2 Mutations in the GAA gene cause absence or deficiency of the enzyme leading to an accumulation of glycogen in the lysosomes of several tissues particularly cardiac, skeletal, and smooth muscle.3
Patients are classified as infantile (IOPD) or adult onset Pompe disease (AOPD) depending on the age at onset of symptoms.3 Classic IOPD patients have a rapidly progressive disorder characterized by hypotonia, muscle weakness, respiratory insufficiency, and hypertrophic cardiomyopathy that lead patients to death before the first year of life if untreated.4 AOPD patients have a more heterogeneous clinical picture ranging from asymptomatic HyperCKemia to weakness involving limb, axial, and respiratory muscles.5 Natural history studies have shown that AOPD is a slowly progressive disorder in which most of the patients will present moderate to severe motor disability developing respiratory insufficiency and needing noninvasive ventilation.6
Enzyme replacement therapy (ERT) with recombinant alpha‐glucosidase has changed natural history of the disease. In infantile patients, the treatment reduces the risk of death improving cardiomyopathy and respiratory function.7, 8 In AOPD patients, ERT improves and stabilizes muscle function for a period of 2–3 years and afterwards motor function slowly deteriorates9. There is an active discussion in the scientific community to know which tests are most sensitive to identify changes in muscle performance in AOPD patients after shorter periods of time10, 11. ERT is not approved for asymptomatic AOPD patients, therefore these patients are being followed up using motor function tests to identify changes in motor performance that can support starting ERT. However it is possible that irreversible muscle degeneration has already started without influencing motor function. The identification of new tools to follow‐up progression of patients is one of the hottest topics in the neuromuscular disorders field. In this sense, quantitative muscle MRI (qMRI) has revealed to be useful in several muscle disorders including Pompe disease.10, 12 However, qMRI is not available in all centers and, experience is needed to analyze it properly. Therefore, other potential biomarkers are needed and serum molecules such as microRNAs (miRNAs) could be useful.
A good biomarker should be detected in a sample that can be obtained by minimally invasive procedures for the patient. In this sense, serum or plasma is the best candidates.13 miRNAs are small noncoding RNAs that exert their effect on gene expression at the posttranscriptional level. They have a length of approximately 22 nucleotides and, in general, they inhibit protein synthesis or favor mRNA degradation by binding to the UTR 3’ regions of those mRNAs.14 Each miRNA regulates several target genes and a given gene can be regulated by multiple miRNAs.15 miRNAs regulate several processes in skeletal muscle such as muscle growth, atrophy, or regeneration. A specific group of miRNAs, known as dystromirs are distinctly expressed in serum of patients with muscle dystrophies. They have been studied in Duchenne muscle dystrophy (DMD), Becker muscle dystrophy of facio‐scapulo‐humeral muscle dystrophy (FSHD), but they have not been analyzed in serum samples of patients with AOPD.16
In this project, we studied the serum expression of 190 miRNAs, including the so called dystromirs, in a group of 35 AOPD patients and 10 controls with the following aims: (1) identify miRNAs that were up‐ or downregulated in AOPD patients; (2) investigate if they could be used as biomarkers of progression of the disease and; (3) understand the physiopathology of muscle degeneration in Pompe disease.
Material and Methods
Patient and study design
This study is part of an ongoing prospective open‐label study in which we are following up a group of symptomatic and nonsymptomatic AOPD patients using muscle function tests, muscle MRI, and blood analysis. This study has been registered in Clinicaltrials.gov (identifier NCT01914536). The study was approved by The Ethics Committee of Hospital de la Santa Creu i Sant Pau (HSCSP) in Barcelona. All participants signed an appropriate informed consent form. All patients included fulfilled the diagnostic criteria of Pompe disease established by the European Pompe Consortium.17
We defined a patient as symptomatic when we identified muscle weakness in clinical examination using the Medical Research Council score (MRC) or when Forced Vital Capacity (FVC) seated was lower than 85%. We included in this study a total of 35 patients: 21 symptomatic patients treated with ERT, nine symptomatic patients nontreated with ERT and five asymptomatic patients. Clinical and genetic data of this Pompe cohort have been previously published.10 Nontreated symptomatic patients were visited before starting ERT. We also studied 10 age and sex‐matched healthy controls.
miRNA isolation in serum sample
Serum miRNAs were isolated from serum with DANAGENE microRNA and Cell free RNA Kit (DANAGEN, Badalona, Spain) using MS2 RNA (Roche, Basel, Switzerland) as a carrier to improve the RNA isolation and exogenous controls (Unisp2,4 and 5). miRNAs were retrotranscribed into a cDNA with the miRCURY LNATM Universal RT microRNA PCR (EXIQON).
Analysis of miRNA expression in serum
miRNA expression studies consisted two different phases: discovery and validation. For the discovery phase, we selected five serum samples from four different groups: controls, asymptomatic AOPD, symptomatic nontreated AOPD and symptomatic AOPD‐treated patients. For the validation phase, we used samples from 10 healthy control and 35 Pompe patients.
Discovery phase
miRNAs were measured by qPCR using Serum/Plasma Focus microRNA PCR 384 wells Panels, (V4.M) (EXIQON). Results were analyzed with software available online by Applied Biosystems (SDS 2.4). Mean and SD were calculated and a statistical analysis of the raw data was performed by ANAXOMICS, a consulting company expert in the analysis of miRNA data. The relative levels of selected serum miRNAs were normalized by using cel‐miR‐39‐3p and Unisp6 as spike‐in18 and the miRNAs has‐miR‐126‐3p, has‐miR‐151a‐3p, and has‐miR‐21‐5p were identified by applying the geNorm algorithm19, a pair‐wise stability‐based method previously described.20
Validation phase
The miRNAs identified in the discovery phase were subsequently studied in the validation phase. Although miR‐206 was not included in the panels used for the discovery phase but was also included in this phase because it is one of the already described dystromirs, together with miR‐1‐3p and miR‐133a‐3p.21 miR‐126‐3p was used as an endogenous normalizer. ExiLENT SYBR Green master mix (QIAGEN, Venlo, Netherlands) and miRCURY LNA miRNA PCR Assays (QIAGEN) to study each miRNAs. We studied 21 miRNA: 14 problem miRNAs, one normalizer, two hemolysis controls and four exogenous controls. Relative miRNA expression was analyzed using comparative Ct method with the SDS 2.4 software.22
Analysis of miRNA expression in muscle
We analyzed the expression levels of miRNAs in muscles obtained from AOPD patients (n = 4) and healthy controls (n = 5). Muscle miRNAs were isolated using DANAGENE microRNA Kit (DANAGEN). For this technique, we also used MS2 (MS2 RNA, Roche) and exogenous controls for the RNA isolation. For miRNA expression analysis, ExiLENT SYBR Green master mix (QIAGEN) and different miRCURY LNA miRNA PCR Assays (QIAGEN) were used for each of the miRNAs. We used U6 snRNA (RNU6‐2) as an endogenous control to normalize the data.23
Muscle function tests
The following muscle function tests were performed: 6 min walking test (6MWT), time to walk 10 meters, timed up‐and‐go test, time to climb up and down four steps, and motor function measure‐20 items scale (MFM‐20). Muscle strength was studied using both MRC and hand‐held myometry. ACTIVLIM, INQoL, and SF‐36 were used as patient reported outcome measures. Forced vital capacity seated and in lying position was obtained with a spirometer Carefusion Microlab ML 3500 MK8 (Carefusion, Yorba Linda, CA, USA).
Muscle imaging
All patients were examined in a Philips Achieva XR 1.5 Teslas located at HSCSP. Three‐dimensional 3‐point Dixon images were acquired and ROIs were manually drawn in the areas of interest as previously described10. Mean thighs fat fraction was calculated from the data acquired and used for the statistical analysis.
Data analysis
Discovery phase
ANOVA tests were performed to identify differences in miRNA expression levels between cohorts. P‐values were adjusted for multiple testing using Tukey‐Kramer correction. Dataset with all the variables from the individual patients was analyzed using a data science strategy based on data mining approach, in which, the following methods were used: as feature selection and extraction algorithms CHOW‐LIU, MRMR, RELIEFF, RFE‐SVM, SFFS, and Wilcoxon with correlation and as the base classifier GLM_Binomal and Naïve Bayes.24, 25, 26, 27, 28, 29 In order to prioritize the generalization capability of the conclusion, a K‐fold validation analysis was performed, yielding cross‐validated quality measures (such as accuracy) for each biomarker. The study was carried out with and without imputed missing values. The imputation method applied used the k‐nearest neighbor averaging, using Euclidean metric in the space of each miRNA by cohort to impute the missing values elements.18
Validation phase
We used nonparametric tests for the statistical analysis of the variables. We used the Mann–Whitney U test to investigate whether there were significant differences in the variables between groups (asymptomatic vs symptomatic/controls vs Pompe).
Clinical correlation
We used Spearman's rank correlation (coefficient reported as) to investigate whether there was a correlation between serum miRNA expression and the clinical results. As we run multiple correlations, a Bonferroni test was performed to avoid type 1 error. The results of all statistical studies were considered significant if P was lower than 0.05. Statistical studies were performed using IBM SPSS® Statistics software version 21.
Results
Description of the cohort
We included in the study 35 AOPD patients. Thirty patients were symptomatic and five were asymptomatic. Twenty‐one out of the 30 symptomatic patients were already receiving ERT when the first sample was obtained. In the remaining nine symptomatic patients, blood samples were obtained before ERT was started. Demographic and clinical data of the patients are described in Table 1. We also included samples from 10 age and sex‐matched controls.
Table 1.
Clinical characteristics of the control group and the Pompe patients included in the study. Mean and standard deviation are provided for the quantitative variables.
| Clinical characteristics | Control n = 10 | Asymptomatic n = 5 | Nontreated n = 9 | Treated n = 21 |
|---|---|---|---|---|
| Age | 41.2 ± 16.4 | 24 ± 8.2 | 50.6 ± 11.4 | 50.81 ± 8.6 |
| Sex (male/female) | 6/4 | 2/3 | 6/3 | 7/15 |
| Time from onset of symptoms | – | – | 9.4 ± 6.6 | 15 ± 9.1 |
| Time on ERT | – | – | – | 5.1 ± 2.5 |
| Aids for walking | – | – | 0 | 10 |
| Ventilation | – | – | 2 | 9 |
miRNA expression in the discovery phase
We observed different expression levels between the groups of study in nine of the 185 miRNAs included in the discovery assay (ANOVA, P < 0.05). Expression levels of these nine miRNAs were further analyzed in the validation phase. These miRNAs included: miR‐106b‐5p, miR‐199a‐5p, miR‐652‐3p, miR‐423‐5p, miR‐1‐3p, miR‐133a‐3p, miR‐186‐5p, miR‐338‐3p, and miR‐23a‐3p. We also included in the validation phase a group of miRNAs that have been involved in the muscle degeneration process: miR‐206, miR‐145‐5p, miR‐181‐5p, miR‐486 and miR‐29a‐3p.21
miRNA expression in the validation phase
We observed a statistically significant increase in the concentration of miR‐1‐3p and miR‐206 between AOPD patients and controls (Mann–Whitney U test, P < 0.05) (Fig. 1A and C). We also analyzed if there were differences between the different subgroups of study: asymptomatic AOPD, symptomatic nontreated AOPD, symptomatic‐treated AOPD and controls. We observed significant differences between symptomatic‐treated AOPD patients and controls in the expression of miR‐1‐3p, miR‐133a‐3p and miR‐206 (Mann–Whitney U test, P < 0.05), (Fig. 1). In miR‐206, we also observed significant differences between asymptomatic and symptomatic‐treated AOPD patients (Mann–Whitney U test, P < 0.05) (Fig. 1C). Although we identified other potential miRNAs in the discovery phase none of them showed significant differences in the validation phase (Fig. 2).
Figure 1.
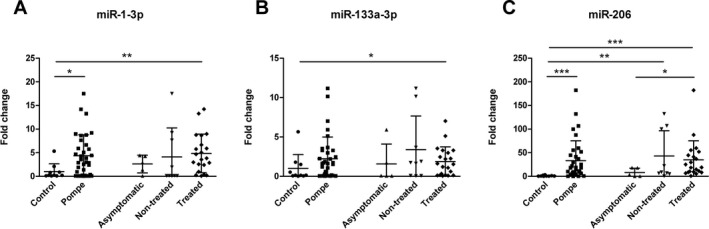
Normalized relative expression levels of validated serum miRNAs in Pompe patients. Data are presented as mean ± SD. Mann–Whitney U test *P < 0.05, **P < 0.01 and ***P < 0.001.
Figure 2.
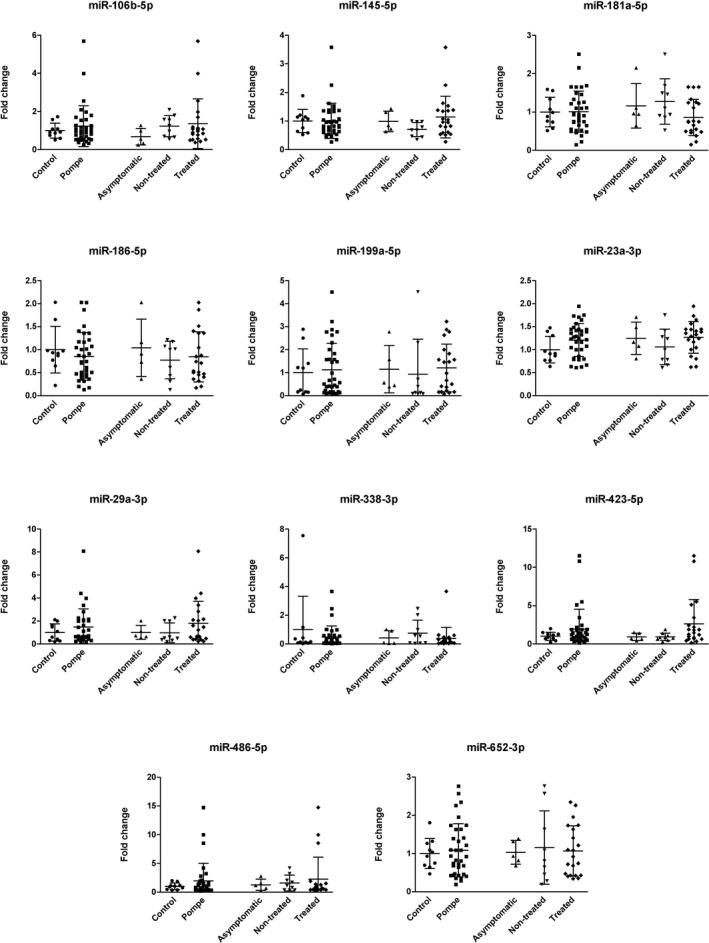
Normalized relative expression levels of serum miRNAs in Pompe patients. Data are presented as mean ± SD. Mann–Whitney U test.
Expression of dystromirs in the progression of the disease
We decided to study if starting ERT treatment modified serum levels of dystromirs (miR‐1‐3p, miR‐133a‐3p and miR‐206). We studied samples for three patients that were obtained before and 6 months to 1 year after starting ERT treatment. Dystromirs serum levels increased in two patients while they decreased in one of the patients (Fig. 3). However, we did not apply any statistical study due to the small size of our cohort.
Figure 3.
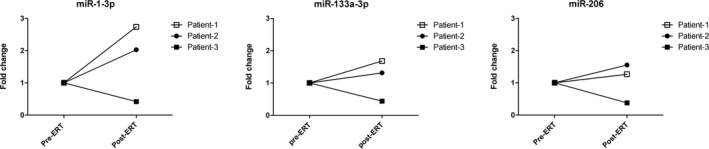
Dystromir expression changes in Pompe patients in response to the treatment. Data are presented as fold change values in post‐ERT patients regarding their pre‐ERT samples.
miRNA expression in muscle
We observed a statistically significant increase in the concentration of miR‐206 in muscle biopsies from AOPD patients taken before the treatment was started (n = 4) compared to controls (n = 5) (Fig. 4C). Although the differences observed in miR‐1‐3p and miR‐133a‐3p muscle expression were not statistically significant, there was a trend to be increased in patients compared to controls (Fig. 4A and B).
Figure 4.
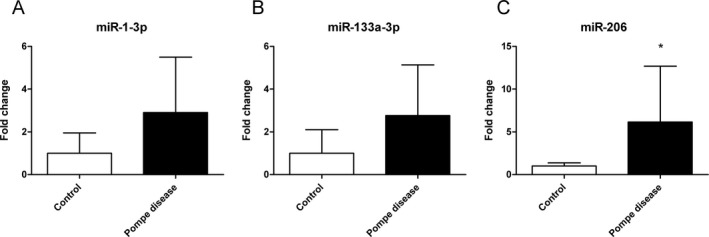
Normalized relative expression levels of muscle miRNAs in Pompe patients. Data are presented as mean ± SD. Mann–Whitney U test *P < 0.05.
Correlations between clinical features and miRNAs serum levels
We identified a statistically significant correlation between serum levels of miR‐1‐3p, miR‐133a‐3p, and miR‐206a in AOPD patients (Spearman test, P < 0.05 (Table 2). We also investigated if there were correlations between serum levels dystromirs and clinical features of the patients. We did not identify any correlation between age at study, age at start of ERT, time on ERT or delay in the start of ERT, and serum levels of miRNAs. In contrast, we identified significant correlations (Spearman test, P < 0.05) between dystromirs' serum levels and some of the muscle function tests (Table 2). However, the correlation coefficients were lower than 0.6. There was a correlation between mean thighs fat fraction analyzed using quantitative muscle MRI and miR‐206 serum levels, although as it happened with muscle function tests, the correlation coefficient was lower than 0.
Table 2.
Correlation between microRNAs serum levels and clinical features.
| miR‐1‐3p | miR‐133a‐3p | miR‐206 | ||||
|---|---|---|---|---|---|---|
| Sig | CC | Sig | CC | Sig | CC | |
| miR‐1‐3p | – | – | 0.007 | 0.457** | 0 | 0.761** |
| miR‐133a‐3p | 0.007 | 0.457** | – | – | 0.003 | 0.493** |
| miR‐206 | 0.000 | 0.761** | 0.003 | 0.493** | – | – |
| MRC score | 0.089 | – | 0.094 | – | 0.035 | −0.385* |
| Myometry score | 0.899 | – | 0.803 | – | 0.337 | – |
| 6MWT | 0.803 | – | 0.073 | – | 0.584 | – |
| Time to walk 10 m | 0.514 | – | 0.043 | 0.378* | 0.151 | – |
| Timed up‐and‐go test | 0.446 | – | 0.061 | – | 0.589 | – |
| Time to climb up 4 steps | 0.386 | – | 0.053 | – | 0.119 | – |
| Time to climb down 4 steps | 0.855 | – | 0.12 | – | 0.211 | – |
| MFM‐20 | 0.388 | – | 0.039 | −0.373* | 0.064 | – |
| CVF seated | 0.465 | – | 0,025 | −0.416* | 0.243 | – |
| CVF lying | 0.286 | – | 0.057 | – | 0.535 | – |
| Activlim | 0.628 | – | 0.986 | – | 0.462 | – |
| qMRI fat fraction | 0.107 | – | 0.205 | – | 0.013 | 0.420* |
Sig, bilateral significance; CC, correlation coefficient.
Spearman Rho test P < 0.05.
Spearman Rho test P < 0.01.
Discussion
The aim of this work was to study miRNA profiles in Pompe disease patients and evaluate their role as potential biomarkers. We studied the expression of 190 miRNAs in AOPD patients and controls and we found increased expression levels in AOPD patients in three miRNAs: miR‐1‐3p, miR‐133a‐3p, and miR‐206. These differences were more robust when we compared serum levels of symptomatic patients with controls. Moreover, serum levels of miR‐206 were higher in symptomatic than presymtomatic patients, suggesting that it could be a valid biomarker of disease progression. Despite this finding, we identified weak correlations between muscle function tests and serum levels of miRNAs.
Several reports have demonstrated that miRNAs are potential biomarkers of different diseases, such as cancer, liver injury, or heart failure.30, 31, 32 The expression profile of miRNAs has also been studied in different muscle diseases such as DMD, myotonic dystrophy, and FSHD.16, 33, 34, 35, 36 In these studies, several miRNAs have a different pattern of expression when compared to controls. Three of them, the miR‐1‐3p, miR‐133a‐3p and miR‐206, are persistently increased in muscular dystrophies and for this reason are known as dystromirs. Dystromirs are only expressed by heart and skeletal muscle and, therefore, they can potentially be a valuable tool to follow muscle pathology in patients with muscle disorders. The function of dystromirs in the process of muscle degeneration and regeneration has been investigated. While miR‐133 enhances proliferation of myoblasts, miR‐1 and miR‐206 promote differentiation from myoblasts to myotubes. Dystromirs expression is regulated by MyoD, Myf5, MEF2, SRF, and other transcription factors involved in muscle development.37, 38 The process of muscle degeneration in Pompe disease is not completely understood. Reduced expression of alpha‐glucosidase leads to a progressive accumulation of glycogen inside the lysosomes of muscle fibers disrupting the physiologic process of autophagy. As a result, glycogen charged lysosomes and autophagic vesicles are found in the sarcoplasm, disrupting the contractile properties of skeletal muscle fibers. Eventually, the muscle fiber dies and is replaced by fat and fibrous tissue resembling what happens in muscle dystrophies. In view of this fact, it is reasonable to find increased serum levels of dystromirs in AOPD patients, as happens in other muscle dystrophies, reinforcing the idea that Pompe disease and muscle dystrophies may share a pathologic process of muscle degeneration.
As dystromirs are related with the muscle regeneration process, it is not surprising that they are increased in muscle disorders, especially in those patients in whom the process of muscle degeneration is active. We observed that dystromirs were more expressed in symptomatic than asymptomatic AOPD patients. Specifically, the expression of miR‐206 was significantly higher in serum samples of symptomatic compared to asymptomatic AOPD patients. Therefore, miR‐206 serum levels may have a role as a potential biomarker to follow‐up presymptomatic patients over the years in order to identify patients in whom the process of muscle degeneration has started and are at risk of developing symptoms of muscle weakness. We did not observe differences in the serum levels of dystromirs between treated and nontreated symptomatic patients. However, a higher number of patients should be analyzed to draw conclusions on the effect of ERT on the muscle regeneration process in symptomatic Pompe disease. ERT has been shown to decrease glycogen accumulation in muscle fibers of preclinical animal models of the disease and also in patients. However, recent studies demonstrate that muscle weakness of treated AOPD patient progresses despite the treatment and that muscle fatty replacement, which is related to muscle degeneration process, increases over the years.9, 10 Our finding supports that the process of muscle degeneration remains active despite the starting of the treatment in Pompe patients. In accordance, we did not observe a significant reduction in miRNA expression levels in serum samples obtained before and after the treatment. Recently, Tarallo et al studied the profile of miRNAs in Pompe disease and observed high expression levels of miR‐133a‐3p both in IOPD and AOPD patients.39 These results are in accordance with our investigations, and suggest that miRNAs could be potential biomarkers of Pompe disease.
Other miRNAs have a role in skeletal muscle homeostasis both in health and in disease‐regulating myogenesis, muscle atrophy or structural and metabolic changes related to aging or exercise21. These miRNAs include miR‐23a, miR‐29, miR‐146a, miR‐486, and miR‐181 among others. We have studied serum expression levels of these miRNAs, but we did not find significant differences between AOPD patients and controls. However, although our cohort included 35 patients, it is possible that higher number of samples are needed to identify subtle changes in miRNA expression in serum. In this sense, analysis of miRNA expression using skeletal muscle biopsies should show higher differences. Nevertheless, muscle biopsies of patients are not always available. We studied miRNA expression of muscle biopsies and we confirmed higher expression levels of miR‐206 in AOPD patients than in controls. Although miR‐1‐3p and miR‐133a‐3p levels were increased in AOPD muscle biopsies, we did not find significant differences with controls, mainly due to a high variability of miRNA expression.
In conclusion, we have identified a group of miRNAs with increased levels of expression in blood samples of patients with Pompe disease. The role of these miRNAs in muscle regeneration has been previously reported and suggests that there is a common pathological pathway between Pompe disease and muscle dystrophies.
Author Contributions
ACR performed the experimental part, analyzed the results, performed the statistical analysis and prepared the figures, and wrote the paper. EFS and CL designed the protocol, analyzed the results, and reviewed the paper. IB, IP. and EM designed the protocol, visited the patients, and acquired motor function data. CNP and JL visited the patients and reviewed the paper. SS coordinated the study, visited the patients and reviewed the paper. NDL and XSC designed the protocol and reviewed the paper. II designed and discussed the protocol. JDM designed the protocol, visited the patients, analyzed the images, performed the statistical analysis, and wrote the paper. EG designed the protocol, analyzed the results, wrote the paper, and obtained funding for the study.
Conflict of Interest
This study (GZ‐2015‐11342) has been performed with funding from SANOFI/Genzyme.
Acknowledgments
We thank the people of the Neuromuscular Disorders Unit for their comments and continuous support. This study was supported by a grant from Sanofi‐Genzyme (GZ‐2015‐11342) to Gallardo and has been registered in Clinicaltrials.gov (identifier NCT03045042).
Carrasco‐Rozas A, Fernández‐Simón E, Lleixà C, et al. Identification of serum microRNAs as potential biomarkers in Pompe disease. Ann Clin Transl Neurol. 2019;6:1214–1224. 10.1002/acn3.50800
Funding information
This study was supported by a grant from Sanofi‐Genzyme (GZ‐2015‐11342) to Dr. Gallardo and has been registered in Clinicaltrials.gov (identifier NCT03045042).
Coinvestigators – The Spanish Pompe Study Group: Miguel Angel Barba‐Romero (Hospital General de Albacete, Albacete, Spain), Joseba Barcena (Hospital Universitario Cruces, Baracaldo, Spain), María Rosario Carzorla (Hospital Puerta de Hierro, Majadahonda, Spain), Carlota Creus (Hospital Virgen de las Nieves, Granada, Spain), Jaume Coll‐Cantí (Hospital Germans Tries i Pujol, Badalona, Spain), Noemí de Luna (Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Spain), Manuel Díaz (Hospital de Cabueñes, Gijón, Spain), Cristina Domínguez (Hospital 12 de Octubre, Madrid & Insituto de Investigación i+12, Madrid, Spain), Roberto Fernández Torrón (Hospital Universitario Donostia, Spain), María José García Antelo (Hospital Universitario A Coruña, A Coruña, Spain), Josep María Grau (Hospital Clínic, Barcelona, Spain), María Teresa Gómez Caravaca (Hospital de Córdoba, Spain), Juan Carlos León Hernández (Hospital Universitario Nuestra Señora de la Candelaria, Tenerife, Spain), Adolfo López de Munáin (Hospital Universitario Donostia, Spain), Francisco Antonio Martínez‐García (Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain), Yolanda Morgado (Hospital Universitario Virgen de Valme, Sevilla, Spain), Antonio Moreno (Hospital Universitario Morales Meseguer, Murcia, Spain), Germán Morís (Hospital Universitario de Asturias, Oviedo, Spain), Miguel Angel Muñoz‐Blanco (Hospital Gregorio Marañón, Madrid, Spain), Andres Nascimento (Hospital Virgen del Rocío, Sevilla, Spain), Carmen Paradas (Hospital Virgen del Rocío, Sevilla, Spain), José Luis Parajuá Pozo (Hospital de Can Mises, Ibiza, Spain), Luis Querol (Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Spain), Arturo Robledo‐Strauss (Hospital Juan Ramón Jiménez, Huelva, Spain), Ricard Rojas García (Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, Centro de Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Íñigo Rojas‐Marcos (Hospital Virgen de Macarena, Sevilla, Spain), Jose Antonio Salazar (Hospital Regional Universitario de Málaga, Spain), Mercedes Usón (Hospital de Son Llátzer, Palma de Mallorca, Spain).
Funding Statement
This work was funded by Sanofi‐Genzyme grant GZ-2015-11342.
Contributor Information
Jordi Díaz‐Manera, Email: jdiazm@santpau.cat.
Eduard Gallardo, Email: egallardo@santpau.cat.
Pompe Spanish Study group:
Miguel Angel Barba‐Romero, Joseba Barcena, María Rosario Carzorla, Carlota Creus, Jaume Coll‐Cantí, Manuel Díaz, Cristina Domínguez, Roberto Fernández Torrón, María José García Antelo, Josep María Grau, María Teresa Gómez Caravaca, Juan Carlos León Hernández, Adolfo López de Munáin, Francisco Antonio Martínez‐García, Yolanda Morgado, Antonio Moreno, Miguel Angel Muñoz‐Blanco, Andres Nascimento, Carmen Paradas, José Luis Parajuá Pozo, Luis Querol, Arturo Robledo‐Strauss, Ricard Rojas García, Íñigo Rojas‐Marcos, Jose Antonio Salazar, and Mercedes Usón
References
- 1. van der Ploeg AT, Reuser AJ. Pompe's disease. Lancet 2008;372:1342–1353. [DOI] [PubMed] [Google Scholar]
- 2. Nascimbeni AC, Fanin M, Masiero E, et al. The role of autophagy in the pathogenesis of glycogen storage disease type II (GSDII). Cell Death Differ 2012;19:1698–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Dasouki M, Jawdat O, Almadhoun O, et al. Pompe disease: literature review and case series. Neurol Clin 2014;32:751–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Manganelli F, Ruggiero L. Clinical features of Pompe disease. Acta Myol 2013;32:82–84. [PMC free article] [PubMed] [Google Scholar]
- 5. Desnuelle C, Salviati L. Challenges in diagnosis and treatment of late‐onset Pompe disease. Curr Opin Neurol 2011;24:443–448. [DOI] [PubMed] [Google Scholar]
- 6. van der Beek NA, de Vries JM, Hagemans ML, et al. Clinical features and predictors for disease natural progression in adults with Pompe disease: a nationwide prospective observational study. Orphanet J Rare Dis 2012;7:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nicolino M, Byrne B, Wraith JE, et al. Clinical outcomes after long‐term treatment with alglucosidase alfa in infants and children with advanced Pompe disease. Genet Med 2009;11:210–219. [DOI] [PubMed] [Google Scholar]
- 8. Katzin LW, Amato AA. Pompe disease: a review of the current diagnosis and treatment recommendations in the era of enzyme replacement therapy. J Clin Neuromuscul Dis 2008;9:421–431. [DOI] [PubMed] [Google Scholar]
- 9. Kuperus E, Kruijshaar ME, Wens SCA, et al. Long‐term benefit of enzyme replacement therapy in Pompe disease: a 5‐year prospective study. Neurology 2017;89:2365–2373. [DOI] [PubMed] [Google Scholar]
- 10. Figueroa‐Bonaparte S, Segovia S, Llauger J, et al. Muscle MRI findings in childhood/adult onset Pompe disease correlate with muscle function. PLoS ONE 2016;11:e0163493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schoser B, Laforet P, Kruijshaar ME, et al. 208th ENMC International Workshop: formation of a European Network to develop a European data sharing model and treatment guidelines for Pompe disease Naarden, The Netherlands, 26–28 September 2014. Neuromuscul Disord 2015;25:674–678. [DOI] [PubMed] [Google Scholar]
- 12. Figueroa‐Bonaparte S, Llauger J, Segovia S, et al. Quantitative muscle MRI to follow up late onset Pompe patients: a prospective study. Sci Rep 2018;8:10898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cox TM. Biomarkers in lysosomal storage diseases. 2006. [PubMed]
- 14. Chen JF, Callis TE, Wang DZ. microRNAs and muscle disorders. J Cell Sci 2009;122(Pt 1):13–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guller I, Russell AP. MicroRNAs in skeletal muscle: their role and regulation in development, disease and function. J Physiol 2010;588(Pt 21):4075–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Matsuzaka Y, Kishi S, Aoki Y, et al. Three novel serum biomarkers, miR‐1, miR‐133a, and miR‐206 for Limb‐girdle muscular dystrophy, Facioscapulohumeral muscular dystrophy, and Becker muscular dystrophy. Environ Health Prev Med 2014;19:452–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10‐year experience. Eur J Neurol 2017;24:768‐e31. [DOI] [PubMed] [Google Scholar]
- 18. Marabita F, de Candia P, Torri A, et al. Normalization of circulating microRNA expression data obtained by quantitative real‐time RT‐PCR. Brief Bioinform 2016;17:204–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Perkins JR, Dawes JM, McMahon SB, et al. ReadqPCR and NormqPCR: R packages for the reading, quality checking and normalisation of RT‐qPCR quantification cycle (Cq) data. BMC Genom 2012;13:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Vandesompele J, De Preter K, Pattyn F, et al. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sharma M, Juvvuna PK, Kukreti H, McFarlane C. Mega roles of microRNAs in regulation of skeletal muscle health and disease. Front Physiol 2014;5:239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 2001;25:402–408. [DOI] [PubMed] [Google Scholar]
- 23. Sauer E, Babion I, Madea B, Courts C. An evidence based strategy for normalization of quantitative PCR data from miRNA expression analysis in forensic organ tissue identification. Forensic Sci Int Genet 2014;13:217–223. [DOI] [PubMed] [Google Scholar]
- 24. Peng H, Long F, Ding C. Feature selection based on mutual information: criteria of max‐dependency, max‐relevance, and min‐redundancy. IEEE Trans Pattern Anal Mach Intell 2005;27:1226–1238. [DOI] [PubMed] [Google Scholar]
- 25. Kira K, Rendell LA. The feature selection problem: traditional methods and a new algorithm. Proc of AAAI‐92 1992:122–126.
- 26. Guyon I, Weston J, Bamhill S, Vapnik V. Gene selection for cancer classification using support vector machines. Machine Learn 2002;46:389–422. [Google Scholar]
- 27. Ververidis D, Kotropoulos C. Fast and accurate sequential floating forward feature selection with the Bayes classifier applied to speech emotion recognition. Signal Process 2008;88:2956–2970. [Google Scholar]
- 28. Christin C, Hoefsloot HC, Smilde AK, et al. A critical assessment of feature selection methods for biomarker discovery in clinical proteomics. Mol Cell Proteomics 2013;12:263–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Russell SJ, Norvig P. Artificial intelligence: a modern approach. Pearson. Education 2003. [Google Scholar]
- 30. Romero‐Cordoba SL, Salido‐Guadarrama I, Rodriguez‐Dorantes M, Hidalgo‐Miranda A. miRNA biogenesis: biological impact in the development of cancer. Cancer Biol Ther 2014;15:1444–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McDaniel K, Herrera L, Zhou T, et al. The functional role of microRNAs in alcoholic liver injury. J Cell Mol Med 2014;18:197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Katz MG, Fargnoli AS, Williams RD, et al. MiRNAs as potential molecular targets in heart failure. Future Cardiol 2014;10:789–800. [DOI] [PubMed] [Google Scholar]
- 33. Zaharieva IT, Calissano M, Scoto M, et al. Dystromirs as serum biomarkers for monitoring the disease severity in Duchenne muscular Dystrophy. PLoS ONE 2013;8:e80263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Perfetti A, Greco S, Cardani R, et al. Validation of plasma microRNAs as biomarkers for myotonic dystrophy type 1. Sci Rep 2016;6:38174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Koutsoulidou A, Kyriakides TC, Papadimas GK, et al. Elevated muscle‐specific miRNAs in serum of myotonic dystrophy patients relate to muscle disease progress. PLoS ONE 2015;10:e0125341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Perfetti A, Greco S, Bugiardini E, et al. Plasma microRNAs as biomarkers for myotonic dystrophy type 1. Neuromuscul Disord 2014;24:509–515. [DOI] [PubMed] [Google Scholar]
- 37. Kim HK, Lee YS, Sivaprasad U, et al. Muscle‐specific microRNA miR‐206 promotes muscle differentiation. J Cell Biol 2006;174:677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rosenberg MI, Georges SA, Asawachaicharn A, et al. MyoD inhibits Fstl1 and Utrn expression by inducing transcription of miR‐206. J Cell Biol 2006;175:77–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tarallo A, Carissimo A, Gatto F, et al. microRNAs as biomarkers in Pompe disease. Genet Med 2019;21:591–600. [DOI] [PubMed] [Google Scholar]