

SUMMARY
Proliferating cells must cross a point of no return before they replicate their DNA and divide. This commitment decision plays a fundamental role in cancer and degenerative diseases and has been proposed to be mediated by phosphorylation of retinoblastoma (Rb) protein. Here, we show that inactivation of the anaphase-promoting complex/cyclo-some (APCCdh1) has the necessary characteristics to be the point of no return for cell-cycle entry. Our study shows that APCCdh1 inactivation is a rapid, bistable switch initiated shortly before the start of DNA replication by cyclin E/Cdk2 and made irreversible by Emi1. Exposure to stress between Rb phosphorylation and APCCdh1 inactivation, but not after APCCdh1 inactivation, reverted cells to a mitogen-sensitive quiescent state, from which they can later re-enter the cell cycle. Thus, APCCdh1 inactivation is the commitment point when cells lose the ability to return to quiescence and decide to progress through the cell cycle.
In Brief
Live cell imaging of cell-cycle reporters, reveals that cells commit to cell-cyclem, entry much later than the restriction point, and that there’s a window of time during, which a cell can return to quiescence, rather than moving forward through the, cycle.
Graphical Abstract
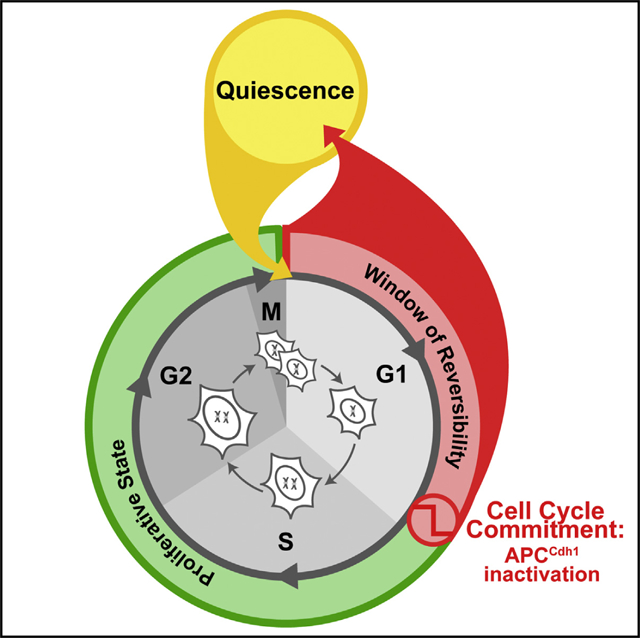
INTRODUCTION
Many mammalian cells spend much of their time in a quiescent state in which they retain the potential to proliferate (Hsu et al., 2014). The decision of quiescent cells to enter the cell cycle must be tightly regulated to ensure that tissue homeostasis is maintained. Dysregulation of this fundamental decision causes cancer and degenerative diseases (Hanahan and Weinberg, 2000). Quiescent cells enter the proliferating state in G1 before DNA replication starts (S) and also exit the proliferating state to go back to quiescence in G1 after completion of mitosis (M) (Pardee, 1974). Cells must already commit in G1 to complete a round of DNA replication and mitosis to prevent damage and ensure a faithful replication.
A long-standing question in cell biology has therefore been how quiescent cells make this decision to enter the cell cycle and commit to complete S and M phase (Planas-Silva and Weinberg, 1997). Pardee proposed over 40 years ago that a specific time point must exist until which cells can reverse their trajectory and return to the quiescent state and after which they cannot return to quiescence and will replicate their DNA and divide (Pardee, 1974) (Figure 1A). By pulsing external proliferation-promoting stimuli (mitogens), the study defined a “restriction point” early in G1 long before DNA replication begins when cells lose their need for mitogens and still complete the cell cycle. This and other studies proposed that the restriction point, characterized by mitogen sensitivity, might also constitute the commitment decision or point of no return for cell-cycle entry (Pardee, 1974; Zetterberg and Larsson, 1985). The molecular basis for the restriction point has been proposed to be the hyperphosphorylation of the tumor suppressor retinoblastoma protein (Rb) and the consequent liberation of the cell-cycle transcription factor E2F (referred to as pRb-E2F activation) (Narasimha et al., 2014; Yaoetal., 2008).
Figure 1. Rapid and Near-Complete APCCdh1 Inactivation Shortly before S Phase Entry.
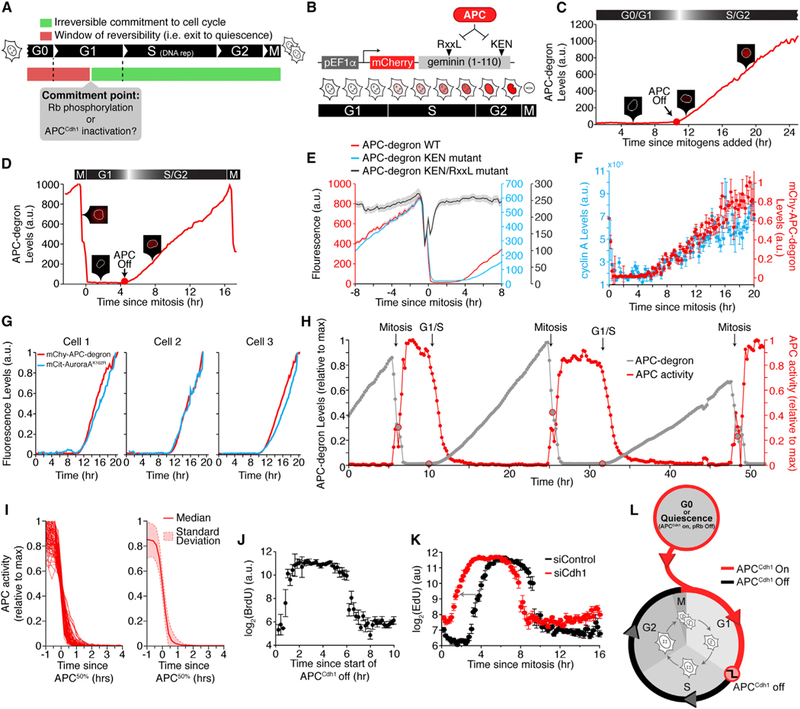
(A) Schematic diagram of the cell-cycle commitment model.
(B) Schematic diagram of the APC-degron reporter (Geminin: aa1–110).
(C) Single-cell trace of APC-degron reporter levels in a representative cell released from mitogen starvation. Inset: snapshots of the APC-degron reporter.
(D) Single-cell trace of the APC-degron reporter in a representative cycling cell as in (C).
(E) MCF10A cells expressing mVenus-APC-degron wild-type and either mChy-APC-degron KEN mutant or mChy-APC-degron KEN/RxxL mutant. Lines are median traces ± SEM. (n = 205 cells, wild-type; n = 800, KEN; n = 600, RxxL).
(F) Cells were imaged for ~4 hr then fixed and stained with α-cyclin A2. Cells were binned by the time since mitosis. Data represent median intensity ± SEM of either cyclin A2 or APC-degron reporter.
(G) HeLa cells transfected with the APC-degron reporter and mCitrine-Aurora-A K162R (three representative cells shown).
(H) APC-degron reporter levels and the derived APC activity for a single cell. Time of mitosis and the G1/S transition are computationally identified.
(I) Left: Single-cell traces of APCCdh1 activity computationally aligned to 50% APCCdh1 activity (random selection of 91 out of 431 cells analyzed). Right: Median APCCdh1 activity trace ± SD (n = 861).
(J) Scatterplot of BrdU levelsversusthetimesinceAPCCdh1 started to inactivate. Fixed cells were mapped back to live-cell data. Single-cell data were binned and data points are median ± SEM (n = 1100).
(K) MCF10A cells were treated with either control siRNA or Cdh1 siRNA. Fixed cells were mapped back to live-cell data. Single-cell data were binned and data points are median ± SEM. n > 30,000 cells per condition. Arrow highlights the shift in timing of EdU incorporation after Cdh1 siRNA.
(L) Schematic when APCCdh1 is on and off during the cell cycle. See also Figure S1.
In addition to mitogen signaling, different stress signals including osmotic stress or UV/ionizing irradiation are known to inhibit or delay proliferation before cells start S phase (Deckbar et al., 2011; Erol, 2011). Several studies also observed that cells could be arrested in G1 even after Rb phosphorylation has already been induced (Dulić et al., 1994; Vousden and Prives, 2009). This raises the question of whether cells that experience stress after Rb phosphorylation are already committed to the cell cycle but can pause before entering S phase or whether cells can return to quiescence even after Rb has been phosphorylated. The latter would argue that the point of no return must be after Rb phosphorylation.
If cells are able to return to quiescence after pRb-E2F activation, it is conceivable that inactivation of the E3 ubiquitin ligase anaphase-promoting complex/cyclosome-Cdh1 (APCCdh1), which is believed to occur after pRb-E2F activation and before DNA replication, may represent the true commitment point for cell-cycle entry. If correct, such a hypothesis predicts that stress perturbations would cause a reversible exit to quiescence until the inactivation of APCCdh1, but not after APCCdh1 inactivation. APCCdh1 is an interesting candidate for a commitment mechanism, as it functions to degrade many proteins necessary for S phase. Cycling cells activate APCCdh1 at the end of mitosis, and quiescent cells maintain active APCCdh1 to prevent re-entry into the cell cycle (Eguren et al., 2011). Cells lacking Cdh1 have a shortened G1 phase, accumulate DNA damage, and undergo apoptosis (Peters, 2006). Based on biochemical bulk-cell analysis, cells exiting mitosis have been proposed to gradually inactivate APCCdh1 prior to S phase entry through the autonomous degradation of the E2 ligase UBCH10 (Rape and Kirschner, 2004). However, this inactivation mechanism has been called into question (Chang et al., 2014; Walker et al., 2008) and alternative inactivation mechanisms have been proposed (Huang et al., 2011; Lukas et al., 1999; Miller et al., 2006; Wang and Kirschner, 2013). This leaves the question open of how APCCdh1 is inactivated and when it occurs in respect to the restriction point, pRb-E2F activation, the start of DNA replication, and cell-cycle commitment.
Our study shows that, in three cell types and in nearly every one out of thousands of individual cells analyzed, APCCdh1 inactivates rapidly within <1 hr to <1% of its previous G1 activity shortly before cells start to replicate their DNA. APCCdh1 inactivation is initiated by cyclin E/Cdk2 before being greatly accelerated and made irreversible by the APCCdh1-inhibitor Emi1. Strikingly, we found that stress stimuli can induce reversible exit from the cell cycle to a mitogen-sensitive quiescent state long after cells have phosphorylated Rb and crossed the restriction point but only until APCCdh1 inactivation. APCCdh1 inactivation exhibits marked hysteresis only in the presence of Emi1, with stress stimuli preventing APCCdh1 inactivation before, but not restoring the activity after APCCdh1 has been inactivated. Thus, APCCdh1 is inactivated by a bistable irreversible switch long after the restriction point and shortly before the G1/S boundary and exhibits all necessary characteristics to be the commitment point when cells decide to progress through the cell cycle and divide.
RESULTS
Rapid, Near-Complete Inactivation of APCCdh1 Is Triggered Shortly before the Start of DNA Replication
Our study focused on two questions: (1) can stress inputs trigger a return to quiescence after the restriction point and pRb-E2F induction and (2) does APCCdh1 inactivation, instead of pRb-E2F induction, function as the point of no return after which cells commit to replicate their DNA and divide (Figure 1A). To measure APCCdh1 inactivation kinetics in single cells, we used non-transformed human MCF10A breast epithelial cells stably expressing one half of the Fucci reporter system, comprised of mCherry conjugated to a peptide from Geminin containing its APC degron motif (APC-degron; Figure 1B) (Sakaue-Sawano et al., 2008). We analyzed changes in the concentration of the APC-degron reporter during the cell cycle by tracking thousands of single living cells over 24–48 hr using automated 96-well microscopy (Figure S1A).
Control experiments showed that, in mitogen-starved cells and in newly born cells after mitosis, two situations where APCCdh1 is known to polyubiquitinate substrates and cause their degradation, APC-degron reporter levels were very low (Figures 1C, 1D, S1A, and S1B). Treatment with either Cdh1 small interfering RNA(siRNA) or the small molecule APC inhibitor proTAME caused buildup of the APC-degron reporter (Figures S1C–S1H). Furthermore, while mutations of the APC degron motifs showed that the two KEN boxes in the reporter were largely dispensable (Geminin: amino acid [aa]13–15 and aa87–89), an additional mutation of the RxxL motif to alanines (AxxA; Geminin: aa23–26) completely prevented reporter degradation, arguing that the degradation of the reporter is mediated by APCCdh1 activity (Figure 1E). We further found a close match between the reporter levels and the levels of endogenous cyclin A2 (Figure 1F), endogenous Geminin (Figure S1I), fluorescently tagged Dbf4 (Figure S1J) (Ferreira et al., 2000), and kinase-dead Aurora A (Figure 1G) (Floyd et al., 2008), arguing that changes in the reporter concentration are a direct reflection of APCCdh1 activity against different known APCCdh1 substrates. Finally, treatment of cells with the proteasome inhibitor MG132 did not change the accumulation rate of the reporter in S and G2, arguing that the reporter is synthesized during the cell cycle at a near constant rate and also that APCCdh1 is nearly completely inactive during S and G2 (Figures S1K and S1L).
Since polyubiquitinated substrates are rapidly degraded (Xu and Qu, 2012), changes in the expression of the APCCdh1 reporter can therefore be used to derive a time course of the activity of APCCdh1 (Figure 1H; Movie S1). This conversion to activity is possible since (1) the promoter region of the reporter construct is unregulated (Figures S1M and S1N), (2) the reporter degradation in quiescence, G1, S, and G2 is primarily regulated by APCCdh1, and (3) the noise in our measurement of reporter intensity changes is very low (Figure S2A; Experimental Procedures).
Because the time of inactivation of APCCdh1 after mitosis or mitogen stimulation was highly variable within the cell population, we aligned APCCdh1 activity time courses from each cell in silico. Markedly, the aligned time courses showed that APCCdh1 was turned off in each cell with stereotypic rapid kinetics with 90% to 10% inactivation within ~50 min (Figure 1I). By comparing the maximal attained fluorescence intensity after 24 hr of buildup (Figure S2B) and the minimal steady-state intensity in G1 (Figure S2A), APCCdh1 must be inactivated >99% during S and G2 compared to its active state during quiescence and G1. We find an identical rapid and near-complete inactivation of APCCdh1 at the end of G1 in MCF10A, HeLa, and non-trans-formed fibroblast cells (Bj-5Ta; Figure S2C) as well as in cells that were already cycling rather than coming out of quiescence (Figure S2D).
To determine when the inactivation of APCCdh1 occurs relative to DNA replication, we exposed cells to either BrdU or EdU (markers for DNA replication) for 5 min at the end of a time-lapse image series, fixed cells, and then automatically mapped each fixed cell back to its own live-cell time course (Figure S2E). We found that cells incorporated EdU only after APCCdh1 had been inactivated to less than 10% of its maximal activity (Figures 1J and S2F). There was an almost perfect correlation between cells inactivating APCCdh1 and an immediate start of DNA replication. This temporal correlation suggests that APCCdh1 inactivation is rate limiting for DNA replication. In support of this hypothesis, Cdh1 knockdown causes premature APCCdh1 inactivation and premature DNA replication as has been reported in other cell types (Figure 1K) (Sigl et al., 2009; Yuan et al., 2014). To show how APCCdh1 inactivation fits into the cell-cycle timeline, Figure 1L illustrates that APCCdh1 is turned on in every cell at the end of mitosis, stays on during quiescence and in G1, and only turns off just prior to DNA replication.
APCCdh1 Inactivation Is Irreversible Once Completed
Since previous in vitro studies found that the APCCdh1 component Cdh1 can be phosphorylated and inactivated by cyclin A/Cdk2 (Kramer et al., 2000; Lukas et al., 1999), we simultaneously measured Cdk2 activation (Figure 2A) (Spencer et al., 2013) and APCCdh1 inactivation in the same cell. Markedly, each cell starts to gradually increase Cdk2 activity and only 3 to 5 hr later, when Cdk2 activity reaches a critical intermediate level, will APCCdh1 inactivation be triggered (Figures 2B–2D). The tight correlation between the increase in Cdk2 activity and APCCdh1 inactivation in thousands of individual cells together with the earlier finding that Cdh1 can be inactivated by Cdk2, suggests that Cdk2 buildup is a rate-limiting step for APCCdh1 inactivation. However, when testing for a potential role of cyclin A, we found no significant change in the kinetics of APCCdh1 inactivation after cyclin A2 knockdown in either MCF10A or HeLa cells (Figures S2G and S2H). The knockdown of cyclin A2 was effective since it diminished the increase in Cdk2 activity in S phase (Figures 2E–2G). However, when we used siRNA to knockdown cyclin E1 and E2 or a small molecule Cdk1/2 inhibitor to inhibit Cdk2 activity (Figure 2F), APCCdh1 inactivation was in both cases abolished (Figures 2H, 2I, and S2I), arguing that cyclin E/Cdk2 is required for the inactivation of APCCdh1.
Figure 2. APCCdh1 Inactivation Is Irreversible and Requires Cyclin E/Cdk2.
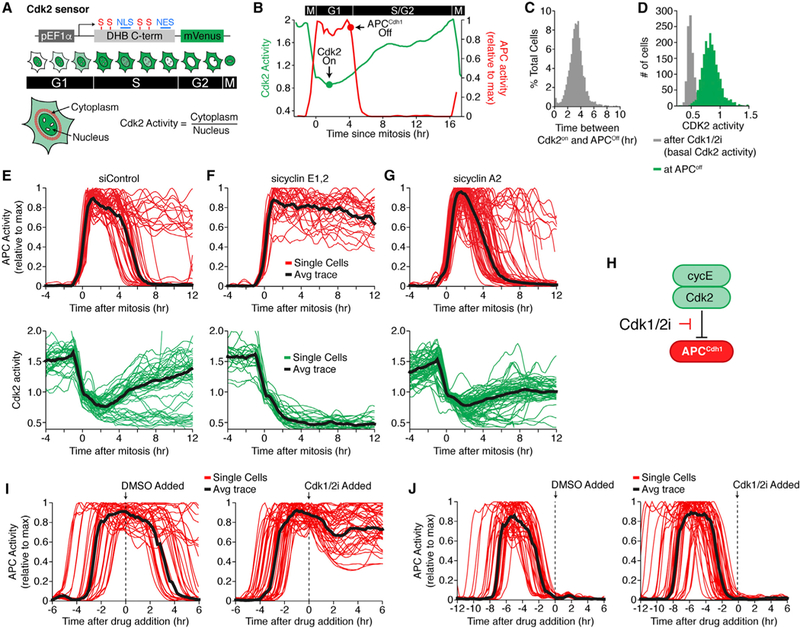
(A) Schematic of the Cdk2 sensor. Cdk2-mediated phosphorylation controls export of the reporter from the nucleus. NLS, nuclear localization signal; NES, nuclear export signal; S, CDK consensus site on serine.
(B) APCCdh1 and Cdk2 activities in a representative single cycling cell.
(C) Histogram of the time between the initial rise of Cdk2 activity and APCCdh1 inactivation. n > 1000 cells.
(D) Histogram of Cdk2 activity when APCCdh1 inactivates (green). Basal Cdk2 activity after treatment with a Cdk1/2 inhibitor (gray) is added for reference.
(E-G) APC and Cdk2 activity aligned in silico to mitosis treated with either control (E), cyclin E1 and E2 (F), or cyclin A2 siRNA(G). Grey line is median trace. n = 54, 26, and 48, respectively.
(H) Schematic diagram showing that cyclin E/Cdk2 mediates inactivation of APCCdh1 and that Cdk2 inhibition is expected to prevent APCCdh1 inactivation.
(I and J) APC activity traces treated with either DMSO or Cdk1/2 inhibitor. Time of treatment is indicated by the dashed line.(I) Cells treated while in G1 phase (n = 41, DMSO; n = 45, Cdk1/2i).(J) Cells treated while in S phase (n = 47, DMSO; n = 45, Cdk1/2i). Black line is median trace. See also Figure S2.
Since rapid and identical APCCdh1 inactivation kinetics in every cell is a hallmark of a bistable switch mechanism (Figure 1I), we next tested whether APCCdh1 inactivation shows hysteresis, the other required characteristic of an irreversible switch. Strikingly, when the same Cdk1/2 inhibitor that prevents APCCdh1 inactivation is added after APCCdh1 inactivation, APCCdh1 activity remained off, demonstrating that APCCdh1 inactivation shows hysteresis and has the characteristic of an irreversible switch mechanism (Figure 2J).
Rapid and Irreversible APCCdh1 Inactivation Is Mediated by the APCCdh1 Inhibitor Emi1
We next determined whether additional factors control the rapid and irreversible characteristic of APCCdh1 inactivation. One candidate is Skp2, which is part of a cullin SCFSkp2 E3 ligase important for S phase fidelity and has been suggested to mediate APCCdh1 inactivation (Barr et al., 2016; Fukushima et al., 2013). However, application of a pan-cullin inhibitor that blocks Skp2-mediated degradation of Cdh1 did not change the kinetics of APCCdh1 inactivation. The same experiment also excludes a range of cullin-containing E3 ligases as potential regulators (Figures S3A–S3D) (Soucy et al., 2009). Also, knockdown of the deubiquitinating protein Usp37, another regulator of APCCdh1 (Huang et al., 2011), had only a small effect on the inactivation kinetics of APCCdh1 (Figures 3A and 3B). In contrast, when we used siRNAto knockdown the early mitotic inhibitor 1 (Emi1, Fbxo5), a previously identified inhibitor of APCCdh1 (Miller et al., 2006), we found that the rate of APCCdh1 inactivation was markedly slowed (Figure 3C). Furthermore, the APCCdh1 inactivation rate became bimodal (Figures 3C, S3E, and S3F) despite a continuous distribution of siRNA knockdown efficiency (Figures S3G and S3H), suggesting a threshold level of Emi1 protein is needed to trigger rapid APCCdh1 inactivation. Conversely, overexpression of wild-type Emi1 almost immediately inactivated APCCdh1 after mitosis (Figure S3I) at an even faster rate compared to control cells (Figures 3D and 3E). Together, this shows that Emi1 has a fundamental role in controlling the rate of APCCdh1 inactivation.
Figure 3. Emi1 Is Required for Rapid and Irreversible APCCdh1 Inactivation.
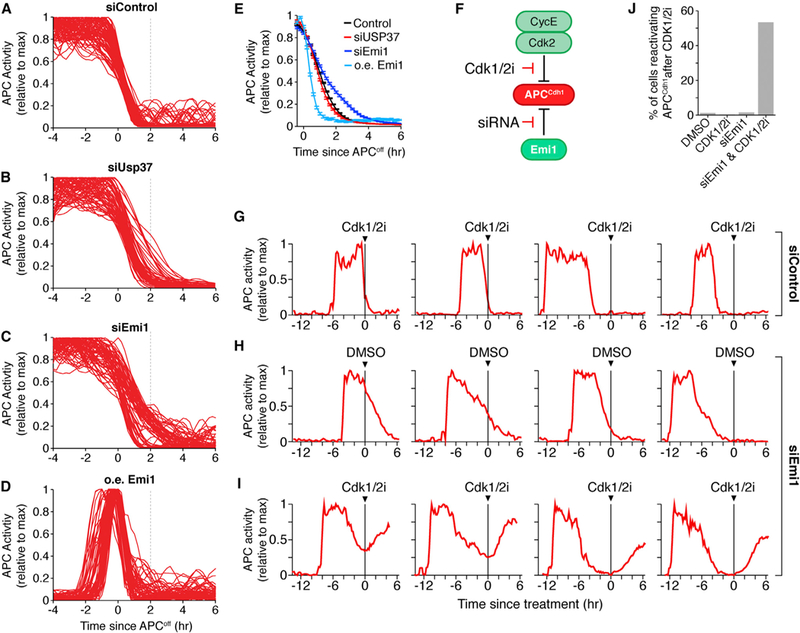
(A-D) Single-cell APC activity traces aligned to the time that APCCdh1 starts to turn off in cells treated with either control (A), Usp37 (B), or Emi1 siRNA (C) or cells overexpressing (oe) wild-type Emi1 (D). Dashed line added for reference between panels.
(E) Median APC activity trace ± SEM from cells in (A-D). n = 276, 201, 214, 152, respectively.
(F) Schematic of experimental design for (G-I).
(G-I) Single-cell APC activity traces treated with either control (G) or Emi1 siRNA (H and I). Cells were exposed to either DMSO (H) or Cdk1/2 inhibitor (G and I) at the indicated time. Data from all cells is in Figure S3J.
(J) Bar graph of the percent of cells that reactivate APCCdh1 upon Cdk1/2 inhibitor addition. n≈ 250 cells per condition. See also Figure S3.
We next explored the possibility that Emi1 not only speeds up APCCdh1 inactivation but is also required to render APCCdh1 inactivation irreversible. We knocked down Emi1 using siRNA and then added a Cdk1/2 inhibitor and monitored APCCdh1 activity (Figure 3F). Strikingly, while Cdk1/2 inhibition in control cells does not reactivate APCCdh1 once it has been turned off, Cdk1/2 inhibition in Emi1 knockdown cells caused an immediate reactivation of APCCdh1 (Figures 3G–3J and S3J). We observed this APCCdh1 reactivation when cells were in the process of turning off APCCdh1 during the G1/S transition as well as when cells were in S or G2 phase and the APCCdh1 was completely off. Thus, while Cyclin E/Cdk2 is necessary to initiate the inactivation of APCCdh1, Emi1 has a dual role to make APCCdh1 inactivation rapid as well as irreversible, ensuring that APCCdh1 activity does not turn back on once cells have started to replicate their DNA.
Single-Cell Time-Course Analysis Shows a Large Time Gap from the Restriction Point and Rb Phosphorylation to APCCdh1 Inactivation
Our finding of a rapid stereotypic inactivation of APCCdh1 shortly before DNA replication was unexpected based on the previous, more gradual inactivation seen in bulk-cell measurements. The existence of an irreversible regulatory step close before DNA replication suggested that APCCdh1 inactivation might be the commitment point for cell-cycle entry or, alternatively, it was conceivable that APCCdh1 inactivation represents merely a temporary cell-cycle checkpoint in which cells pause after the decision to commit to the cell cycle has already been made with the activation of pRb-E2F. In a third plausible model, APCCdh1 inactivation might be triggered simultaneously along with the restriction point and pRb-E2F activation. To validate or exclude this third model, we performed single-cell analysis to measure when mitogen sensitivity is lost and when APCCdh1 is inactivated. We starved cells of mitogens for 48 hr and released them with pulses of mitogens at variable durations. Rather than observing a sharp transition, the probability of cells in the population entering the cell cycle gradually increased the longer mitogens were present, with a pulse of 8 hr resulting in half of the cells entering the cell cycle while the other half stayed quiescent (Figure 4A). Inhibition of the MAPK pathway, a pathway known to mediate the mitogen signaling response, showed the same time dependence as mitogen withdrawal (Figure 4A). When we monitored the timing of APCCdh1 inactivation in the same cells, we found that individual cells rapidly inactivated APCCdh1 at different times from 8 hr to >24 hr after mitogen stimulation, with half of the cells inactivating APCCdh1 16 hr after release from mitogen starvation (Figures 4B and 4C). This argues that a long time gap separates the restriction point from APCCdh1 inactivation.
Figure 4. A Large Time Gap Separates the Restriction Point and pRb-E2F Induction from APCCdh1 Inactivation.
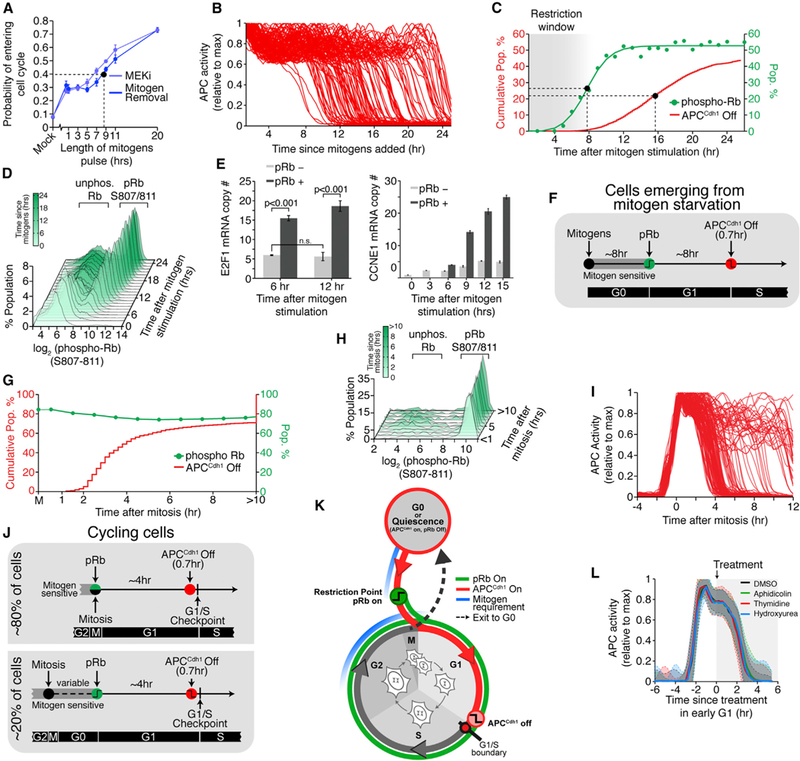
(A) Probability of entering the cell cycle (i.e., EdU incorporation after 24 hr) as a function of the length of mitogen or MEKi pulse after release from starvation. Dashed line indicates when 50% of cells enter the cell cycle. Error bars are SD from four experiments, n > 3000 cells per data point.
(B) APC activity in cells released from mitogen-starvation (n = 114).
(C) Cumulative distribution function (CDF) of the time cells inactivated APCCdh1 after release from mitogen-starvation (red; n = 9,068). Scatterplot of the percent of cells with phospho-Rb (S807/S811) as a function of time since mitogen stimulation (n > 15,000 cells per data point). Green line is sigmoidal best-fit line. Dashed lines indicate when half max is reached.
(D) Histograms of pRb807/811 levels taken at 1 hr intervals after release from mitogen starvation (n > 15,000 cells per histogram).
(E) Single-cell mRNA FISH of E2F1 and cycE1 after release from mitogen starvation. Cells were separated into pRb negative and positive based on bimodal histogram as in (D). Error bars are SEM from four experiments, n > 1000 cells per condition.
(F) Schematic diagram of the timing of cell-cycle events in cells released from mitogen starvation.
(G) Scatterplot of the percent of cells with pRb807/811 at each bin relative to mitosis (green; n « 400 cells per data point). CDF of the time cells inactivated APCCdh1 relative to mitosis (red; n = 1042).
(H) Histograms of pRb807/811 levels divided into bins since mitosis. (n ≈ 400 cells per histogram).
(I) Single-cell traces of APC activity in asynchronous cells, aligned to mitosis (n = 140).
(J) Schematic of the timing of cell-cycle events in cycling cells. Following mitosis, cells either maintain Rb hyper-phosphorylation and stay in the cell cycle or they lose Rb phosphorylation and enter a transient G0-like state.
(K) Schematic showing when cells are sensitive to mitogens relative to Rb phosphorylation and APCCdh1 inactivation.
(L) Median traces ± SD of APC activity in cells treated with the indicated drug in G1 phase. n = 113, control; n = 107, aphidicolin; n = 116, thymidine; n = 84, hydroxyurea. See also Figure S4.
During a similar time window when cells lost their need for mitogens, the fraction of cells that transitioned to a state where Rb was phosphorylated at a dual Serine 807 and 811 site (hereafter Rb phosphorylation) increased from 5–12 hr (Figure 4C), again long before APCCdh1 was inactivated ~8 hr later. The bimodal distributions in histograms of single-cell Rb phosphorylation levels in thousands of cells over time suggested that Rb phosphorylation rapidly increases to a maximal level in every cell but in each cell after a different delay time (Figure 4D), consistent with the previously shown rapid transition to hyperphosphory- lated Rb (Havens et al., 2006). Also, since Rb has multiple phosphorylation sites that may in principle be regulated at different times, we directly tested whether the rapid maximal phosphorylation of S807/S811 is reflective of the activation of the transcription factor E2F (Weinberg, 1995; Yao et al., 2008; Zetterberg et al., 1995). Single-molecule RNA fluorescence in situ hybridization (FISH) analysis of the expression of the E2F target genes E2F1 and CCNE1 (cyclin E1) shows that both mRNA transcripts are induced only in cells with maximal pRb pS807/S811, but not in cells with low pRb S807/S811, regardless of the time since mitogen release and before the APCCdh1 inactivation switch (Figure 4E). Taken together, our data suggest a timeline in which a typical MCF10A cell entering the cell cycle out of quiescence crosses the restriction point, rapidly hyperphosphorylates Rb, and induces E2F after ~8 hr and then triggers the APCCdh1 inactivation switch after another 8 hr delay (Figure 4F). This demonstrates that, when cells come out of quiescence, pRb-E2F activation and APCCdh1 inactivation are each rapidly executed as sequential events separated by a long time gap.
Since the situation in cycling cells might be different, we also investigated whether a similar time gap exists in asynchronous cycling cells. Using live-cell tracking software, we automatically called the point of mitosis when the condensed chromosomes separate into two daughter nuclei at anaphase. Approximately 80% of cells kept Rb phosphorylated while in mitosis and in early G1 (Figures 4G, 4H, S4A, and S4B). This population of cycling cells that never loses Rb phosphorylation inactivates APCCdh1 and starts S phase on average 4 hr after anaphase (Figure 4I). The other 20% of cells go through a variable length, quiescent-like state without phosphorylated Rb before later crossing the restriction point, phosphorylating Rb, and re-entering the cell cycle (Figure 4J). Consistent with previous studies (Spencer et al., 2013; Stacey, 2003), we conclude that cells have two distinct time windows when they are sensitive to mitogens, a window early in G0 and G1 when they come out of quiescence and a window in the previous G2, M, and early G1 phase when they are proliferating (Figure 4K). Thus, while the timing greatly varied between individual cells and stimulation conditions, in all cases, the restriction point and pRb-E2F activation was separated by a long time gap after APCCdh1 inactivation, excluding the model that pRb-E2F induction and APCCdh1 inactivation are part of a single commitment mechanism.
Irreversible Inactivation of APCCdh1 Precedes Checkpoint Arrest at the G1/S Boundary
Our finding that APCCdh1 inactivation is an irreversible step is consistent with APCCdh1 inactivation being either a commitment point or a checkpoint mechanism that allows cells to wait for completion of the buildup in Cdk2 activity before progressing with the cell cycle. Particularly, we considered that APCCdh1 inactivation might be related to a previously described DNA replication stress-mediated checkpoint arrest at the G1/S boundary that can be triggered by aphidicolin, thymidine, or hydroxyurea (Davis et al., 2001). Markedly, we found that cells inactivated APCCdh1 with the same kinetics in the presence of these compounds, arguing that cells undergo a checkpoint arrest at the G1/S boundary only after inactivating APCCdh1 and without altering the APCCdh1 inactivation kinetics (Figure 4L). When cells became arrested at the G1/S boundary and DNA replication stopped, APCCdh1 activity remained off and Cdk2 activity stopped increasing but maintained an intermediate amount of activity, arguing that cells cannot exit to quiescence any more once they reach the G1/S boundary (Figure S4C). Thus, APCCdh1 inactivation is an irreversible bistable switch that is triggered independently and before the previously described cell-cycle checkpoint arrest at the G1/S boundary.
Stress Inputs Can Trigger Reversible Exit to Quiescence until APCCdh1 Is Inactivated, but Not after
With the temporal framework for mitogen sensitivity and pRb- E2F activation, APCCdh1 inactivation, and the G1/S boundary in place, we focused on our main question of whether pRb-E2F activation or APCCdh1 inactivation represents the point of no return for cell-cycle entry. Particularly, we tested how different stresses applied after pRb-E2F activation alter the cell-cycle trajectories of cells. Several studies reported that S phase entry can be prevented by stresses that do not require DNA replication (Deckbar et al., 2011; Havens et al., 2006). While these studies were consistent with a temporary checkpoint arrest mechanism that allows for repair before cells will proceed with the cell cycle, we considered that these arrested cells might instead be able to exit back to a mitogen-sensitive quiescent state. Such a return to quiescence after pRb-E2F activation would argue that the restriction point is not the point of no return for cell-cycle entry and therefore not the commitment point (Figure 5A).
Figure 5. Stress Signaling Can Induce Cell-Cycle Exit to Mitogen-Regulated Quiescence after pRb-E2F Induction but Only until APCCdh1 Inactivation.
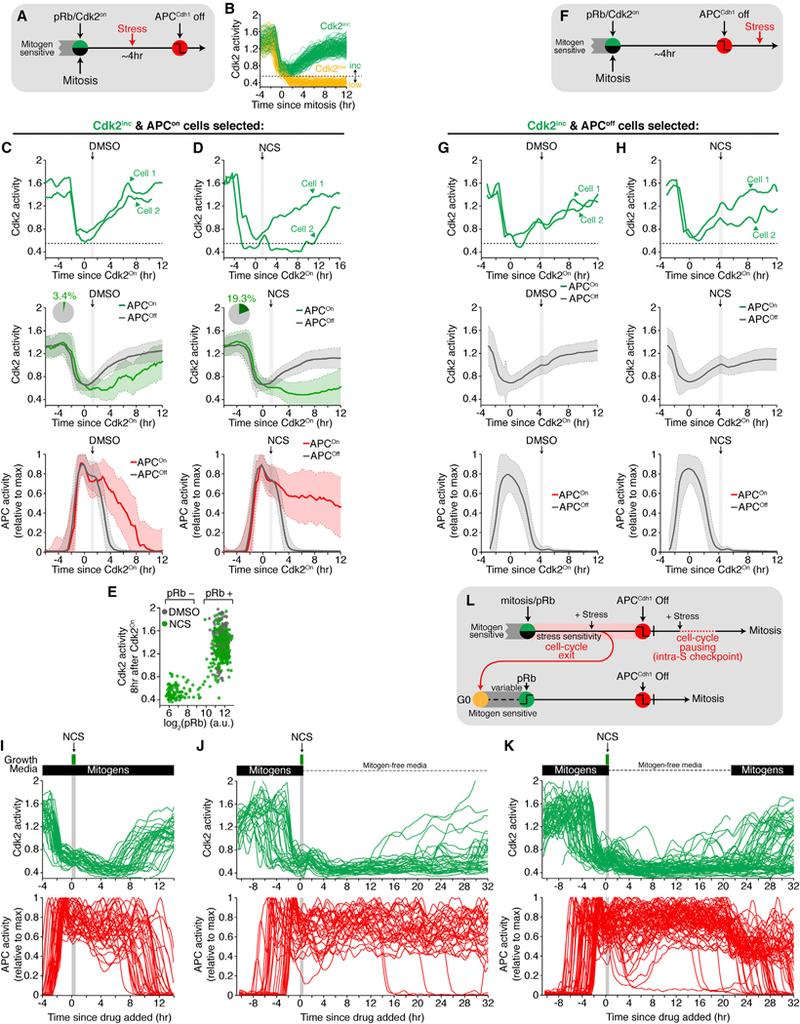
(A) Experimental setup for stress administration after the restriction point but before APCCdh1 inactivation.
(B) Schematic showing cells that have increasing Cdk2 activity (Cdk2lnc) or basal Cdk2 activity (Cdk2low) 2 hr after mitosis. Horizontal dashed line indicates the threshold level of Cdk2 activity used to categorize a cell as either Cdk2inc or Cdk2low. Only Cdk2inc cells were considered in Figures 5C–5H.
(C and D) Cdk2 and APCCdh1 activities in cells exposed to NCS after Cdk2 activity started to rise. Light gray band represents time when cells were exposed to stress. Top: two individual cells are shown. Middle: median Cdk2 activity ± SD. Bottom: median APCCdh1 activity. Traces are colored if Cdk2 inactivated after initially turning on or are gray if Cdk2 stayed active and APCCdh1 inactivated. Inset: percentage of cells that inactivated Cdk2 activity despite initially turning on. (C) DMSO or (D) 40ng/mL Neocarzinostatin (NCS) (n = 270, DMSO; n = 554, NCS; see Figure S5E).
(E) Scatterplot of pRb807/811 levels versus Cdk2 activity 8 hr after the initial rise in Cdk2 activity.
(F) Experimental setup for stress administered after APCCdh1 inactivation. (G and H) Cdk2 and APCCdh1 activities exposed to stress after APCCdh1 inactivation. Top: two cells are highlighted. Middle: median Cdk2 activity ± SD. Bottom: median APCCdh1 activity.
(G) DMSO or (H) 40ng/mL Neocarzinostatin (NCS). Traces are colored if Cdk2 inactivated despite initially turning on or are gray if Cdk2 stayed active and APCCdh1 inactivated. Note no traces were colored (n = 587, DMSO; n = 1284).
(I) Cdk2 and APCCdh1 activity traces of cells treated with NCS after crossing the restriction point. These cells inactivated Cdk2, only to re-activate Cd-k2 and reenter the cell cycle several hours later (n = 35).
(J) Cdk2 and APCCdh1 activity traces treated with 200 ng/mL NCS for 12 min, then washed with mitogen-free media, and monitored for an additional 32 hr. (n = 33).
(K) Cdk2 and APC traces treated with 200 ng/mL NCS for 12 min, then washed with mitogen-free media, and monitored for an additional 21 hr. After 21 hr, full- growth media was added and cells were monitored for an additional 10 hr. Cells re-entered the cell cycle after stimulation with mitogens (n = 61).
(L) Cells stressed in G1 enter a quiescent state while cells stressed in S or G2 pause before continuing through the cell cycle. See also Figure S5.
We made use of a previous finding that Cdk2 activity bifurcates after mitosis and stays elevated in those cells that will enter the cell cycle (Figure 5B). This allows for a Cdk2-activity threshold to be selected after mitosis, with cells above the threshold maintaining high Rb phosphorylation and Cdk2 activity with >96% of them inactivating APCCdh1 4 hr later (Figures 5C, S5A, and S5B; Spencer et al., 2013). We first tested for stress-mediated cell-cycle exit using Neocarzinostatin (NCS), a commonly used drug to generate DNA double-stranded breaks. When we added a 20 min pulse of NCS to cells in G1 that we selected by live-cell analysis to have a Cdk2 activity above the threshold (Figures 5A and 5B), 20% of the NCS- treated cells now inactivated Cdk2 and failed to inactivate APCCdh1 (Figures 5D, S5D, and S5E) compared to 4% in control cells (Figures 5C, S5C, and S5E). To confirm that these cells lost Rb phosphorylation, we fixed cells 8 hr after NCS addition and measured Rb phosphorylation at S807/S811 by mapping fixed to live cells. We found that cells with low Cdk2 activity also had low levels of phosphorylated Rb and kept APCCdh1 activity on, suggesting that they have entered quiescence (Figure 5E).
Markedly, cells that were exposed to the same pulse of NCS after APCCdh1 inactivation (Figure 5F) showed hysteresis with each cell either continuing to build up Cdk2 activity or maintaining an elevated level of Cdk2 activity. In no case in hundreds of cells did we observe cells that responded by re-activating APCCdh1 (Figures 5F–5H, S5F, and S5G). Cells exposed to NCS after APCCdh1 inactivation went on to complete mitosis several hours later (Figure S5H), indicating these cells were committed to divide. Of note, cells exposed to NCS in late G2 delayed mitosis for several hours but were still able to enter mitosis and divide, suggesting that these committed cells paused to fix damage before completing the cell cycle (Figure S5I).
In the presence of mitogens, NCS-treated cells in G1 that exited to quiescence often again increased Cdk2 activity and re-entered the cell cycle approximately 10 hr later (Figure 5I). However, when we removed mitogens at the same time that we added NCS, cells kept Cdk2 activity low and stayed quiescent (Figure 5J). When we tested whether these cells still responded to mitogens at a later time, they still could be activated by mitogens and re-enter the cell cycle (Figure 5K). Together, these data show that even after passing the restriction point and activating pRb-E2F, but only up until the point when APCCdh1 inactivates, can cells return to a mitogen-sensitive quiescent state (Figure 5L).
We initially considered that stress-mediated exit to quiescence might be a rare event as a majority of cells still entered the cell cycle after NCS addition in late G1. However, when we analyzed cells with increasing Cdk2 activity in G1 phase that were exposed to hyper-osmotic stress, 96% of cells reversibly exited to quiescence and 63% of cells exited to quiescence in response to reactive oxygen stress (H2O2 application; Figures 6A–D and S6A-S6C). We again found that cells exposed to these stresses after APCCdh1 inactivation showed hysteresis, maintained Cdk2 activity, and kept APCCdh1 inactivated in response to the same stresses (Figures 6E–6G, S6D, and S6E). Furthermore, stress-mediated exit from the cell cycle in G1 was not restricted to cycling cells as we observed the same stress sensitivity after cells build up Cdk2 activity in cells coming out of quiescence (Figure S6F). Thus, diverse stresses that induce distinct stress pathways mediate a reversible return to a mitogen-sensitive quiescent state even after cells have crossed the restriction point as long as APCCdh1 is not inactivated.
Figure 6. Multiple Types of Stresses Can Cause Cell-Cycle Exit until, but Not after APCCdh1 Inactivation.
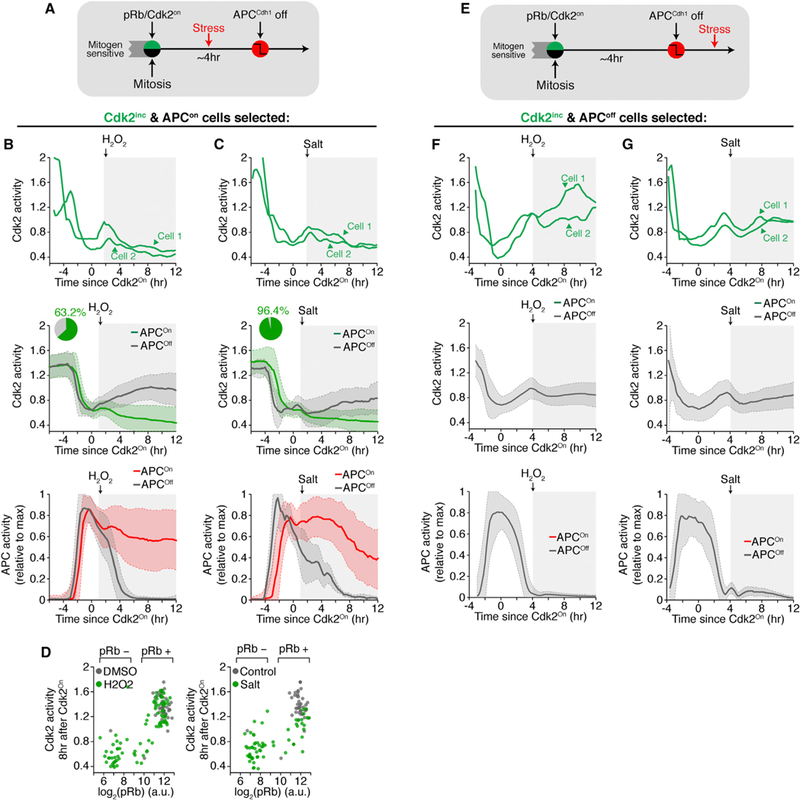
(A) Experimental setup for stress administration after the restriction point but before APCCdh1 inactivation.
(B and C) Representative examples and median traces ± SD of Cdk2 and APC activities in cells exposed to stress after Cdk2 activity started to rise. Light gray band represents time when cells were exposed to stress. Top: two cells are highlighted. Middle: median Cdk2 activity. Bottom: median APC activity. Traces are colored ifCdk2 inactivated despite initially turning on or are gray ifCdk2 stayed active and APCCdh1 inactivated. Inset: percentage of cells that inactivated Cdk2 activity despite initially turning on. (B) 200 μM Hydrogen peroxide (H2O2) or (C) 100 mM NaCl (salt) (n = 821, H2O2; n = 223, salt). See Figure S6C.
(D) Scatterplot of pRb807/811 levels versus Cdk2 activity 8 hr after the initial rise in Cdk2 activity (n = 1152, H2O2; n = 194, salt).
(E) Experimental setup for stress administered after APCCdh1 inactivation.
(F and G) Median traces ± SD of Cdk2 and APC activity exposed to stress afterAPCCdh1 inactivation. Top: two cells are highlighted. Middle: median Cdk2 activity. Bottom: median APC activity. (F) 200 μM Hydrogen peroxide (H2O2) or(G) 100mM NaCl (salt).Traces are colored ifCdk2 inactivated despite initially turning on or are gray if Cdk2 stayed active and APCCdh1 inactivated. Note no traces were colored. See also Figure S6.
Emi1-Mediated APCCdh1 Inactivation Suppresses Stress Sensitivity and Irreversibly Commits Cells to Progress through the Cell Cycle
To further test the hypothesis that APCCdh1 inactivation mediates cell-cycle commitment, we determined whether Emil renders APCCdh1 inactivation irreversible not only in respect to Cdk2 activity, but also in respect to stress signaling. We knocked down Emil using siRNA and then exposed cells to NCS in G1 phase. Cells treated with Emil siRNA were slightly more susceptible to NCS, as a higher percentage of cells exited the cell cycle prior to APCCdh1 inactivation (Figures 7A and S7A–S7C). However, cells that continued on to S phase failed to keep APCCdh1 inactivated and lost Cdk2 activity when the same stress was applied (Figure 7A). In contrast, cells treated with control siRNA and were exposed to the same stress were able to maintain APCCdh1 inactivation and enter the next mitosis several hours later. Thus, APCCdh1 inactivation becomes reversible in respect to stress application only in the absence of Emil. This argues that APCCdh1 inactivation is an Emil-regulated bistable switch that results in a loss of a cell’s ability to exit to the quiescent state in response to stress. Together, our data argue that the Emi1-mediated irreversible inactivation of APCCdh1 is triggered shortly before the G1/S boundary and long after pRb-E2F activation and that APCCdh1 inactivation has all the needed characteristics to be the commitment point after which cells must complete the cell cycle and can no longer return to quiescence before they divide by passing through mitosis.
Figure 7. APCCdh1 Inactivation Is a Rapid, Irreversible, and Bistable Switch that Commits Cells to Progress through the Cell Cycle.
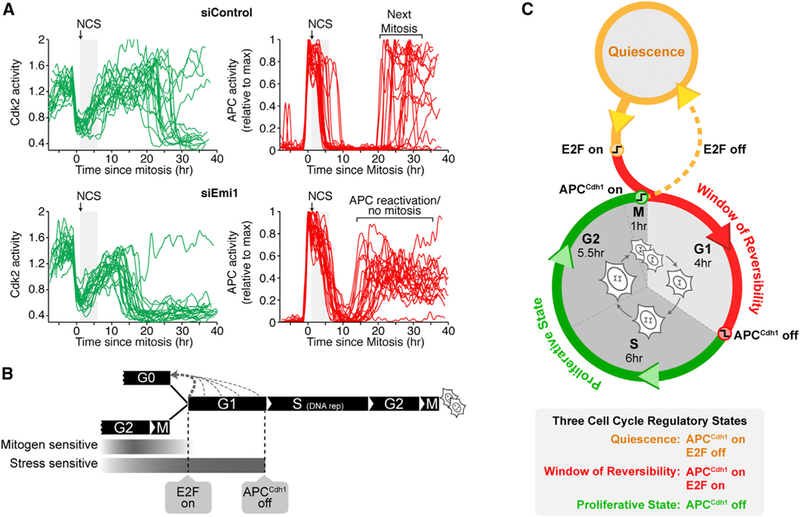
(A) Cdk2 and APCCdh1 activities in cells exposed to NCS and treated with either control (top) or Emil siRNA (bottom). Cells treated with Emil siRNA re-activate APCCdh1 and inactivate Cdk2. Control siRNA treated cells re-activate APCCdh1 much later during mitosis (n = 28, si Control; n = 23, siEmil).
(B) Windows of mitogen and stress sensitivity during cell-cycle entry and exit. Timing of cell-cycle entry and exit shows that mitogens and stress regulate entry during different time windows. Approximate cell-cycle phase durations for MCF10Aare highlighted to place APCCdh1 inactivation and cell-cycle commitment into the overall cell-cycle context.
(C) Cell-cycle entry and exit is characterized by three regulatory states: quiescence, a window of reversibility, and a proliferative state, defined by changes in APCCdh1 and E2F activities. The scheme shows the relationship of these states with the cell-cycle phases. See also Figure S7.
DISCUSSION
APCCdh1 Inactivation Is a Rapid and Irreversible Switch that Commits Cells to Start DNA Replication
Our study introduces a live-cell analysis approach using geminin-and helicase-B-derived peptide reporters (Sakaue-Sawano et al., 2008; Spencer et al., 2013) to quantitatively compare changes in APCCdh1 and Cdk2 activities in hundreds of individual cells throughout the human cell cycle. Our study provides three main lines of arguments that APCCdh1 inactivation underlies the cell-cycle commitment point. First, inactivation is rapid, bistable, and shows complete hysteresis with respect to Cdk2 inhibition, all critical characteristics of an irreversible decision process. The APCCdh1 inactivation kinetics is the same in every cycling cell and in cells coming out of quiescence and also the same in the three cell models we tested, arguing for a general bistable switch mechanism. Second, in a time window of reversibility that lasts from pRb-E2F until APCCdh1 inactivation, stress perturbations can cause cells to exit to quiescence. Only after APCCdh1 inactivation do cells lose their stress sensitivity, demonstrating a key hysteresis characteristic required for a point of no return or commitment point (Figure 7B). This marked rapid irreversibility is lost upon knockdown of the APCCdh1 inhibitor Emil, arguing that Emil is responsible for the speed, bistability, and hysteresis of the switch mechanism. Third, previous studies already showed that the inactivation of APCCdh1 is a rate-limiting step for entering S phase as knockdown of Cdh1 causes premature DNA replication (Sigl et al., 2009; Yuan et al., 2014). Our single-cell analysis confirmed this data and further shows that APCCdh1 inactivation is followed in nearly every cell by DNA replication within an hour. Thus, APCCdh1 inactivation has the necessary characteristics to be a general commitment point for cell-cycle entry distinct from the much earlier restriction point.
Cyclin E/Cdk2 Initiates APCCdh1 Inactivation and Emi1 Makes APCCdh1 Inactivation Irreversible
Of the proteins previously identified to regulate APCCdh1, we found that cyclin E/Cdk2 and Emil were the most relevant to trigger APCCdh1 inactivation before the G1/S boundary. In support of this model, Cdh1 has nine potential Cdk consensus phosphorylation sites that, when mutated, prevent Cdh1 from binding the APC (Kramer et al., 2000). Our proposed model of APCCdh1 inactivation implies that cyclin E and Cdk2 are both necessary for cell-cycle progression, and our data from siRNA knockdown experiments of cyclin E1 and E2 confirm this essential role. However, complete ablation of cyclin E or Cdk2 in mice did not prevent cell proliferation, in apparent conflict with our model. In the case of cyclin E knockout mice (Geng et al., 2003), our experiments in which we overexpress Emil give a plausible explanation of how cells can compensate for the loss of cyclin E (Figures 3D and 3E). We and others observed that high levels of Emil can inhibit APCCdh1 without an increase in cyclin E/Cdk2 activity. In the case of Cdk2 knockout mice, the discrepancy is likely due to compensation for the loss of Cdk2 by upregulation of Cdk1 activity. Indeed, it has been shown that cyclin E can bind and activate Cdk1 in cells derived from a Cdk2 knockout mouse (Aleem et al., 2005), indicating that in the absence of Cdk2, cyclin E/Cdk1 is capable of phosphorylating Cdk2 substrates. Consistent with cyclin E activating Cdk1 as a compensation mechanism, we found that addition of the Cdk1 inhibitor R0–3306 to wild-type mouse embryonic fibroblasts (MEFs) only prevented a small fraction of cells from accumulating the APCCdh1 substrates geminin and cyclin A (Figure S7D), while the Cdk1 inhibitor had a larger effect in Cdk2−/− MEFs (Figure S7E). Thus, while our model suggests that cyclin E/Cdk2 and Emil are the primary mediators of APCCdh1 inactivation, in the absence of cyclin E or Cdk2, there is likely compensation with cyclin E/Cdk1, cyclin A/Cdk2, and upregulated Emil also contributing to the inactivation of APCCdh1.
While we did not find a significant role for cyclin A/Cdk2 (Lukas et al., 1999) or SCFSkp2 (Barr et al., 2016; Fukushima et al., 2013) for the rapid inactivation of APCCdh1 in normal cells, it is likely that these APCCdh1 regulators are still functionally important at later points in the cell cycle when Cdh1 also becomes at least partially degraded (Fukushima et al., 2013). We also observed that knockdown of the APCCdh1-regulator Usp37 (Huang et al., 2011) had a small but significant effect on the kinetics of APCCdh1 inactivation and may therefore function as an additional regulator of APCCdh1 inactivation.
Different Stresses Can Cause Exit to Quiescence until APCCdh1 Inactivation or, Alternatively, Checkpoint Arrest Immediately after APCCdh1 Inactivation at the G1/S Boundary
Our study tested osmotic, hydrogen peroxide, and double strand DNA break stresses and showed that all three can cause cells to exit to quiescence after pRB-E2F activation until APCCdh1 inactivation. It is likely that many other stresses and signaling pathways known to inhibit proliferation can cause a similar reversible exit to quiescence during this time window. We also show that addition of thymidine, aphidicolin, or hydroxyurea in G1 has no effect on the inactivation kinetics of APCCdh1 but all result in a DNA-damage-mediated arrest at the G1/S boundary with Cdk2 activity remaining elevated and APCCdh1 remaining off. This argues that these G1/S-arrested cells merely pause at the onset of S phase and do not return to quiescence start of DNA replication (Borel et al., 2002).
Thus, two mechanisms have to be distinguished that prevent S-phase entry after the restriction point: (1) immediately acting stress and other signals in G1 that can prevent APCCdh1 inactivation and induce an exit to quiescence versus (2) stress perturbations that require DNA replication that lead to a checkpoint arrest only after APCCdh1 inactivation at the G1/S boundary without causing exit to quiescence.
Choreography of Cell-Cycle Entry and Exit Is Regulated by Mitogens and Stress
Our study reveals an overall timeline of when cells are sensitive to mitogens and stress during the cell cycle of MCF10A cells (Figure 7B). In cells coming out of quiescence, the time window for mitogen sensitivity is primarily before Rb phosphorylation. In cycling cells, the time window for mitogen sensitivity is primarily during G2 and M of the preceding cell cycle (Spencer et al., 2013; Stacey, 2003). In the latter case, as a corresponding molecular mechanism, we found that the same fraction of cells that lose Rb phosphorylation during mitosis exit the cell cycle after mitosis (Figure 7B, bold dashed gray line), while the remaining cells keep Rb phosphorylated, with most of the cells increasing Cdk2 activity and re-entering the cell cycle (Spencer et al., 2013).
Even though cells with Rb phosphorylated and E2F activated are on a trajectory to enter the cell cycle, they still respond to stress and can reverse Rb phosphorylation, E2F induction, and Cdk2 activation to return back to a mitogen-sensitive quiescent state (Figure 7B, dashed gray lines). Our results argue that mitogen signals are counteracted by diverse stress signals during a window of reversibility that lasts from pRB-E2F activation until APCCdh1 inactivation, after which stress signals can only mediate checkpoint arrest and not a return to quiescence.
From a regulatory perspective, this breaks the cell cycle into three states marked by their E2Fand APCCdh1 activities (Figure 7C): (1) quiescence, defined as a state when E2F activity is off and APCCdh1 is on, (2) a window of reversibility, defined as a state when E2F and APCCdh1 are both on, during which cells prepare for proliferation but can still exit to quiescence, and (3) A proliferative state, defined as a state when APCCdh1 is off. We propose that the window of reversibility reflects a competition between slower-acting mitogen signaling and faster-acting stress signaling pathways that can trigger throughout G1 an exit to quiescence until APCCdh1 is inactivated. Such a competition is well suited as a general safety control circuit by giving cells sufficient time to express the E2F target genes needed for proliferation while also providing a mechanism to remain sensitive to inhibitory stimuli and prevent damaged or challenged cells from entering the cell cycle.
EXPERIMENTAL PROCEDURES
Cell Lines
All experiments were done using MCF-10A cells, a non-transformed human mammary epithelial cell line, unless otherwise noted, and were obtained from ATCC (CRL-10317). In addition to MCF10A cells, APC activity was also measured in both HeLa (ATCC, CCL-2) and BJ-5ta cells (ATCC, CRL-4001).
Time-Lapse Microscopy
Cells were plated ~24 hr prior to imaging in full-growth media in a 96-well dish (Costar #3904) such that the density would remain sub-confluent until the end of the imaging period. Time-lapse imaging was performed in 290 μL full-growth media. Images were taken in CFP, YFP, and RFP channels every 12 min on an IXMicro microscope (Molecular Devices) with a 10× 0.3NA objective. Total light exposure time was kept under 600 ms for each time point. Cells were imaged in a humidified, 37°C chamber in 5% CO2. Cell tracking and data analysis was done using custom MATLAB scripts.
Immunofluorescence
Cells were fixed in 4% paraformaldehyde, washed three times in PBS, permeabilized with 0.2% triton, and stained overnight at 4°C with anti-phospho-Rb (807/811; Cell Signaling Technology, #8516), anti-cyclin A (Santa Cruz Biotechnology, sc-751), anti-p21 (BD PharMingen, 556430), anti-Geminin (Sigma-Aldrich, HPA049977), or anti-BrdU (Abcam, #ab6326). Primary antibodies were visualized using a secondary antibody conjugated to Alexa Fluor-647 and imaged with a Far Red filter. Cells were treated with 10 mM BrdU for 15 min prior to fixation. For figures showing EdU staining, cells were treated with 10 mM EdU for 15 min and fixed and processed according to manufacturer’s instructions (Invitrogen, #C10356).
Inhibitors
The inhibitors used in this study were: proTAME (10 mM, APC inhibitor, Boston Biochem, I-440), MLN-4924 (3 mM, neddylation inhibitor, Active Biochem, #A-1139), Cdk1/2i III (3 mM, EMD Biosciences #217714), neocarzinostatin (40 ng/mL, Sigma-aldrich, N9162), aphidicolin (2 mM, Sigma-aldrich, A0781), thymidine (2mM, Sigma-aldrich, T9250), and hydroxyurea (500 mM, Sigma- aldrich, H8627).
Supplementary Material
Highlights.
Rapid inactivation of APCCdh1 is triggered minutes before, DNA replication starts
Cyclin E/Cdk2 starts and Emi1 executes rapid and., irreversible APCCdh1 inactivation
A large time gap separates pRb-E2F activation from APCCdh1, inactivation
Cells can reverse back to quiescence until APCCdh1, inactivation, but not after it
ACKNOWLEDGMENTS
We thank Hee Wong Yang, Jia-yun Chen, Sean Collins, Feng-Chiao Tsai, Sam Bandara, Nalin Ratnayeke, Roy Wollman, Karlene Cimprich, James Ferrell, and Charles Sherr for helpful discussions, Philipp Kaldis for supplying the Cdk2−/− MEFs, and the Stanford Shared FACS Facility for cell sorting. S.D.C. and S.L.S. were supported by the Damon Runyon Cancer Research Foundation (DRG-2141-12 and DRG-2043-10). S.L.S. was also supported by an American Cancer Society Robert and Mary Ann Forsland Postdoctoral Fellowship (PF-13-304-01-CCG). T.M. was supported by NIH grants GM118377, GM030179, and P50GM107615.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and two movies and can be found with this article online at http://dx.doi.org/10.1016Zj.cell.2016.05.077.
REFERENCES
- Aleem E, Kiyokawa H, and Kaldis P (2005). Cdc2-cyclin E complexes regulate the G1/S phase transition. Nat. Cell Biol. 7, 831–836. [DOI] [PubMed] [Google Scholar]
- Barr ARH, Heldt FS, Zhang T, Bakal C, and Novák B (2016). A dynamical framework for the all-or-none G1/S transition. Cell Syst. 2, 27–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borel F, Lacroix FB, and Margolis RL (2002). Prolonged arrest of mammalian cellsattheG1/S boundary results in permanent S phase stasis. J. Cell Sci. 115, 2829–2838. [DOI] [PubMed] [Google Scholar]
- Chang L, Zhang Z, Yang J, McLaughlin SH, and Barford D (2014). Molecular architecture and mechanism of the anaphase-promoting complex. Nature 513, 388–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis PK, Ho A, and Dowdy SF (2001). Biological methods for cell-cycle synchronization of mammalian cells. BioTechniques 30, 1322–1326, 1328, 1330–1331. [DOI] [PubMed] [Google Scholar]
- Deckbar D, Jeggo PA, and Löbrich M (2011). Understanding the limitations of radiation-induced cell cycle checkpoints. Crit. Rev. Biochem. Mol. Biol. 46, 271–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dulić V, Kaufmann WK, Wilson SJ, Tlsty TD, Lees E, Harper JW, El-ledge SJ, and Reed SI (1994). p53-dependent inhibition of cyclin-dependent kinase activities in human fibroblasts during radiation-induced G1 arrest. Cell 76, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Eguren M, Manchado E, and Malumbres M (2011). Non-mitotic functions of the Anaphase-Promoting Complex. Semin. Cell Dev. Biol. 22, 572–578. [DOI] [PubMed] [Google Scholar]
- Erol A (2011). Genotoxic stress-mediated cell cycle activities forthe decision of cellular fate. Cell Cycle 10, 3239–3248. [DOI] [PubMed] [Google Scholar]
- Ferreira MF, Santocanale C, Drury LS, and Diffley JF (2000). Dbf4p, an essential S phase-promoting factor, is targeted for degradation by the anaphase-promoting complex. Mol. Cell. Biol. 20, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Floyd S, Pines J, and Lindon C (2008). APC/C Cdh1 targets aurora kinase to control reorganization of the mitotic spindle at anaphase. Curr. Biol. 18, 1649–1658. [DOI] [PubMed] [Google Scholar]
- Fukushima H, Ogura K, Wan L, Lu Y, Li V, Gao D, Liu P, Lau AW, Wu T, Kirschner MW, et al. (2013). SCF-mediated Cdh1 degradation defines a negative feedback system that coordinates cell-cycle progression. Cell Rep. 4, 803–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Yu Q, Sicinska E, Das M, Schneider JE, Bhattacharya S, Rideout WM, Bronson RT, Gardner H, and Sicinski P (2003). Cyclin E ablation in the mouse. Cell 114, 431–443. [DOI] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2000). The hallmarks of cancer. Cell 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Havens CG, Ho A, Yoshioka N, and Dowdy SF (2006). Regulation of late G1/S phase transition and APC Cdh1 by reactive oxygen species. Mol. Cell. Biol. 26, 4701–4711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu YC, Li L, and Fuchs E (2014). Transit-amplifying cells orchestrate stem cell activity and tissue regeneration. Cell 157, 935–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Summers MK, Pham V, Lill JR, Liu J, Lee G, Kirkpatrick DS, Jackson PK, Fang G, and Dixit VM (2011). Deubiquitinase USP37 is activated by CDK2 to antagonize APC(CDH1) and promote S phase entry. Mol. Cell 42, 511–523. [DOI] [PubMed] [Google Scholar]
- Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, and Peters JM (2000). Mitotic regulation oftheAPC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11, 1555–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas C, Sørensen CS, Kramer E, Santoni-Rugiu E, Lindeneg C, Peters JM, Bartek J, and Lukas J (1999). Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase-promoting complex. Nature 401, 815–818. [DOI] [PubMed] [Google Scholar]
- Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A, and Jackson PK (2006). Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev. 20, 2410–2420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimha AM, Kaulich M, Shapiro GS, Choi YJ, Sicinski P, and Dowdy SF (2014). Cyclin D activates the Rb tumor suppressor by monophosphorylation. eLife 3, e02872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardee AB (1974). A restriction point for control of normal animal cell proliferation. Proc. Natl. Acad. Sci. USA 71, 1286–1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell Biol. 7, 644–656. [DOI] [PubMed] [Google Scholar]
- Planas-Silva MD, and Weinberg RA (1997). The restriction point and control of cell proliferation. Curr. Opin. Cell Biol. 9, 768–772. [DOI] [PubMed] [Google Scholar]
- Rape M, and Kirschner MW (2004). Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432, 588–595. [DOI] [PubMed] [Google Scholar]
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. [DOI] [PubMed] [Google Scholar]
- Sigl R, Wandke C, Rauch V, Kirk J, Hunt T, and Geley S (2009). Loss of the mammalian APC/C activator FZR1 shortensG1 and lengthens S phase but has little effect on exit from mitosis. J. Cell Sci. 122, 4208–4217. [DOI] [PubMed] [Google Scholar]
- Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S, et al. (2009). An inhibitorofNEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736. [DOI] [PubMed] [Google Scholar]
- Spencer SL, Cappell SD, Tsai FC, Overton KW, Wang CL, and Meyer T (2013). The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 155, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stacey DW (2003). Cyclin D1 serves as a cell cycle regulatory switch in actively proliferating cells. Curr. Opin. Cell Biol. 15, 158–163. [DOI] [PubMed] [Google Scholar]
- Vousden KH, and Prives C (2009). Blinded by the Light: The Growing Complexity of p53. Cell 137, 413–431. [DOI] [PubMed] [Google Scholar]
- Walker A, Acquaviva C, Matsusaka T, Koop L, and Pines J (2008). UbcH10 has a rate-limiting role in G1 phase but might not act in the spindle checkpoint oras part of an autonomous oscillator. J. Cell Sci. 121, 2319–2326. [DOI] [PubMed] [Google Scholar]
- Wang W, and Kirschner MW (2013). Emi1 preferentially inhibits ubiquitin chain elongation by the anaphase-promoting complex. Nat. Cell Biol. 15, 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg RA (1995). The retinoblastoma protein and cell cycle control. Cell 81, 323–330. [DOI] [PubMed] [Google Scholar]
- Xu L, and Qu Z (2012). Roles of protein ubiquitination and degradation kinetics in biological oscillations. PLoS ONE 7, e34616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao G, Lee TJ, Mori S, Nevins JR, and You L (2008). A bistable Rb-E2F switch underlies the restriction point. Nat. Cell Biol. 10, 476–482. [DOI] [PubMed] [Google Scholar]
- Yuan X, Srividhya J, De Luca T, Lee JH, and Pomerening JR (2014). Uncovering the role of APC-Cdh1 in generating the dynamics of S-phase onset. Mol. Biol. Cell 25, 441–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, and Larsson O (1985). Kinetic analysis of regulatory events in G1 leading to proliferation or quiescence of Swiss 3T3 cells. Proc. Natl. Acad. Sci. USA 82, 5365–5369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetterberg A, Larsson O, and Wiman KG (1995). What is the restriction point? Curr. Opin. Cell Biol. 7, 835–842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.