

ABSTRACT
Epigenetic modulators play pivotal roles in directing gene expression for the maintenance of normal cellular functions. However, when these modulators are aberrantly regulated, this can result in a variety of disease states, including cancer. One class of epigenetic regulators, protein arginine methyltransferases (PRMTs), have been shown to play critical roles in disease through methylation of arginine residues (R) on histone or non-histone proteins. Quite different from PRMTs, microRNAs (miRNAs) belong to the family of modulators known as noncoding RNAs (ncRNA) that act to regulate gene expression via RNA-mediated gene silencing. Importantly, miRNAs are frequently dysregulated and contribute to the progression of cancer and other conditions, including neurological and cardiovascular diseases. Recently, numerous studies have shown that miRNAs and other epigenetic enzymes can co-regulate each other. This review highlights multiple nodes of interaction between miRNAs and PRMTs and also discusses how this interplay might open up promising opportunities for drug development for the treatment of cancer and other diseases.
KEYWORDS: Cancer, disease, miRNA, PRMTs
I. Introduction
It is widely appreciated that disruption of processes governed by epigenetic factors can alter gene function, leading to malignant cellular transformation and a host of other disease states. Protein arginine methylation is an important type of post-translational modification (PTM) mediated by PRMTs and is commonly associated with normal cellular processes including DNA repair, cell cyle regulation, transcription, mRNA splicing and signal transduction. Given these important roles, it is unsurprising that frequent dysregulation of this PTM is linked to the initiation and progression of cancer, cardiovascular and neurological dysfunction, among other diseases [1–3].
PRMTs catalyze the transfer of a methyl group from S-adenosylmethionine (SAM) to arginine’s guanidino group [4] and are structurally and functionally conserved from yeast to human [5]. Briefly, the mammalian PRMT family can be categorized as type I-III that contain 9 members according to their methyl-arginine products. Type I enzymes (PRMT1-4, PRMT6 and PRMT8) catalyze the formation of ω-NG-monomethylarginine (MMA), ω-NG, and NG-asymmetric dimethylarginine (ADMA), while type II (PRMT5, PRMT7, PRMT9) enzymes catalyze the formation of ω-MMA, ω-NG, and N’G-symmetric dimethylarginine (SDMA) [6]. The type III enzyme (PRMT7) only catalyzes the formation of ω-MMA [7,8]. The catalytic process of each type of PRMT is summarized in Figure 1 [9]. Importantly, methylation of many protein substrates catalyzed by PRMTs has been linked to multiple cancer processes such as initiation, progression, and metastasis [10]. Additionally, as summarized by Yang et al, altered expression of several PRMTs has been linked to cancers, including PRMT1, 2, 3 and 5 which were overexpressed in breast cancer, leukaemia, lung cancer, and colorectal cancer compared with normal cells [11].
Figure 1.
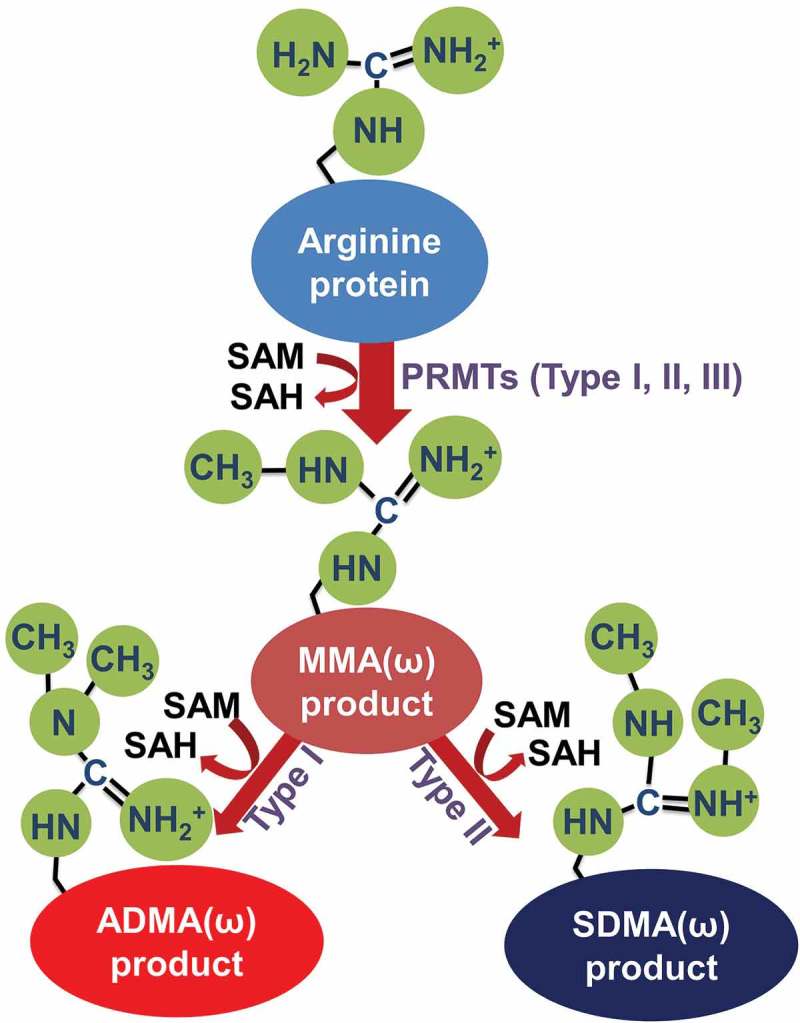
Types of arginine protein methylation.
Types I, II, and III protein arginine methyltransferases (PRMTs) produce monomethylated arginine (MMA) on a guanidino nitrogen atom using S-adenosyl-L-methionine (SAM) as a methyl donor. After the reaction, S-adenosyl-L-homocysteine (SAH) is released, and the formation of asymmetric dimethylated arginine (ADMA) is catalyzed by Type I PRMTs (PRMT1-4, 6, and 8), whereas symmetric dimethylated arginine (SDMA) is catalyzed by Type II enzymes (PRMT5, 7, 9). Additionally, PRMT7 functions as a Type III enzyme. Type III enzymes monomethylate instead of dimethylating the arginine residues on the protein substrates. This figure is adapted from [9].
Quite different from PRMTs, miRNAs constitute another important class of regulators that play critical roles in various biological processes. miRNAs, 20–23 nucleotide small noncoding RNAs, act by regulating mRNA expression of targeted genes via translational repression or degradation. The biogenesis of miRNA is a well-known stepwise process described in Figure 2 [12]. Briefly, in the nucleus, miRNAs are initially transcribed by RNA polymerase II as primary-miRNAs (pri-miRNAs), which can be several hundreds of nucleotides in length. Second, after cutting the 5ʹ-cap and 3ʹ-A tailing of pri-miRNAs by a nuclear RNase III (Drosha), pri-miRNAs turn into the precursor miRNA (pre-miRNA). Third, the exportin-5 (Exp5) protein helps export pre-miRNA from the nucleus to the cytoplasm. In the cytoplasm, RNase III Dicer cleaves pre-miRNA and produces a miRNA/miRNA* duplex (miRNA* represents the carrier miRNA which can be eventually degraded). The RNA-induced silencing complex (RISC) eventually produces one mature miRNA that inhibits targeted mRNA expression by binding its 3ʹ-UTR or degrading targeted mRNA [13]. Like PRMTs, miRNAs are also frequently implicated in playing a role in diseases such as cancer. For instance, miRNAs have been shown to influence cancer cell proliferation, invasion, migration, apoptosis, and cell cycle regulation [14,15]. Consequently, miRNAs have emerged as useful as important disease biomarkers [13].
Figure 2.
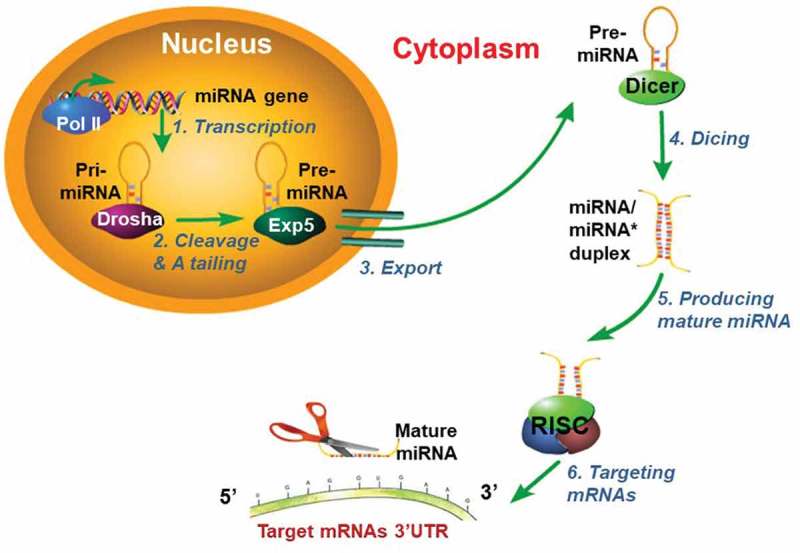
Model for miRNAs biogenesis.
The biogenesis of miRNAs occurs through 6 general steps [1]. Transcription: miRNA genes are initially transcribed by RNA polymerase II (Pol II) as primary-miRNAs (pri-miRNAs) transcripts (hundreds of nucleotides), which has an imperfectly double-strand region with a hairpin structure where miRNA sequences are embedded [2]. Cleavage and A tailing: A nuclear RNase III named Drosha cleaves the stem-loop of pri-miRNAs to generate the precursor miRNAs (pre-miRNAs, about 60 nucleotides) [3]. Export: pre-miRNAs are exported from the nucleus to the cytoplasm by Exportin-5 [4]. Dicing: Dicer cleaves pre-miRNAs and generates a miRNA/miRNA* duplex (about 20 nucleotides) in the cytoplasm. The miRNA/miRNA* duplex is made up of a biologically active strand called miRNA, which is usually converted into a mature RNA, and an inactive form called miRNA*, which is a “carrier” miRNA that is usually degraded during processing into a mature RNA [5]. Production of mature miRNAs: The miRNA/miRNA* duplex is incorporated in the RNA-induced silencing complex (RISC) which acts to load and unwind genes [6]. Targeting of mRNAs: One miRNA strand (miRNA*) degrades and the other mature strand (miRNA) guides RISC complex to target mRNAs in order to inhibit their expression [9]. This figure is adapted from [13].
Lately, numerous studies have shown that there is a functional cooperativity between miRNAs and various epigenetic enzymes, including DNA methyltransferases, histone deacetylases and more recently, PRMTs [16]. Importantly, this mutual regulation results in coordinated control of several aspects of disease progression including abnormal gene expression, cell migration, invasion, cell stemness maintenance, among others [17]. Here, we emphasize multiple mechanisms of interaction between miRNAs and PRMTs with a specific focus on two main mechanisms of co-regulation. On one hand, miRNAs can be regulated by PRMTs via methylation of histones (e.g. H4R3, H3R8) in the promoters that suppress miRNA transcription as well as direct methylation of proteins of the miRNA processing machinery. On the other hand, PRMTs themselves are also subject to regulation by miRNAs at multiple levels, including targeting the 3‘-untranslated regions (3ʹ-UTRs) of various PRMTs for degradation or indirectly via silencing of proteins of the PRMT interactome (Figure 3). Finally, we review potential opportunities that the interplay between PRMTs and miRNAs may present for drug development for the treatment of cancer and other diseases and suggest some pertinent questions that remain to be answered.
Figure 3.
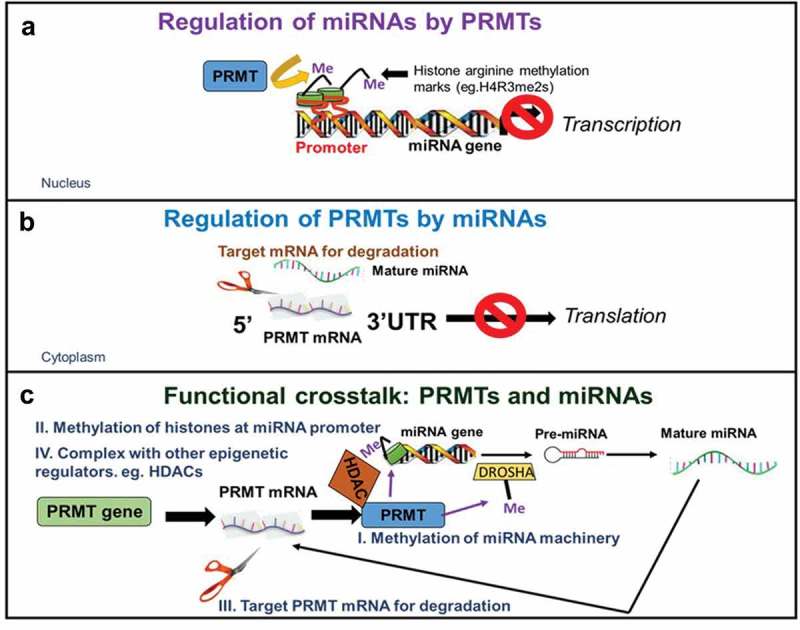
Mechanisms of interaction between miRNAs and PRMTs.
Multiple mechanisms of interaction between miRNAs and PRMTs including, a. Regulation of miRNAs by PRMTs in which histones in the promoter regions of miRNAs are methylated such as H3R8me2s, H4R3me2s, etc. resulting in transcriptional silencing of these miRNAs; b. Regulation of PRMTs by miRNAs involves targeting of 3ʹ-UTRs of PRMTs for degradation, ultimately blocking translation of the PRMT protein; c. Example of the reciprocal interplay between PRMTs and miRNAs at multiple levels in normal and cancerous states: during physiologically normal conditions, (I) miRNAs target PRMTs at their 3ʹ-UTRs for degradation, keeping their levels in check. However, in cancer, decrease of miRNA levels via epigenetic mechanisms including (II) PRMT-catalyzed methylation of histones at the promoters of miRNAs can act to silence these same miRNAs. (III) Additionally, PRMTs can methylate proteins of the miRNA processing machinery such as Drosha, which results in deregulation of miRNA biogenesis. Furthermore, another level of complexity is introduced when (IV) certain regulatory loops involve other epigenetic components such as HDACs which can form repressive complexes to further suppress miRNA expression. The net effect is upregulation of PRMTs that promote cancer processes.
II. Interplay between PRMTs and miRNAs
1. Multi-layered regulation of miRNAs by PRMTs in solid tumors and hematological malignancies
Although the number of reported deregulated miRNAs in cancer is rapidly expanding, the underlying mechanisms of aberrant miRNA expression are still poorly studied. Fortunately, knowledge regarding the multi-layered relationship between PRMTs and miRNAs has shed important light on some of these mechanisms, thus presenting new avenues for controlling aberrant miRNA regulation. Here, we discuss some of the most up-to-date findings regarding the regulation of miRNAs by PRMTs, whether directly via methylation of histones associated with miRNA promoters or indirectly by methylation of proteins involved in miRNA processing. Additionally, we will highlight another level of cooperativity between PRMTs and other epigenetic regulators at the promoters of miRNAs.
Methylation of histones that suppress miRNA transcription
It is widely appreciated that miRNAs contribute to the pathogenesis of cancer via the misregulated control of gene expression. However, the mechanisms underlying dysregulation of miRNA genes themselves remains to be fully understood. To date, several correlative reports suggest that certain miRNA genes are frequently silenced by aberrant histone modifications in their promoter regions, particularly histone arginine methylation. Importantly, these methylation events may promote cancer progression due to the subsequent accumulation of certain oncogenic molecules normally targeted by these miRNAs. Here, we discuss current evidence of PRMT-catalyzed silencing of miRNAs and how this in turn contributes to tumor initiation and progression in solid tumors and hematological malignancies.
Targhat et al first reported a tumor-promoting role for PRMT5-mediated symmetric dimethylation of H4R3 (H4R3me2s) in the regulation of miRNA expression in acute myeloid leukemia (AML). The authors demonstrated that H4R3me2s within the promoter region of the miR-29b correlated with its suppressed expression (Table 1). This in turn led to the activation of Fms-like tyrosine kinase 3 (FLT3) transcription and enhanced cell survival and growth of AML cell lines [18]. In contrast, PRMT4-mediated suppression of miR-223 transcription had the opposite effect by hampering myeloid differentiation (Table 1). Another study by Bueno et al. showed that epigenetic silencing of miR-203 enhanced the expression of BCR-ABL1, a well-known fusion gene involved in the progression of chronic myelogenous leukemia (CML) [19]. Importantly, a possible mechanistic link between histone methylation-mediated silencing of the miR-203 and upregulation BCR-ABL1 expression was later established by Yanli Jin et al. This group reported a mutual positive feedback loop between PRMT5 and BCR-ABL1 in which PRMT5 depletion led to decreased levels of H3R8me2s and H4R3me2s within the miR-203 promoter region, thus in turn, increased miR-203 expression, leading to reduced BCR-ABL1 gene transcription [20]. This study reveals yet another complex node in the multi-layered regulation of miRNAs by PRMTs via concerted de-repression and thus upregulation of critical oncogenes such as BCR-ABL1.
Table 1.
List of known interplay between PRMTs and miRNAs in cancer.
| PRMTs | miRNAs | Cancers involved | Cell line discovered | References |
|---|---|---|---|---|
| PRMT1 | miR-503 | Hepatoma | HepG2 cells | [36] |
| PRMT4 | miR-223 | Acute myeloid leukemia (AML) | Patient blood samples | [21] |
| miR-195-5p | Colorectal cancer (CRC) | HCT-15, SW480 cells | [37] | |
| miR-195-5p | CRC | HCT-116, HT-29 cells | [38] | |
| PRMT5 | miR-92b, 96 | Mantle cell lymphoma (MCL) | Human tonsil transformed lymphoid cells | [30] |
| miR-19a, 25, 32, 92, 92b, 96 | Transformed B-cell chronic lymphocytic leukemia (B-CLL) | WaC3CD5, Mec1, Mec2 cells | [31] | |
| miR-29b | AML | MV4-11, THP-1 cells | [18] | |
| miR-203 | Chronic myelogenous leukemia (CML) | KBM5, KBM5-T315I, K562 cells | [20] | |
| miR-4518 | Glioma | U251, H4, SW1783, LN229 cells | [32] | |
| miR-99a-5p, 99b-5p, 100-5p | Lung cancer | A549, H1299, H345, H446, H520, H460, H358 cells | [27] | |
| miR96 | Epstein-Barr virus (EBV) -driven lymphoma | Lymphoma-derived lymphoblastoid cells | [29] | |
| PRMT9 | miR-21 | Melanoma, prostate cancer, glioblastoma | MT330, SJ-G2, B16, DU145 cells | [22] |
| miR-621 | Breast cancer | MCF-7, MDA-MB-231, ZR-75–1 cells | [34] | |
| miR-543 | Osteosarcoma (OS) | Saos2, MNNG/HOS, U2OS, MG63 cells | [33] | |
| miR-26a-5p | Hepatoma | HepG2, MHCC97H, HCCLM3 cells | [35] |
Another pertinent example is the recent identification of PRMT5 as an important regulator of the miR-99 family/fibroblast growth factor receptor 3 (FGFR3) signaling axis in lung cancer. Specifically, these findings demonstrated that enrichment of H4R3me2s at the promoter of miR-99b led to its repressed transcription which in turn increased FGFR3 expression and activation of the Erk1/2/Akt pathway. The net effect of this regulation was the increase of cell growth and metastasis of lung cancer cells. Conversely, loss of PRMT5 inhibited lung cancer progression via inhibiting methylation-mediated silencing of the miR-99 family of ncRNAs [27].
Taken together, these findings point to the detrimental consequences of disrupting miRNAs’ tumor-suppressive functions through arginine methylation-mediated histones silencing associated with miRNAs’ promoters.
Regulation of miRNA protein machinery by arginine methylation
As outlined in Figure 2, the miRNA biogenesis pathway is a multistep, finely-tuned process mediated by various complexed proteins at each step. Importantly, alteration of overall levels or activity of these proteins has been linked to the development of a variety of pathologies, including cancer due to the resultant changes in global miRNA levels. For example, increased or decreased expression of Drosha and Dicer has been shown to inversely correlate with advanced tumor stages and poor patient outcome. However, a recent study highlights yet another important mechanism of regulation of miRNA biogenesis machinery via arginine methylation. Bonaldi et. al demonstrated that 70% of Large Drosha Complex (LDC) proteins were extensively methylated on approximately 82 distinct sites, of which 61 occurred on arginine sites [28]. Moreover, the study revealed that pharmacological blockade of PRMT1 activity resulted in widespread alteration of the arginine methylation state of the complex including proteins such as interleukin enhancer binding factor 3 (ILF3), thus impairing the pri-to-pre-miRNA processing step. This in turn led to a global decrease of miRNA expression [28]. Overall, this study suggests an important role for arginine methylation in modulating the interaction of LDC components with pri-miRNAs, thus uncovering another aspect of the complex, multi-layered regulation of miRNA expression by PRMTs at the level of biogenesis. This study suggests the potential for a novel therapeutic avenue for cancer treatment via modulation of aberrant miRNA expression using PRMTs inhibitors. Further investigation into the existence of arginine methylation of other miRNA biogenesis machinery components frequently dysregulated in cancer such as Dicer, is warranted.
PRMTs cooperate with other epigenetic regulators at the promoters of miRNAs
The cooperation between various epigenetic enzymes such as histone methyltransferases and deacetylases in the establishment of specific gene expression signatures is frequently observed (Figure 3). Interestingly, it has also been demonstrated that genes encoding for miRNAs undergo a similar regulatory process in the context of cancer. For example, in a model of Epstein-Barr virus (EBV)–induced B-cell lymphomagenesis, Alinari et al. demonstrated that the expression of PRMT5 was associated with a malignant phenotype in which PRMT5 inhibited miR-96 transcription by forming a PRMT5/p65/histone deacetylase 3(HDAC3)-repressive complex at the miR-96 promoter, therefore inhibiting miR-96 expression. Logically, when a PRMT5 specific inhibitor was applied, it blocked the recruitment of this repressive complex to miR-96 promoter, and thus restored miR-96 expression [29]. Intriguingly, in a reciprocal manner, another study previously demonstrated that overexpressed miR-96 and miR-92b could repress PRMT5 levels whereas downregulation of these miRNAs enhanced PRMT5 mRNA in various B cell lymphoma types [30]. These studies arguably suggest a putative negative feedback loop in which PRMT5 promotes its own expression through direct blockade of specific miRNA programs. Collectively, these findings depict an important yet frequently observed mutual interplay in which miRNAs and PRMTs coordinately regulate each other to maintain cancerous phenotypes.
2. Targeting of 3ʹ-UTRs of PRMTs for degradation or via blocking translation initiation
Over the past decade, advanced technologies such as high-throughput deep sequencing and computational analyses, have facilitated the discovery of thousands of genes as potential miRNA targets, among which, various epigenetic enzymes constitute an important class of targets. Unfortunately, the discovery of PRMTs as bona fide miRNA targets still lags given the relative paucity of reports on PRMTs whose expression are in turn regulated by miRNAs. Nonetheless, we delineate some of the contextual published reports on the regulation of PRMTs by miRNAs, with specific emphasis on where and how these histone methyltransferases form networks with miRNAs in various cancer types as well as cardiovascular, neurological, and inflammatory conditions. We will also highlight the importance of the reciprocal regulation between PRMTs and miRNAs.
Cancer
At the molecular level, evidence has been provided for a role of miRNA-mediated targeting of PRMTs in cancer. For instance, Sif and colleagues reported an interplay between PRMT5 and miR-92 or miR-96 in various lymphomas (eg. mantle cell lymphoma (MCL)), in which overexpression of miR-92b or miR-96 inhibited the translation of PRMT5 by binding to the 3ʹ-UTR of its mRNA in vivo and in vitro [30]. Furthermore, in B-CLL cell lines, electroportation of PRMT5-specific miRNAs (miR-19a, miR-25, miR-32, miR-92b, and miR-96) reduced the protein expression of PRMT5, further inhibiting cancer cell proliferation [31]. In terms of the regulation of PRMT5 in solid tumors, miR-4518 was found to target the 3ʹ-UTR region of PRMT5 and negatively regulate its expression in normal cells, whereas in their cancer counterparts, the lncRNA SNHG16 was found to function as an oncogene by sponging miR-4518, thus leading to the upregulation of PRMT5 expression in glioma cells [32].
Another arginine methyltransferase, namely PRMT9, has also garnered much attention in terms of the functional networks it forms with several miRNAs as well as other transcription factors. For example, Zhang et al showed that the levels of miR-543 were significantly increased and inversely correlated with PRMT9 levels in ostoecarcoma (OS) cells. Moreover, mechanistically, miR-543 was shown to target the 3ʹ-UTR of PRMT9 mRNA to inhibit its translation, thus hampering PRMT9-enhanced cell oxidative phosphorylation to promote OS cell proliferation. On the other hand, miR-543 depletion promoted PRMT9-mediated hypoxia-inducible factor −1α (HIF-1α) instability, which in turn inhibited glycolysis. Overall, these findings represent a potential new therapeutic strategy to impede OS cell proliferation via targeting the miR-543/PRMT9/HIF-1α axis [33]. Meanwhile, another study revealed a novel miR-621/PRMT9/p53 axis in which overexpression of miR-621, a miRNA that also targets PRMT9 3ʹ-UTR, enhanced p53 activity, leading to increased apoptosis of breast cancer cells exposed to paclitaxel plus carboplatin chemotherapeutics (Table 1) [34]. Furthermore, Ma et al. showed that not only did PRMT9 overexpression play a positive role in disease progression in hepatocellular carcinoma (HCC) patients, but also miR-26a could directly target the 3ʹ-UTR region of PRMT9 to suppress its expression in HCC cells [35].
Other PRMTs such as PRMT1 and 4 have also been reported as being regulated by miRNAs. For instance, Li et al. suggested that miR-503 and PRMT1 had antagonistic effect with each other in clinical HCC samples. Particularly, overexpression of miR-503 could significantly inhibit the invasion and migration of HCC cells as well as repress epithelial-mesenchymal transition (EMT) by targeting the 3ʹ-UTR region of PRMT1 [36]. Similarly, another study reported that ectopic expression of miRNA-195-5p decreased the expression of endogenous PRMT4 protein in colorectal cancer (CRC) cells, resulting in a sharp reduction of their proliferative and colony-formative capabilities [37]. Additionally, Zheng et al reported that ectopic expression of miR-195 enhanced the radiosensitivity and reduced cell colonies number by suppressing PRMT4 in HCT-116 and HT-29 CRC cells when compared with normal control cells that were treated with radiotherapy [38]. In summary, these findings suggest a highly complicated network of reciprocal interconnections between miRNAs and PRMTs.
Cardiovascular diseases
Recently, there is increasing evidence that miRNAs may have clinical utility as diagnostic biomarkers for several cardiovascular disorders [39]. Furthermore, several reports point to a role for miRNAs in the regulation of PRMTs in this context. One primary example is a regulatory loop formed by miR-15a and PRMT4. In this study, Liu et al. reported that in the pathogenesis of acute coronary syndrome (ACS), both mRNA and protein levels of PRMT4 were found to be elevated in the peripheral blood mononuclear cells (PBMCs) of ACS patients whereas miR-15a levels were coordinately decreased. Moreover, the expression of PRMT4 itself was further negatively regulated by miR-15a through its binding to the 3ʹ-UTR of PRMT4. Importantly, PRMT4 was shown to co-activate NF-κB-dependent chemokines such as interferon-inducible protein-10 (IP-10), monocyte chemoattractant protein 1 (MCP-1), and interleukin-8 (IL-8) (Table 2) [40]. Overall, this report represents another example of the multi-layered regulation of PRMTs by miRNAs that involves cooperation with transcription factors such as NF-κB and subsequent activation of certain pro-inflammatory gene signatures, albeit these findings require further validation in animal models and large-scale patient cohorts.
Table 2.
List of known interplay between PRMTs and miRNAs in other conditions.
| PRMTs | miRNAs | Conditions involved | Cell line discovered | References |
|---|---|---|---|---|
| PRMT1 | let-7 | Development in C. elegans | Worm | [23] |
| miR-19a | Asthma | Human airway smooth muscle cells | [43] | |
| PRMT3 | aae-miR-2940 | Replication of bacteria Wolbachia in mosquito | Mosquito Aag2 cells | [24] |
| PRMT4 | Myogenic microRNAs | Muscle development and differentiation | Mouse NIH 3T3 cells | [45] |
| miR-181 family | Human embryonic stem cells (hESC) differentiation | hESC X-01 cells (Lab made) | [25,44] | |
| miR92a | Neuronal development | hESC BG01V cells | [41] | |
| miR-15a | Acute coronary syndrome | Patient peripheral blood | [40] | |
| PRMT7 | miR-24-3p and miR-24–2-5p | Mouse embryonic stem cells (mESC) self-renewal and pluripotency | mESC V6.5 cells | [7] |
| miR-221 | mESC differentiation | mESC V6.5, R1 cells | [26] |
Neurological conditions
Altered expression of both miRNAs and PRMTs in neurodegenerative diseases suggests these modulators could have a potentially crucial regulatory role in these disorders. However, there are currently no known reports of the cooperativity between the two in this context. Nonetheless, the impact of these alterations along with other mechanisms on gene expression is likely to contribute to transcriptional dysregulation in the degenerative brain. Hence, an understanding of the interplay between miRNAs and PRMT5 in normal neuronal development processes is paramount to understanding how changes in this interconnection could influence pathogenesis. We will highlight a few examples related to PRMT4 in the context of neuronal lineages.
Selvi et al recently demonstrated that H3R17me2a mediated by PRMT4 is required for both the establishment and the maintenance of the astroglial population. Moreover, they further reported that absence of H3R17me2a downregulated miR-92a levels, which was previously known to participate in neural development. Interestingly, H3R17me2a enhanced the binding of the Nanog protein to miR-92 promoter, contributing to the maintenance of normal gene expression programs affecting the glial lineage. However, once PRMT4 was inhibited, Nanog binding to the miR-92 promoter was decreased, resulting in abnormal gene expression and altered glial lineage (Table 2)[41]. Although this study provides evidence regarding the interplay between PRMTs and miRNAs in various neuronal lineages, emerging insights into how imbalances in the levels of either could result in catastrophic degenerative outcomes, are necessary.
Inflammatory conditions
Inflammatory mechanisms are closely related to many diseases, including cancer, neurodegeneration, respiratory illnesses, among others. Importantly, miRNAs are key regulators of the immune response and affect many aspects of immune cells such as differentiation, proliferation, and release of inflammatory mediators [42].
For instance, during the pathogenesis of asthma, in which dysregulation of airway smooth muscle cells (ASMCs) plays a critical role, miR-19a was shown to be significantly down-regulated in ASMCs isolated from asthmatic patients. This study revealed that reduced miR-19a expression upregulated the expression of PRMT1 and several other important inflammatory signaling mediators such as extracellular signal-regulated kinases 1/2 (ERK1/2), mitogen-activated protein kinases (MAPKs), signal transducer and activator of transcription 1 (STAT1) to form a novel ERK1/2-MAPK-STAT1-PRMT1 axis. The net result was enhanced ASMC cell proliferation and migration, suggesting that PRMT1 could potentially be an attractive target to limit airway wall remodeling in ASMCs with constitutively increased levels of PRMT1 expression in asthmatic patients (Table 2) [43].
Collectively, the above study suggests that the functional cooperation between PRMT1 and miRNAs may potentially play a significant role in the activation and maintenance of pro-inflammatory signaling pathways. However, considering the inadequacy of reports in this field, more experimental evidence of the role that other PRMTs and miRNAs play in the pathogenesis of inflammatory diseases need to be forthcoming.
3. Other gene programs influenced by reciprocal interplay between PRMTs and miRNAs
In addition to the findings summarized above, several recent reports also implicate the interplay between PRMTs and miRNAs in other cell type specific gene programs. For example, several studies about the co-regulation between PRMTs and miRNAs and their functional importance in human and mouse embryonic stem cell (hESCs and mESCs, respectively) have been described. Briefly, PRMT4 was found to upregulate the expression of pluripotency genes and induce differentiation in hESCs X-01 cells, a process that could be mitigated by miR-181c-mediated repression of PRMT4 via 3ʹ-UTR targeting [44]. In mESCs however, PRMT7 was reported to be involved in the self-renewal and pluripotency gene programs in which a feedback loop between PRMT7 and miR-24–2 was identified [7]. Specifically, miR-24-3p and miR-24–2-5p, both derived from the same precursor, miR-24–2, were found to target the 3ʹ-UTRs of PRMT7. Conversely, PRMT7 epigenetically repressed the expression of miR-24-3p and miR-24–2-5p by symmetrically dimethylating H4R3, thus maintaining the “stemness” of mESCs [7].
PRMT-miRNA interactions have also been implicated in muscle development and differentiation which involves co-regulation of the expression of certain “myogenic miRNAs”. Both PRMT4 and 5 were found to be required for myogenic miRNA induction during muscle differentiation where PRMT4 was found to regulate myogenic miRNAs via H3R17me2a marks at upstream regulatory miRNA sequences, a process that requires the recruitment of myogenin and the switch/sucrose non-fermentable (SWI/SNF) complex. PRMT5 however, was found to play a more indirect role via regulation of myogenin expression [45]. In summary, these studies once again highlight the importance of the cooperation of PRMTs with other factors to induce the activation of cell-specific miRNA programs.
III. Therapeutic implications of the coordinated actions between PRMTs AND miRNAs
Although other aspects of the complex layers of the interplay between miRNAs and PRMTs and their coordinated control of gene expression have yet to be fully elucidated, it is clear that this interplay is an essential event in several pathological processes, especially cancer. As such, the cooperation between miRNAs and PRMTs represents a promising area for novel therapeutic interventions in these related diseases.
Currently, the existence of several epigenetic-based agents has been solely based on the knowledge of their regulation of conventional protein-coding genes. Furthermore, such therapies inhibit epigenetic modulators that work globally, resulting in unintended changes in gene expression that could potentially further aid cancer progression. In fact, very few inhibitors against PRMTs have made it to the clinical phase stage and none have been US Food and Drug Administration (FDA)-approved due to their off-target effects. In this respect, the advent of epigenomic editing to alter locus-specific chromatin structure is likely to become a critical approach for developing effective therapies and has the potential to be the next breakthrough in cancer treatment. Hence, with the knowledge of PRMTs that specifically target the promoter regions of miRNAs, one could theoretically devise clustered regularly interspaced short palindromic repeats (CRISPR)-dCas9-based editing agents that selectively alter these locus-specific PRMTs and in turn, the corresponding PRMT-catalyzed marks. The ideal result would be targeted restoration or repression of miRNAs with known tumor suppressive or oncogenic functions, respectively.
Additionally, although still in its infancy, efforts to utilize miRNA mimics and inhibitors as adjunctive therapeutic approaches via controlling expression of cancer-related genes, including PRMTs, represent another promising therapeutic approach. Moreover, given the important feedback circuitries between miRNAs and PRMTs, from a clinical standpoint, a correlative assessment of circulating miRNAs and methylated histones has the potential to be a useful diagnostic and prognostic tool. This could also be potentially applied to monitoring patient response as well as the development of drug resistance to standard treatment regimens.
IV. Conclusions
The evidence discussed in this review indicates a strong interplay between miRNAs and PRMTs in the context of cancer and other diseases. Not only are miRNAs epigenetically silenced by PRMT-catalyzed histone methylation, but many of these miRNAs are involved in regulatory circuits that lead to miRNA-mediated targeted post-transcriptional silencing of the very PRMTs that regulate them. Moreover, the discovery that these miRNA/PRMT circuits can also involve transcription factors or other effectors of the epigenetic machinery introduces new layers of complexity in the gene regulation pertinent to the maintenance of various disease states. Therefore, an important question under current investigation is whether similar indirect targeting of PRMTs by miRNA via 3ʹ-UTR-mediated degradation of other PRMT co-regulators and/or transcriptional activators occur. Furthermore, are there any transcription factors cooperating with PRMTs to co-regulate the expression of miRNAs? Are there specific stimuli that act to disrupt this tightly controlled interplay? And if so, how do these disruptions exert a combinatorial effect in pathogenesis? This series of questions all necessitate more studies in the future. Moreover, considering the reversible nature of methylation, it will also be critical to further investigate, to what extent, protein arginine demethylation occurs in this context and whether there is a balance between methylation and demethylation in the regulation of miRNAs by PRMTs. A better understanding of these mechanisms and of the intertwined relationship between miRNAs, epigenetic regulators and other effectors of gene transcription is imperative for devising the most effective therapeutics.
In conclusion, we anticipate that more miRNA-based therapies will be developed in the near future for the treatment of cancer, cardiovascular, neurodegenerative, and inflammatory, diseases. Until then, continuous efforts should be made to address the above-mentioned questions which will help us to understand the functional cooperation between PRMTs and miRNAs at a much deeper level.
Funding Statement
This work was supported by the National Cancer Institute [1R03CA223906-01]; National Institute of General Medical Sciences [1R01GM120156-01A1]; 100 voices of hope (US) [2987613]; V Foundation for Cancer Research [4486242].
Acknowledgments
We thank Ms. Lisa King from Department of Pharmacology and Toxicology at Indiana University School of Medicine for her professional help with editing this review. This publication is made possible, in part, with support from the Indiana Clinical and Translational Sciences Institute (CTSI) funded from the National Institutes of Health (NIH), National Center for Advancing Translational Sciences (NCATS)’s Clinical and Translational Sciences Award (CTSA) (to TL), V foundation Kay Yow Cancer Fund (Grant 4486242 to TL), NIH-NIGMS Grant (#1R01GM120156-01A1 (to TL), and 100 VOH Grant (#2987613 to TL), as well as NIH-NCI Grant (#1R03CA223906-01).
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Blanc RS, Richard S.. Arginine methylation: the coming of age. Mol Cell. 2017;65(1):8–24. [DOI] [PubMed] [Google Scholar]
- [2].Wei H, Mundade R, Lange K, et al. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle. 2014;13(1):32–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Prabhu L, Wei H, Chen L, et al. Adapting AlphaLISA high throughput screen to discover a novel small-molecule inhibitor targeting protein arginine methyltransferase 5 in pancreatic and colorectal cancers. Oncotarget. 2017;8(25):39963–39977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wei H, Wang B, Miyagi M, et al. PRMT5 dimethylates R30 of the p65 subunit to activate NF-κB. Proc Natl Acad Sci U S A. 2013;110(33):13516–13521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bachand F. Protein arginine methyltransferases: from unicellular eukaryotes to humans. Eukaryot Cell. 2007;6(6):889–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cook JR, Lee J-H, Yang Z-H, et al. FBXO11/PRMT9, a new protein arginine methyltransferase, symmetrically dimethylates arginine residues. Biochem Biophys Res Commun. 2006;342(2):472–481. [DOI] [PubMed] [Google Scholar]
- [7].Lee S-H, Chen T-Y, Dhar SS, et al. A feedback loop comprising PRMT7 and miR-24-2 interplays with Oct4, Nanog, Klf4 and c-Myc to regulate stemness. Nucleic Acids Res. 2016;44(22):10603–10618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bedford MT, Clarke SG. Protein arginine methylation in mammals: who, what, and why. Mol Cell. 2009;33(1):1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Stouth DW, vanLieshout TL, Shen NY, et al. Regulation of skeletal muscle plasticity by protein arginine methyltransferases and their potential roles in neuromuscular disorders. Front Physiol. 2017;8:870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Baldwin RM, Morettin A, Cote J. Role of PRMTs in cancer: could minor isoforms be leaving a mark? World J Biol Chem. 2014;5(2):115–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013;13(1):37–50. [DOI] [PubMed] [Google Scholar]
- [12].Romano G, Veneziano D, Acunzo M, et al. Small non-coding RNA and cancer. Carcinogenesis. 2017;38(5):485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hayes J, Peruzzi PP, Lawler S. MicroRNAs in cancer: biomarkers, functions and therapy. Trends Mol Med. 2014;20(8):460–469. [DOI] [PubMed] [Google Scholar]
- [14].Tang J, Ahmad A, Sarkar FH. The role of microRNAs in breast cancer migration, invasion and metastasis. Int J Mol Sci. 2012;13(10):13414–13437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lynam-Lennon N, Maher SG, Reynolds JV. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009;84(1):55–71. [DOI] [PubMed] [Google Scholar]
- [16].Iorio MV, Piovan C, Croce CM. Interplay between microRNAs and the epigenetic machinery: an intricate network. Biochim Biophys Acta. 2010;1799(10–12):694–701. [DOI] [PubMed] [Google Scholar]
- [17].Datta J, Kutay H, Nasser MW, et al. Methylation mediated silencing of MicroRNA-1 gene and its role in hepatocellular carcinogenesis. Cancer Res. 2008;68(13):5049–5058. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [18].Tarighat SS, Santhanam R, Frankhouser D, et al. The dual epigenetic role of PRMT5 in acute myeloid leukemia: gene activation and repression via histone arginine methylation. Leukemia. 2016;30(4):789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bueno MJ, Pérez de Castro I, Gómez de Cedrón M, et al. Genetic and epigenetic silencing of microRNA-203 enhances ABL1 and BCR-ABL1 oncogene expression. Cancer Cell. 2008;13(6):496–506. [DOI] [PubMed] [Google Scholar]
- [20].Jin Y, Zhou J, Xu F, et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J Clin Invest. 2016;126(10):3961–3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Vu LP, Perna F, Wang L, et al. PRMT4 blocks myeloid differentiation by assembling a methyl-RUNX1-dependent repressor complex. Cell Rep. 2013;5(6):1625–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Yang CH, Pfeffer SR, Sims M, et al. The oncogenic microRNA-21 inhibits the tumor suppressive activity of FBXO11 to promote tumorigenesis. J Biol Chem. 2015;290(10):6037–6046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Hunter SE, Finnegan EF, Zisoulis DG, et al. Functional genomic analysis of the let-7 regulatory network in Caenorhabditis elegans. PLoS Genet. 2013;9(3):e1003353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhang G, Hussain M, Asgari S. Regulation of arginine methyltransferase 3 by a Wolbachia-induced microRNA in Aedes aegypti and its effect on Wolbachia and dengue virus replication. Insect Biochem Mol Biol. 2014;53:81–88. [DOI] [PubMed] [Google Scholar]
- [25].Wu Z, Li H, Rao L, et al. Derivation and characterization of human embryonic stem cell lines from the Chinese population. J Genet Genomics. 2011;38(1):13–20. [DOI] [PubMed] [Google Scholar]
- [26].Chen T-Y, Lee S-H, Dhar SS, et al. Protein arginine methyltransferase 7-mediated microRNA-221 repression maintains Oct4, Nanog, and Sox2 levels in mouse embryonic stem cells. J Biol Chem. 2018;293(11):3925–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jing P, Zhao N, Ye M, et al. Protein arginine methyltransferase 5 promotes lung cancer metastasis via the epigenetic regulation of miR-99 family/FGFR3 signaling. Cancer Lett. 2018;427:38–48. [DOI] [PubMed] [Google Scholar]
- [28].Spadotto V, Giambruno R, Massignani E, et al. PRMT1-mediated methylation of the Large Drosha Complex regulates microRNA biogenesis. BioRxiv. [Preprint] November 9, 2018. [cited 2019 Feb 5]. doi: 10.1101/466813 [DOI] [Google Scholar]
- [29].Alinari L, Mahasenan KV, Yan F, et al. Selective inhibition of protein arginine methyltransferase 5 blocks initiation and maintenance of B-cell transformation. Blood. 2015;125(16):2530–2543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Pal S, Baiocchi RA, Byrd JC, et al. Low levels of miR-92b/96 induce PRMT5 translation and H3R8/H4R3 methylation in mantle cell lymphoma. Embo J. 2007;26(15):3558–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Wang L, Pal S, Sif S. Protein arginine methyltransferase 5 suppresses the transcription of the RB family of tumor suppressors in leukemia and lymphoma cells. Mol Cell Biol. 2008;28(20):6262–6277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lu Y-F, Cai X-L, Li -Z-Z, et al. LncRNA SNHG16 functions as an oncogene by sponging MiR-4518 and up-regulating PRMT5 expression in Glioma. Cell Physiol Biochem. 2018;45(5):1975–1985. [DOI] [PubMed] [Google Scholar]
- [33].Zhang H, Guo X, Feng X, et al. MiRNA-543 promotes osteosarcoma cell proliferation and glycolysis by partially suppressing PRMT9 and stabilizing HIF-1α protein. Oncotarget. 2017;8(2):2342–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Xue J, Chi Y, Chen Y, et al. MiRNA-621 sensitizes breast cancer to chemotherapy by suppressing FBXO11 and enhancing p53 activity. Oncogene. 2016;35(4):448–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Ma Y, Deng F, Li P, et al. The tumor suppressive miR-26a regulation of FBXO11 inhibits proliferation, migration and invasion of hepatocellular carcinoma cells. Biomed Pharmacother. 2018;101:648–655. [DOI] [PubMed] [Google Scholar]
- [36].Li B, Liu L, Li X, et al. miR-503 suppresses metastasis of hepatocellular carcinoma cell by targeting PRMT1. Biochem Biophys Res Commun. 2015;464(4):982–987. [DOI] [PubMed] [Google Scholar]
- [37].Zhang M, Wu W, Gao M, et al. Coactivator-associated arginine methyltransferase 1 promotes cell growth and is targeted by microRNA-195-5p in human colorectal cancer. Tumour Biol. 2017;39(3):1010428317694305. [DOI] [PubMed] [Google Scholar]
- [38].Zheng L, Chen J, Zhou Z, et al. miR-195 enhances the radiosensitivity of colorectal cancer cells by suppressing CARM1. Onco Targets Ther. 2017;10:1027–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhou -S-S, Jin J-P, Wang J-Q, et al. miRNAS in cardiovascular diseases: potential biomarkers, therapeutic targets and challenges. Acta Pharmacol Sin. 2018;39(7):1073–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Liu X, Wang L, Li H, et al. Coactivator-associated arginine methyltransferase 1 targeted by miR-15a regulates inflammation in acute coronary syndrome. Atherosclerosis. 2014;233(2):349–356. [DOI] [PubMed] [Google Scholar]
- [41].Selvi BR, Swaminathan A, Maheshwari U, et al. CARM1 regulates astroglial lineage through transcriptional regulation of Nanog and posttranscriptional regulation by miR92a. Mol Biol Cell. 2015;26(2):316–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Baulina N, Kulakova O,Favorova O. MicroRNAs: the role in autoimmune inflammation. Acta Naturae. 2016;8(1):21–33. [PMC free article] [PubMed] [Google Scholar]
- [43].Sun Q, Liu L, Wang H, et al. Constitutive high expression of protein arginine methyltransferase 1 in asthmatic airway smooth muscle cells is caused by reduced microRNA-19a expression and leads to enhanced remodeling. J Allergy Clin Immunol. 2017;140(2):510–524.e3. [DOI] [PubMed] [Google Scholar]
- [44].Xu Z, Jiang J, Xu C, et al. MicroRNA-181 regulates CARM1 and histone aginine methylation to promote differentiation of human embryonic stem cells. PLoS One. 2013;8(1):e53146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mallappa C, Hu Y-J, Shamulailatpam P, et al. The expression of myogenic microRNAs indirectly requires protein arginine methyltransferase (Prmt)5 but directly requires Prmt4. Nucleic Acids Res. 2011;39(4):1243–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]