


Abstract
DNA methylation is an important epigenetic mark but how its locus-specificity is decided in relation to DNA sequence is not fully understood. Here, we have analyzed 34 diverse whole-genome bisulfite sequencing datasets in human and identified 313 motifs, including 92 and 221 associated with methylation (methylation motifs, MMs) and unmethylation (unmethylation motifs, UMs), respectively. The functionality of these motifs is supported by multiple lines of evidence. First, the methylation levels at the MM and UM motifs are respectively higher and lower than the genomic background. Second, these motifs are enriched at the binding sites of methylation modifying enzymes including DNMT3A and TET1, indicating their possible roles of recruiting these enzymes. Third, these motifs significantly overlap with “somatic QTLs” (quantitative trait loci) of methylation and expression. Fourth, disruption of these motifs by mutation is associated with significantly altered methylation level of the CpGs in the neighbor regions. Furthermore, these motifs together with somatic mutations are predictive of cancer subtypes and patient survival. We revealed some of these motifs were also associated with histone modifications, suggesting a possible interplay between the two types of epigenetic modifications. We also found some motifs form feed forward loops to contribute to DNA methylation dynamics.
INTRODUCTION
DNA methylation plays crucial roles in many biological processes and aberrant DNA methylation patterns are often observed in diseases. There are three DNA methyltransferases (DNMTs) in human that are responsible for de novo or maintaining methylation of cytosine. Although these enzymes themselves do not show strong sequence preference in vivo, DNA methylation is highly locus-specific such as hypo-methylation of active promoters and enhancers. An urging question is how such a locus-specific DNA methylation pattern is established. One of the possible mechanisms is that DNA binding proteins or non-coding RNAs recognize specific DNA motifs and their binding recruits DNMTs to a particular locus to methylate cytosines in the region. These factors can be specifically active in a cell type or state such that to provide the cell type- and locus-specificity. Accumulating evidence suggests that protein binding such as CTCF and other proteins can create low methylated regions in the regulatory sites and introducing specific nucleotide sequences can establish DNA methylation (1,2). These observations suggest the importance of DNA sequence in shaping methylation state. Several studies have illustrated the relationship between sequence features and DNA methylation (3–14) but the DNA motifs recognized by the DNA methylation associated proteins have not been well characterized. Therefore, cataloging these motifs would pave the way towards understanding the mechanism of the locus-specificity of DNA methylation.
Cataloging DNA methylation associated motifs requires a comprehensive set of methylomes and whole-genome bisulfite sequencing (WGBS) is a common technology to map DNA methylation in the entire human genome. The NIH Roadmap Epigenomics Project (15) has generated WGBS data in 34 cell lines or tissues, which provides an opportunity to discern motifs associated with DNA methylation. We reasoned that contrasting regions that are commonly methylated across cells/tissues to those commonly unmethylated would increase the signal-to-noise ratio to identify the motifs most relevant to DNA methylation. Furthermore, to consider the impact of cell type and cell state on DNA methylation, we also need to uncover motifs associated with variable methylation levels across cells/tissues; a caveat is that these motifs can be confounded by those only related to cell specificity. To this end, we have defined commonly methylated (unmethylated) regions across the 34 cells (CMR/CUR) as well as variably methylated (unmethylated) regions (VMR/VUR) that show cell-specific methylation (unmethylation). We have found the DNA motifs that are discriminative of these regions.
To confirm the association with methylation, we overlapped the motifs with DNMT and TET ChIP-seq peaks and observed strong enrichment. We also used TCGA (The Cancer Genome Atlas) (27) dataset to further assess the importance of these motifs in shaping DNA methylation. Interestingly, we found that, if these are somatic mutations occurring in the motifs, the methylation levels in the nearby CpGs are significantly altered, i.e. perturbation to a MM (UM) motif in a highly (lowly) methylated region would decrease (increase) the local methylation level. This observation strongly supports the functionality of the identified motifs in establishing or maintaining locus-specific DNA methylation. Furthermore, we observed “somatic QTLs” (quantitative trait loci) of methylation and expression are enriched in the found motifs. We showed that the combination of somatic mutations and the found motifs can significantly improve the prediction accuracy of cancer type and patient survival than using somatic mutations alone, which further supported the functionality of these DNA methylation associated motifs. Additional analyses also revealed the potential interplay between DNA methylation and histone modification as well as their contribution to DNA methylation dynamics.
MATERIALS AND METHODS
De novo motif discovery
11.5 million CpG sites common across all 34 human methylomes were collected from the NIH Roadmap Epigenomics Project (16). Methylation regions were defined by segments merged with two or more CpGs with a maximal distance of 400 bp apart (i.e. CpGs and only CpGs within 400 bp of each other were merged into a methylation region) and the region methylation level was defined by the mean CpG beta values. Each region was then assigned mean and standard deviation of methylation across all 34 tissues and cells. We used a normalized score to measure the overall methylation level of a methylation region across 34 methylomes in comparison to the whole genome methylation distribution:
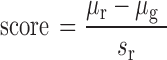 |
where 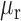 and
and 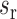 are the mean and standard deviation of the methylation of the region,
are the mean and standard deviation of the methylation of the region, 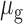 is the mean of methylation genome-wide. We used the ranking of this score in our analysis to select the methylation regions, i.e. commonly methylated regions (CMRs) are the CpGs with the top 0.5% score and commonly unmethylated regions (CURs) bottom 0.5% score, while variably methylated regions (VMRs) are defined by the top 20% standard deviation (Figure 1A, B). For common motifs MM and UM, we performed Epigram (3) contrasting CMRs and CURs. In short, Epigram looks for enriched motifs that best differentiate the foreground from the background sequences. In both sets of the input sequences, Epigram iterates through all possible k-mers to calculate their occurrences, enrichment over genomic background and enrichment over shuffled input. These values are combined to determine the enrichment of k-mers. Position weight matrices (PWMs) are then generated by first picking a top k-mer and enriched k-mers similar to itself to construct a ‘seed’ PWM, which is then extended by adding more enriched k-mers that are a few base pairs shifted from the original one. The motifs are then further ranked and filtered based on how well they differentiate the foreground from the background using LASSO (least absolute shrinkage and selection operator) logistic regression. The final set of motifs is then evaluated by random forest.
is the mean of methylation genome-wide. We used the ranking of this score in our analysis to select the methylation regions, i.e. commonly methylated regions (CMRs) are the CpGs with the top 0.5% score and commonly unmethylated regions (CURs) bottom 0.5% score, while variably methylated regions (VMRs) are defined by the top 20% standard deviation (Figure 1A, B). For common motifs MM and UM, we performed Epigram (3) contrasting CMRs and CURs. In short, Epigram looks for enriched motifs that best differentiate the foreground from the background sequences. In both sets of the input sequences, Epigram iterates through all possible k-mers to calculate their occurrences, enrichment over genomic background and enrichment over shuffled input. These values are combined to determine the enrichment of k-mers. Position weight matrices (PWMs) are then generated by first picking a top k-mer and enriched k-mers similar to itself to construct a ‘seed’ PWM, which is then extended by adding more enriched k-mers that are a few base pairs shifted from the original one. The motifs are then further ranked and filtered based on how well they differentiate the foreground from the background using LASSO (least absolute shrinkage and selection operator) logistic regression. The final set of motifs is then evaluated by random forest.
Figure 1.

Defining methylated regions and searching for methylation associated motifs. (A) The strategy of identifying DNA methylation associated motifs. (B) WGBS CpG sites are merged within 400bp regions. Based on average CpG beta values of the region, we defined commonly methylated (CMR), commonly unmethylated (CUR) and variably methylated regions (VMR). (C) Identification of DNA methylation associated motifs in 34 cells and tissues. Example motifs are shown on the right (if matched to a known motif, the known motif logo is shown on the top).
For tissue-specific VMM and VUM, we contrasted top 6000 most methylated and unmethylated regions in each methylome. In total, we identified 5172 motifs from 35 Epigram runs (34 methylome + 1 common) with default parameters of Epigram (3) (Figure 1C). For each run, Epigram found DNA motifs that discriminate enrichment peaks of the high methylation region under consideration (e.g. CMR) from a background of low methylation region (e.g. CUR). Importantly, the background has equal GC content, number of regions and sequence lengths as the foreground to avoid inflated prediction results caused by simple features or unbalanced data set.
Motif curation and defining motif occurrence site
Following our previous study (3), we matched motifs to the 1156 known motifs documented by the HOCOMOCO ChIP-seq consortium (17) using an E-value cutoff of 0.05 with Tomtom (18). Next, we merged the similar motifs to remove redundancy. We calculated a pairwise motif distance using weighted Jensen-Shannon Divergence:
 |
 |
where 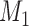 ,
, 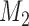 are PWMs of the two motifs, respectively,
are PWMs of the two motifs, respectively, 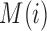 represents the
represents the 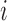 th column in the matrix,
th column in the matrix, 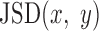 is Jensen-Shannon divergence,
is Jensen-Shannon divergence, 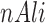 and
and 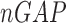 are respectively the lengths of the aligned sequence and gaps. Gap penalty function
are respectively the lengths of the aligned sequence and gaps. Gap penalty function 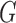 has
has 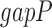 as weight parameter set at 0.1. To ensure high similarity within the motif cluster, the gap penalty function is set to quadratic which is more stringent compared to traditional linear function to prevent having excessive gaps and hangovers. Motifs were hierarchically clustered with UPGMA (19) algorithm and clusters were chosen using a distance cutoff of 0.1. As a result, we obtained 3226 clusters and selected the motif closest to the centroid of the cluster to represent all the motifs in that cluster. We combine the P-value of motifs in the cluster using Fisher's combined probability test. Enrichment of each cluster was combined by geometric mean. Each unique motif is named by its group (MM or UM), combined P-value (log), combined enrichment, number of similar motifs in the cluster, followed by a short descriptive string. This string is either its best aligned known motif (e.g. UM_180.0_3.14_0.56_7_known-CTCF) matched by Tomtom described previously, or a consensus sequence (e.g. MM_10.2_2.16_0.54_1_ATKGCGSCA) determined by a minimal information loss method (20). The strongest 313 motifs were filtered by volcano test with combined P < 1e–10 and enrichment > 2 (Supplementary Figure S1B). Finally, motif occurrence sites were determined by a P < 1e–5 calculated by FIMO (21).
as weight parameter set at 0.1. To ensure high similarity within the motif cluster, the gap penalty function is set to quadratic which is more stringent compared to traditional linear function to prevent having excessive gaps and hangovers. Motifs were hierarchically clustered with UPGMA (19) algorithm and clusters were chosen using a distance cutoff of 0.1. As a result, we obtained 3226 clusters and selected the motif closest to the centroid of the cluster to represent all the motifs in that cluster. We combine the P-value of motifs in the cluster using Fisher's combined probability test. Enrichment of each cluster was combined by geometric mean. Each unique motif is named by its group (MM or UM), combined P-value (log), combined enrichment, number of similar motifs in the cluster, followed by a short descriptive string. This string is either its best aligned known motif (e.g. UM_180.0_3.14_0.56_7_known-CTCF) matched by Tomtom described previously, or a consensus sequence (e.g. MM_10.2_2.16_0.54_1_ATKGCGSCA) determined by a minimal information loss method (20). The strongest 313 motifs were filtered by volcano test with combined P < 1e–10 and enrichment > 2 (Supplementary Figure S1B). Finally, motif occurrence sites were determined by a P < 1e–5 calculated by FIMO (21).
Normalized motif occurrence and center-to-edge enrichment at DNMTs and TETs ChIP-seq peaks.
DNMTs and TETs occurrences were downloaded from the published studies (22–25), including the ChIP-seq peaks of TET1 in HuES8 (a human embryonic stem cell line) from Verma et al. (25), TET2 in HEK293T (a human embryonic kidney cell line) from Suzuki et al. (22), TET2/TET3 in HEK293T from Deplus et al. (23), and DNMT1/3A/3B in NCCIT (a human embryonic carcinoma cell line) from Jin et al. (24). The 5000 bp neighbor regions around the ChIP-seq peaks were included as the background or edge. Normalized motif occurrence was calculated using the following formula.
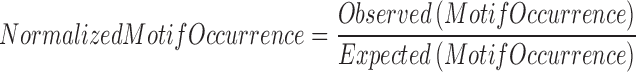 |
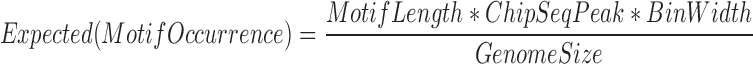 |
where 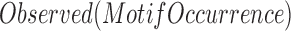 is the observed occurrence number of a motif in a 100 bp bin,
is the observed occurrence number of a motif in a 100 bp bin,  is the total length of genome-wide motif occurrences defined by FIMO (see the above section),
is the total length of genome-wide motif occurrences defined by FIMO (see the above section), 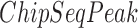 is the total number of ChIP-seq peaks,
is the total number of ChIP-seq peaks,  is 100 bp and
is 100 bp and 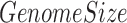 is the genome size of 3.14x109 bp for the human genome hg19. We did this calculation for each of the 313 top enriched motifs in each 100 bp bin. We also downloaded 6251 differential CpGs (dCpGs) with P < 0.05 defined by Kemp et al. (26), which were CpGs showing destabilized methylation level when CTCF contains point mutation or copy number aberrations. Center-to-edge enrichment of motif occurrences in the 500 bp around these reported dCpGs was performed the same as described above. Results are plotted in Figure 2C and Supplementary Figure S2B.
is the genome size of 3.14x109 bp for the human genome hg19. We did this calculation for each of the 313 top enriched motifs in each 100 bp bin. We also downloaded 6251 differential CpGs (dCpGs) with P < 0.05 defined by Kemp et al. (26), which were CpGs showing destabilized methylation level when CTCF contains point mutation or copy number aberrations. Center-to-edge enrichment of motif occurrences in the 500 bp around these reported dCpGs was performed the same as described above. Results are plotted in Figure 2C and Supplementary Figure S2B.
Figure 2.

Identified motifs mark methylation level. (A) Example motifs are shown with average CpG methylation level calculated in 50 bp bins around all motif sites, determined by FIMO at 10–5P-value cutoff. The examples are chosen from MM, UM, de novo motifs, matched known TFs, common region and sorted variable regions. Upper panel, from left to right: UM_180.0_3.14 (matched to CTCF); UM_106.1_4.08 (de novo); UM_238.2_3.88 (matched to WT1); lower panel, from left to right: MM_65.9_2.90 (matched to TOPORS); MM_814.4_2.02 (matched to PAX5); MM_206.3_2.16 (de novo). (B) DNA methylation levels in the ROADMAP (left) and TCGA (right) data sets over the gene body. Each gene body was split into ten equal bins and the Beta values of all CpGs in the same bin were averaged over all genes. Lower panel shows the correlation between the motif occurrences and CpG methylation in ROADMAP (WGBS data from H1, mesoderm, and liver) and TCGA (450K methylation of CpGs averaged in patients from PAAD, LUAD, and BRCA) around TP53 (chr17:7 540 000–7 650 000). (C) Normalized motif occurrence of UM, MM and known TFs (excluding matched) from HOCOMOCO (17) at 5000 bp windows centering ChIP-seq peaks of TET1, DNMT3A and DNMT3B collected from various studies (22,24,25). The lower panel shows the clustered heatmap of normalized z-score. (D). Center-to-edge enrichment of UMs and MMs in comparison with TF NR6A1 and CTCF, which were reported to recruit DNMT and TET to specific loci, at the ChIP-seq peaks of DNMTs and TETs.
Further, center-to-edge enrichment was calculated by 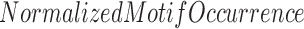 in the center 100 bp ChIP-seq bin divided by the average of
in the center 100 bp ChIP-seq bin divided by the average of 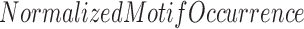 at the bins 2500 bp upstream and downstream. Average enrichment and standard deviation were calculated across all MMs or UMs, followed by a two-tailed two-sample t-test, with P < 0.01 marked as significant. Results are plotted in Figure 2D.
at the bins 2500 bp upstream and downstream. Average enrichment and standard deviation were calculated across all MMs or UMs, followed by a two-tailed two-sample t-test, with P < 0.01 marked as significant. Results are plotted in Figure 2D.
Somatic quantitative trait loci (QTL) enrichment analysis with TCGA
We downloaded the processed data (level 3) of 36 TCGA cancers from the Firebrowse service (http://firebrowse.org/) including patient survival, somatic mutations, 450K methylation array, and RNA-seq data. All the somatic mutations taken from TCGA were first detected in Affymetrix Genome-Wide Human SNP Array 6.0, and determined by contrasting variants in cancer primary tissues with germline tissues, according to the TCGA Consortium (27). Matrix eQTL (28) linear model was used to identify somatic QTL of methylation and expression co-variating with methylation and transcript RNA-seq level, with 5000 bp distance cutoff from somatic mutation to CpG and transcript TSS, respectively. We used a conservative P-value cutoff of 0.01 on top of an FDR cutoff of 10%. Then we calculated the number of somatic mQTL or eQTL out of all somatic mutations in 10 bins of gene body, i.e. 0–10%, 10–20%, …, 90–100% of the mRNA transcript length, defined in Gencode v19 (29). We performed such analysis on all genes and repeated it with the UM and MM occurrence sites (Figure 2A). To determine the significance of enrichment, a chi-square test was carried out in each of the 10 bins of gene body, with the null hypothesis that somatic mQTL% or eQTL% occurring at motif sites are the same as the rest of all genes, with P < 0.01 marked as significant.
Methylation quantitative trait loci (mQTL) enrichment analysis with three independent datasets
Three human methylome studies with independently called mQTLs were collected, i.e. human life course study (30), GenCord Cohort study (31) and a Schizophrenia study (32). We took the mQTL SNPs identified from the original studies and these can be either somatic or germline mutations, which were not distinguished in the publication work. In total, there are around 16 000–30 000 identified mQTLs collected from these published studies. We defined an enrichment score using the following formula.
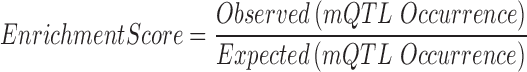 |
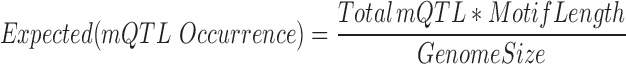 |
where 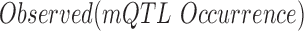 is the observed occurrences of mQTLs in the occurrence sites of the 313 motifs genome-wide,
is the observed occurrences of mQTLs in the occurrence sites of the 313 motifs genome-wide, 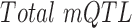 is the total number of mQTLs identified in each study,
is the total number of mQTLs identified in each study, 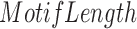 is the total length of genome-wide motif occurrences, and
is the total length of genome-wide motif occurrences, and 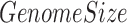 is the genome size of 3.14E9 bp for the human genome hg19. The occurrences of motifs have been defined by FIMO (see the above section).
is the genome size of 3.14E9 bp for the human genome hg19. The occurrences of motifs have been defined by FIMO (see the above section).
We repeated this process in all samples from all three studies and calculated the standard deviation. Specifically, (i) five life stages from birth, childhood, adolescence, pregnancy and middle age in human life course study (blood samples from 1018 mother–child pairs), (ii) three tissues from fibroblasts, LCLs and T-cells in GenCord cohort by Maria el al (204 newborn umbilical cord samples) and (iii) three regions from prefrontal cortex, striatum and cerebellum of adult brain regions in the Schizophrenia study (173 fetal brain samples ranging from 56 to 169 days post-conception). Finally, we used a single-tail one-sample t-test to determine the statistical significance (P < 0.01, Supplementary Figure S3A).
Predicting TCGA cancer type with somatic mutation and motif
For each of the 32 TCGA cancers (in total 7120 patients), we trained two gradient boosting models (33) (mutation and mutation + motif) to distinguish one specific cancer from the other cancers. We chose gradient boosting implemented in Scikit-learn (34) and tuned its parameter based on a recent study (35), which showed that this decision-tree-based model is robust and performs well. Note that TCGA has four aggregated cancer types (GBMLGG, COADREAD, KIPAN and STES) that combine individual cancers such as GBMLGG combining GBM and LGG; we excluded them from the 32 TCGA datasets to avoid inflating the performance due to using the same patients in both the training and testing sets. In a mutation-only model, the cancer subtype of each patient was predicted only by somatic mutations as features. Because the input features are large (1.3 million unique somatic mutations for 7120 patients), we first reduced feature number. Each feature was assigned a score by the gradient boosting out-of-bag importance and averaged in 5-fold cross-validation to avoid overfitting. Features with negative importance scores were removed. The optimal number of features were determined as we observed the best model performance at around 500 features (Supplementary Figure S4A, left panel). Top 500 somatic mutations ranked by the average score were used while assuring equal or better performance compared to the full model (Supplementary Figure S4A, right panel).
After feature selection, we obtained 500 selected somatic mutations (from here referred simply as mutation). We used a series (length 500) of 0s and 1s to indicate which mutations a patient has. For example, 1, 1, 0, 1, … indicates patient have the first, second and fourth mutation. For a mutation + motif model, each patient was represented not only by these 500 selected mutations but also by whether each of the 313 motifs is disrupted by mutations. We used a series (length 313) of integers to indicate how many mutations (without feature selection) are harbored in the occurrence sites for each of the 313 motifs. For example, 10, 20, 0, … indicates there are 10 mutations in all the occurrence sites of the first motif, 20 in the second and none in the third. The performances of the two models were evaluated by auROC and auPRC with 5-fold cross-validations for each cancer (Figure 4A). Feature importance was determined by the default out-of-bag (OOB) important scores using the mean decrease of Friedman squared error over all cross-validated predictions in mutation+motif models. We filtered features with importance score >0.01 within the enriched 313 motif groups and mutation located in the well-studied driver genes identified by the IntOGen Consortium (36). To reduce false positives of selecting predictive features, we only considered 26 out of 32 TCGA cancers that showed auPRC > 0.3 (Figure 4B).
Figure 4.

Combining motif and mutation improves the prediction of cancer diagnosis and patient survival. (A) auROC and auPRC for cancer type prediction. Classification model of each cancer built with gradient boosting. Performance evaluated with auROC (area under the receiver operating characteristic, good for an overall evaluation.) and auPRC (area under the precision-recall curve, good for an unbalanced dataset where the positive label is scarce). Label: mutation: using somatic mutations as features. mutation+motif: using both somatic mutations and collective disruption of motif site as features (see Materials and Methods for details). * Adjusted P < 0.05. (B) Results of top predictive features (score > 0.01) using gradient boosting out-of-bag estimation. Twenty six cancers with auPRC > 0.3 are shown. (C) Survival analysis with gradient boosting with mutation and mutation + motif as models. Left: multivariate survival analyses for all solid TCGA cancers. Forest plots showing log2 hazard ratio (95% confidence interval) of the predicted high-risk group by both models. *Adjusted P < 0.05 (blue for the mutation model and red for the mutation+motif model). Right: Kaplan–Meier survival estimation (95% confidence interval) in the high-risk group versus low-risk group predicted by both models. (D) Multivariate survival analysis showing factors correlating with patient survival (P < 0.05) with the log2 hazard ratio (95% confidence interval).
Predicting TCGA patient survival with somatic mutation and motif
All patients in 22 TCGA cancers with patient survival and mutation information were dichotomized based on 5-year survival to train two gradient boosting models (mutation and mutation+motif). We used the same 500 mutation features and 813 mutation+motif features from the diagnosis analysis and cross-validations were performed the same way as described above. The model performance was evaluated by the log2 hazard ratio and Kaplan–Meier estimator of the patient 5-year survival rate in the R package survival (37) (Figure 4C). Multivariate survival analysis was performed to show factors significantly (P < 0.05) correlated with patient survival with 95% confidence interval (Figure 4D).
Feedforward loop analysis
We built a network with three types of nodes: motifs, TET1/DNMT3A, genes. We defined promoters as the region –1000 bp and +500 bp from the transcription start sites (TSS) of protein-coding genes (including TET1 and DNMT3A) from Gencode v19 (29), as previously described. A directed edge was defined if the source node has an occurrence site at the promoter of the target nodes. For TET1 and DNMT3A, occurrence site was defined by ChIP-seq data previously measured in hESC and NCCIT cells, respectively. For motifs, the occurrence site was defined by FIMO with P < 10–5. When a coding gene is a target, we first check if the gene is a known transcription factor, then define its binding site by FIMO with P < 10–5. Finally, tracks were visualized in integrated genome viewer and the methylation tracks were provided by WGBS from The Epigenomics Roadmap Project (15) and 450K array from the TCGA consortium (27).
RESULTS
Defining DNA methylation regions and the de novo motif discovery
We aimed to identify DNA motifs associated with DNA methylation and thus started with searching for methylation regions that have the strongest signals. We collected whole genome bisulfite sequencing (WGBS) data of 34 human methylomes generated by the NIH Roadmap Epigenomics Project (16,38) (Figure 1A). We took an approach similar to the Ziller et al. study (39) and defined 1.55 million methylation regions containing 11.5 million CpG sites in the 34 methylomes. Because the methylome data is noisy, we only considered regions containing two or more CpGs within 400 bp apart, which covers 29.2% of the human genome.
Methylation level is associated with different functions. For example, low methylated regions (LMRs) are important in hematopoiesis and leukemia development (40), DNA methylation valleys (DMVs) are long hypomethylated regions involved in embryonic development and tissue-specific regulation (41,42); focal hypermethylation and long-range hypomethylation are found in cancer (43); variably methylated regions (VMR) are associated with histone modification and enhancer (44). In this study, we defined three types of methylation regions based on the mean and standard deviation of the CpG methylation level in each region (Figure 1A, B): (i) Top 0.5% (or 7726) commonly methylated regions (CMR) which have the highest methylation level across 34 methylomes; (ii) Top 0.5% (or 7726) commonly unmethylated regions (CUR) with the lowest methylation levels; (3) Top 20% (or 309 040) variably methylated regions (VMR) with the highest standard deviation and this percentage is consistent with the previously reported 21.8–22.6% VMRs in the methylome (39,44). We are aware that these regions can vary upon the data sets used to define them. Because the 34 methylomes are derived from diverse cells and tissues, we argue the derived motifs are still reasonable starting points of revealing DNA binding proteins recruiting DNA methylation enzymes.
Defining commonly and variably methylated/unmethylated regions allow identification of motifs that are associated with DNA methylation independent of cell type or cell-type specific. CMRs and CURs are regions that show consistent methylation pattern across a diversity of 34 cells and tissues, and therefore they likely harbor motifs associated with methylation/demethylation in a cell-type independent manner. GREAT (45) analysis showed CMRs are strongly (P < 1e–30) linked to DNA repair and mitosis and are mostly (68%) found in introns (Supplementary Figure S1A) (46). CURs prefer promoters (66%) associated with (P < 10–30) cell differentiation, development, and morphogenesis, indicating the important roles of demethylation in these processes (47,48) (Supplementary Figure S1A). By contrasting CMRs to CURs, we identified 55 CMR and 87 CUR motifs using a motif finding algorithm Epigram (3) (Figure 1A, C). A 5-fold cross-validation using Epigram (3) successfully discriminated CMRs from CURs using the motifs (AUC = 0.97) (Figure 1C). Note that Epigram balances the GC content, sequence number, and length in the foreground and background, which avoids identification of trivial sequence motifs (see details in Materials and Methods and (3)). Because these motifs are associated with high or low methylation regions commonly shared by diverse cell types, it is reasonable to argue that they are important or even casual for establishing, maintaining or removing DNA methylation.
Similar to TFs whose binding motifs are defined but their activities are specific, the usage of DNA methylation associated motifs is determined by the cellular state. The VMRs show cell type-specific methylation patterns, which provides an opportunity to identify motifs active in particular cell types. We contrasted top 6000 methylated and unmethylated VMRs sorted in each cell type and discovered average 63 methylation- and 85 unmethylation-associated motifs in each methylome, with an average AUC of 0.79 (Figure 1C).
In total, 5172 motifs were identified from 35 Epigram runs (1 common + 34 cell-specific). Because the same or similar motifs could be found in multiple cells, we clustered these motifs into 3226 unique ones using motif similarity measurement based on Jensen-Shannon divergence (see Materials and Methods). To control false discovery rate (FDR), we further conducted a robust volcano test (49) with a stringent requirement (P-value < 10–10 and enrichment > 2), resulting in 313 methylation motifs for the follow-up analysis (Figure 1A, Supplementary Figure S1B), including 221 unmethylation motifs (UM) and 92 methylation motifs (MM). Among them, 36 (16.2%) and 14 (17.1%) are matched to 50 known motifs in the latest version of HOCOMOCO (17). The matched included previously confirmed factors to influence methylation levels such as CTCF (2) and PAX5 (50) as well as factors KLF4, SP4 and EGR1 that have been reported to regulate gene expression by binding to CpG rich promoters (51). Furthermore, we also found 22 (24%) top enriched MMs were matched with the 657 reported methyl-specific motifs (52). In addition, we have profiled the binding of 845 known TFs with ChIP-seq experiments documented in the latest GTRD (Gene Transcription Regulation Database) (53) in the motif occurrence sites (Supplementary Figure S2C). These TFs can collaborate with the MMs/UMs to define the local methylation state. All motifs, their alignment results, and the TF occupancy profile can be found on our website (http://wanglab.ucsd.edu/star/MethylMotifs). The majority of the motifs are novel and showed strong sequence preference. UMs are more similar to each other and have higher GC content (e.g. CCGCCGCCG) than MMs (Supplementary Figures S1C, D). Note that these motifs were found by Epigram after sequence balancing which removes GC content bias (3). While high GC content and CpG-rich sequences are known to be associated with hypomethylation in regions such as CG-islands (54) and in specific cells (55–57), our analysis revealed specific DNA motifs with sophisticated patterns that may be recognized by proteins or ncRNAs.
Identified motifs are associated with the local DNA methylation deviated from the background
We first investigated the DNA methylation levels around the identified motif occurring sites (determined by FIMO (21) using P < 10−5, the same parameters were used for all the relevant analyses thereinafter). We did observe hypomethylation and hypermethylation in the neighbor CpGs of the UM and MM motifs, respectively. Several representative examples are shown in Figure 2A. It is obvious that DNA methylation levels around the motif sites show a sharp ‘dip’ or ‘peak’, suggesting the association is highly locus-specific. Interestingly, this trend remains the same in different cell types despite that the methylation levels in the surrounding regions vary. For example, motif UM_238.2_3.88_0.53_5 (matched to the WT1 motif) was identified from VMRs in the right ventricle tissues; the methylation level at its occurring sites decreases in all the cell types although the methylation level ranges from 0.6 to 0.8 in the surrounding regions (Figure 2A). This observation confirms the functionality of individual UM and MM motifs even though the local environment is overall hyper- or hypo-methylated.
We further examined the impact of these motifs on methylation in the gene coding regions. UM and MM consistently mark lower and higher local CpG methylation levels in the gene coding regions (Figure 2B). In the Roadmap dataset, we observed a significant impact of UMs or MMs on DNA methylation level around the transcription start sites (TSS) (Figure 2B, left panel). DNA methylation in the promoters is important for regulating gene expression (58) and thus itself is likely under active regulation. We observed the same trend in the TCGA DNA methylation data of 9037 patients from 32 cancers measured by Illumina 450K array (27) (Figure 2B, right panel). On average, CpG methylation decreases from the beta value of 0.81 in the Roadmap dataset, dominated by normal cell lines and tissues, to 0.59 in the TCGA cancer patients across 20,260 protein-coding genes. This observation is consistent with the global hypomethylation in cancer cells that have been reported in the literature (41,47,59). However, the MM and UM occurring bins still showed respectively higher and lower methylation levels than the background. As an example, UM and MM occurrence sites are characterized by lower and higher methylation in the gene coding region of TP53 (chr17:7 540 000–7 650 000) in both TCGA and Roadmap data. Collectively, our results on two separate data sets generated by different technologies support that the identified DNA motifs play critical roles in influencing the local CpG methylation.
Identified motifs are significantly enriched at TETs and DNMTs binding sites
Locus-specific DNA demethylation or methylation depends on the recruitment of specific enzymes such as TET (60) and DNMTs (61) to particular genomic regions (62–64). We reasoned that, if the identified motifs are important for recruiting the enzymes, these motifs would be enriched around the binding sites of the recruited enzymes. To this end, we have collected all the available ChIP-seq experiments of TET and DNMT enzymes (22–25). Indeed, at the center of TET1 ChIP-seq peaks in hESC H1 cells (25), the UM sites occur 26.7 times of expected counts (see details in Materials and Methods), whereas MM motifs occur roughly same (1.4 times) as the expected counts (Figure 2C). This observation is consistent with the previous reports that TETs can be recruited to specific locus by DNA binding factors (60,64). Interestingly, the wide distribution of UM around TET peaks compared to MM-DNMT overlap is consistent with the previously reported role of TET in protecting spanned low-methylation regions termed methylation canyons against hypermethylation (65). Furthermore, TET prefers CpG-rich patterns such as CpG island which spans several kilobases (66) and can bind CpG-rich DNA sequences (62) in mammalians to maintain stable demethylation (67); consistently, UMs have significantly higher GC content than MMs and known motifs (P < 0.05, Supplementary Figure S1C).
We observed different motif occurring patterns around the binding sites of different DNMT enzymes. DNMT3A and DNMT3B are responsible for de novo methylation (68). At the center of DNMT3A ChIP-seq peaks in the human NCCIT cells (24), we observed a peak of the MM motif occurrence compared to the known and UM motifs (Figure 2C). Interestingly, the MMs are enriched at the shoulder regions of the DNMT3B binding sites but depleted at the center (Figure 2C). Note that only 2.2% of DNMT3A and 3.8% of DNMT3B peaks overlap with each other (24) (Supplementary Figure S2A). Several studies have demonstrated some distinct roles of DNMT3A and DNMT3B, showing that DNMT3B preferentially targets gene bodies marked with H3K36me3 (69–72); in fact, H3K36me3 is 4.27 times enriched at the DNMT3B compared to DNMT3A peaks in gene coding regions (Supplementary Figure S2A). These observations suggest that the MMs are likely recognized by DNA binding factors involved in actively recruiting DNMT3A, whereas DNMT3B may be recruited by flanking sequences containing MMs and together with chromatin marks and/or other factors such as H3K36me3. Interestingly, DNMT1, an enzyme involved in DNA methylation maintenance and recognizing hemimethylation (73), shows a different profile from DNMT3A/B (Figure 2C). This difference may have resulted from the different mechanisms or factors involved in active and passive DNA methylation.
To further validate if the observed co-occurrence around methylation enzyme is significant, we also compared the center-to-edge enrichment of UM and MM with TFs known to regulate DNA methylation (Figure 2D, method). Previous studies have reported that introducing a CTCF binding site at a particular locus leads to local DNA demethylation and enrichment of TET (2). NR6A1 has also been confirmed to recruit DNMT to methylate at target genes (74). Here, we show that at the center of TETs binding sites, UMs are significantly more enriched than MMs, and have even higher enrichment than CTCF (Figure 2D, left panel). Similarly, MMs are significantly more enriched than UMs at the center of DNMT3A binding sites, surpassing that of NR6A1 (Figure 2D, right panel). The enrichment of MMs and UMs were further compared with the known TFs such as PAX5, TOPORS, WT1 and PPARG that are most enriched at the TETs and DNMT3A sites. Furthermore, we downloaded the most confident (P < 0.05) differential CpGs (dCpGs) defined by Kemp et al. (26), i.e. CpGs showing destabilized methylation level when CTCF contains point mutation or copy number aberrations in several human cancers. The CTCF’s critical role in affecting the local DNA methylation in these loci was confirmed and we indeed found that CTCF and UMs were even more enriched at these loci (Supplementary Figure S2B). These results demonstrated that the identified motifs can be recognized by particular DNA binding factors that in turn recruit the methylation modifying enzymes in a locus-specific manner. Given that the majority of MMs (71.4%) and UMs (83.9%) are de novo motifs, our findings pave the way towards identifying particular factors involved in locus-specific methylation regulation.
Genetic variants at identified DNA motif sites are associated with altered methylation level
To validate the functionality of the identified motifs, we investigated the enrichment of quantitative trait loci of expression (eQTL) and methylation (mQTL) at motif occurrence sites. Note that we only took somatic mutations identified by the TCGA consortium in this analysis. We analyzed the relationship between somatic mutation and methylation level using the TCGA data (27) and identified “somatic” methylation quantitative trait loci (mQTL), which are somatic mutations correlating with CpG variation within 5000 bp. Using Matrix eQTL (28), we identified 26 341 mutation-CpG pairs, corresponding to 17 038 unique mutations and 20 043 CpGs, from a total of 1.3 million somatic mutations in 9037 patients of 32 cancers. We observed an average 11.7% mQTL discovery rate at the motif sites compared to 2.3% in the background (Figure 3A, left panel). This enrichment difference is most prominent around the transcription start site, suggesting that the identified motifs have a stronger impact on methylation at TSS (Figure 2B) (75–77). Enrichment of mQTL in both MM and UM sites was also found in three additional human methylome datasets using the reported mQTLs in the original studies (30–32) (Supplementary Figure S3A), which confirms the generality of this observation. Because DNA methylation is associated with gene expression (39,78), it is not surprising that MMs and UMs significantly overlap with somatic expression quantitative trait loci (eQTL), which are mutations correlated with gene expression level (Figure 3A, right panel).
Figure 3.

Somatic mutation at motif sites co-occur with local methylation alteration. (A) Distribution of somatic quantitative trait loci corresponding to methylation (mQTL) and gene expression (eQTL) over gene body (see details in Materials and Methods). Each gene body is split into ten equal bins. (B) Methylation level change of CpG sites nearby TET1-UM sites (TET1 binding peaks containing UM motifs) overlapping with somatic mutations. Asterisks indicate P < 0.01 calculated with paired one-tail t-test, pairing foreground observed methylation change to the corresponding background expected methylation change. Foreground (FG), somatic mQTL at TET1-UM sites. Background (BG), somatic mQTL at TET1 binding peaks (22,24,25). To ensure the statistical significance, we only considered the 15 cancers with >100 CpGs within 5000 bp of TET1-UM sites (see details in Methods). (C) An example showing disruption of a UM motif (no match with known motifs) by a C→T somatic mutation at chr16:68002415 significantly increases the methylation level of the four nearby CpGs in the LUAD patients.
To investigate the causality between these motifs and DNA methylation level, we analyzed whether disrupting these motifs would lead to DNA methylation change. We chose to focus on the possible binding sites of TET1 and DNMT3A containing these motifs because the significant enrichment of the found motifs in the enzyme-binding regions implies that the active methylation/demethylation is most likely mediated by DNA binding factors to recruit TET1/DNMT3A. Despite the ChIP-seq experiment of TET1/DNMT3A was done in one particular cell type, the sequence features, i.e. the motif composition in these regions, do not change and thus the mechanism of the active methylation regulation. The methylation change is decided by which factors are expressed and active in a specific cell type or state. Disrupting these motifs would lead to methylation change in the nearby CpGs.
Using the TCGA data, we first identified 5372 CpG sites from 15 cancers within 5000 bp of the TET1 binding peaks that also contain mutations overlapping with UMs in at least one patient. Because we did not have TET1 ChIP-seq data in the cancer patients, we used the published data measured in hESC (see Figure 2C, D). We compared the methylation change of these CpGs between patients with and without the mutation in each cancer. Thirteen out of 15 cancers showed significant (P < 0.01) increased methylation level of with-mutation compared to the without-mutation patients (background) (Figure 3B, see Methods for details). One example is given in Figure 3C for a UM motif UM_91.0_3.11_0.56_2. This motif is within a TET1 peak and is disrupted by a C→T somatic mutation at chr16:68002415 on the first exon of SLC12A4 in one LUAD cancer patient. All 4 CpGs within 500 bp upstream of the mutation showed increased methylation (beta value increased from 6.2% to 52%, 8.8% to 55%, 6.2% to 44% and 17% to 56%, respectively). Hypomethylation in the SLC12A4 promoter is related to resistance to platinum-based chemotherapy in ovarian cancer (79); the four CpGs affected by the mutation are located in the SLC12A4 promoter, suggesting a mechanism of how the mutation may affect response to chemotherapy through regulation of local DNA methylation. More examples of mutation-induced methylation change through disrupting UMs are shown in Supplementary Figure S3B.
Overlapping MM and mutations with DNMT3A peaks only resulted in <100 CpGs sites in two cancers. Although we observed decreased methylation level of DNMT3A-MM overlapping with somatic mQTL as predicted, the analysis did not have enough statistical power. Because the methylation was measured by 450K array and mutations were detected and called from Affymetrix Genome-Wide Human SNP Array 6.0, it is reasonable to expect that more sites can be observed with whole methylome and whole genome sequencing data.
Combining Motifs and somatic mutation Shows Diagnosis and Prognosis Power
DNA methylation has been shown to be predictive for cancer diagnosis and patient survival prospective (80,81). Since we have shown motif disruption is associated with methylation change, we hypothesized that combining motifs with mutations can improve prediction for cancer diagnosis and patient survival. To evaluate this, we trained gradient boosting models (33) using mutation and mutation+motif as features in 32 TCGA cancers from 7120 patients (see Materials and Methods for details). We calculated both auROC and auPRC (a metric for an imbalanced dataset to avoid inflated evaluation of the performance) (82). The inclusion of the motifs in the models resulted in increased auROC and auPRC in all the 32 cancers. On average, auROC increased from 0.78 to 0.92 and auPRC from 0.45 to 0.56, whereas 26 (for auROC) and 13 (for auPRC) improvement are statistically significant (P < 0.01) (Figure 4A). Notably, several cancers showed drastic improvement, including ovarian cancer (OV, auPRC from 0.41 to 0.79), thyroid carcinoma (THCA, auPRC from 0.49 to 0.82), acute myeloid leukemia (LAML, auPRC from 0.6 to 0.88), pheochromocytoma and paraganglioma (PCPG, auPRC 0.49–0.75) (Supplementary Figure S4B). These cancers all have reported aberrant methylome and have methylation associated diagnosis and therapeutic targets (83–86).
For 26 cancers with auPRC >0.3, the 67 most predictive features (score > 0.01) determined by the gradient boost estimator are shown in Figure 4B (see Materials and Methods for details), including 13 mutations, 20 MMs, and 34 UMs. Only two MMs are matched to known motifs (RXRB and PAX5), whereas seven UMs to AP2B, BTD, PLAL1, GLIS2, WT1, CNOT3 and GTF3A. The predictive mutations include those occurring on the cancer driver genes such as BRAF (in 16 cancers), TP53 (in 14 cancers), IDH1 (in 14 cancers), PIK3CA (in 13 cancers) and KRAS (in 12 cancers). Strikingly, we found numerous MMs and UMs very predictive in multiple cancers. Notably, MM_814.4_2.02_0.62_8 (PAX5) that has been shown to strongly impact local methylation level (Figure 3C) is important in 12 cancers. The five UMs predictive in >10 cancers are UM_78.3_2.97_0.58_2 (BTD), UM_13.5_2.17_0.53_2, UM_195.4_2.88_0.56_5 (GTF3A), UM_35.4_2.56_0.54_3 and UM_61.9_2.40_0.56_4 (Figure 4B).
To evaluate the prognosis power of the motifs, we trained two gradient boosting models (mutation and mutation+motif) to discriminate low-risk from high-risk patients. We evaluated the performance using the survival hazard ratio of the predicted high-risk group (higher ratio means better performance). The mutation-only model found 6 out of 22 cancers having significant (P < 0.05) hazard ratio. In comparison, the mutation+motif model achieved 16 out of 22 cancers having significant (P < 0.05) hazard ratio (Figure 4C, left panel, see Materials and Methods for details). Kaplan–Meier test showed a better separation of patient survival between the predicted low-risk and high-risk groups by considering motifs (P = 3.6x10–43 for the mutation-only model and P = 3.2x10–270 for the mutation+motif model, Figure 4C, right panel). Multivariate survival analysis on the full model revealed important factors correlated with patient survival (P < 0.05), including 6 mutations, 7 MMs and 20 UMs (Figure 4D). These results further confirmed the functionality of the discovered motifs and highlighted the potential for clinical application.
Motifs involved in both DNA methylation and histone modifications
Both DNA methylation and histone modification play important roles in regulating gene expression and their interplay has been well recognized (87,88). In a separate study, we identified 361 histone motifs (89) that are associated with 6 (H3K4me1, H3K4me3, H3K27ac, H3K27me3, H3K9me3, H3K36me3) histone modifications from 110 diverse human cell types/tissues. By comparing the 313 methylation motifs with these 361 histone motifs, we found that 56.5% MMs (52 out of 92) overlap with them (e-value cutoff of 0.05 using Tomtom) (Figure 5A). Among these, 35 MMs are aligned to H3K36me3 motifs as H3K36me3 can recruit DNMT3A/3B through their PWWP domain (90,91). In contrast, 74.2% (164 out of 221) UMs found no match to histone motifs. 57 UMs are matched to motifs associated with the active promoter or enhancer marks: 12 UMs matched to H3K27ac, an active promoter and enhancer mark; another 12 UMs matched to the promoter mark H3K4me3. As active enhancers and promoters tend to have low methylation (16), this observation is not unexpected. Interestingly, we observed another 12 UMs matched to the motifs associated with the poised promoter markers H3K4me3+H3K27me3. Previous studies also suggested the colocalization of H3K4me3 and H3K27me3 marks DNA hypomethylation in pre-implantation embryos (92).
Figure 5.

Methylation motifs interplay with TET1, DNMT3A, and histone modification. (A) Methylation motifs matched to histone motifs (89). Motifs are aligned with Tomtom with e < 0.05. Lower panel showing several examples. (B) Feedforward loop targeting TET1 and DNMT3A. C. Feedforward loop via TET1 and DNMT3A.
Regulatory loops on DNA methylation
DNA methylation is dynamically regulated in response to the cell state change. We analyzed the putative regulatory connectivity between the identified motifs, transcription factors and the modifying enzymes of TET1 and DNMT3A. We only considered TET1 and DNMT3A here because their binding peaks are significantly enriched with UMs and MMs, respectively (Figure 2C). It is well accepted that a known TF motif occurring in the promoter of a gene suggests a possible regulation of the gene expression by the TF. Similarly, we infer the occurrence of a UM or MM in a gene's promoter indicates putative regulation on the DNA methylation level and thus affecting gene expression.
We first analyzed the promoters of TET1 and DNMT3A. We found 19 UMs in the promoters of both TET1 and DNMT3A. We also found these UMs appearing in the promoters of 25 TFs that also have motifs in the promoters of both TET1 and DNMT3A and presumably regulate the two enzymes (Figure 5B). Such a topology forms a feed-forward loop (FFL) (93) that involves three nodes: two regulator nodes (motifs and TFs), one regulates the other (motifs regulates TFs), and both jointly regulating a target (TET1 or DNMT3A) (see Materials and Methods). UMs induce demethylation of TET1/DNMT3A and their regulator TFs, which forms positive FFLs to enhance the expression of both TET1 and DNMT3A once the motifs are activated. We also found two and five MMs occurring in the promoters of TET1 and DNMT3A, respectively. These MMs appear in the promoters of 14 TFs as the other regulator of TET1 or DNMT3A, of which one TF only regulates TET1, seven TFs only regulates DNMT3A and six TFs regulate both (Figure 5B); these FFLs form enhanced dynamic regulation to repress TET1 and DNMT3A expressions. Overall, there are many more activating than repressive FFLs on regulating TET1 and DNMT3A.
Previous reports have also shown TET1 and DNMT3A have competitive binding to regulate promoters in mouse embryonic stem cells (94). In addition, in honey bees, Dnmt and Tet (homolog of vertebrate DNMTs and TETs) were found to target memory-associated genes sequentially, while Dnmt3 was found in a negative feedback loop for DNA methylation (95). We found six genes targeted by UMs and also by both TET1 and DNMT3A (as indicated by their ChIP-seq peaks in hESC and NCCIT cells, respectively) (Figure 5C). Interestingly, four of them (KLHL3, C1orf61, ACVR1C, PTPRO) are also targeted by MMs and either TET1 or DNMT3A (Figure 5C). One of them, PTPRO, a cancer suppressor and therapeutic target of a variety of solid and liquid tumors, is silenced by promoter hypermethylation (96). In fact, we observed higher methylation at the promoter of the first TSS of PTPRO (TSS1, chr12:15 474 979–15 476 332) in the TCGA patients (beta value average at 0.15) compared to the Roadmap methylomes (beta value averaged at 0.05) (Figure 5C). PTPRO has multiple TSSs and alternative splicing forms (97), and each TSS has a TET1 or DNMT3A ChIP-seq peak (Figure 5C). As competitive binding of activator and repressor can lead to sharp turn on/off of the gene expression (98–100), we speculate the competitive FFLs formed by the motifs and modifying enzymes would thus allow dynamic regulation of the methylation and presumably the expression levels of these genes.
DISCUSSION
In this study, we present a comprehensive catalog of the DNA motifs associated with DNA methylation. We did observe coincident higher and lower methylation levels around the MM and UM occurring sites, respectively. Furthermore, the motif sites are also enriched with functional mutations, including somatic mQTL and eQTL. We also showed that combining DNA motifs and mutations can achieve accurate prediction of diagnosis and prognosis in TCGA cancer patients, which supports the importance of these motifs.
Our analysis suggested that these motifs are most likely involved in recruiting TET and DNMT3A for active demethylation and methylation, as indicated by their significant enrichment in the binding sites of these enzymes. The passive or maintenance methylation mediated by DNMT1 seems to be regulated by mechanisms other than DNA binding co-factors because we did not observe an enrichment of the found motifs in the DNMT1 binding sites.
Interestingly, some of these motifs may also play roles in histone modifications as they were also found associated with histone modifications, particularly those relevant to DNA methylation such as H3K36me3 that were reported to recruit DNMT3A/B through their PWWP domains. Furthermore, these motifs can form feed-forward loops (FFLs) with TFs to regulate TET1 and DNMT3A or regulate genes together with TET1/DNMT3A. These FFLs allow possible regulation of the DNA methylation dynamics and presumably the gene expression dynamics. The interplay between DNA and epigenetic signatures is central to TF recruitment and eukaryotic gene expression regulation. Binding sites of TFs are determined by combined factors including DNA sequence, methylation (101), histone modification (102) and nucleosome landscape (103). Our motif analysis suggests putative mechanisms for experimental test.
We have shown multiple lines of evidence to support that the identified motifs are involved in regulating DNA methylation. To confirm the causal relationship between TF-DNA binding and methylation, additional experimental tests are needed such as mutating the found motifs in a specific locus and measuring its impact on the local DNA methylation change. We have made all the motifs and their occurrence sites available, which will allow designing particular experiments for testing the functions of these motifs in disease or other biological contexts. These experiments are still challenging nowadays because it requires to simultaneously mutate multiple short motifs. Given the fast advancement of the genome editing technology, it will become feasible to perform such a test in a high-throughput fashion of the predicted motifs in the future. There exist more than one mechanism of establishing and maintaining locus-specific DNA methylation patterns (63,101), which may require different combinatorial interactions between different factors. Our study establishes a catalog of the possible participating motifs, which provides a starting point towards fully deciphering the grammar of regulating the locus-specific DNA methylation.
DATA AVAILABILITY
Methylation motifs are available at the website (http://wanglab.ucsd.edu/star/MethylMotifs).
Supplementary Material
ACKNOWLEDGEMENTS
Author contributions: M.W. performed the computational analyses and interpreted the results. K.Z., V.N. and J.W.W. helped discovered the de novo motifs. K.Z. designed the motif merging and contributed to finding motif occurrence. S.F., Y.C. and L.Z. contributed to TCGA methylation analysis. V.N. and J.W. contributed to mutation analysis. C.L. and Z.C. contributed to functional annotation of motifs. W.W. conceived the study, supervised the analyses and interpreted the results. M.W. and W.W. wrote the paper with contribution from all authors.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [U54HG006997, R01HG009626 to W.W.]; CIRM [RB507012 to W.W.]; NSFC [61872063 to S.F.].
Conflict of interest statement. None declared.
REFERENCES
- 1. Lienert F., Wirbelauer C., Som I., Dean A., Mohn F., Schübeler D.. Identification of genetic elements that autonomously determine DNA methylation states. Nat. Genet. 2011; 43:1091–1097. [DOI] [PubMed] [Google Scholar]
- 2. Stadler M.B., Murr R., Burger L., Ivanek R., Lienert F., Schöler A., van Nimwegen E., Wirbelauer C., Oakeley E.J., Gaidatzis D. et al.. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011; 480:490–495. [DOI] [PubMed] [Google Scholar]
- 3. Whitaker J.W., Chen Z., Wang W.. Predicting the human epigenome from DNA motifs. Nat. Methods. 2015; 12:265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wu C., Yao S., Li X., Chen C., Hu X.. Genomewide prediction of DNA methylation using DNA composition and sequence complexity in human. Int. J. Mol. Sci. 2017; 18:E420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Das R., Dimitrova N., Xuan Z., Rollins R.A., Haghighi F., Edwards J.R., Ju J., Bestor T.H., Zhang M.Q.. Computational prediction of methylation status in human genomic sequences. Proc. Natl. Acad. Sci. U.S.A. 2006; 103:10713–10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Feng P., Chen W., Lin H.. Prediction of CpG island methylation status by integrating DNA physicochemical properties. Genomics. 2014; 104:229–233. [DOI] [PubMed] [Google Scholar]
- 7. Angermueller C., Lee H.J., Reik W., Stegle O.. DeepCpG: accurate prediction of single-cell DNA methylation states using deep learning. Genome Biol. 2017; 18:67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Edwards J.R., O’Donnell A.H., Rollins R.A., Peckham H.E., Lee C., Milekic M.H., Chanrion B., Fu Y., Su T., Hibshoosh H. et al.. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010; 20:972–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamada Y., Satou K.. Prediction of genomic methylation status on CpG islands using DNA sequence features. WSEAS Trans. Biol. Biomed. 2008; 5:153–162. [Google Scholar]
- 10. Su J., Shao X., Liu H., Liu S., Wu Q., Zhang Y.. Genome-wide dynamic changes of DNA methylation of repetitive elements in human embryonic stem cells and fetal fibroblasts. Genomics. 2012; 99:10–17. [DOI] [PubMed] [Google Scholar]
- 11. Wang Y., Liu T., Xu D., Shi H., Zhang C., Mo Y.-Y., Wang Z.. Predicting DNA methylation state of CpG dinucleotide using genome topological features and deep networks. Sci. Rep. 2016; 6:19598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Wrzodek C., Büchel F., Hinselmann G., Eichner J., Mittag F., Zell A.. Linking the epigenome to the genome: correlation of different features to DNA methylation of CpG islands. PLoS One. 2012; 7:e35327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zeng H., Gifford D.K.. Predicting the impact of non-coding variants on DNA methylation. Nucleic Acids Res. 2017; 45:e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ngo V., Wang M., Wang W.. Finding de novo methylated DNA motifs. Bioinformatics. 2019; doi:10.1093/bioinformatics/btz079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Amin V., Harris R.A., Onuchic V., Jackson A.R., Charnecki T., Paithankar S., Lakshmi Subramanian S., Riehle K., Coarfa C., Milosavljevic A.. Epigenomic footprints across 111 reference epigenomes reveal tissue-specific epigenetic regulation of lincRNAs. Nat. Commun. 2015; 6:6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roadmap Epigenomics Consortium Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J. et al.. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A. et al.. HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 2018; 46:D252–D259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bailey T.L., Boden M., Buske F.A., Frith M., Grant C.E., Clementi L., Ren J., Li W.W., Noble W.S.. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res. 2009; 37:W202–W208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sokal R.R., Michener C.D.. A statistical method for evaluating systematic relationship. Univ. Kansas Sci. Bull. 1958; 28:1409–1438. [Google Scholar]
- 20. Wang M., Wang D., Zhang K., Ngo V., Fan S., Wang W.. Motto: representing motifs in consensus sequences with minimum information loss. 2019; bioRxiv doi: 10.1101/607408, 13 April 2019, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed]
- 21. Grant C.E., Bailey T.L., Noble W.S.. FIMO: scanning for occurrences of a given motif. Bioinformatics. 2011; 27:1017–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Suzuki T., Shimizu Y., Furuhata E., Maeda S., Kishima M., Nishimura H., Enomoto S., Hayashizaki Y., Suzuki H.. RUNX1 regulates site specificity of DNA demethylation by recruitment of DNA demethylation machineries in hematopoietic cells. Blood Adv. 2017; 1:1699–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Deplus R., Delatte B., Schwinn M.K., Defrance M., Méndez J., Murphy N., Dawson M.A., Volkmar M., Putmans P., Calonne E. et al.. TET2 and TET3 regulate GlcNAcylation and H3K4 methylation through OGT and SET1/COMPASS. EMBO J. 2013; 32:645–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jin B., Ernst J., Tiedemann R.L., Xu H., Sureshchandra S., Kellis M., Dalton S., Liu C., Choi J.-H., Robertson K.D.. Linking DNA methyltransferases to epigenetic marks and nucleosome structure genome-wide in human tumor cells. Cell Rep. 2012; 2:1411–1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Verma N., Pan H., Doré L.C., Shukla A., Li Q.V., Pelham-Webb B., Teijeiro V., González F., Krivtsov A., Chang C.-J. et al.. TET proteins safeguard bivalent promoters from de novo methylation in human embryonic stem cells. Nat. Genet. 2018; 50:83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kemp C.J., Moore J.M., Moser R., Bernard B., Teater M., Smith L.E., Rabaia N.A., Gurley K.E., Guinney J., Busch S.E. et al.. CTCF haploinsufficiency destabilizes DNA methylation and predisposes to cancer. Cell Rep. 2014; 7:1020–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Cancer Genome Atlas Research Network Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R.M., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Shabalin A.A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics. 2012; 28:1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Harrow J., Frankish A., Gonzalez J.M., Tapanari E., Diekhans M., Kokocinski F., Aken B.L., Barrell D., Zadissa A., Searle S. et al.. GENCODE: the reference human genome annotation for The ENCODE Project. Genome Res. 2012; 22:1760–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Gaunt T.R., Shihab H.A., Hemani G., Min J.L., Woodward G., Lyttleton O., Zheng J., Duggirala A., McArdle W.L., Ho K. et al.. Systematic identification of genetic influences on methylation across the human life course. Genome Biol. 2016; 17:61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gutierrez-Arcelus M., Lappalainen T., Montgomery S.B., Buil A., Ongen H., Yurovsky A., Bryois J., Giger T., Romano L., Planchon A. et al.. Passive and active DNA methylation and the interplay with genetic variation in gene regulation. Elife. 2013; 2:e00523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hannon E., Spiers H., Viana J., Pidsley R., Burrage J., Murphy T.M., Troakes C., Turecki G., O’Donovan M.C., Schalkwyk L.C. et al.. Methylation QTLs in the developing brain and their enrichment in schizophrenia risk loci. Nat. Neurosci. 2016; 19:48–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Friedman J.H. Greedy function approximation: A gradient boosting machine. Ann. Stat. 2001; 29:1189–1232. [Google Scholar]
- 34. Pedregosa F., Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., Prettenhofer P., Weiss R., Dubourg V. et al.. Scikit-learn: Machine Learning in Python. J. Mach. Learn. Res. 2011; 12:2825–2830. [Google Scholar]
- 35. Olson R.S., Cava W.L., Mustahsan Z., Varik A., Moore J.H.. Data-driven advice for applying machine learning to bioinformatics problems. Pac. Symp. Biocomput. 2018; 23:192–203. [PMC free article] [PubMed] [Google Scholar]
- 36. Gonzalez-Perez A., Perez-Llamas C., Deu-Pons J., Tamborero D., Schroeder M.P., Jene-Sanz A., Santos A., Lopez-Bigas N.. IntOGen-mutations identifies cancer drivers across tumor types. Nat. Methods. 2013; 10:1081–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Therneau T.M., Lumley T.. Package ‘survival’. R. Top. Doc. 2015; 128:https://cran.r-project.org/web/packages/survival/survival.pdf. [Google Scholar]
- 38. Schultz M.D., He Y., Whitaker J.W., Hariharan M., Mukamel E.A., Leung D., Rajagopal N., Nery J.R., Urich M.A., Chen H. et al.. Human body epigenome maps reveal noncanonical DNA methylation variation. Nature. 2015; 523:212–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ziller M.J., Gu H., Müller F., Donaghey J., Tsai L.T.-Y., Kohlbacher O., De Jager P.L., Rosen E.D., Bennett D.A., Bernstein B.E. et al.. Charting a dynamic DNA methylation landscape of the human genome. Nature. 2013; 500:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jeong M., Sun D., Luo M., Huang Y., Challen G.A., Rodriguez B., Zhang X., Chavez L., Wang H., Hannah R. et al.. Large conserved domains of low DNA methylation maintained by Dnmt3a. Nat. Genet. 2014; 46:17–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Witte T., Plass C., Gerhauser C.. Pan-cancer patterns of DNA methylation. Genome Med. 2014; 6:66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hovestadt V., Jones D.T.W., Picelli S., Wang W., Kool M., Northcott P.A., Sultan M., Stachurski K., Ryzhova M., Warnatz H.-J. et al.. Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing. Nature. 2014; 510:537–541. [DOI] [PubMed] [Google Scholar]
- 43. Berman B.P., Weisenberger D.J., Aman J.F., Hinoue T., Ramjan Z., Liu Y., Noushmehr H., Lange C.P.E., van Dijk C.M., Tollenaar R.A.E.M. et al.. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat. Genet. 2011; 44:40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gu J., Stevens M., Xing X., Li D., Zhang B., Payton J.E., Oltz E.M., Jarvis J.N., Jiang K., Cicero T. et al.. Mapping of variable DNA methylation across multiple cell types defines a dynamic regulatory landscape of the human genome. G3. 2016; 6:973–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G.. GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 2010; 28:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Maunakea A.K., Chepelev I., Cui K., Zhao K.. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013; 23:1256–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Swami M. Epigenetics: demethylation links cell fate and cancer. Nat. Rev. Cancer. 2010; 10:740. [DOI] [PubMed] [Google Scholar]
- 48. Bagci H., Fisher A.G.. DNA demethylation in pluripotency and reprogramming: the role of tet proteins and cell division. Cell Stem Cell. 2013; 13:265–269. [DOI] [PubMed] [Google Scholar]
- 49. Kumar N., Hoque M.A., Sugimoto M.. Robust volcano plot: identification of differential metabolites in the presence of outliers. BMC Bioinformatics. 2018; 19:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Giambra V., Volpi S., Emelyanov A.V., Pflugh D., Bothwell A.L.M., Norio P., Fan Y., Ju Z., Skoultchi A.I., Hardy R.R. et al.. Pax5 and linker histone H1 coordinate DNA methylation and histone modifications in the 3′ regulatory region of the immunoglobulin heavy chain locus. Mol. Cell. Biol. 2008; 28:6123–6133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Maag J.L.V., Kaczorowski D.C., Panja D., Peters T.J., Bramham C.R., Wibrand K., Dinger M.E.. Widespread promoter methylation of synaptic plasticity genes in long-term potentiation in the adult brain in vivo. BMC Genomics. 2017; 18:250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Xuan Lin Q.X., Sian S., An O., Thieffry D., Jha S., Benoukraf T.. MethMotif: an integrative cell specific database of transcription factor binding motifs coupled with DNA methylation profiles. Nucleic Acids Res. 2019; 47:D145–D154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yevshin I., Sharipov R., Kolmykov S., Kondrakhin Y., Kolpakov F.. GTRD: a database on gene transcription regulation-2019 update. Nucleic Acids Res. 2019; 47:D100–D105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Deaton A.M., Bird A.. CpG islands and the regulation of transcription. Genes Dev. 2011; 25:1010–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cuozzo C., Porcellini A., Angrisano T., Morano A., Lee B., Di Pardo A., Messina S., Iuliano R., Fusco A. et al.. DNA damage, homology-directed repair, and DNA methylation. PLoS Genet. 2007; 3:e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. de la Rica L., Deniz Ö., Cheng K.C.L., Todd C.D., Cruz C., Houseley J., Branco M.R.. TET-dependent regulation of retrotransposable elements in mouse embryonic stem cells. Genome Biol. 2016; 17:234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Luu P.-L., Schöler H.R., Araúzo-Bravo M.J.. Disclosing the crosstalk among DNA methylation, transcription factors, and histone marks in human pluripotent cells through discovery of DNA methylation motifs. Genome Res. 2013; 23:2013–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jones P.A. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13:484–493. [DOI] [PubMed] [Google Scholar]
- 59. Cancer Genome Atlas Research Network Weinstein J.N., Collisson E.A., Mills G.B., Shaw K.R., Ozenberger B.A., Ellrott K., Shmulevich I., Sander C., Stuart J.M.. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013; 45:1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kohli R.M., Zhang Y.. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013; 502:472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Robertson K.D. DNA methylation and human disease. Nat. Rev. Genet. 2005; 6:597–610. [DOI] [PubMed] [Google Scholar]
- 62. Rasmussen K.D., Helin K.. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016; 30:733–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Blattler A., Farnham P.J.. Cross-talk between site-specific transcription factors and DNA methylation states. J. Biol. Chem. 2013; 288:34287–34294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ravichandran M., Jurkowska R.Z., Jurkowski T.P.. Target specificity of mammalian DNA methylation and demethylation machinery. Org. Biomol. Chem. 2018; 16:1419–1435. [DOI] [PubMed] [Google Scholar]
- 65. Wiehle L., Raddatz G., Musch T., Dawlaty M.M., Jaenisch R., Lyko F., Breiling A.. Tet1 and Tet2 protect DNA methylation canyons against hypermethylation. Mol. Cell Biol. 2016; 36:452–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Elango N., Yi S.V.. Functional relevance of CpG island length for regulation of gene expression. Genetics. 2011; 187:1077–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Zhang L., Gu C., Yang L., Tang F., Gao Y.Q.. The sequence preference of DNA methylation variation in mammalians. PLoS One. 2017; 12:e0186559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Okano M., Bell D.W., Haber D.A., Li E.. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999; 99:247–257. [DOI] [PubMed] [Google Scholar]
- 69. Baubec T., Colombo D.F., Wirbelauer C., Schmidt J., Burger L., Krebs A.R., Akalin A., Schübeler D.. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015; 520:243–247. [DOI] [PubMed] [Google Scholar]
- 70. Morselli M., Pastor W.A., Montanini B., Nee K., Ferrari R., Fu K., Bonora G., Rubbi L., Clark A.T., Ottonello S. et al.. In vivo targeting of de novo DNA methylation by histone modifications in yeast and mouse. Elife. 2015; 4:e06205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Duymich C.E., Charlet J., Yang X., Jones P.A., Liang G.. DNMT3B isoforms without catalytic activity stimulate gene body methylation as accessory proteins in somatic cells. Nat. Commun. 2016; 7:11453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Challen G.A., Sun D., Mayle A., Jeong M., Luo M., Rodriguez B., Mallaney C., Celik H., Yang L., Xia Z. et al.. Dnmt3a and Dnmt3b have overlapping and distinct functions in hematopoietic stem cells. Cell Stem Cell. 2014; 15:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jones P.A., Liang G.. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009; 10:805–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Sato N., Kondo M., Arai K.-I.. The orphan nuclear receptor GCNF recruits DNA methyltransferase for Oct-3/4 silencing. Biochem. Biophys. Res. Commun. 2006; 344:845–851. [DOI] [PubMed] [Google Scholar]
- 75. Rapkins R.W., Wang F., Nguyen H.N., Cloughesy T.F., Lai A., Ha W., Nowak A.K., Hitchins M.P., McDonald K.L.. The MGMT promoter SNP rs16906252 is a risk factor for MGMT methylation in glioblastoma and is predictive of response to temozolomide. Neuro. Oncol. 2015; 17:1589–1598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Dyrvig M., Qvist P., Lichota J., Larsen K., Nyegaard M., Børglum A.D., Christensen J.H.. DNA methylation analysis of BRD1 promoter regions and the schizophrenia rs138880 Risk Allele. PLoS One. 2017; 12:e0170121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chuang T.-J., Chen F.-C., Chen Y.-Z.. Position-dependent correlations between DNA methylation and the evolutionary rates of mammalian coding exons. Proc. Natl. Acad. Sci. U.S.A. 2012; 109:15841–15846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Razin A., Cedar H.. DNA methylation and gene expression. Microbiol. Rev. 1991; 55:451–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. de Leon M., Cardenas H., Vieth E., Emerson R., Segar M., Liu Y., Nephew K., Matei D.. Transmembrane protein 88 (TMEM88) promoter hypomethylation is associated with platinum resistance in ovarian cancer. Gynecol. Oncol. 2016; 142:539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Hao X., Luo H., Krawczyk M., Wei W., Wang W., Wang J., Flagg K., Hou J., Zhang H., Yi S. et al.. DNA methylation markers for diagnosis and prognosis of common cancers. Proc. Natl. Acad. Sci. U.S.A. 2017; 114:7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Guo S., Diep D., Plongthongkum N., Fung H.-L., Zhang K., Zhang K.. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017; 49:635–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Saito T., Rehmsmeier M.. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One. 2015; 10:e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Koukoura O., Spandidos D.A., Daponte A., Sifakis S.. DNA methylation profiles in ovarian cancer: implication in diagnosis and therapy (Review). Mol. Med. Rep. 2014; 10:3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Ellis R.J., Wang Y., Stevenson H.S., Boufraqech M., Patel D., Nilubol N., Davis S., Edelman D.C., Merino M.J., He M. et al.. Genome-wide methylation patterns in papillary thyroid cancer are distinct based on histological subtype and tumor genotype. J. Clin. Endocrinol. Metab. 2014; 99:E329–E337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kroeger H., Jelinek J., Estécio M.R.H., He R., Kondo K., Chung W., Zhang L., Shen L., Kantarjian H.M., Bueso-Ramos C.E. et al.. Aberrant CpG island methylation in acute myeloid leukemia is accentuated at relapse. Blood. 2008; 112:1366–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. de Cubas A.A., Korpershoek E., Inglada-Pérez L., Letouzé E., Currás-Freixes M., Fernández A.F., Comino-Méndez I., Schiavi F., Mancikova V., Eisenhofer G. et al.. DNA methylation profiling in pheochromocytoma and paraganglioma reveals diagnostic and prognostic markers. Clin. Cancer Res. 2015; 21:3020–3030. [DOI] [PubMed] [Google Scholar]
- 87. Cedar H., Bergman Y.. Linking DNA methylation and histone modification: patterns and paradigms. Nat. Rev. Genet. 2009; 10:295–304. [DOI] [PubMed] [Google Scholar]
- 88. Rose N.R., Klose R.J.. Understanding the relationship between DNA methylation and histone lysine methylation. Biochim. Biophys. Acta. 2014; 1839:1362–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ngo V., Chen Z., Zhang K., Whitaker J.W., Wang M., Wang W.. Epigenomic analysis reveals DNA motifs regulating histone modifications in human and mouse. Proc. Natl. Acad. Sci. U.S.A. 2019; 116:3668–3677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Rondelet G., Dal Maso T., Willems L., Wouters J.. Structural basis for recognition of histone H3K36me3 nucleosome by human de novo DNA methyltransferases 3A and 3B. J. Struct. Biol. 2016; 194:357–367. [DOI] [PubMed] [Google Scholar]
- 91. Rinaldi L., Datta D., Serrat J., Morey L., Solanas G., Avgustinova A., Blanco E., Pons J.I., Matallanas D., Von Kriegsheim A. et al.. Dnmt3a and Dnmt3b Associate with Enhancers to Regulate Human Epidermal Stem Cell Homeostasis. Cell Stem Cell. 2016; 19:491–501. [DOI] [PubMed] [Google Scholar]
- 92. Liu X., Wang C., Liu W., Li J., Li C., Kou X., Chen J., Zhao Y., Gao H., Wang H. et al.. Distinct features of H3K4me3 and H3K27me3 chromatin domains in pre-implantation embryos. Nature. 2016; 537:558–562. [DOI] [PubMed] [Google Scholar]
- 93. Mangan S., Alon U.. Structure and function of the feed-forward loop network motif. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:11980–11985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Gu T., Lin X., Cullen S.M., Luo M., Jeong M., Estecio M., Shen J., Hardikar S., Sun D., Su J. et al.. DNMT3A and TET1 cooperate to regulate promoter epigenetic landscapes in mouse embryonic stem cells. Genome Biol. 2018; 19:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Biergans S.D., Giovanni Galizia C., Reinhard J., Claudianos C.. Dnmts and Tet target memory-associated genes after appetitive olfactory training in honey bees. Sci. Rep. 2015; 5:16223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Jacob S.T., Motiwala T.. Epigenetic regulation of protein tyrosine phosphatases: potential molecular targets for cancer therapy. Cancer Gene Ther. 2005; 12:665–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Aguiar R.C., Yakushijin Y., Kharbanda S., Tiwari S., Freeman G.J., Shipp M.A.. PTPROt: an alternatively spliced and developmentally regulated B-lymphoid phosphatase that promotes G0/G1 arrest. Blood. 1999; 94:2403–2413. [PubMed] [Google Scholar]
- 98. Wang W., Cherry J.M., Nochomovitz Y., Jolly E., Botstein D., Li H.. Inference of combinatorial regulation in yeast transcriptional networks: a case study of sporulation. Proc. Natl. Acad. Sci. U.S.A. 2005; 102:1998–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Miller J.A., Widom J.. Collaborative competition mechanism for gene activation in vivo. Mol. Cell Biol. 2003; 23:1623–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Darieva Z., Clancy A., Bulmer R., Williams E., Pic-Taylor A., Morgan B.A., Sharrocks A.D.. A competitive transcription factor binding mechanism determines the timing of late cell cycle-dependent gene expression. Mol. Cell. 2010; 38:29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Zhu H., Wang G., Qian J.. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016; 17:551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Xin B., Rohs R.. Relationship between histone modifications and transcription factor binding is protein family specific. Genome Res. 2018; doi:10.1101/gr.220079.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Zhu F., Farnung L., Kaasinen E., Sahu B., Yin Y., Wei B., Dodonova S.O., Nitta K.R., Morgunova E., Taipale M. et al.. The interaction landscape between transcription factors and the nucleosome. Nature. 2018; 562:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Methylation motifs are available at the website (http://wanglab.ucsd.edu/star/MethylMotifs).