

Abstract
Focal cortical dysplasia (FCD) causes drug‐resistant epilepsy and is associated with pathogenic variants in mTOR pathway genes. How germline variants cause these focal lesions is unclear, however a germline + somatic “2‐hit” model is hypothesized. In a boy with drug‐resistant epilepsy, FCD, and a germline DEPDC5 pathogenic variant, we show that a second‐hit DEPDC5 variant is limited to dysmorphic neurons, and the somatic mutation load correlates with both dysmorphic neuron density and the epileptogenic zone. These findings provide new insights into the molecular and cellular correlates of FCD determining drug‐resistant epilepsy and refine conceptualization of the epileptogenic zone.
Introduction
Type II focal cortical dysplasia (FCD) is a major cause of drug‐resistant epilepsy with surgical resection often the only effective treatment. It is characterized by cortical dyslamination with dysmorphic neurons (FCDIIA) or cortical dyslamination with dysmorphic neurons + balloon cells (FCDIIB). Dysmorphic neurons have been described as the “seizure generators” in cortical dysplasia1. Previous studies of resected FCD and hemispheric malformations identified low‐level mosaic somatic pathogenic variants in mTOR pathway genes AKT3, PIK3CA, MTOR, TSC1, and TSC2 2, 3, 4, 5. It was assumed these lesions were due to postzygotic alterations in neuronal precursors during cortical development, with earlier mutations leading to larger malformations6. Aside from tuberous sclerosis complex (TSC), familial FCD is extremely rare. It has been associated with germline pathogenic variants in the mTOR GATOR1 complex genes DEPDC5, NPRL2, and NPRL3 7, 8, 9, yet how these germline variants cause focal lesions is unclear. In TSC, second somatic mutations have been identified in brain tissue in up to 35% of cases10. In 2015, a single case of FCDIIA associated with germline + somatic mutations in DEPDC5 was reported, however, it could not be determined if these were biallelic11. The first definitive evidence of biallelic pathogenic germline + somatic mutations in non‐TSC FCD was recently demonstrated in a single case of DEPDC5‐associated FCDIIA12. Here, we combine deep sequencing, stereology, and laser capture microdissection to show a correlation between somatic mutation load, dysmorphic neuron density, and the epileptogenic zone in a child with FCD due to biallelic germline + somatic mutations in DEPDC5. Our findings suggest that seizures in FCD are generated from regions of maximal density of dysmorphic neurons harboring a second‐hit somatic mutation.
Methods
Clinical data
This study was approved by the Royal Children’s Hospital Ethics Committee (HREC 29077) with written informed consent obtained from the individual’s parents. Clinical data were obtained by patient assessment and medical records.
Sample preparation
Cortex was preserved as FFPE blocks from middle temporal gyrus posteriorly (#1), middle temporal gyrus anteriorly (#2), temporal pole (#3), inferior temporal gyrus (#4), and uncus (#5). Tissue from the middle temporal gyrus was fresh frozen at −80°C.
Detection of germline and somatic pathogenic variants in DEPDC5
Barcoded libraries were prepared and analyzed with a custom HaloPlexHS panel13. A clonal assay14 of 252 clones was performed to determine if the variants were in cis‐ or trans‐ configuration.
Droplet digital PCR
The somatic variant was detected using a Custom TaqMan® Assay (Assay ID ANPRNP2; Thermo Fisher Scientific) according to the manufacturer’s protocol. Limit of detection assay was performed according to published protocols15.
Immunostaining
FFPE tissue sections underwent citrate‐based antigen retrieval method (VectorLab, #3300) and were incubated overnight at 4°C with anti‐Neurofilament SMI‐311R antibody (1:1000, BioLegend, #837801), anti‐Phospho‐S6 Ribosomal Protein (Ser235/236) antibody (1:100, Cell‐Signalling, #2211), or anti‐NeuN clone A60 antibody (1:100, Millipore, MAB377) according to published methods16. Biotinylated secondary antibodies were applied for 1 h at room temperature. Visualization of immunoreactivity was performed using the ABC kit (VectorLab, #PK‐4000) and DAB kit (VectorLab, #SK‐4100) according to manufacturer’s instructions.
Stereology
Fifteen micron FFPE sections (1:4 series) from the temporal pole (#3) and inferior temporal gyrus (#4) were immunostained with anti‐Neurofilament SMI‐311R antibody. Stereological analysis was conducted using a Zeiss Axio M2 with the StereoInvestigator (MBF Bioscience) module. We followed ILAE’s criteria for identification of dysmorphic neurons; immunoreactivity for neurofilament, whole cell size >20 μm, and nuclei >15 μm17. Dysmorphic neuron number was quantitated using the optical fractionator method18 and parameters we described recently in a study of TSC 19.
Laser capture microdissection
Fifteen micron FFPE sections from region #4 were mounted onto PEN membrane slides (Thermo Fisher Scientific, #LCM0522) and immunostained to identify dysmorphic neurons (SMI‐311R) or normal neurons (NeuN). Pools of 10–15 target cells were captured using Arcturus Veritas Laser Capture Microdissection. Neurons were captured within a small region (normal neurons) or selected individually (dysmorphic neurons). DNA was extracted using QIAamp DNA Micro Kit (Qiagen, #56304), followed by polymerase chain reaction (PCR) preamplification and ddPCR.
Results
Case report
The seven‐year‐old boy is the second child of healthy, unrelated parents with no relevant family history. At two months, he developed asymmetric infantile spasms and developmental arrest, going undiagnosed until he presented at the age of four months. EEG showed right temporal slowing, right > left‐sided epileptiform discharges, and a cluster of epileptic spasms with subtle chin and arm movements. Brain 3T MRI was normal, and spasms ceased on high‐dose prednisolone but recurred at eight months before ceasing with topiramate. Right temporal and generalized epileptiform activity persisted.
At two years of age, he developed events of staring, eye rolling, and chin trembling. Video EEG monitoring at 2.5yr showed prominent right temporal focal slowing and subclinical electrographic seizures from the right anterior midtemporal region. The clinical events of concern were captured but were not associated with an electrographic correlate. 3Tesla (3T) MRI showed poor gray–white differentiation of the right temporal pole and cortical thickening of the inferior temporal sulcus (Fig. 1A). FDG PET showed right anterior temporal hypometabolism (Fig. 1B). By 3 years he showed delay in language (single words only) and fine motor skills, repetitive behaviors and impaired social interaction, and was formally diagnosed with autism spectrum disorder. At 3.5 years, a right anterior temporal lobectomy was performed, including the uncus but sparing the hippocampus (Fig. 1C). Intraoperative electrocorticography with 4‐contact strip and depth electrodes showed attenuation of background fast activity, excess slowing, bursts of gamma fast activity, and spike‐wave discharges over the right temporal pole and uncus. Spike‐waves were higher voltage, more frequent, and associated with fast ripples in contacts recording from the uncus (Fig. 1D). Histopathology showed FCDIIA (Fig. 1E). Seizures ceased, anticonvulsants were weaned, and follow‐up EEGs showed only right temporal slowing. Postoperatively there was acceleration in development with improvements in receptive and expressive language, motor skills, and social interaction, although autistic features have persisted.
Figure 1.
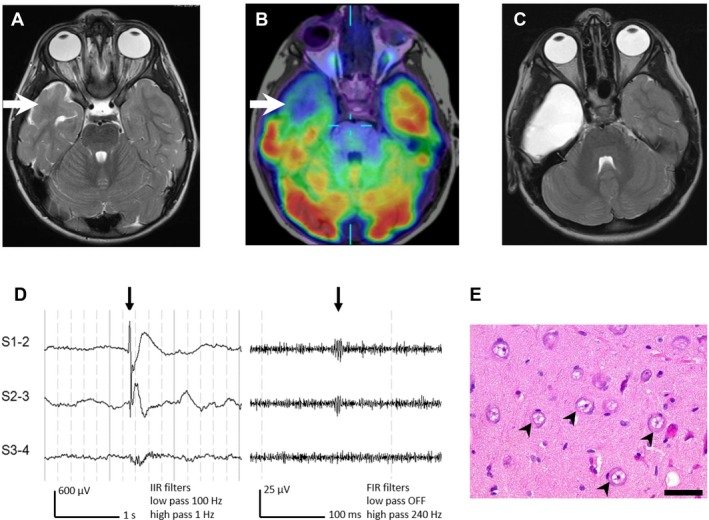
Imaging, electrocorticography and histopathology data. (A) is an axial T2‐weighted MRI (3T) at 3 years showing a dilated temporal horn and poor gray–white differentiation of the right anterior temporal pole. (B) is an interictal FDG PET coregistered with axial T1‐weighted MRI showing right anterior temporal hypometabolism. (C) is a postoperative axial T2‐weighted MRI (1.5T) at 5 years. (D) shows intraoperative electrocorticography using a 4‐contact (10 mm spacing) subdural strip electrode shown in bipolar montage with standard (left) and high‐frequency oscillations (right) display settings and filters, showing spike‐associated fast ripples in the uncus (#5). (E) is an H&E‐stained section at high power from the uncus showing multiple large, dysmorphic neurons with prominent Nissl substance (black arrowheads). Scale bar = 50 μm.
Detection of germline and somatic pathogenic variants in DEPDC5
A pathogenic de novo heterozygous germline DEPDC5 variant (NM_001242896.1: c.2390delA p.Q797Rfs*18) was previously reported in this individual20. Deep sequencing of brain (595x) and blood (525x)‐derived gDNA confirmed this variant. Additionally, we identified a pathogenic truncating variant in DEPDC5 (c.3994C > T p.R1332*), present only in brain‐derived DNA with an allele frequency of 3.9% (22/569 reads). This variant has been previously reported as germline pathogenic9. To determine if the germline and the somatic variants were in cis‐ or in trans‐ configuration, we performed a clonal assay13. RNA was isolated from the fresh frozen middle temporal gyrus posteriorly (region #1) was converted tocDNA using SuperScript III (Invitrogen, #18080400) and a 2137 bp fragment, spanning the two variants, was amplified by PCR. The PCR product was subcloned into PCR2.1 TOPO TA cloning vector (Thermo Fisher Scientific, #450641) and 252 clones analyzed using qPCR. The somatic and germline variants were not identified in the same clone, confirming the variants were in trans and suggesting biallelic inactivation of DEPDC5 in a subset of cells.
Somatic mutation gradient in affected cortex
We quantified mutation load in the five tissue blocks (Fig. 2A) using ddPCR and detected a mutation gradient from mean allele frequency of 0.2%–2.5% (Fig. 2B). The greatest mutation load was in the uncus (#5), where maximal epileptic activity was recorded during intraoperative electrocorticography. Immunohistochemistry showed abundant phospho‐S6‐positive neurons, indicating upregulation of the mTOR pathway (Fig. 2C). The discordant allele frequency in region #2 of 3.9% measured by HaloPlexHS panel sequencing compared to 1.8% measured by ddPCR is likely attributable to PCR amplification using the HaloPlexHS protocol, which can result in less precision in determination of allele frequency. The allele frequency determined by ddPCR is considered the more accurate measure.
Figure 2.
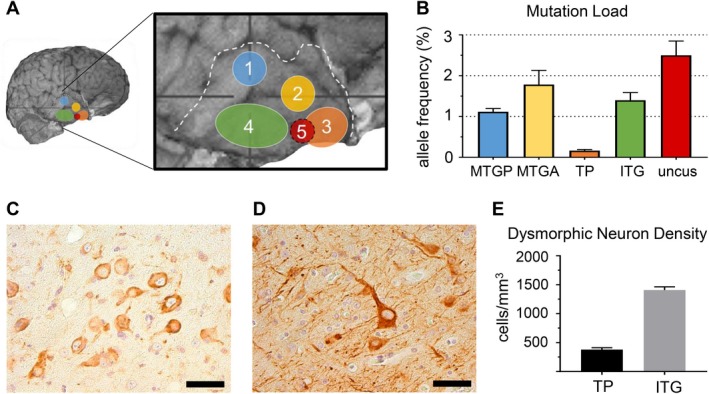
Droplet digital PCR, immunostaining and dysmorphic neuron density stereology results. (A) shows the five regions from which tissue samples were available. (B) shows the variations in somatic pathogenic variant allele frequency across the five sampled lesion regions. The highest allele frequency (2.5%) was seen at the uncus (#5) and the lowest (0.2%) was seen at the temporal pole (#3). Regions analyzed are MTGP: medial temporal gyrus posteriorly (#1), MTGA: medial temporal gyrus anteriorly (#2), TP: temporal pole (#3), ITG: inferior temporal gyrus (#4) and uncus (#5). Error bars represent SEM. (C) shows immunostaining with anti‐phospho‐S6 showing highest immunoreactivity in the dysmorphic neurons suggestive of mTOR dysregulation. Scale bar = 50 μm. (D) shows immunostaining with anti‐Neurofilament SMI‐311R showing highest immunoreactivity in dysmorphic neurons. Scale bar = 50 μm. (E) shows the results of quantification of dysmorphic neuron density by stereology from temporal pole (#3) and inferior temporal gyrus (#4). Dysmorphic neuron density is significantly higher in the inferior temporal gyrus (temporal pole: 380.4 ± 30.4 cells/mm3 vs. inferior temporal gyrus: 1408.3 ± 56.3 cells/mm3, mean ± SEM, P < 0.001).
Quantification of dysmorphic neuron density
To determine if somatic mutation load correlated with dysmorphic neuron density, we stained sections with anti‐Neurofilament (Fig. 2D) and performed stereological analysis in the temporal pole (#3, somatic mutation mean allele frequency 0.2%) and inferior temporal gyrus (#4, somatic mutation mean allele frequency 1.4%). Analysis was not performed in other regions due to limited tissue. The estimated dysmorphic neuron density was 380.43 cells/mm3 (coefficient of error = 0.08) in temporal pole and 1408.27 cells/mm3 (coefficient of error = 0.04) in inferior temporal gyrus (Fig. 2E), which suggested a correlation between somatic mutation load and dysmorphic neuron density.
Detection of cell‐specific somatic pathogenic variants in DEPDC5
Using laser capture microdissection, we isolated multiple independent pools of 10–15 neurons, restricted to either morphologically normal (Fig. 3A) or dysmorphic neurons (Fig. 3B). SNP genotyping using ddPCR detected the somatic pathogenic variant in none of the normal neuron captures (n = 0/12) (Fig. 3C) and in 50% of the dysmorphic neuron captures (n = 5/10) (Fig. 3D). Our finding suggests the somatic pathogenic variant was restricted to dysmorphic neurons.
Figure 3.
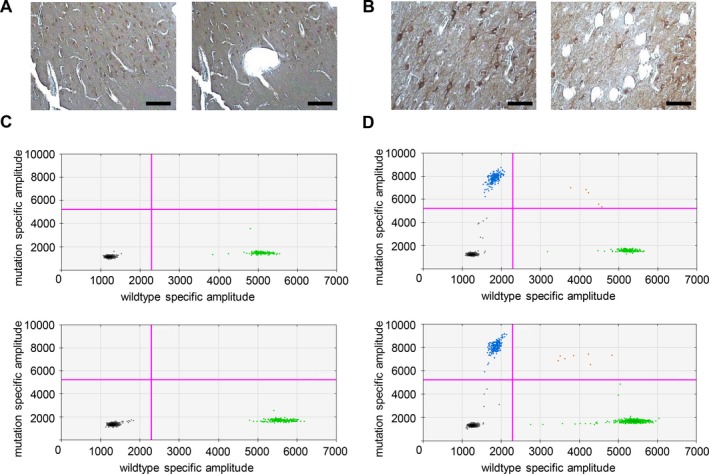
Genetic analysis of dysmorphic and morphologically normal neurons. (A and B) show before and after images from laser capture microdissection of normal neurons (A) and dysmorphic neurons (B). Scale bar = 100 μm. Equivalent number of neurons (10–15) were captured within a small region (normal neurons) or selected individually (dysmorphic neurons). DNA prepared from laser capture microdissected cells was analyzed with ddPCR using SNP‐specific probes designed to identify both the DEPDC5 reference allele (c.3994C, green) and the somatic pathogenic variant (c.3994C > T, blue). (C) shows representative results of ddPCR from normal neurons showing the amplification of wild‐type alleles (green). (D) shows representative results of ddPCR from dysmorphic neurons showing amplification of both wild‐type (green) and mutant (blue) alleles. The black dots are negative droplets, where no gDNA was captured for amplification and the red dots are droplets where multiple templates were captured in a single droplet.
Discussion
“mTORopathies” have emerged as increasingly important causes of epilepsy with a spectrum from mild focal epilepsy with normal imaging to multifocal or hemispheric malformations with drug‐resistant epilepsy and encephalopathy. Knudson’s “two‐hit” hypothesis21, regarding the genetic basis of many neoplastic disorders including familial cancers, posits that focal pathology is the consequence of a pathogenic germline variant and a somatic second‐hit. This hypothesis also provides a plausible explanation as to why individuals with germline pathogenic variants develop focal lesions, such as FCD. Finding the elusive second somatic hit has been difficult, presumably due to technical limitations of identifying variants in only a small fraction of cells within malformed tissue. Application of deep sequencing to resected brain tissue is allowing advances in detection of very low‐level mosaicism showing similarities in the pathogenesis of neoplastic and non‐neoplastic disorders of cell growth and differentiation.
Here, we confirm the two‐hit model for DEPDC5‐associated FCD and demonstrate that a “mutation gradient” correlates with a “dysmorphic neuron density gradient”, and the highest mutation load correlates with the epileptogenic region for seizure onset. We build on previous work showing a “mutation gradient” in FCD22, by demonstrating, for the first time, that the gradient is due to the somatic second‐hit being limited to dysmorphic neurons, the critical epileptogenic cell type in type II FCD. Presumably this is secondary to an early postzygotic pathogenic variant affecting only a subset of neuronal precursors during cortical development.
Our findings expand the concept of the epileptogenic zone, which is the minimum amount of brain tissue needed to be resected to control seizures. Previously, it was determined by clinical, imaging, electrophysiological, and histological data. There has been much debate as to whether the epileptogenic zone is at the periphery of lesions such as FCD and the tubers of TSC, or within the centre of such lesions. Our findings of a correlation between mutation gradient, dysmorphic neuron density, and epileptogenicity support the likelihood of the epileptogenic zone being intralesional and not at the periphery, and this has also been confirmed by our electrophysiological studies in TSC23. Our findings, and that of others who have recently identified a mutation gradient within lesions, refine the concept of the epileptogenic zone, to now include mutation load, neuronal cell density, and cell type‐specific genetic data. These findings may have direct clinical implications for epilepsy treatment by expanding the evidence base for highly focussed surgical resections aimed at removing only a nidus containing a high density of mutant dysmorphic neurons within a larger lesion, guided by preoperative imaging, electrophysiological data, and, perhaps one day, intraoperative mutational testing, as is under investigation for brain tumor surgery24. Further studies are required to determine how often this two‐hit mechanism applies to other cases of FCD, or indeed to other focal epilepsies with normal imaging.
Author Contributions
WSL: conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures.
SEMS: conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures.
KBH: conception and design of the study and acquisition and analysis of data.
KP: conception and design of the study and acquisition and analysis of data.
GG: conception and design of the study and acquisition and analysis of data.
AW: conception and design of the study and acquisition and analysis of data.
WM: conception and design of the study and acquisition and analysis of data.
SAM: conception and design of the study and acquisition and analysis of data.
SFB: conception and design of the study and drafting a significant portion of the manuscript.
IES: conception and design of the study and drafting a significant portion of the manuscript.
DM: acquisition and analysis of data and drafting a significant portion of the manuscript or figures.
ASH: conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures.
PJL: conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures.
RJL: conception and design of the study, acquisition and analysis of data, and drafting a significant portion of the manuscript or figures.
Conflict of Interest
The authors have no conflicts of interest to report.
Acknowledgments
The authors thank Michael Wilson, Gemma L Carvill, and Heather C Mefford for their contributions. This work was supported by the Australian Government National Health and Medical Research Council GNT1128933, the Murdoch Children’s Research Institute, the Campbell Edwards Trust, and the Brain Foundation. Additional funding was provided by the Independent Research Institute Infrastructure Support Scheme and the Victorian State Government Operational Infrastructure Program. RJL and KBH are supported by the Melbourne Children’s Clinician Scientist Fellowship scheme. We thank the family for agreeing to be part of this research study.
Funding Statement
This work was funded by Victorian State Government grant ; Brain Foundation grant N/A; Campbell Edwards Trust grant N/A; National Health and Medical Research Council grant GNT1128933; Murdoch Children's Research Institute grant N/A.
Contributor Information
Paul J. Lockhart, Email: paul.lockhart@mcri.edu.au.
Richard J. Leventer, Email: richard.leventer@rch.org.au.
References
- 1. Cepeda C, Andre VM, Vinters HV, et al. Are cytomegalic neurons and balloon cells generators of epileptic activity in pediatric cortical dysplasia? Epilepsia 2005;46(Suppl 5):82–8. [DOI] [PubMed] [Google Scholar]
- 2. Lee JH, Huynh M, Silhavy JL, et al. De novo somatic mutations in components of the PI3K‐AKT3‐mTOR pathway cause hemimegalencephaly. Nat Genet 2012;44:941–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Poduri A, Evrony GD, Cai X, et al. Somatic activation of AKT3 causes hemispheric developmental brain malformations. Neuron 2012;74:41–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Lim JS, Kim WI, Kang HC, et al. Brain somatic mutations in MTOR cause focal cortical dysplasia type II leading to intractable epilepsy. Nat Med 2015;21:395–400. [DOI] [PubMed] [Google Scholar]
- 5. Lim JS, Gopalappa R, Kim SH, et al. Somatic mutations in TSC1 and TSC2 cause focal cortical dysplasia. Am J Human Genet 2017;100:454–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Poduri A, Evrony GD, Cai X, Walsh CA. Somatic mutation, genomic variation, and neurological disease. Science 2013;341:1237758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Scheffer IE, Heron SE, Regan BM, et al. Mutations in mammalian target of rapamycin regulator DEPDC5 cause focal epilepsy with brain malformations. Ann Neurol 2014;75:782–7. [DOI] [PubMed] [Google Scholar]
- 8. Sim JC, Scerri T, Fanjul‐Fernandez M, et al. Familial cortical dysplasia caused by mutation in the mammalian target of rapamycin regulator NPRL3. Ann Neurol 2016;79:132–7. [DOI] [PubMed] [Google Scholar]
- 9. Ricos MG, Hodgson BL, Pippucci T, et al. Mutations in the mammalian target of rapamycin pathway regulators NPRL2 and NPRL3 cause focal epilepsy. Ann Neurol 2016;79:120–31. [DOI] [PubMed] [Google Scholar]
- 10. Martin KR, Zhou W, Bowman MJ, et al. The genomic landscape of tuberous sclerosis complex. Nat Commun 2017;8:15816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Baulac S, Ishida S, Marsan E, et al. Familial focal epilepsy with focal cortical dysplasia due to DEPDC5 mutations. Ann Neurol 2015;77:675–83. [DOI] [PubMed] [Google Scholar]
- 12. Ribierre T, Deleuze C, Bacq A, et al. Second‐hit mosaic mutation in mTORC1 repressor DEPDC5 causes focal cortical dysplasia‐associated epilepsy. J Clin Invest 2018;128:2452–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Marsh AP, Heron D, Edwards TJ, et al. Mutations in DCC cause isolated agenesis of the corpus callosum with incomplete penetrance. Nat Genet 2017;49:511–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Leventer RJ, Scerri T, Marsh AP, et al. Hemispheric cortical dysplasia secondary to a mosaic somatic mutation in MTOR. Neurology 2015;84:2029–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hildebrand MS, Harvey AS, Malone S, et al. Somatic GNAQ mutation in the forme fruste of Sturge‐Weber syndrome. Neurol Genet 2018;4:e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Baybis M, Yu J, Lee A, et al. mTOR cascade activation distinguishes tubers from focal cortical dysplasia. Ann Neurol 2004;56:478–87. [DOI] [PubMed] [Google Scholar]
- 17. Blumcke I, Mühlebner A. Neuropathological work‐up of focal cortical dysplasias using the new ILAE consensus classification system ‐ practical guideline article invited by the Euro‐CNS Research Committee. Clin Neuropathol 2011;164–77. [DOI] [PubMed] [Google Scholar]
- 18. West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in the subdivisions of the rat hippocampus using the optical fractionator. Anat Rec 1991;231:482–97. [DOI] [PubMed] [Google Scholar]
- 19. Stephenson SEM, Owens HG, Richards KL, et al. Dysmorphic neuron density is highest in the centre of epileptogenic cortical tubers. bioRxiv 2019:621607. [Google Scholar]
- 20. Carvill GL, Crompton DE, Regan BM, et al. Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol Genet 2015;1:e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer 2001;1:157–62. [DOI] [PubMed] [Google Scholar]
- 22. Mirzaa GM, Campbell CD, Solovieff N, et al. Association of MTOR mutations with developmental brain disorders, including megalencephaly, focal cortical dysplasia, and pigmentary mosaicism. JAMA Neurol 2016;73:836–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kannan L, Vogrin S, Bailey C, et al. Centre of epileptogenic tubers generate and propagate seizures in tuberous sclerosis. Brain 2016;139(Pt 10):2653–67. [DOI] [PubMed] [Google Scholar]
- 24. Shankar GM, Francis JM, Rinne ML, et al. Rapid intraoperative molecular characterization of glioma. JAMA Oncol 2015;1:662–7. [DOI] [PMC free article] [PubMed] [Google Scholar]