


Abstract
Xenobiotic nucleic acids (XNA) are nucleic acid analogues not present in nature that can be used for the storage of genetic information. In vivo XNA applications could be developed into novel biocontainment strategies, but are currently limited by the challenge of developing XNA processing enzymes such as polymerases, ligases and nucleases. Here, we present a structure-guided modelling-based strategy for the rational design of those enzymes essential for the development of XNA molecular biology. Docking of protein domains to unbound double-stranded nucleic acids is used to generate a first approximation of the extensive interaction of nucleic acid processing enzymes with their substrate. Molecular dynamics is used to optimise that prediction allowing, for the first time, the accurate prediction of how proteins that form toroidal complexes with nucleic acids interact with their substrate. Using the Chlorella virus DNA ligase as a proof of principle, we recapitulate the ligase's substrate specificity and successfully predict how to convert it into an XNA-templated XNA ligase.
INTRODUCTION
Xenobiotic nucleic acids (XNA) are chemical analogues of natural nucleic acids, modified in at least one of their three main chemical moieties: nucleobase, sugar or phosphate backbone. Modifications in the sugar-phosphate backbone can enhance resistance against alkali hydrolysis or against nucleases compared to natural nucleic acids. Early XNA research focused on enhanced chemical and biological stability for the development of nucleic acid-based therapeutics such as antisense, RNA silencing and aptamers. More recently, XNAs are explored in synthetic biology as an alternative carrier of genetic information that can be used to generate novel robust platforms for biocontainment. Full replacement of a natural nucleic acid with an XNA in vivo is a challenging goal. More feasible is the development of an episome capable of co-existing, but not interfering with the natural machinery. The development of such ‘orthogonal’ episome requires XNA processing enzymes not only to manipulate XNA in vitro but also to enable the maintenance of the XNA information in vivo.
To construct an XNA episome, XNA oligonucleotides are needed beyond what is currently possible with conventional solid-phase synthesis (∼30-mers). An efficient enzyme to join chemically synthesized XNA fragments would bring synthetic genetics to a next level. Some natural enzymes are able to recognize and ligate specific XNA fragments in optimized experimental conditions (1,2).These enzymes are useful tools for in vitro manipulation but also show limited specificity for XNA. Ideally, a fully orthogonal XNA should be exclusively processed by highly specific XNA processing enzymes. Methods based on in vitro evolution and engineering are described to obtain efficient XNA polymerases and replicases (3–5). However, structure-based design of XNA processing enzymes is underexplored. Many structures of nucleic acid processing enzymes bound to their native substrate have been determined by experimental methods (6). Independent of their catalytic role, most enzymes involved in DNA metabolism wrap around their substrate, adopting a ring-shaped structure with a central hole (‘toroid’) accommodating the DNA with extensive intermolecular contacts to the nucleobases and the sugar phosphate backbone (7–9). In these closed complexes, the flexible DNA adopts typically a structure that deviates from canonical A and B type helices. Known experimental XNA structures (10) diverge significantly from the distorted natural nucleic acids in the central hole of nucleic acid processing enzymes. Therefore, molecular modeling cannot simply replace DNA or RNA by XNA in the available crystal structures.
We focused on ligation of oligonucleotide fragments composed of 2′-O-methyl-modified ribonucleotides (2′OMeRNA) and hexitol-based XNA nucleotides (HNA) (Figure 1) as model (xenobiotic) nucleic acid polymers since they have experimental structures for homoduplexes (11–13) and duplexes with natural nucleic acid (14–16) that do not resemble the typical B-type DNA helix. Therefore, they were considered to be well positioned as test cases.
Figure 1.
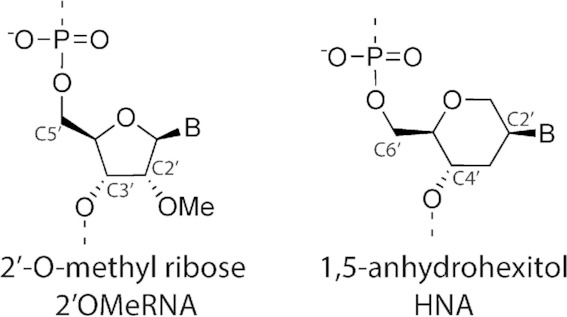
Chemical structure of 2′O-methylated ribose (left) and 1,5-anhydrohexitol (right).
DNA ligases are a class of enzymes that catalyze the joining of breaks in the phosphodiester backbone (nicks) of duplex DNA in nature. They assist in forming a covalent bond between the terminal 5′ monophosphate of a donor strand and the 3′ hydroxyl group of an adjacent acceptor strand that are both bound to a template. All DNA ligases have a nucleotidyl-transferase (NTase) domain and an oligonucleotide binding domain (OB) that constitute the catalytic core of the enzyme. ATP-dependent ligases from eukaryotes and archaea have a third DNA-binding domain (DBD) in their N-terminus that allows the three-domain ligases to fully encircle the nicked double-stranded DNA substrate (17). For the structure-based design of an XNA ligase, we chose to focus on the Chlorella virus DNA ligase (ChVLig), a well-characterized enzyme with multiple crystal structures available, including in complex with DNA (18). The ChVLig is a viral ATP-dependent ligase and one of the smallest ligases characterized to date, with the DBD domain replaced by a short surface loop forming a ‘latch’ domain (19,20).
Compared to molecular modelling on protein–protein complexes, computational approaches on the protein–nucleic acid prediction and calculation (21–24) is lagging behind. Most software that is applicable to calculate models for DNA-protein complexes was originally developed for proteins and later adapted to accept nucleic acids as input structures (25). Two main categories of docking algorithms exist: first, machine learning algorithms to predict molecule interactions based on sequence-based and/or structure-based information; and second, data-driven algorithms to calculate interactions with information from known crystal structures (26). Both methods have different respective strengths and weaknesses (27). Data-driven methods are capable of integrating heterogeneous experimental data and are usually quite computationally efficient. Therefore, this approach was selected to generate models for unbound nucleic acids encircled by a protein. A limited number of docking algorithms for protein–DNA docking and their web servers such as HADDOCK (28), NPDock (29) and HDOCK (30) have been developed and made available for public access but they are mostly restricted to natural nucleic acids. The HADDOCK software for protein–nucleic acids docking (31) allows to implement synthetic nucleic acids and enables data-driven docking with biochemical and biophysical information on the target system to guide the docking process. Nevertheless, docking of the near-toroidal protein–DNA complex from the protein–DNA benchmark (PDB ID: 3bam) (32) cannot readily recapitulate the known crystal structure since it is classified by HADDOCK as ‘difficult’. NPDdock can model the docking of the complex from an unbound protein state but the resulting prediction has an RMSD of 19 Å compared to the crystal structure, limiting its predictive power using unbound docking approaches.
In this work, a docking strategy is introduced that enables the accurate modelling of toroidal protein complexes bound to nucleic acids, which is an essential step in their engineering. We further demonstrate the power of our method engineering a ChVLig variant capable of XNA-dependent XNA ligation, the first of its kind and a key milestone in the development of XNA molecular biology.
MATERIALS AND METHODS
Solvated protein–nucleic acid docking
Split-docking protocol
The coordinate file of dsDNA bound to a toroid protein was retrieved from the RCSB Protein Data Bank (PDB) (33). The coordinate file was split into distinct pdb-files for defined protein domains to generate multiple docking bodies. A standard ambiguous interaction restraints (AIR) file was generated by the HADDOCK server and edited accordingly. All docking partners were subjected to initial HADDOCK (high ambiguity driven docking) refinement. Python scripts derived from ARIA were used for automated structure calculations.
During docking runs, the active residues were set to be semi-flexible. Each subunit was then surface docked on the refined duplex using the default protocol for protein–DNA docking (28) executed by HADDOCK version 2.2 using CNS version 1.3 for all docking simulations. Although the structures of the three subunits and of their interface were already geometry-optimized, the side chains and backbone atoms at the interface were still allowed to move during the torsion angle dynamics (TAD) simulated annealing and the water refinement process. The TAD-factor was set to be 8. The dielectric constant was set to 78.0 instead of the default 10.0 to dampen the electrostatic contribution of DNA in vacuum. The respective position and orientation of the four docking partners were first randomized. After rigid body energy minimization (1000 conformations), semi-flexible simulated annealing in torsion angle space (200 conformations) and final refinement in explicit solvent was carried out. The quality of the first docking run was assessed by calculating the interface RMSD (iRMSD).
HADDOCK scoring was performed according to the weighted sum (HADDOCK score) of different energy terms, which include van der Waals energy, electrostatic energy, distance restraints energy, inter-vector projection angle restraints energy, diffusion anisotropy energy, dihedral angle restraints energy, symmetry restraints energy, binding energy, desolvation energy and buried surface area.
A cluster analysis was performed on the final docking output using a minimum cluster size of 5 and cut-off Root-Mean-Squared-Deviation (RMSD) of 4 Å. After the docking run, the 10 top structures based on the highest affinities were manually curated to identify the best models (visual inspection using Pymol). These 10 structures were uploaded to the 3D-DART server (34) for DNA analysis and custom DNA structure model generation. Because the 3D-DART server cannot recognize XNA nucleotides the analysis for protein-XNA docking after the first docking run, was carried out using the HADDOCK score of the 10 best structures within the top cluster, after manual curation of the calculated complexes. The output was uploaded as an ensemble for the second docking run.
Solvated protein–DNA docking
The split-docking protocol described above was run for the regeneration of the crystal structure of the ChVLig in complex with a nicked DNA duplex (PDB ID: 2q2t) and BamHI restriction endonuclease in complex with dsDNA (PDB ID: 3bam).
For ChVLig - dsDNA modeling, a nicked DNA duplex with canonical B-type structure (5′-ATTGCGACC▾CCACTATCGGAA-3′ and 5′-TTCCGATAGTGGGGTCGCAAT-3′) was built using a combination of the 3D-DART software to generate a canonical B-type duplex and the AMBER16 forcefield to introduce the nick indicated by ‘▾’. The NTase, OB and latch domain of ChVLig were extracted from the coordinate file (PDB ID: 2q2t) and separated into three distinct pdb-files, generating three docking bodies with original residue numbering 1-188, 189-201/232-293 and 202–231 for the NTase, OB and latch domain respectively. HADDOCK was set up with restraints of 3 Å in the AIR file. Standard error margins of 0.5 Å in the AIR file were reduced to 0.1 Å error range for residues at the cutting sides of docking bodies and for restraints involving catalytic residues of ChVLig (11) since correct positioning of the catalytic site is crucial for enzyme activity (Supplementary information S5.1). The DNA duplex and the three ligase subdomains were defined as the four docking bodies in the multibody docking approach described above. The iRMSD was then calculated with the crystal structure as a reference and plotted with the HADDOCK score.
For the dimeric BamHI endonuclease – dsDNA docking, the same protocol was used as described for the ChVLig, using a canonical B-type DNA duplex (5′-ATGGATCCATA-3′) with three BamH1 subdomains as docking partners (1.A-192.A, 193.A-206.A, 1.B-209.B) and the ambiguous interactions defined as described in SI (Supplementary information S5.2).
Solvated protein-XNA docking
To generate the nicked HNA with the same sequence as the dsDNA substrate for ChVLig mentioned above, simulated annealing by CNS (35) version 1.3 was used including hydrogen bond restraints and planarity for base pairing in addition to dihedral angle restraints on the backbone and hexitol rings as described in literature for HNA structures (11,12). The nicked 2′OMeRNA duplex with identical sequence was generated in AMBER using simulated annealing including backbone dihedral angles from available structures, hydrogen bond restraints and planarity for base pairing. Atom types and backbone parameters of XNA residues were added to DNA/RNA restraints definition file (Supplementary information S5.1). The nick in both XNA duplexes was introduced with AMBER16. For XNA docking purposes in HADDOCK, the restraint definition file for DNA was adjusted to fit 2′OMeRNA and HNA definitions. CNS runfile was adjusted to incorporate the custom topology definition and parameters. AIR restraints used in the ChVLig–dsDNA docking with an increased error range of 2.0 Å (except for AIR involving catalytic residues) were applied during protein-XNA docking to buffer for altered XNA-duplex binding. A cluster analysis was performed on the final docking output using a minimum cluster size of 5 and cut-off RMSD of 4 Å. Because no crystal structure is available for ChVLig binding dsXNA, the target for RMSD calculations is set to be the top-ranked calculated conformation with optimal HADDOCK score. The second docking run was initiated with the 10 top-ranked calculated structures based on their combined HADDOCK score—iRMSD values. After the second water-refinement run, the top-ranked calculated complex was accepted as the final complex and subjected to Molecular Dynamics (MD) simulation.
Molecular dynamics
All preparative and base pair-restrained molecular dynamics simulations in explicit solvent were run using the GPU version PMEMD engine provided with AMBER16 (36) package. The ff14SB force field (37) was used to describe the protein in the generated complex. As in a previously described protocol for 2′OMeRNA parameters (38) in AMBER, the restrained electrostatic potential (RESP) (39) was used for force-field parametrization of HNA residues. Accordingly, additions were made to the updated AMBER DNA.OL15 force-field (40) to incorporate the XNA residues (Supplementary information S2–S3). Missing hydrogens and counter ions for neutralization were added through the LEAP module, together with the definition of terminal phosphate in residues on 5′- residues at donor side of the nick (DCn, MCn and HCn).
Prior to the set-up, all systems were immersed into an octahedral box with TIP3P (41) water molecules, spacing the atoms of the protein-NA complex 12 Å from the boundary of the simulation box edges. The system included 15424 atoms. SHAKE algorithm was applied to all systems to constrain all bond lengths involving hydrogen atoms allowing a 2 fs time-step. The cut-off distance for van der Waals interaction was set to be 12 Å. A system of Particle Mesh Ewald (PME) method was used to treat long-range electrostatic interactions. To remove steric clashes, a seven-step minimization procedure involving 1000 steps of steepest descent energy minimization was first performed followed by 4000 steps of conjugate gradient minimization at each step. The system was then slowly heated to 300 K after which the complexes were equilibrated by 2 ns position restraint MD simulations with 10.0 kcal/mol/Å2 constant force on the heavy atoms of protein and substrate under NPT condition (1 atm). After this equilibration protocol, base pairing restrained MD was performed on all solvated systems under control of a Berendsen thermostat (1 atm) and a Langevin thermostat (300 K) using periodic boundary conditions applied in all three cartesian directions to mimic the infinity of the system. The nonbonded list was updated every 25 steps, with new random number seeds chosen every 25 ns for each simulation to prevent simulation synchronization of the trajectories. The trajectories were sampled every 100 ps for analysis in production dynamics. 200 ns MD simulations without any AIR restraints on the three binding domains were finally carried out under NPT conditions.
For trajectories of all calculated complexes, the cpptraj module (42) of AmberTools was used to measure distances between two sets of amino-acids involved in clamp closing (Phe215 and Tyr217 of the latch versus Phe44 and Lys5 of the NTase domain). The same module was used to calculate root-mean-squared deviation (RMSD) on Cα atoms from the average structure. Root mean square fluctuation per residue (RMSF) on Cα atoms from the average structure was also determined with the cpptraj module.
The collective motion of the alpha carbon (Cα) atoms of both wild-type and mutant enzyme in complex with DNA, 2′OMeRNA and HNA was investigated using essential dynamics (ED) analysis (43), often called principal component analysis (PCA) of the trajectory. The eigenvectors and eigenvalues of the covariance matrix were calculated, thereby describing large-scale domain movements. Each of the eigenvectors describes a collective motion of particles, where the values of the vector indicate how much the corresponding atom participates in the motion. The PCA analysis of the Cα-atoms was plotted taking the projection of the first eigenvector with respect to the projection of the second eigenvector to represent the total phase space the protein is able to occupy.
Cloning
Enzymes required for cloning were purchased from New England Biolabs (NEB) unless otherwise stated. All reagents for media were bought from Fisher Scientific (Pittsburgh, PA). PCR primers were purchased from IDT (Leuven). In-FusionTM assembly (44) (ClonTech, Takara) was used to construct the recombinant pET16-ChVLig vector. The ChVLig gene (45) (896 base pairs (bp), PBCV1_A544R) was used as insert DNA sequence, ordered as a gBlock fragment from IDT. The 5.6 kb pET16b (Novagen®) was used as vector DNA template. Sense and antisense PCR primers were designed with 15-bp overlap-regions with pET16b (underlined) next to 22- and 23-bp overlap-regions with the insert sequence (bold) (RV: AGCCGGATCCTCGAGCTAACGGTCTTCCTCGTGACGA, FW: CATATCGAAGGTCGTATGGCAATCACAAAGCCATTGCT). The same bold format was used in for complementary sequences in the gBlock fragment (Supplementary information S1). pET16 was linearized with NcoI restriction enzyme. The insert DNA fragment was amplified using the designed primers with the highly accurate PrimeSTAR GXL DNA polymerase (Takara-Bio) by polymerase chain reaction (PCR). The amplified insert was purified using Nucleospin®, gel and PCR cleanup (ClonTech, Takara) following agarose gel electrophoresis. The restriction enzyme-digested vector was mixed with the amplified, gel-purified DNA insert and the In-Fusion HD Enzyme Premix (Clontech, Takara) to a total volume of 10 μl. The mix was incubated for 15 min at 50°C, transferred to ice, after which the resulting plasmid was transformed into Stellar competent cells (Invitrogen) plated on LB agar plates containing ampicillin and incubated at 37°C for 12–16 h. After sequencing-based validation, the plasmid was further transformed into Rosetta2 pLysS chemocompetent cells (Novagen).
Site directed mutagenesis
The glycine insertion was achieved using the Q5 site-directed mutagenesis kit (New England BioLabs, NEB). Reactions were carried out according to the manufacturer's recommendations. To insert a glycine at position 189 (further coded as 189insG) the pET16(b)-ChVLig construct was PCR amplified with forward primer (insert codon underlined) 5′-GGCCAGTTCAAAGATGCAGAG-3′ and reverse primer 5′- TTTCATCTTCAGTAGAATACC-3′ after which the construct was blunt-end ligated using T4 DNA ligase (New England Biolabs). The resulting pET16(b)-189insG construct was transformed into Stellar competent cells. After sequencing-based validation, the plasmid was further transformed into Rosetta2 pLysS chemocompetent cells (Novagen).
Expression and purification
Starting from plated Rosetta2 pLysS cells containing recombinant pET16(b)-ChVLig plasmid, inocula were grown to saturation overnight in Luria Broth medium (LB, Sigma). For plate selection ampicillin was used at 100 μg/ml and chloramphenicol at 30 μg/ml. The pET16-based construct was over-expressed using the auto-inducing medium ZYP-5052 (57) (20 g Tryptone, 10 g yeast extract, 1860 mL sterile water, 2 mM MgSO4, 500 μl 1000× Trace metals mix, 1× 5052, 1× NPS, 100 μg/ml Ampicillin, 10 μg/ml Chloramphenicol). All culture growth was done at 25°C for 24–26 h and subsequently at 18°C for another 24 h. Large-scale expression was conducted in a 3 l glass culture flask containing 1 l of culture medium (shaking at 121 rpm, orbital diameter 26 mm). High density cultures (OD600 > 4) were pelleted by ultracentrifugation (13 000g, 12 min, 4°C) after which dry pellet was stored overnight at −80°C. The pellet was then thawed in a beaker of iced water for 2 h. The harvested cells from 1 liter of culture were resuspended in 10 ml lysis buffer (50 mM Tris–HCl (pH 7.5), 50 mM NaCl, 10 mM MgCl2, ATP 0.5 mM, imidazole 5 mM, glycerol 10% (v/v)) supplemented with DNaseI, placed on ice and disrupted using a Hielscher sonicator (70% duty; 15 min on; 3 min off; five cycles). Hereafter, 2 ml of a 50% slurry of Ni-NTA resin (Qiagen) was washed twice with 10 ml Milli-Q water and further equilibrated with binding buffer (10 mM imidazole, 50 mM NaH2PO4, 500 mM NaCl, pH 8,0). The supernatant was then mixed with 2 ml of pre-equilibrated Ni-NTA resin (Qiagen) and incubated with gentle agitation for 2 h at 4°C. The resin was packed by gravitational flow and washed four times with wash buffer (50 mM Tris–HCl, pH 8.0, 200 mM NaCl, 10 mM MgCl2, 20 mM imidazole, 10 mM ATP). Elution of HIS-tagged ChVLig and its 189insG mutant was done in 10 ml elution buffer (50 mM Tris–HCl, pH 8.0, 200 mM NaCl, 10 mM MgCl2, 300 mM imidazole, 10 mM ATP). The purity of enzymes was assessed by SDS-PAGE analysis using current-limited electrophoresis conditions set at 25 mA for 90 min with acrylamide concentrations of 15% and 4% for separation gel and stacking gel, respectively.
A prominent 34 kDa recombinant His-tagged product was detectable by SDS-PAGE in whole-cell extracts of auto-induced BL21 Rosetta2 pLysS bacteria. This polypeptide was not present when bacteria containing the pET16b vector alone were induced. After lysis by sonication followed by centrifugal separation of the crude lysate, ChVLig was recovered from the soluble supernatant fraction and concentrated in a 15-kDa cutoff filter in 1× SplintR ligase reaction buffer (50 mM Tris–HCl (pH 7.5), 10 mM MgCl2, 1 mM ATP, 10 mM DTT). The 34.5 kDa recombinant mutant 189insG was readily detectable by SDS-PAGE in whole-cell extracts of auto-induced BL21 Rosetta pLysS bacteria. After lysis and centrifugal separation of the cell-debris, the protein of interest was recovered and concentrated in a 15-kDa cutoff filter in 1× SplintR ligase reaction buffer.
XNA ligation assay
Ligation activity of ChVLig and mutant ligase was tested on both 2′OMeRNA and HNA nicked duplexes, with DNA ligation as a positive control. 6-FAM (6-carboxyfluorescein) was used as fluorescent dye attachment in all acceptor oligonucleotides. C3Sp (C3 Spacer phosphoramidite) was used to block 3′ends of template and donor sequences. All substrate sequences (acceptor, donor, template oligo) are listed numerically in Supplementary information S7.1. Custom DNA-oligonucleotides were ordered from IDT (acceptor oligo 3a, donor oligo 1a, template oligo 5a). Custom 2′OMeRNA-oligonucleotides were ordered from IDT (acceptor oligo 3b, donor oligo 1b, template oligo 5b). Custom HNA oligonucleotides were prepared in-house by phosphoramidite oligonucleotide synthesis on ABI Expedite® 8909 Nucleic Acid Synthesis System (acceptor oligo 3c, donor oligo 1c, template oligo 5c). All the oligonucleotides were purified by 15% denaturing PAGE after which they were eluted from the gel using 0.3 M sodium acetate (pH 5.4) shaking overnight at 37°C. All sodium acetate eluates were desalted using Illustra NAP-25 columns, followed by ethanol precipitation. Ligation assay was carried out by mixing 5′-6-FAM-labeled acceptor- and 3′-blocked-donor-oligo's with complementary 3′-blocked-template in a 1:3:3 molar ratio, with enzyme (50 nM). Ligation efficiency on nicked DNA and 2′OMeRNA duplexes was tested across a range of reaction times (1 h, 3 h, ON), and different temperatures (25, 37°C) (data not shown). Overnight ligation at 37°C provided the highest ligation efficiency after which the combination of these conditions (37°C, ON) were used in further experiments. All ligation assays were carried out in duplicate (technical replicates) and the data presented are the result of four independent experiments (biological replicates, n = 4).
A 15% polyacrylamide–urea gel (20 cm × 30 cm) was made with 1 mm wide wells. Before the samples were loaded onto PAGE for analysis, the wells were flushed with approximately 1 ml of 1× TBE buffer (90 mM Trizma, 90 mM boric acid, pH 8.3 and 2 mM EDTA) in a syringe to remove polymerized debris and urea after which a pre-run was applied to equilibrate the gel with TBE buffer. To 11 μl of the sample, 1 μl of proteinase-K (800 units/ml, New England Biolabs) was added and incubated at 50°C. After 30 min, the sample was mixed with 12 μl of a 2× loading buffer (0.025% SDS, 18.75 mM EDTA, 0.02% bromophenol blue in 95% formamide) and heated to 94°C for 10 min after which it was loaded on PAGE. The electrophoresis was carried out using 1× TBE as running buffer with a 25 mA constant current. Typhoon™ FLA9500 (GE Healthcare Life Sciences) was used for fluorescence visualization. Ligation efficiency quantification was done by measurement of relative intensity between 5′-6-FAM-labeled ssXNA or ssDNA acceptor fragments and donor–acceptor ligated fragments using ImageQuant TL 1D v8.1 software.
Mutant characterization
Ligase specificity
To probe sequence dependency of the mutant ligase, four near-identical nicked substrates were used (combinations 2a, 4, 6a; 2b, 4, 6b; 2c, 4, 6c and 2d,4, 6d; Supplementary information S7.1) substituted with 2′OMeRNA chemistry. More specifically, the donor sequences were varied by substituting the bases flanking the nick at the donor-side with all possible canonical base-pairs together with complementary template sequences. Moreover, these oligos differ in sequence, length (34-mer versus 38-mer) and fraction of xenonucleotides (14/14 versus 6/18 substituted in acceptor, 12/20 versus 6/20 substituted in donor) compared to the oligos used in general XNA ligation assays. Ligation reaction was carried out using 100 nM 189insG mutant ligase at 37°C for 16 h and subsequently quenched using quenching solution. Ligated and non-ligated acceptor fragments were separated using polyacrylamide gel electrophoresis, following the method described in the XNA ligation assay.
Concentration dependency
A dilution series of the 189insG mutant ligase (100, 20, 10, 5, 2 and 1 nM) was prepared in six aliquots of ligation assay mixtures containing reaction buffer supplemented with acceptor–donor–template mixture in 1:3:3 molar fraction (Supplementary Information S7.1, substrate 1c, 3c, 5a). Ligation reaction was carried out at 37°C for 16 h. Polyacrylamide gel electrophoresis was used to separate ligated and non-ligated acceptor fragments.
Estimation of kcat and KM
To measure the Michaelis–Menten parameters kcat and KM, a series of steady state assays were performed with the same oligo combination as described in the ins189G concentration dependency assay. A typical aliquoted assay mixture (12 μl) consisted of 2.4 μl in-house prepared 5× ligase buffer (330 mM Tris–HCl, 50 mM MgCl2, 5 mM DTT, 5 mM ATP, pH 7.4), supplemented with a relevant acceptor–donor–template mixture in 1:3:3 molar fraction in a volume corresponding to the final substrate concentration. The components were mixed by gentle pipetting and incubated at 37°C for 10 min. All ligation assay aliquots were initiated by the addition of 189insG mutant ligase (500 nM, 10×) in storage buffer (50 mM KCl, 10 mM Tris–HCl, 0.1 mM EDTA, 1 mM DTT, 50% glycerol) to the pre-incubated reaction mixtures followed by gentle mixing and incubation at 37°C. Final concentrations of XNA/DNA were 250 nM, 500 nM, 750 nM, 1 μM, 2 μM and 4 μM in the FAM label (acceptor). In short-run kinetics experiments, time points (12 μl) were collected after 0.5, 1, 2, 3, 5, 7 and 10 min and quenched with 12 μl of 2× quenching solution (0.025% SDS and 18.75 mM EDTA in 95% formamide). In long-run kinetics experiments, time points (12 μl) were collected after 0.25, 0.5, 1, 3, 6 and 16 h and quenched with 12 μl of 2× quenching solution. To estimate kcat and KM values, a final enzyme concentration of 100 nM was used. Polyacrylamide gel electrophoresis was used to separate ligated and non-ligated acceptor fragments. Ligation efficiencies were estimated by relative intensity between unconsumed and ligated substrate from densitometric gel analysis and expressed relative to time to determine enzyme velocities. The data were fit using GraphPad Prism over the full time course and concentration series. The experiment was repeated two times for each substrate concentration tested, and the reported values are the mean of two experiments. The average of the data sets is presented with a simulation using these averaged fit constants.
Stability of the 189insG ligase
To asses enzyme stability, four aliquots of ligation assay mixture were prepared containing ligase reaction buffer supplemented with 100 nM 189insG ligase mutant. Ligation assay aliquots were pre-incubated at 37°C for 10 min. Each pre-incubated ligation assay aliquot was supplemented with a 1:3:3 molar fraction 10× acceptor–donor–template mix (Supplementary Information S7.1, substrates 1c, 3c, 5a) at a different time point, resulting in a final substrate concentration of 1 μM in the FAM label (acceptor). Each final mixture was incubated for another 16 h following the timepoint of substrate addition (1, 3, 6 and 16 h) and subsequently quenched using 2× quenching solution. Polyacrylamide gel electrophoresis was used to separate ligated and non-ligated acceptor fragments.
RESULTS
Docking strategy starting from unbound nucleic acid duplexes
The default protocol for protein–DNA docking in HADDOCK is not suitable to reproduce accurately the structure of an endonuclease wrapped around its dsDNA substrate (3bam) (23). We decided to a ‘divide and conquer’ strategy, developed based on multi-body docking and molecular dynamics as schematically depicted in Figure 2. The protein partner in the complex was split into its constituting domains as described in the literature. After initial minimization of separated protein domains and dsDNA, each domain was docked as an individual body onto the dsDNA substrate. At this stage, ambiguous interaction restraints (AIR) between dsDNA and protein derived from the available crystal structure were applied to correctly position different domains relative to the nick in the DNA duplex. We used the available crystal structures of protein–nucleic acid complexes to derive interaction restraints (AIR) in modelling. Nevertheless, it is also possible to obtain those restraints from mutational studies or chemical shift changes in NMR that identify the protein binding surface.
Figure 2.

Schematic representation of the molecular modeling strategy followed to obtain protein–DNA complexes starting from unbound dsDNA. DNA from the 10 best structures obtained in an initial docking stage are used in ensemble docking. Final MD is performed on the structure with the best HADDOCK score.
Although the original protein subunits were already in the bound state, simulated annealing and refinement in explicit solvent allowed the side chains and backbone atoms at the protein–nucleic acid interface to be flexible. Based on the geometry of the calculated DNA duplexes after this docking run, 10 coordinate files for the dsDNA binding partner were created and used in the second ‘ensemble-based’ docking round. The structure obtained with the best HADDOCK score in most populated cluster after the second split-docking stage was submitted to an extensive base pair-restrained molecular dynamics (MD) with explicit water, during which interdomain bonds were restored.
ChVLig—dsDNA and BamHI-dsDNA crystal structures are accurately reproduced
The docked complex, including all domains, with dsDNA was compared to the corresponding crystal structure by their alignment RMSD before and after simulation. These results (Supplementary information S5.5) demonstrate that split-domain docking followed by MD simulation provides an accurate reproduction of the crystal structures. Figure 3C shows the simulated complex of dsDNA and ChVLig generated by HADDOCK (green) superimposed with the crystal structure (dark blue), illustrating high similarity. The protein encircles the broken and intact DNA strands with extensive DNA backbone contacts. The overall bending of the dsDNA in crystallized and calculated complexes displayed comparable angles of 12.3° and 11.2° respectively. The distortion of B-type unbound dsDNA at the base-pairs close to the break in the phosphodiester backbone between C31 and C32 towards an A-type helix is observed in the model (Supplementary information S5.3) and is in agreement with the crystal structure. As visible in the close-ups of Figure 3C, the spatial orientation of amino acids at the DNA nick was reproduced in the calculated structure with stacking interaction between Phe44 and Phe215 and the hydrogen bond interaction between Lys5 and Tyr217 despite the flexibility of these residues during calculations (Supplementary information S4).
Figure 3.

(A) HADDOCK score vs iRMSD from crystal structure (PDB ID: 2q2t). (B) HADDOCK score versus RMSD from lowest energy structure. For detailed statistics on top HADDOCK clusters for bound and unbound docking see Supplementary information S5.4C) Superimposition of the simulated docked complex (green) with the crystal structure (dark blue) together with close-ups. Upper close-up shows the spatial conservation of the nick, lower close-up shows the conserved stacking (Phe44-Phe215) and hydrogen bond (Lys5-Tyr217) interaction (Supplementary information S4).
The complex of BamHI restriction endonuclease and its target dsDNA from the protein–DNA benchmark (PDB ID: 3bam) was generated according to the same split-docking approach in two steps followed by MD for comparison with performance results reported in the literature for HADDOCK, NPDOCK and HDOCK. Our approach achieved a significant improvement compared to earlier attempts, as elaborated in Supplementary information S5.6. Results demonstrate the ability of our combined split-docking and MD strategy to generate a reliable docking solution starting from apoprotein in a bound conformation and canonical B-type dsDNA helices.
From ChVLig—dsXNA models towards a mutant with altered substrate specificity
Nicked ds2′OMeRNA and dsHNA with sequences matching dsDNA in the crystal structure of ChVLig were built. The two-step docking strategy described above was applied to calculate structures for the three ChVLig domains bound to a 2′OMeRNA duplex, resulting in clusters with average scores per cluster between –388 and –275 kcal mol−1 with the top-ranked calculated ds2′OMeRNA complex displaying a score of –399 kcal mol−1 (Figure 5A, gray). The same docking approach on the nicked HNA duplex yielded clusters with average HADDOCK scores per cluster ranging between –326 and –298 kcal mol−1 with the top-ranked calculated dsHNA complex displaying a score of –336 kcal mol−1 (Figure 5B, gray). The observed lower docking score for the complex with HNA in comparison to 2′OMeRNA is in line with HNA being more rigid and structurally more divergent from native DNA compared to 2′OMeRNA. Increased docking energy compared to dsDNA docking (between 80 and 150 kcal mol−1) originates primarily from AIR violations, and can be traced to the structural differences between dsDNA and dsXNA.
Figure 5.

Plot (A) (2’OMeRNA) and plot (B) (HNA) showing the overlay between the ‘open’ WT-dsXNA (gray) and closed 189insG–dsXNA complexes (green with orange insert). Position of the latch domain is indicated by an arrow. The evolutionary conserved clamp-closing in green complexes is highlighted in a dashed circle. RMSF plots in panel (C) (2’OMeRNA) and panel (D) (HNA), different domains are depicted below the graph. Red asterisk indicates the insert position.
The OB and NTase domains are positioned primarily over the XNA minor groove and make extensive backbone contacts. Because the minor groove in dsXNA is different from dsDNA, a slight displacement of both domains relative to each other is observed in comparison to the ChVLig–dsDNA structure. Striking differences occur for the latch that does not show close contacts with dsXNA and NTase domain resulting in an open conformation of the ChVLig.
With the exception of minimal Lig E-type DNA ligases, a clade of phylogenetically distinct ATP-dependent DNA ligases found almost exclusively in proteobacteria, the general paradigm is that DNA ligases engage their DNA substrate through full encirclement of the duplex, completed by inter-domain kissing contacts via loops or additional domains (46). Studies on recombinant ΔLatch-ChVLig reported that loss of the latch decreased specific activity in nick sealing in dsDNA by ten-fold, compared with wild-type ligase (18) Therefore, we considered the disruption of the evolutionarily conserved clamp closure in ChVLig-dsXNA complexes as a possible reason why ChVLig lacks ligation efficiency towards 2′OMeRNA and HNA fragments (Figure 4A and C).
Figure 4.

Ligation of DNA, 2′OMeRNA and HNA fragments by ChVLig (panel A) and its 189insG mutant (panel B) on different complementary templates. Panels a and b display typical PAGE gels for ligation efficiency. On both PAGE, lane L displays a ladder marked at 18 and 36 oligonucleotide length (Supplementary information S7.1, substrates 3a and 7b), lanes indicated with C are negative controls for template independent ligation (lane C1), enzyme independent DNA ligation in the presence of a DNA template (lane C2), enzyme independent 2′OMeRNA ligation in the presence of a 2′OMeRNA template (lane C3) and enzyme independent HNA ligation in the presence of a HNA template (lane C4). The ligation reactions are shown in panel D, where DNA, 2′OMe-RNA and HNA segments are colored in blue, gold and red respectively. The diagram (panel C) summarizes results quantitatively (ChVLig – gray and 189insG –green). Minimum and maximum ligation efficiency are shown as interval bars (n = 4). Representative points of ligation efficiencies from panels a and b are marked with x. Almost full 2′OMeRNA ligation on both DNA and 2′OMeRNA templates occurs compared to a virtually non-existing activity for ChVLig. HNA ligation efficiency reaches on average 63 (±19)% and 53 (±18)% on DNA and HNA templates respectively, compared to non-measurable HNA ligation by the wild-type enzyme.
We noticed that after the second round of split-docking of CHVLig to dsXNA, which is meant to position individual domains in an optimal conformation, Lys188 (NTase) and Gln189 (OB) remained ≈4.0 Å apart compared to the ChVLig–dsDNA complex. During subsequent base pair-restrained MD, this distance reduced to the length of a covalent bond while the relative position of protein domains drifted away from the crystal structure. Fluctuation of protein Cα atoms about their average position during MD (RMSF), demonstrates that the latch emanating from the OB domain undergoes the strongest fluctuation (Figure 5C and D). In the complex including dsHNA the fluctuation of the OB domain deviates also in comparison to the calculations for the ChVLig–dsDNA complex. These results are linked to the increased diameter of dsXNA compared to dsDNA that cannot be spanned by the ligase clamp.
As described in the supplementary material (Supplementary information S6), geometric calculations based on the increased helix diameter of the studied dsXNA compared to dsDNA suggested that about 3.9 and 4.6 Å increase in the spanning width of the clamp in ChVLig is required to form a closed complex with intimate interactions between the latch and dsHNA and ds2′OMeRNA respectively. However, this approximation does not take into account possible induced fit mechanisms upon binding.
Since after the second docking round the observed distance between last and first residues in respectively NTase and OB domain approaches the length of a stretched amino acid (3.6 Å) (47), the hinge between OB and NTase was considered as the ‘hot spot’ for an insert mutation to increase the spanning size of the clamp. The position of a single amino acid insert between residues Lys188 (NTase) and Gln189 (OB) was selected to minimize potential interactions of the extra residue with OB, NTase and dsXNA. Insertion of glycine was chosen as commonly used in protein engineering experiments (48).
Templated ligation efficiency of wild-type ChVLig and its 189insG mutant on DNA, 2′OMeRNA and HNA was tested on duplexes that correspond to those in the modeled structures. PAGE gels demonstrate that insertion of glycine in the hinge between OB and NTase domains boosted ligation of both XNA on a DNA as well as on a corresponding XNA template (Figure 4).
No stability issues were observed when the mutant ligase was incubated in ligase buffer for 16 h prior to spiking the solution with substrate and subsequent 16 hours ligation reaction (Supplementary information S7.2). The mutant ligase shows little discrimination between ligating DNA substrates against DNA, 2′OMe-RNA and HNA templates with ligation efficiencies exceeding 70% (Figure 4C, lanes 1, 2 and 3). Near-complete ligation of 2′OMe-RNA donor and acceptor substrates against both DNA and 2′OMeRNA templates can be achieved with the mutant (Figure 4C, lanes 4 and 5). Varying the sequence of the 2′OMe-RNA donor (when ligating it to a 2′OMe-RNA acceptor against a DNA template) has negligible impact on reaction efficiency, suggesting that if there are sequence dependencies they are not in the immediate vicinity of the ligation site (Supplementary information S7.3). For dsHNA/DNA hybrid duplexes, an average of 63% ligation is seen (Figure 4C, lane 5). However, more than 80% HNA ligation can be achieved at higher enzyme and substrate concentrations (Supplementary information S7.4) and longer incubation time. dsHNA with its six-membered ‘sugar’ rings in the backbone, being a wider and more rigid duplex than ds2′OMe-RNA, remains a poorer substrate but on average 53% ligation efficiency can be achieved (when acceptor, donor and template are HNA) (Figure 4C, lane 6). The wild-type enzyme has no detectable HNA ligation even in circumstances where the template is DNA, but in those conditions, it is possible to fit the activity of the mutant to a simple Michaelis–Menten model with kcat of 0.20 ± 0.01 s−1 and KM of 0.19 ± 0.06 μM (Supplementary information S7.5). With an apparent kcat/KM ≈ 1.02 × 106 s−1 M−1, the ins189G ligase exhibits less catalytic efficiency then previously reported kcat/KM parameters for PBCV-1 DNA ligase on RNA-splinted DNA ligation (49).
Insights on increased dsXNA ligation efficiency
The split-docking and MD were repeated with the 189insG mutant to gain more insight into how the insertion could be contributing to the XNA ligation activity. The mutant was predicted to bind both dsHNA and ds2′OMe-RNA substrates more stable than the wild-type. The top-ranked ds2′OMe-RNA in complex with the mutant (Figure 5A, green) shows an increase in affinity of 100 or 10 kcal mol−1, compared to the wild-type enzyme binding ds2′OMeRNA or dsDNA respectively (HADDOCK score of –489 kcal mol−1 and average score per cluster between –446 and –367 kcal mol−1). For dsHNA, calculated affinity for the mutant improved about 60 kcal mol−1 (Figure 5B, green) compared to wild-type ChVLig (HADDOCK score of –363 kcal mol−1 and average score per cluster between –360 and –287 kcal mol−1).
In contrast to the wild-type enzyme in complex with dsXNA, clamp closing persists in both complexes of the 189insG mutant during a 200ns MD simulation (Figure 5). Fluctuation of protein Cα atoms about their average position during MD (RMSF) indicates that the increased mobility in the latch that is present in ChVLig-dsXNA complexes does not occur in complexes of dsXNA with mutant ligase (Figure 5C and D). RMSF plots of complexes with 189inG become similar to those of ChVLig in complex with dsDNA.
In addition to the RMSF calculations, domain movement during simulation was analyzed and visualized by essential dynamics. Using the Cα as representative points for each residue, principal component analysis (PCA) was used to assess the key differences between enzymes bound to different substrates. Figure 6 demonstrates that ChVLig in complex with dsXNA (black) covers a larger region of phase space compared to the complex with dsDNA (orange), indicating a less ordered structure. However, motion of the mutant–XNA complexes (green) clearly converges towards the native complex.
Figure 6.

PCA results obtained for ChVLig (black) and its 189insG mutant (green) in complex with ds2′OMe-RNA (plots A and B) and dsHNA (plots C and D). For comparison, results on the wild-type ligase complexed with dsDNA is superimposed in orange on all plots. The motion of the mutant-dsXNA complexes (plots B and D, green) clearly converges towards the native state, in comparison to ChVLig–dsXNA complexes (plots A and C, black).
During MD on the 189insG mutant, stable interactions at binding interface between the tip of the latch and NTase domain remain. Distance measurements in time demonstrate that the ionic interaction between Lys45 and Asp213 and stacking between Phe44 and Phe215 at this binding interface becomes comparable to the wild-type enzyme in complex with dsDNA when a glycine is inserted in the hinge region between OB and NTase domains (Figure 7).
Figure 7.

Interaction analysis for the evolutionary conserved stacking (Phe44–Phe215) (left) and ionic (Lys45–Asp213) interactions (right) for ChVLig–dsXNA (black), ChVLig–dsDNA (orange) and 189insG–dsXNA (green). Results on 2′OMeRNA and HNA are depicted in top and bottom graphs respectively. All plots show the distance in time measurements between respective amino acids during a 200 ns MD. Close-ups illustrate the interactions occurring in 189insG simulated complex with ds2′OMeRNA (top) and dsHNA (bottom).
DISCUSSION
We developed a split-docking approach starting from unbound nucleic acids followed by MD to obtain adequate models for proteins encircling double stranded nucleic acids. Our method is the first available method to model, with high accuracy, nucleic-acid-binding proteins that make extensive contacts with their substrate in a closed clamp. Previous approaches started from unbound protein and bound nucleic acids. In this work we followed an opposite strategy starting from unbound DNA and isolated domains of the bound protein within the scope of structure-based enzyme design to generate an XNA ligase.
Despite the broad application of ligases in molecular biology, only few examples are described on DNA ligases that have been engineered, always focused on improving DNA ligation (50–52). Some wild-type DNA ligases can join non-canonical substrates on a DNA template, generally in the presence of crowding agents and additional co-solutes (2,53). Efficiency of ligation is dependent on the XNA being used and on whether donor and/or acceptor are modified. We have focused on XNA templated ligation of 2′OMe-RNA and HNA given the availability of high-resolution structures, their high chemical and biological resistance and the inherently low ligation efficiency of natural ligases for double-stranded XNA templates.
Starting from nicked dsXNA that was generated using structural characteristics described in literature and the bound crystal structure of ChVLig, an accurate model was generated that allowed structure-based engineering. A single glycine was introduced in the hinge region connecting both domains of the enzyme that are involved in its catalytic activity (NTase and OB domains). In silico experiments on the mutant enzyme bound to dsXNA revealed that the insertion of this glycine results in stable complexes that keep a closed conformation during molecular dynamics. In vitro experiments demonstrated that the insert of this glycine introduced high ligation efficiency of XNA fragments on an XNA template. Also DNA templated ligation of XNA fragments was boosted for the 189insG mutant from very low (2.13% on 2′OMeRNA) or non-existent (HNA) for wild-type ChVLig to 95% and 60% respectively, significantly outperforming results on T4Lig in optimized conditions reported in literature. No sequence dependency was observed for the ins189G mutant which is in agreement with previous reports on ligases (54). The KM value (0.19 ± 0.06 μM) for the mutant ligating HNA on a DNA template is significantly higher compared to previously reported KM values for native ligases. Strikingly, the low kcat (0.20 ± 0.01 s−1) indicates slow catalytic conversion of acceptor to product which leaves a margin for further improvement of the catalytic step of the mutant ligase.
CONCLUSION
This work describes a new in silico approach to generate reliable models for nucleic acids bound within a protein clamp. The strategy was applied for the structure-based design of the first ligase that efficiently joins XNA fragments on an XNA template. Obtained experimental data clearly demonstrate XNA ligation that is induced by a single residue insert in case of ChVLig. The 189insG mutant of ChVLig is an important new tool for synthetic genetics which will enable the synthesis of longer, gene size XNA through ligation, supplementing the function of current XNA polymerases.
Supplementary Material
ACKNOWLEDGEMENTS
Authors are grateful to Prof. Dr A.M.J.J. (Alexandre) Bonvin from the University of Utrecht and the WeNMR institute for his expert contribution. We have greatly benefited from discussions and help from numerous postdocs over the years (in particular, Dr E. Groaz, Dr E. Eremeeva, Dr J. Masschelein, Dr S. Xiaoping and Dr M. Renders) as well as graduate student D. Kestemont and undergraduate student M. Abdel Fattah Ismail. We express our gratitude to L. Margamuljana for helpful discussions and excellent technical assistance on in vitro experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
European Research Council under the European Union's Seventh Framework Program (FP7/2007-2013)/ERC [ERC-2012-ADG 20120216/320683]; Research Fund KU Leuven [OT/14/128]; Biotechnology and Biosciences Research Council [BB/N01023X/1, BB/N010221/1]. Funding for open access charge: university budget.
Conflict of interest statement. None declared.
REFERENCES
- 1. McCloskey C.M., Liao J.-Y., Bala S., Chaput J.C.. Ligase-mediated threose nucleic acid synthesis on DNA templates. ACS Synth. Biol. 2019; 8:2282–2286. [DOI] [PubMed] [Google Scholar]
- 2. Kestemont D., Renders M., Leonczak P., Abramov M., Schepers G., Bernardes Pinheiro V., Rozenski J., Herdewijn P.. XNA ligation using T4 DNA ligase in crowding conditions. Chem. Commun. 2018; 54:6408–6411. [DOI] [PubMed] [Google Scholar]
- 3. Pinheiro V.B., Taylor A.I., Cozens C., Abramov M., Renders M., Zhang S., Chaput J.C., Wengel J., Peak-Chew S.-Y., McLaughlin S.H. et al.. Synthetic genetic polymers capable of heredity and evolution. Science. 2012; 336:341–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chen T., Hongdilokkul N., Liu Z., Adhikary R., Tsuen S.S., Romesberg F.E.. Evolution of thermophilic DNA polymerases for the recognition and amplification of C2′-modified DNA. Nat. Chem. 2016; 118:6072–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Larsen A.C., Dunn M.R., Hatch A., Sau S.P., Youngbull C., Chaput J.C.. A general strategy for expanding polymerase function by droplet microfluidics. Nat. Commun. 2016; 7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Luscombe N.M., Laskowski R.A., Thornton J.M.. NUCPLOT: A program to generate schematic diagrams of protein–nucleic acid interactions. Nucleic Acids Res. 1997; 25:4940–4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pascal J.M., O’Brien P.J., Tomkinson A.E., Ellenberger T.. Human DNA ligase I completely encircles and partially unwinds nicked DNA. Nature. 2004; 432:473–478. [DOI] [PubMed] [Google Scholar]
- 8. Daitchman D., Greenblatt H.M., Levy Y.. Diffusion of ring-shaped proteins along DNA: case study of sliding clamps. Nucleic Acids Res. 2018; 1:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hingorani M.M., O’Donnell M.. Toroidal proteins: running rings around DNA. Curr. Biol. 1998; 8:R83–R86. [DOI] [PubMed] [Google Scholar]
- 10. Anosova I., Kowal E.A., Dunn M.R., Chaput J.C., Horn W.D.V., Egli M.. The structural diversity of artificial genetic polymers. Nucleic Acids Res. 2016; 44:1007–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lescrinier E., Esnouf R.M., Schraml J., Busson R., Herdewijn P.. Solution structure of a hexitol nucleic acid duplex with four consecutive T-T base pairs. Helv. Chim. Acta. 2000; 83:1291–1310. [Google Scholar]
- 12. Declercq R., Aerschot A. Van, Read R.J., Herdewijn P.. Crystal structure of double helical hexitol nucleic acids. JACS. 2002; 124:1–6. [DOI] [PubMed] [Google Scholar]
- 13. Adamiak D.A., Rypniewski W.R., Milecki J., Adamiak R.W.. The 1.19 angstrom X-ray structure of 2 ′-O-Me(CGCGCG)(2) duplex shows dehydrated RNA with 2-methyl-2,4-pentanediol in the minor groove. Nucleic Acids Res. 2001; 29:4144–4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lescrinier E., Esnouf R., Schraml J., Busson R., Heus H.A., Hilbers C.W., Herdewijn P.. Solution structure of a HNA-RNA hybrid. Chem. Biol. 2000; 7:719–731. [DOI] [PubMed] [Google Scholar]
- 15. Maier T., Przylas I., Strater N., Herdewijn P., Saenger W.. Reinforced HNA backbone hydration in the crystal structure of a decameric HNA/RNA hybrid. J. Am. Chem. Soc. 2005; 127:2937–2943. [DOI] [PubMed] [Google Scholar]
- 16. Tereshko V., Portmann S., Tay E.C., Martin P., Natt F., Altmann K.H., Egli M.. Correlating structure and stability of DNA duplexes with incorporated 2′-O-modified RNA analogues. Biochemistry. 1998; 37:10626–10634. [DOI] [PubMed] [Google Scholar]
- 17. Martin I.V., MacNeill S.A.. ATP-dependent DNA ligases. Genome Biol. 2002; 3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Nair P.A., Nandakumar J., Smith P., Odell M., Lima C.D., Shuman S.. Structural basis for nick recognition by a minimal pluripotent DNA ligase. Nat. Struct. Mol. Biol. 2007; 14:770–778. [DOI] [PubMed] [Google Scholar]
- 19. Piserchio A., Nair P.A., Shuman S., Ghose R.. Solution NMR Studies of Chlorella Virus DNA Ligase-adenylate. J. Mol. Biol. 2010; 395:291–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Samai P., Shuman S.. Structure-function analysis of the OB and latch domains of Chlorella virus DNA ligase. J. Biol. Chem. 2011; 286:22642–22652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blanco J.D., Radusky L., Climente-González H., Serrano L.. FoldX accurate structural protein–DNA binding prediction using PADA1 (Protein Assisted DNA Assembly 1). Nucleic Acids Res. 2018; 46:3852–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Linge J.P., Williams M.A., Spronk C.A.E.M., Bonvin A.M.J.J., Nilges M.. Refinement of protein structures in explicit solvent. Proteins Struct. Funct. Genet. 2003; 50:496–506. [DOI] [PubMed] [Google Scholar]
- 23. Van Dijk M., Visscher K.M., Kastritis P.L., Bonvin A.M.J.J.. Solvated protein–DNA docking using HADDOCK. J. Biomol. NMR. 2013; 56:51–63. [DOI] [PubMed] [Google Scholar]
- 24. Miller B.R., Parish C.A., Wu E.Y.. Molecular dynamics study of the opening mechanism for DNA Polymerase I. PLoS Comput. Biol. 2014; 10:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Krüger A., Zimbres F.M., Kronenberger T., Wrenger C.. Molecular modeling applied to nucleic acid-based molecule development. Biomolecules. 2018; 8:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Si J., Cui J., Cheng J., Wu R.. Computational prediction of RNA-binding proteins and binding sites. Int. J. Mol. Sci. 2015; 16:26303–26317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Xue L.C., Dobbs D., Bonvin A.M.J.J., Honavar V.. Protein-protein interface predictions by data-driven methods: a review. FEBS Lett. 2016; 30:1627–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Van Zundert G.C.P., Rodrigues J.P.G.L.M., Trellet M., Schmitz C., Kastritis P.L., Karaca E., Melquiond A.S.J., Van Dijk M., De Vries S.J., Bonvin A.M.J.J.. The HADDOCK2.2 web server: user-friendly integrative modeling of biomolecular complexes. J. Mol. Biol. 2016; 428:720–725. [DOI] [PubMed] [Google Scholar]
- 29. Tuszynska I., Magnus M., Jonak K., Dawson W., Bujnicki J.M.. NPDock: A web server for protein–nucleic acid docking. Nucleic Acids Res. 2015; 43:W425–W430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yan Y., Zhang D., Zhou P., Li B., Huang S.Y.. HDOCK: A web server for protein–protein and protein–DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017; 45:W365–W373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Van Dijk M., Van Dijk A.D.J., Hsu V., Rolf B., Bonvin A.M.J.J.. Information-driven protein–DNA docking using HADDOCK: It is a matter of flexibility. Nucleic Acids Res. 2006; 34:3317–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. van Dijk M., Bonvin A.M.J.J.. A protein–DNA docking benchmark. Nucleic Acids Res. 2008; 36:e88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Berman H.M., Westbrook J., Feng Z., Gilliland G., Bhat T.N., Weissig H., Shindyalov I.N., Bourne P.E.. The protein data bank helen. Nucleic Acids Res. 2000; 28:235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. van Dijk M., Bonvin A.M.J.J.. 3D-DART: a DNA structure modelling server. Nucleic Acids Res. 2009; 37:235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brunger A.T. Version 1.2 of the crystallography and NMR system. Nat. Protoc. 2007; 2:2728–2733. [DOI] [PubMed] [Google Scholar]
- 36. Case D.A., Cheatham T.E., Darden T., Gohlke H., Luo R., Merz K.M., Onufriev A., Simmerling C., Wang B., Woods R.J.. The Amber biomolecular simulation programs. J. Comput. Chem. 2005; 26:1668–1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lindorff-Larsen K., Piana S., Palmo K., Maragakis P., Klepeis J.L., Dror R.O., Shaw D.E.. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins Struct. Funct. Bioinforma. 2010; 78:1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Aduri R., Psciuk B.T., Saro P., Taniga H., Schlegel H.B., SantaLucia J.. AMBER force field parameters for the naturally occurring modified nucleosides in RNA. J. Chem. Theory Comput. 2007; 3:1464–1475. [DOI] [PubMed] [Google Scholar]
- 39. Dupradeau F.Y., Cézard C., Lelong R., Stanislawiak É., Pêcher J., Delepine J.C., Cieplak P.. R.E.DD.B.: a database for RESP and ESP atomic charges, and force field libraries. Nucleic Acids Res. 2008; 36:360–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pérez A., Marchán I., Svozil D., Sponer J., Cheatham T.E., Laughton C.A., Orozco M.. Refinement of the AMBER force field for nucleic acids: improving the description of alpha/gamma conformers. Biophys. J. 2007; 92:3817–3829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L.. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983; 79:926. [Google Scholar]
- 42. Roe D.R., Cheatham T.E. III. PTRAJ and CPPTRAJ: software for processing and analysis of molecular synamics trajectory data. J. Chem. Theory Commun. 2013; 9:3084–3095. [DOI] [PubMed] [Google Scholar]
- 43. Amadei A., Linssen A.B.M., Berendsen H.J.C.. Essential dynamics of proteins. Proteins Struct. Funct. Genet. 1993; 17:412–425. [DOI] [PubMed] [Google Scholar]
- 44. Zhu B., Cai G., Hall E.O., Freeman G.J.. In-Fusion® assembly: seamless engineering of multidomain fusion proteins, modular vectors, and mutations. BioTechniques. 2007; 43:354–359. [DOI] [PubMed] [Google Scholar]
- 45. Odell M., Malinina L., Sriskanda V., Teplova M., Shuman S.. Analysis of the DNA joining repertoire of Chlorella virus DNA ligase and a new crystal structure of the ligase-adenylate intermediate. Nucleic Acids Res. 2003; 31:5090–5100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Williamson A., Grgic M., Leiros H.-K.S.. DNA binding with a minimal scaffold: structure–function analysis of Lig E DNA ligases. Nucleic Acids Res. 2018; 46:8616–8629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pauling L., Corey R.B.. The pleated sheet, a new layer configuration of polypeptide chains. Proc. Natl. Acad. Sci. U.S.A. 1951; 37:251–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Priyanka V., Chichili R., Kumar V., Sivaraman J.. Linkers in the structural biology of protein–protein interactions. Protein Sci. 2013; 22:153–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lohman G.J.S., Zhang Y., Zhelkovsky A.M., Cantor E.J., Evans T.C.. Efficient DNA ligation in DNA-RNA hybrid helices by Chlorella virus DNA ligase. Nucleic Acids Res. 2014; 42:1831–1844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tanabe M., Ishino Y., Nishida H.. From structure-function analyses to protein engineering for practical applications of DNA ligase. Archaea. 2015; 2015:267570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wilson R.H., Morton S.K., Deiderick H., Gerth M.L., Paul H.A., Gerber I., Patel A., Ellington A.D., Hunicke-Smith S.P., Patrick W.M.. Engineered DNA ligases with improved activities in vitro. Protein Eng. Des. Sel. 2013; 26:471–478. [DOI] [PubMed] [Google Scholar]
- 52. Tanabe M., Ishino S., Yohda M., Morikawa K., Ishino Y., Nishida H.. Structure-based mutational study of an archaeal DNA ligase towards improvement of ligation activity. ChemBioChem. 2012; 13:2575–2582. [DOI] [PubMed] [Google Scholar]
- 53. Chaput J.C., Szostak J.W.. TNA synthesis by DNA polymerases. J. Am. Chem. Soc. 2003; 125:9274–9275. [DOI] [PubMed] [Google Scholar]
- 54. Liu P., Burdzy A., Sowers L.C.. DNA ligases ensure fidelity by interrogating minor groove contacts. Nucleic Acids Res. 2004; 32:4503–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.