

Abstract
Tridegin is a potent and specific 66mer peptide inhibitor of coagulation factor XIIIa with six cysteines involved in three disulfide bonds. Three of the fifteen possible 3-disulfide-bonded isomers have been identified, which share a bridge between cysteines 19 and 25. We synthesized the three possible 2-disulfide-bonded analogs using a targeted protecting-group strategy to investigate the impact of the C19-C25 bond on tridegin’s folding, stability, and function. The FXIIIa inhibitory activity of the analogs was retained, which was shown by in vitro fluorogenic activity and whole blood clotting assays. Molecular dynamics simulations of wild type tridegin and the analogs as well as molecular docking studies with FXIIIa were performed to elucidate the impact of the C19-C25 bond on conformational stability and binding mode. The strategy of selectively reducing disulfide bonds to facilitate large-scale synthesis, while retaining the functionality of disulfide-bonded peptides, has been demonstrated with our present study.
Keywords: Coagulation factor XIIIa, transglutaminase, peptide inhibitor, cysteine-rich, disulfide connectivity, molecular modeling, molecular dynamics
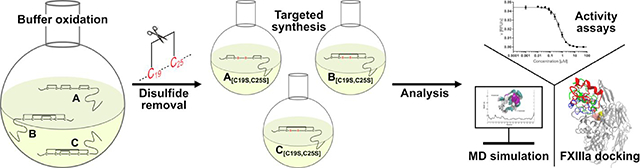
Graphical Abstract
INTRODUCTION
Cysteine-rich peptides and proteins are found in different organisms and possess a large variety of biological functions.1 Representatives, such as hirudin,2 conotoxins,3,4 snake5,6 and scorpion7 toxins produced by different vertebrates and invertebrates, harbor high therapeutic potential. Recent research focused on the blood coagulation factor XIIIa (FXIIIa) inhibitor tridegin, a 66mer 3-disulfide-bonded peptide produced by the giant amazon leech Haementeria ghilianii.8 The peptide represents a therapeutically promising, yet synthetically challenging compound. It was first isolated and described in 1997,8 yet its folding pathway remains to be explored.9,10 Tridegin inhibits FXIIIa with significant potency and high specifity.8 Several efforts have been undertaken to produce this inhibitor by either synthetic or recombinant strategies.9,10 These earlier reports also propose a major role for the three disulfide bonds located in the N-terminal region on the inhibitory activity of the peptide. In addition, tridegin seems to be composed of two functionally deviating segments: The flexible C-terminal fragment (R38-E66) was shown to be responsible for inhibitory activity, whereas the disulfide-linked N-terminal segment (K1-C37) confers conformational stability and contributes to the binding to FXIIIa.9,10 As a result of an oxidative self-folding approach, only three out of fifteen possible disulfide-bridged isomers were identified and their disulfide connectivities confirmed.10 Accordingly, tridegin neither folds in one major isomer like bovine pancreatic trypsin inhibitor (BPTI),11,12 or the conotoxins μ-SIIIA13 and κ-RIIIK3, nor tends to form several of the fifteen possible isomers, such as hirudin,11 and the conotoxins μ-PIIIA4,14 and μ-SmIIIA3. All three of the identified tridegin isomers, however, share the disulfide bond C19-C25, suggesting its conservation as well as its functional importance for the peptide. During synthesis it was also observed that partially-oxidized tridegin species formed a bridge involving C25 and C37 which seemed to trap the peptide in an irreversibly misfolded state.10 One could therefore suppose that the disulfide bond C19-C25 is important for correct oxidative folding in buffer. Structure elucidation by multi-dimensional NMR spectroscopy or X-ray crystallography was not successful so far due to the occurrence of aggregates and different isomers in the mixture.10 Consequently, the three isomer structures were modelled and subjected to docking studies with FXIIIa15 revealing different docking poses for the three isomers.
Targeted synthesis of cysteine-rich peptides with three disulfide bonds is a challenging process.14 Hence, and with respect to a possible future medical application, a simplification of cysteine-rich peptides is highly desirable.16–19 Since both truncation of tridegin and removal of all disulfide bonds resulted in a significant loss of activity,9,10 our present approach was to reduce the complexity of the peptide structure by reducing the number of disulfide bonds. Several studies have elucidated the role of individual disulfide bonds in different cysteine-rich peptides – with different outcome regarding the respective biological activity of the targeted peptide. For example, in the scorpion toxin leiurotoxin I,16 the μ-conotoxin KIIIA,17 the leech carboxypeptidase inhibitor18, and the α-conotoxin Vc1.119 one disulfide bond was removed without a significant loss of inhibitory potential. In contrast, Han et al. showed that in case of μ-GIIIA all three disulfide bridges were essential for bioactivity.20
The aim of the present study was thus to explore the influence of the common disulfide bridge C19-C25 on the structure and function of FXIIIa inhibitor tridegin. In a combined approach of experimental and computational studies, the three 2-disulfide-bonded tridegin isomers (bead, ribbon, and globular fold)21 were synthesized and evaluated for their FXIIIa inhibitory activity and their structural stability based on earlier established models of the 3-disulfide-bonded tridegin.10 Molecular dynamics simulations as well as docking studies with FXIIIa were used additionally to substantiate and also correlate with the in vitro data. Our study defines the role of the C19-C25 disulfide bond for the structure-activity relationships of tridegin, the results of which may also be applicable to other disulfide-rich peptides and proteins.
RESULTS AND DISCUSSION
Design and synthesis of peptides
According to the 3-disulfide-bonded tridegin isomers (A–C) identified in our previous report, three novel disulfide-deficient analogs A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S], respectively, were synthesized by a protecting group strategy (Figure 1, Table S1).10 Herein, the disulfide bond between C19-C25 was omitted by replacing cysteines with serines. In addition, another C19-C25-deficient analog of tridegin (peptide ABC[C19S, C25S]), i.e. an unknown mixture of 2-disulfide-bonded isomers, was synthesized in an oxidative self-folding approach performed in buffer as described earlier for wild type tridegin.9,10 Furthermore, the 3-disulfide-bonded tridegin (ABC)10 as well as a linear tridegin analog (All-Ser)10 with serine replacements for all six cysteines, which have been described in earlier studies,10 were used as controls in the fluorogenic enzyme activity assay. Apart from the All-Ser mutant other peptides still showed acceptable, yet reduced, inhibitory activity in our former study, e.g., four of the peptides derived from the C-terminal segment such as P39-P64.9 These peptides can be synthesized with a higher yield in comparison to the All-Ser mutant due to their shorter length. As the whole-blood assays required considerably more substance, herein P39-P64 was used as a control instead of the All-Ser mutant and the parent peptide ABC. The sequences and the respective disulfide bond patterns of the peptides are summarized in Figure 1A.
Figure 1.

A) Tridegin peptide sequence (cysteines are marked in blue) and disulfide connectivities of analyzed disulfide isomers and analogs. Cys-Ser exchanges are marked in red. Peptides ABC,9,10 All-Ser,10 and P39-P649 were described in previous studies. All peptides were produced as C-terminal amides. B) Synthesis strategy of sequential SPPS and oxidative folding in solution for the tridegin mutants A[C19S, C25S]-C[C19S, C25S]. lin., linear precursor; TFA, trifluoroacetic acid; eq., equivalents.
Synthesis of disulfide-containing peptides can be achieved by a variety of approaches. Depending on the peptide sequence and the number of cysteines, diverse methods, such as buffer oxidation or regioselective synthesis by different protecting-group strategies, can be applied.22 The targeted synthesis of the different 2-disulfide-bonded analogs was performed using a stepwise protecting-group strategy with acetamidomethyl (Acm)- and trityl (Trt)-protecting groups (Figure 1B, Table S1). Partially Cys-protected linear precursors (Figure S1, 1) were synthesized and subjected to stepwise oxidation in the following order according to their side chain protection: 1) oxidation of formerly Trt-protected Cys-pair, 2) deprotection and oxidation of the Acm-protected Cys-pair, and 3) RP HPLC purification. In the process, increasing amounts of iodine were used for selective formation of the first and second disulfide bond (Figure 1B).23 The oxidation reaction was stopped by addition of an excess of ascorbic acid14 or by iodine extraction and monitored via HPLC and MS. The reaction generating the first disulfide bond was carried out more efficiently when iodine extraction was applied (Figure S1). Termination of the reaction by adding ascorbic acid led to by-products (4) besides the desired peptides (3) (Figure S1). The occurrence of by-products could, however, be diminished upon extraction of the iodine from the aqueous reaction mixture in an organic solvent instead of adding ascorbic acid. The observed by-products were supposedly a result of side reactions, such as iodination or oxidation of sensitive residues, which are often associated with iodine-catalyzed oxidation.24–26 Particularly during the lyophilization step after oxidation, a concentration of residual iodine can occur, which may lead to the formation of undesired by-products.24 The efficient removal of excessive iodine by extraction was therefore crucial for the outcome of the synthesis. Thus, an increased yield of the extracted variants was achieved after purification. Analytical HPLC and MS data of the synthesized peptides are shown in Figure S2. The oxidation yield of each peptide ranged from 33 % for B[C19S, C25S], which was the most efficient synthesis, to 14 % for A[C19S, C25S] and 19 % for C[C19S, C25S] (Table S2). These results are comparable to what has been described earlier for other peptides,24,25 yet are remarkably higher compared to the synthesis of the 3-disulfide-bonded tridegin (< 10%).
Analysis of disulfide connectivities of tridegin isomers
In an earlier study we analyzed the elution behavior of different disulfide isomers of μ-conotoxin PIIIA,14 and realized that most of the isomers elute with the same retention time independent of the chromatography set-up used. In order to find out about the elution profile of mixtures of the tridegin isomers of the present study, equimolar amounts of analogs A[C19S, C25S]-C[C19S, C25S] in water were mixed and injected on a C18 column for RP HPLC analysis. The coelution profile (Figure S3) resulted in a single peak at a retention time of 19.3 min. Accordingly, the isomers in the mixture possess similar physicochemical properties, rendering it difficult to separate them by conventional RP HPLC methods. This confirms the hypothesis from our earlier study that these disulfide isomers cannot be separated by HPLC,10 and other methods (e.g. MS/MS, see below) have to be employed in order to confirm the identity of the peptides.
The verification of the correct disulfide-bridge formation of the three different tridegin analogs (A[C19S, C25S] - C[C19S, C25S]) was performed by tandem mass spectrometry analysis of a chymotryptic digest according to an earlier reported protocol.10 Chymotryptic proteolysis of the linear precursors as well as the fully-oxidized analogs and separation of the digested fragments via HPLC were carried out. The fragments were analyzed by LC-ESI-MS and -MS/MS before and after reduction by DTT (Figure 2). In case of the linear peptides, the correct positions of the acetamidomethyl protecting groups were confirmed by MS/MS analysis (Table S3, Figures S4, S5, S6). The evaluation of disulfide-linked fragments confirmed the disulfide connectivity of the different oxidized analogs as well. This is exemplarily shown for analog A[C19S, C25S] (Figure 2). Several linked fragments belonging to the disulfide bonds C5-C17 and C31-C37 were identified in the chymotryptic digest confirming the disulfide connectivity (Table S4).
Figure 2.

Analysis of A[C19S, C25S] during synthesis (A, B) and chymotryptic digest for mass spectrometry analysis (C, D). A) Reaction control during first and second oxidation analyzed by HPLC. B) MS and HPLC analysis of purified A[C19S, C25S]. C) The chymotryptic digest of A[C19S, C25S] was analyzed by HPLC and the collected fractions were subjected to MS analysis. Peptide fragments containing disulfide bonds were identified as exemplified here for the C5-C17 bond (C, D). D) The fractions containing disulfide bonds were reduced with DTT, and the identity of the reduced fragments was confirmed by subsequent tandem mass spectrometry (example: reduced fragment containing C5).
The oxidative self-folding approach peptide ABC[C19S, C25S] was also subjected to chymotryptic digest and subsequent MS and MS/MS analysis. All three possible isomers (A[C19S, C25S] - C[C19S, C25S]) were identified by specific linked fragments as depicted in Table S5.
Fluorogenic enzyme activity assay
In former studies using a chromogenic and a fluorogenic assay, we analyzed the role of the disulfide-stabilized N-terminal and the flexible C-terminal part as well as the general impact of disulfide bonds on the inhibitory potential of tridegin.9,10 A comparison of fully-oxidized tridegin (mixture of isomers A-C, ABC) with the reduced variant as well as with the Cys-mutated version All-Ser revealed a 3- to 4-fold decrease of inhibitory activity, which was explained by the structural changes induced by oxidative folding of the peptide.9,10 Here we investigated the special effect of the disulfide bond C19-C25 on the inhibitory effect of tridegin by testing the three disulfide-deficient isomers A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S] (Figure 1). The earlier reported 3-disulfide-bonded wild type ABC as well as the All-Ser mutant were used as controls.10 All tested inhibitors showed linear progress curves pointing to stable inhibition of FXIIIa (Figure S7). The tridegin analogs A[C19S, C25S], B[C19S, C25S], C[C19S, C25S], and ABC[C19S, C25S] displayed IC50 values of 0.55 ± 0.05 μM (n = 4), 0.50 ± 0.05 μM (n = 3), 0.48 ± 0.06 μM (n = 4), and 0.51 ± 0.02 μM (n = 2), respectively (Figure 3A–E). The control peptides ABC and All-Ser inhibited FXIIIa with IC50 values of 0.45 ± 0.03 μM (n = 3) and 0.72 ± 0.05 μM (n = 4) (Figure 3E). The difference between the control peptide ABC and the tridegin analog A[C19S, C25S] was minor, whereas B[C19S, C25S], C[C19S, C25S], and ABC[C19S, C25S] did not differ significantly from ABC (Figure 3A–E, Tables S6, S7). Our experimental data thus suggest almost full retention of the inhibitory activity upon removal of the disulfide bond C19-C25.
Figure 3.

Activity assays of different tridegin variants. A)–E) In vitro FXIII assay in a purified system: Exemplary IC50 plots of tridegin analogs A[C19S, C25S], B[C19S, C25S], C[C19S, C25S], ABC[C19S, C25S], and corresponding bar chart of averaged IC50 values in comparison to controls All-Ser and ABC. The dashed red line marks the IC50 value of wild type ABC. F)–I) Whole blood clot contraction assay of A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S] in comparison to control P39-P64.
Whole blood clotting assay
We also tested the inhibitory effect of A[C19S, C25S], B[C19S, C25S], C[C19S, C25S] and the previously described variant P39-P649 on the FXIIIa activity in a whole blood milieu. This assay is based on the discovery that FXIIIa promotes red blood cell retention in contracted whole blood clots,27,28 and therefore, is a major determinant of whole blood clot size. A[C19S, C25S], B[C19S, C25S], C[C19S, C25S], and P39-P64 each reduced red blood cell retention in clots and clot weight with IC50 values of 0.7 ± 0.4 μM, 1.1 ± 0.1 μM, 2.3 ± 2.2 μM, and 2.2 ± 2.0 μM, respectively (n = 3, Figure 3F–I, Table S7). Consistent with prior findings,8 the tridegin-derived peptides did not alter the kinetics of fibrin formation or final clot turbidity in plasma (Figure S8). Effects were similar to that seen with the FXIIIa active site covalent inhibitor T101 (IC50 0.5 ± 0.1 μM, data not shown), a smallmolecule imidazolium derivative,28 and within 1.5–4.5-fold of values obtained in the enzyme kinetic studies with FXIIIa. Given the lack of efficient and specific compounds for inhibiting FXIIIa, the novel peptides A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S] represent promising leads for this function.
Conformational analysis using molecular dynamics simulations
Interestingly, A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S] did not show significantly different inhibitory activities towards FXIIIa in both assays (Figure 3, Tables S6, S7). This is surprising at first glance, due to the theoretical impact of disulfide bonds on peptide conformation and thus structure-function properties. In particular, tridegin variant A[C19S, C25S] harbors the shortest distances between linked cysteines and therefore was expected to represent a considerably more flexible structure than variants B[C19S, C25S] and C[C19S, C25S], and thus to differ in its inhibitory potential. The contribution of the different disulfide bridges on the peptide’s inhibitory activity, however, seems to be of a more complex nature, which was analyzed by molecular dynamics and docking studies.
Molecular dynamics (MD) based conformational analysis was conducted on previously reported threaded models for tridegin isomers A-C10 owing to the non-availability of NMR or X-ray crystallographic structures for these peptides. Two independent rounds of MD simulations were conducted for each isomer and its variants. In order to improve the level of equilibration in simulation the final frame from the first 100 ns simulation was used as the starting structure for a second simulation for 300 ns. All results are discussed from this longer 300 ns simulation. The structural variabilities of the simulated isomers, variants with in silico removal of the C19-C25 bond, and their corresponding serine-mutated analogs were compared (Figure 4). Moreover, root mean square deviation (RMSD) as well as per-residue root mean square fluctuation (RMSF) profiles during the initial 100 ns (Figure S9) and an extended 300 ns MD simulations were calculated (Figures S10–S14).
Figure 4.

Comparison of structural variability of tridegin isomers in simulation. A) The structures of the three isomers A, B, and C shown in a (grey) cartoon representation. The disulfide bonds are marked and highlighted. B) Simulated structures of A (blue), B (red), and C (green) superimposed on their respective starting structures (transparent/blown glass representation). C) Simulated structures of A (blue), B (red), and C (green) with in silico removal of the C19-C25 disulfide bond superimposed on their respective starting structures (transparent/blown glass representation). D) Simulated structures of A[C19S, C25S] (blue), B[C19S, C25S] (red), and C[C19S, C25S] (green) superimposed on their respective starting structures (transparent/blown glass representation). Serine mutations of cysteines (C19S and C25S) are depicted in yellow. E) Simulated structures of A[C19S, C25S] (blue), B[C19S, C25S] (red), and C[C19S, C25S] (green) superimposed on their respective parent 3-disulfide bonded isomers (grey cartoons from row A)). All simulated structures presented here refer to the energy-minimized structures of the final snapshot from the 300 ns MD simulation.
Isomer A showed a moderate level of structural deviation from its starting conformation with a time-averaged RMSD value of 3.33 Å computed on the backbone of the whole molecule (Figures 4B, S10, Table S8). The replacement of cysteines C19 and C25 by serines brought this deviation down to 2.59 Å as observed for the novel analog A[C19S-C25S] (Figures 4D, S10, Table S8). The average structural deviation between isomer A and its novel analog A[C19S-C25S] differed by 0.74 Å global RMSD only.
The structure of isomer B displayed minor deviation from its initial conformation in simulation, i.e. average backbone RMSD of 2.70 Å (Figures 4B, S10, Table S8). The highest structural deviation between all three isomers was for the serine mutated novel analog B[C19S, C25S] (Figures 4D, S10, Table S8). The overall backbone RMSD was 10.74 Å of which 8.25 Å was attributed to deviations arising from the unstructured C-terminal region (residues 38–66) (Table S8). The disulfide-linked part of the peptide (residues 1 to 37) on the other hand, remained comparatively stable with a meagre 1.54 Å backbone RMSD (Table S8) in comparison to 1.77 Å in the parent isomer B. Hence, in the case of the analog B[C19S, C25S], though the overall molecule deviates significantly from its 3-disulfide-bonded version, the region constrained by the two disulfide bonds remained almost unaffected, i.e. only 0.23 Å difference (Table S8), on the constrained segment of residues 1–37.
The isomer C displayed the greatest amount of structural rigidity in simulation (Figure 4B). With a backbone RMSD of 1.71 Å, the entire molecule, including the C-terminal region, remained structurally rigid (Table S8, Figure S11G). This could be attributed to the constrained conformation rendered by the cross-linked disulfide bonds C5-C37 and C17-C31 (Figures 1, 4A). Similar to the behavior observed in isomer B, the isomer C[C19S, C25S] deviated significantly in backbone RMSD, i.e. 7.03 Å compared to its 3-disulfide-bonded parent isomer (Table S8). Here again 6.52 Å of the overall structural deviation arose from the conformations sampled by the unstructured C-terminal residues (38–66) (Table S8). The overall structural change between the isomer C and its serine-mutated analog C[C19S, C25S] within the disulfide-stabilized N-terminal region (residues 1–37) was still as low as 1.09 Å.
To gain deeper insight into the regions responsible for the conformational flexibility in isomers A–C, RMSF was related to conformation snapshots throughout the simulation (Figures S12, S14). From the RMSD-based conformational analysis of the three parent isomers and their disulfide-deficient analogs from the present study two clear inferences can be made. Firstly, the removal of the C19-C25 disulfide bonds in all three isomers does not significantly impact the structural integrity of the region bracketed by the two remaining disulfide bonds. Secondly, at the same time a marked impact on the structural conformation of the C-terminal region between residues 38–66 is observed. The distances sampled in simulation between the Sγ atoms of the unbound cysteine residues by in silico disulfide bond removal did not increase significantly (Figure S13). This further confirms that these partially unfolded states do not experience further structural disruption or unfolding. Videos of these simulations as well as additional discussions of the results of the MD studies are available in the supporting information.
Overall, it can be stated that neither the in silico opening of the disulfide bond C19-C25 nor the serine mutations of corresponding cysteine residues seem to promote unfolding or structural disruption of the peptides, which goes in line with the experimental data generated.
Docking of disulfide-deficient isomers to FXIIIa
Blind docking simulations were conducted wherein the resultant structures from the MD simulations of the fully-bridged isomers of wild type tridegin (A, B, and C),10 and the corresponding 2-disulfide-bonded isomers were docked to FXIIIa (PDB 4KTY).15 Blind docking protocols are usually used as a means to scan for possible binding sites and binding modes of peptide ligands over the surface of target proteins.29 Following ensemble docking protocol (see methods), the best docked poses of tridegin-FXIIIa complexes based on the predicted binding energies were chosen and analyzed. It is known from previous studies that the active site of FXIIIa is defined by the catalytic triad C314, H373, and D396.15 The most interesting observation from the docked conformations of the different analogs with in silico removal of the C19-C25 bond and their corresponding serine-mutated analogs (A[C19S, C25S], B[C19S, C25S], and C[C19S, C25S]) is that the best docked conformation among all docked variants binds to approximately the same region on FXIIIa as was observed earlier for wild type isomers A and B.10 For example, in all of the docked complexes the C314 residue of FXIIIa was found in close proximities to the docked tridegin isomer. This result is in agreement with previous studies using the starting structures of the simulations with respect to isomers A and B.10 Regarding isomer C, however, the present study reveals that, with the C19-C25 disulfide bond opened, conformations adopted by the isomer were able to bind close to the active site of FXIIIa. This docked conformation of isomer C deviates from the previously described docking studies wherein the isomer C bound far away from the active site of FXIIIa.10
The docking simulations of the tridegin analogs A[C19S, C25S], B[C19S, 25S], and C[C19S, 25S] showed that a vast majority of the conformations sampled by these analogs bound at close proximity to the active site (Figure 5). This key finding supports the experimental data suggesting comparable inhibitory activities of all three novel tridegin analogs (A[C19S, C25S], B[C19S, C25S], C[C19S, 25S]).
Figure 5.

Docking of isomers with blood coagulation factor XIIIa. FXIIIa (PDB 4KTY) is represented as a grey ribbon structure in all panels. Hydrogen bonds are depicted as dashed lines. Docking of the three tridegin isomers with the in silico opened C19-C25 bond on FXIIIa: A) isomer A (blue), B) isomer B (red), and C) isomer C (green). Interacting residues of the isomers (cyan) and FXIIIa (magenta) are labelled. D) Two 180° rotated orientations showing A[C19S, C25S] (blue ribbon), B[C19S, C25S] (red ribbon) and C[C19S, C25S] (green ribbon) docked in close proximity to active site C314 (pink surface), H373 (yellow surface) and D396 (orange surface). Zoom-in on analog A[C19S, C25S] (E, blue), B[C19S, C25S] (F, red), and C[C19S, C25S] (G, green) docked on FXIIIa with the interacting residues labelled as in D.
Based on the docking experiments and the experimental data, one can hypothesize that the disulfide-deficient tridegin analogs inhibit FXIIIa by blocking the active site, rather than by direct interactions with individual active site residues. The fact that multiple conformations of the three tridegin analogs in this study dock at very similar poses, i.e. close to the catalytic triad in a manner that blocks the active site, may serve as a strong indicator to this suggested mechanism of inhibition. The presented docking simulations provide visual evidence to the experimentally determined consensus that all three isomers synthesized and tested in this study lacking the C19-C25 disulfide bond possess comparable inhibitory activities.
CONCLUSION
In the present study we were able to elucidate the role of the disulfide bridge C19-C25 of the previously identified 3-disulfide-bonded tridegin isomers10. Targeted synthesis of the three different 2-disulfide-bonded tridegin analogs (peptides A[C19S-C25S], B[C19S-C25S], C[C19S-C25S]) was accomplished via a protecting-group strategy. Moreover, a mixture of these isomers (ABC[C19S-C25S]) was produced by an oxidative self-folding approach. The fluorogenic activity assay revealed a similar inhibitory potential of all analogs A[C19S-C25S], B[C19S-C25S], and C[C19S-C25S] towards FXIIIa and a comparable activity in relation to the formerly described fully-oxidized 3-disulfide-bonded isomer mixture (ABC). This points to a minor influence of the central disulfide bond on overall inhibitory activity. Furthermore, the disulfide-deficient analogs were tested in a whole-blood milieu and significantly reduced red blood cell retention in clots and clot weight. These findings hint at the peptides’ possible potential in anticoagulant therapy. Surprisingly, in both assay systems no significant differences in inhibitory potential were observed among the three different 2-disulfide-bonded variants A[C19S-C25S], B[C19S-C25S], and C[C19S-C25S]. This is in line with previous findings10 accrediting the N-terminal disulfide-bridged part of tridegin a stabilization and binding role whereas the flexible C-terminal part is mostly responsible for inhibitory action.
Molecular dynamics studies on models of the variants in comparison with unmodified tridegin support the experimental data suggesting only minor impairment of the N-terminal fragment overall structure. The simulations conducted with the in silico opened C19-C25 disulfide bond enabled the examination of the peptides’ folding behavior while the corresponding simulations with mutated cysteine residues C19 and C25 aided in illustrating the structural properties of the peptides synthesized in this study. The disulfide connectivity of the disulfide-deficient tridegin seems to be of little importance regarding its globular structure and hence its activity. Molecular docking of selected simulation screenshots suggests only slightly different binding modes of the different disulfide-deficient variants towards FXIIIa, which – again – matches the experimental data.
In summary, we thoroughly characterized three different 2-disulfide-bonded tridegin analogs without significant loss of inhibitory potential while facilitating large-scale synthesis. The presented tridegin variants may serve as valuable tools for future research on FXIIIa and as lead structures for inhibitor development. Moreover, the reduction of the number of disulfide bonds might also be applicable to other cysteine-rich molecules rendering them more suitable for drug development.
EXPERIMENTAL SECTION
Materials
Fmoc-amino acids, HBTU, and resins for solid phase peptide synthesis were purchased from Orpegen Peptide Chemicals (Heidelberg, Germany), and IRIS Biotech (Marktredwitz, Germany), respectively. Other chemicals used for peptide synthesis including reagent-grade N-methylmorpholine, piperidine, trifluoroacetic acid (TFA), and N,N-dimethylformamide (DMF), were obtained from Sigma-Aldrich Chemie GmbH (Munich, Germany), Alfa Aesar (Karlsruhe, Germany), abcr GmbH (Karlsruhe, Germany), and VWR International GmbH (Darmstadt, Germany). Solvents (analytical grade: acetonitrile, water, methanol, and reagent grade: ethyl acetate, diethyl ether) and chemicals (iodine, acetic acid) used for purification and oxidation were purchased from VWR International GmbH and Fisher Scientific GmbH (Schwerte, Germany). Fibrogrammin P was kindly supplied by CSL Behring GmbH (Marburg, Germany). Lipidated tissue factor (Innovin) was from Siemens (Newark, DE, USA). Phospholipids were provided by Synapse Research Institute (Maastricht, Netherlands).
Peptide synthesis and purification
All peptides were synthesized according to a standard Fmoc solid-phase peptide synthesis protocol by automated peptide synthesis using a ResPep SL peptide synthesizer from Intavis Bioanalytical Instruments GmbH (Cologne, Germany) as described previously.9 The linear peptides were assembled using Rink-Amide MBHA resin with a loading capacity of 0.53 mmol/g. The following side-chain protected amino acids were employed: Arg(Pbf), Asn(Trt), Asp(OtBu), Cys(Acm), Cys(Trt), Gln(Trt), Glu(OtBu), His(Trt), Lys(Boc), Ser(tBu), Trp(Boc), Tyr(tBu). Peptide cleavage from the resin and side-chain deprotection was accomplished as described earlier.9 The purification of the crude peptides was done by semi-preparative RP-HPLC using a JASCO PV-987 instrument (Gross-Umstadt, Germany) equipped with a Knauer Eurospher column (C18, 250 × 32 mm, 5 μm particle size, 100 Å pore size) and using a gradient elution system employing 0.1% TFA in water (eluent A) and 0.1% TFA in acetonitrile/water (90:10) (eluent B). The peptides were eluted with an increase of eluent B from 10–60% over 120 min at a flow rate of 10 mL/min. The detection was at λ=220 nm. The collected fractions were combined, freeze-dried, and stored at −20°C. The purity of the linear precursors was >95 % as assessed by analytical RPHPLC on a Shimadzu LC-20AD system equipped with a Vydac 218TP column (C18, 250 × 4.6 mm, 5 μm particle size, 300 Å pore size). Gradient elution was carried out with a gradient of 2050% eluent B in 30 min with eluent A: 0.1% TFA in water and eluent B: 0.1% TFA in acetonitrile. The flow rate was 1 mL/min. The detection of the peptides was at λ=220 nm. The yield of linear peptides after purification was in the range of 5 to 10%.
Oxidation of linear precursor peptides
The selective oxidation of the 2-disulfide-bonded isomers proceeded in a one-pot reaction. The linear precursors (1 equiv.) were dissolved in 50% acetic acid/water to yield a final concentration of 0.05 mM. 1.1 equiv. iodine (0.1 M in methanol) was added to the peptide solution and the reaction was stirred at room temperature and under atmosphere (1st oxidation). After 15 min, further 13.9 equiv. iodine (0.1 M in methanol) were added and the reaction was stirred for further 30 min (2nd oxidation). The oxidation reaction was stopped by addition of an excess of ascorbic acid (1 M in water) or by removal of iodine by extraction, respectively. During the extraction procedure the aqueous reaction mixture was extracted 3 times with the same volume of ethyl acetate. The peptide-containing aqueous solutions were combined, freeze-dried and stored at −20 °C.
Oxidation of peptide ABC[C19S-C25S] was accomplished by self-folding of the linear precursor in a buffer solution as described earlier.9,10 The product was freeze-dried and stored at −20 °C. The peptide powder was dissolved in water and purified by semi-preparative RP-HPLC using a Shimadzu LC-8A system (Duisburg, Germany) equipped with a Vydac 218TP1022 column (C18, 250 × 22 mm, 5 μm particle size, 100 Å pore size) using gradient elution employing 0.1% TFA in water as eluent A and 0.1% TFA in acetonitrile/water (90:10) as eluent B. The peptides were eluted with a gradient of 10–60 % of eluent B over 120 min at a flow rate of 10 mL/min. The detection was at λ=220 nm. The collected fractions were combined, freeze-dried, and stored at −20 °C. The purity of the peptides was >95 % as assessed by analytical RP-HPLC on a Shimadzu LC-20AD system equipped with a Vydac 218TP column (C18, 250 × 4.6 mm, 5 μm particle size, 300 Å pore size). The elution was performed with a gradient of 20–50 % eluent B in 30 min. The flow rate was 1 mL/min with eluent A: 0.1% TFA in water and eluent B: 0.1% TFA in acetonitrile. The detection of the peptides was at λ=220 nm.
Peptide analytical characterization
Peptides were characterized by analytical HPLC (see above), mass spectrometry, amino acid analysis, and thin layer chromatography (TLC). The amino acid composition of the peptides was verified by amino acid analysis using a LC 3000 system from Eppendorf-Biotronik (Hamburg, Germany). Full peptide hydrolysis was in 6N HCl at 110 °C in sealed tubes for 24 h. Afterwards the samples were dried in a vacuum concentrator, redissolved, and the concentrations were determined by comparison with an amino acid standard. The results were in the expected range for peptide contents between 50 and 80 %.
For characterization of peptide molar masses matrix-assisted laser desorption/ionization (MALDI) and electrospray ionization (ESI) mass spectrometry were used. MALDI mass spectra were measured on an ultrafleXtreme, an autoflex III smartbeam, and an autoflex II instrument (Bruker Daltonics, Bremen, Germany), ESI mass spectra were produced on a micrOTOF-Q III device (Bruker Daltonics, Bremen, Germany) as earlier described.14
Determination of disulfide connectivity
Disulfide connectivity of the fully oxidized peptides was determined by MS and MS/MS analysis of a chymotryptic digest as earlier described.10
Enzyme activity assay
Enzyme activity was measured using a fluorogenic enzyme activity assay.30 The measurements were carried out with the substrate H-Tyr(3-NO2)-Glu(NH-(CH2)4-NH-Abz)-Val-Lys-Val-Ile-NH2 as described previously.10 IC50 plots were fitted in Origin Pro 8G (Origin Lab Corporation, Northampton, MA, USA) by nonlinear regression using the equation y = vmax / (1 + (x / x0)s) with y = response, vmax = maximum velocity, x = inhibitor concentration, x0 = relative IC50, and s = slope factor. Results were subsequently illustrated in GraphPad v 7.04 (Synergy Software, Reading, PA, USA). IC50 values are shown as the means ± standard deviation. Statistical analysis was performed using an unpaired t-test (Holm-Sidak method) by means of GraphPad v 7.04 (Synergy Software, Reading, PA, USA). P values less than 0.01 were considered statistically significant.
Whole blood and plasma preparation
Phlebotomy was performed on consenting healthy donors in accordance with the Declaration of Helsinki and the University of North Carolina Institutional Review Board. Blood was collected by venipuncture using a 21G butterfly needle (Becton, Dickenson and Company, Franklin Lakes, NJ) into 0.105 M sodium citrate, pH 5.5 (10% v/v, final concentration). Normal pooled plasma (NPP) was prepared from 4 healthy donors. For each donor, the first 5 mL were discarded. Platelet-poor plasma was prepared by sequential centrifugation (150×g for 25 minutes, then 20000×g for 20 minutes), pooled, aliquoted, flash frozen with liquid nitrogen, and stored at −80°C.
Whole blood clot contraction assay
Clotting was triggered in recalcified (10 mM, final concentration) whole blood via addition of tissue factor (Innovin diluted 1:12000, 1 pM, final concentration). Final reaction volumes were 200 μL (85% whole blood; 10% Tridegin variant, T101, or HBS; 2.5% Innovin; 2.5% calcium chloride). Clot contraction proceeded at 37 °C for 120 min in siliconized multi-well plates. Contracted clots were removed and weighed. Analysis of clot weight was performed in GraphPad v 7.02 (Synergy Software, Reading, PA, USA) using the equation y = Bottom + (Top – Bottom) / (1 + (x / IC50) with y = response and x = inhibitor concentration. IC50 values are shown as the means ± standard deviation. Statistical analysis was performed using an unpaired t-test (HolmSidak method) by means of GraphPad v 7.04 (Synergy Software, Reading, PA, USA). P values less than 0.01 were considered statistically significant.
Molecular dynamics (MD) simulations
Since no resolved structures were present for any of the three tridegin isomers, threaded optimized models from an earlier work10 of the same served as starting structures for the simulations. GROMACS 5.1.4 31–34 was used for all the MD simulations in this study. An individual peptide was placed in the center of a cubic box of size 2 × 2 × 2 nm. The TIP3P35 water model was used as the solvent to fill the box. No ions were added to balance charges, since all three isomers had a net neutral charge. Simulations were run using the AMBER99SB-ILDN36 force field. Structures with the opened disulfide bonds were created via the method described in earlier work.12 5000 steps of steepest descents were carried out as the energy minimization protocol. A thermal equilibration at 300 K using velocity rescaling Berendsen thermostat37 and a constant pressure equilibration using the Parrinello-Rahman barostatat38,39 at 1 atm were carried out for 10 ns each, prior to the production MD. During both the constant temperature (NVT) and constant pressure (NPT) ensemble simulations, position restraints on all bonds were applied using the LINCS40 algorithm. Two sets of MD simulations were conducted for each variant of the three tridegin isomers. From the first set of simulations conducted for 100 ns it was decided that the peptides required further unrestrained equilibration, based on the large RMSD deviations observed. Since the observations from these first 100 ns do not directly contribute to the discussions of results, they have been presented in the supplementary information (Figure S9). The final frame from the 100 ns simulation was independently simulated for an additional 300 ns for each isomer with all of the pre-processing steps such as energy minimization, temperature and pressure equilibration applied again. Thus a total of 3.6 μs of MD simulations were conducted during the course of this study. The results are discussed from the observations from the final 300 ns simulation. The production MD for all simulations in this study was done with a 2 fs time. Periodic boundary conditions were applied to the system. Long range electrostatics were accounted by the particle mesh Ewald method.41,42 10000 frames were written to the trajectory simulation per 100 ns of simulation time. The effect of periodic boundary conditions was adjusted by suppression of the center of mass movement from the trajectory prior to analysis. Visualizations of conformations for the analysis was performed using VMD.43
Molecular docking studies
Molecular docking studies of the tridegin analogs were conducted with the dock_runensemble.mcr macro embedded in YASARA44 suite (version 18.4.2). This macro utilizes the Vina45 algorithm to dock a ligand to a receptor ensemble with flexible side-chains. In this ensemble docking approach46 the tridegin analog was docked 400 times on a receptor ensemble created by 20 high scoring side chain orientations of FXIIIa (PDB 4KTY). The procedure was adopted from a previous work47 that employed the same protocol. The activated FXIII structure was optimized first by removing the small molecule peptide like inhibitor (ID: PRD_001125) and filling missing loops and residues on the FREAD loop modeling server (http://opig.stats.ox.ac.uk/webapps/fread/php/).48 The structure was energy minimized in the YASARA suite pre-docking using a combination of steepest descent and simulated annealing minimization runs.
Supplementary Material
ACKNOWLEDGMENT
Authors are grateful to Dr. M. Sylvester (Core Facility Mass Spectrometry, Institute of Biochemistry and Molecular Biology, University of Bonn) and Dr. M. Engeser (Department of Mass Spectrometry, Institute of Chemistry, University of Bonn) for access to the MALDI instruments. We thank Dr. P. Heimer for his support during peptide synthesis and analysis as well as Dr. Maria M. Aleman for analyzing T101 in the whole blood clot clotting assay.
Funding Sources
The Bruker micrOTOF-Q instrument (to D.I.) was funded by the University of Bonn, the Ministry of Innovation, Science and Research of North-Rhine Westphalia and the DFG. This study was financially supported by the German Foundation of Heart Research and by the University of Bonn (to D.I.) as well as the National Institutes of Health (R01HL126974, to A.S.W.).
ABBREVIATIONS
- CID
collision induced decay
- CVD
cardiovascular disease
- DMF
dimethylformamide
- ESI
electrospray ionization
- FXIII
blood coagulation factor XIII
- HPLC
high performance liquid chromatography
- MALDI
matrix-assisted laser desorption/ionization
- RMSD
root mean square deviation
- RMSF
root mean square fluctuation
- TLC
thin-layer chromatography
- NPP
normal pooled plasma
Footnotes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information.
The Supporting Information is available free of charge:
Details of peptide synthesis and analytics (Table S1–S5, Figures S1–S6); details of activity assays (Tables S6–S7, Figures S7–S8), molecular dynamics simulation data (Table S8, Figures S9–S14), additional analysis of molecular dynamics, experimental details turbidity assay. (PDF) 3 videos: dynamics of tridegin isomer A, B, and C with in silico removal of disulfide bond C19-C25. (AVI)
REFERENCES
- (1).Correnti CE; Gewe MM; Mehlin C; Bandaranayake AD; Johnsen WA; Rupert PB; Brusniak MY; Clarke M; Burke SE; De Van Der Schueren W; Pilat K; Turnbaugh SM; May D; Watson A; Chan MK; Bahl CD; Olson JM; Strong RK Screening, Large-Scale Production and Structure-Based Classification of Cystine-Dense Peptides. Nat. Struct. Mol. Biol 2018, 25, 270–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Grütter MG; Priestle JP; Rahuel J; Grossenbacher H; Bode W; Hofsteenge J; Stone SR Crystal Structure of the Thrombin-Hirudin Complex: A Novel Mode of Serine Protease Inhibition. EMBO J. 1990, 9, 2361–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Fuller E; Green BR; Catlin P; Buczek O; Nielsen JS; Olivera BM; Bulaj G Oxidative Folding of Conotoxins Sharing an Identical Disulfide Bridging Framework. FEBS J. 2005, 272, 1727–1738. [DOI] [PubMed] [Google Scholar]
- (4).Tietze AA; Tietze D; Ohlenschläger O; Leipold E; Ullrich F; Kühl T; Mischo A; Buntkowsky G; Görlach M; Heinemann SH; Imhof D Structurally Diverse μ-Conotoxin PIIIA Isomers Block Sodium Channel NaV1.4. Angew. Chemie Int. Ed 2012, 51, 4058–4061. [DOI] [PubMed] [Google Scholar]
- (5).Almeida JR; Mendes B; Lancellotti M; Marangoni S; Vale N; Passos Ó; Ramos MJ; Fernandes PA; Gomes P; Da Silva SL A Novel Synthetic Peptide Inspired on Lys49 Phospholipase A2 from Crotalus Oreganus Abyssus Snake Venom Active against Multidrug-Resistant Clinical Isolates. Eur. J. Med. Chem 2018, 149, 248–256. [DOI] [PubMed] [Google Scholar]
- (6).Moga MA; Dimienescu OG; Arvǎtescu CA; Ifteni P; Pleş L Anticancer Activity of Toxins from Bee and Snake Venom-an Overview on Ovarian Cancer. Molecules 2018, 23, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sarfo-Poku C; Eshun O; Lee KH Medical Application of Scorpion Venom to Breast Cancer: A Mini-Review. Toxicon 2016, 122, 109–112. [DOI] [PubMed] [Google Scholar]
- (8).Finney S; Seale L; Sawyer RT; Wallis RB Tridegin, a New Peptidic Inhibitor of Factor XIIIa, from the Blood-Sucking Leech Haementeria Ghilianii. Biochem. J 1997, 324, 797–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Böhm M; Kühl T; Hardes K; Coch R; Arkona C; Schlott B; Steinmetzer T; Imhof D Synthesis and Functional Characterization of Tridegin and Its Analogues: Inhibitors and Substrates of Factor XIIIa. ChemMedChem 2012, 7, 326–333. [DOI] [PubMed] [Google Scholar]
- (10).Böhm M; Bäuml CA; Hardes K; Steinmetzer T; Roeser D; Schaub Y; Than ME; Biswas A; Imhof D Novel Insights into Structure and Function of Factor XIIIa-Inhibitor Tridegin. J. Med. Chem 2014, 57, 10355–10365. [DOI] [PubMed] [Google Scholar]
- (11).Chang JY Diverse Pathways of Oxidative Folding of Disulfide Proteins: Underlying Causes and Folding Models. Biochemistry 2011, 50, 3414–3431. [DOI] [PubMed] [Google Scholar]
- (12).Paul George AA; Heimer P; Maaß A; Hamaekers J; Hofmann-Apitius M; Biswas A; Imhof D Insights into the Folding of Disulfide-Rich μ-Conotoxins. ACS Omega 2018, 3, 12330–12340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Steiner AM; Bulaj G Optimization of Oxidative Folding Methods for Cysteine-Rich Peptides: A Study of Conotoxins Containing Three Disulfide Bridges. J. Pept. Sci 2011, 17, 1–7. [DOI] [PubMed] [Google Scholar]
- (14).Heimer P; Tietze AA; Bäuml CA; Resemann A; Mayer FJ; Suckau D; Ohlenschläger O; Tietze D; Imhof D Conformational μ-Conotoxin PIIIA Isomers Revisited: Impact of Cysteine Pairing on Disulfide-Bond Assignment and Structure Elucidation. Anal. Chem 2018, 90, 3321–3327. [DOI] [PubMed] [Google Scholar]
- (15).Stieler M; Weber J; Hils M; Kolb P; Heine A; Büchold C; Pasternack R; Klebe G Structure of Active Coagulation Factor XIII Triggered by Calcium Binding: Basis for the Design of Next-Generation Anticoagulants. Angew. Bioinforma 2013, 52, 11930–11934. [DOI] [PubMed] [Google Scholar]
- (16).Zhu Q; Liang S; Martin L; Gasparini S; Ménez A; Vita C Role of Disulfide Bonds in Folding and Activity of Leiurotoxin I: Just Two Disulfides Suffice. Biochemistry 2002, 41, 11488–11494. [DOI] [PubMed] [Google Scholar]
- (17).Han TS; Zhang MM; Walewska A; Gruszczynski P; Robertson CR; Cheatham TE; Yoshikami D; Olivera BM; Bulaj G Structurally Minimized μ-Conotoxin Analogues as Sodium Channel Blockers: Implications for Designing Conopeptide-Based Therapeutics. ChemMedChem 2009, 4, 406–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Arolas JL; Castillo V; Bronsoms S; Aviles FX; Ventura S Designing Out Disulfide Bonds of Leech Carboxypeptidase Inhibitor: Implications for Its Folding, Stability and Function. J. Mol. Biol 2009, 392, 529–546. [DOI] [PubMed] [Google Scholar]
- (19).Yu R; Seymour VAL; Berecki G; Jia X; Akcan M; Adams DJ; Kaas Q; Craik DJ Less Is More: Design of a Highly Stable Disulfide-Deleted Mutant of Analgesic Cyclic α-Conotoxin Vc1.1. Sci. Rep 2015, 5, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Han P; Wang K; Dai X; Cao Y; Liu S; Jiang H; Fan C; Wu W; Chen J The Role of Individual Disulfide Bonds of μ-Conotoxin GIIIA in the Inhibition of Nav1.4. Mar. Drugs 2016, 14, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Wu X; Wu Y; Zhu F; Yang Q; Wu Q; Zhangsun D; Luo S Optimal Cleavage and Oxidative Folding of α-Conotoxin Txib as a Therapeutic Candidate Peptide. Mar. Drugs 2013, 11, 3537–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Akaji K; Kiso Y Synthesis of Cystine Peptides In Synthesis of Peptides and Peptidomimetics; Goodman M, Ed.; Georg Thieme Verlag: Stuttgart, Germany, 2004; pp 101–177. [Google Scholar]
- (23).Mochizuki M; Tsuda S; Tanimura K; Nishiuchi Y Regioselective Formation of Multiple Disulfide Bonds with the Aid of Postsynthetic S-Tritylation. Org. Lett 2015, 17, 2202–2205. [DOI] [PubMed] [Google Scholar]
- (24).Andreu D; Albericio F; Solé NA; Munson MC; Ferrer M; Barany G Formation of Disulfide Bonds in Synthetic Peptides and Proteins In Peptide Synthesis Protocols; Humana Press: New York City, USA, 1995; pp 91–169. [DOI] [PubMed] [Google Scholar]
- (25).Zhang S; Lin F; Hossain MA; Shabanpoor F; Tregear GW; Wade JD Simultaneous Post-Cysteine(S-Acm) Group Removal Quenching of Iodine and Isolation of Peptide by One Step Ether Precipitation. Int. J. Pept. Res. Ther 2008, 14, 301–305. [Google Scholar]
- (26).Chikwana E; Davis B; Morakinyo MK; Simoyi RH Oxyhalogen-Sulfur Chemistry - Kinetics and Mechanism of Oxidation of Methionine by Aqueous Iodine and Acidified Iodate. Can. J. Chem 2009, 87, 689–697. [Google Scholar]
- (27).Aleman MM; Byrnes JR; Wang J-G; Tran R; Lam WA; Di Paola J; Mackman N; Degen JL; Flick MJ; Wolberg AS Factor XIII Activity Mediates Red Blood Cell Retention in Venous Thrombi. J Clin Invest 2014, 124, 3590–3600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Byrnes JR; Duval C; Wang Y; Hansen CE; Ahn B; Mooberry MJ; Clark MA; Johnsen JM; Lord ST; Lam WA; Meijers JCM; Ni H; Ariëns RAS; Wolberg AS Factor XIIIa-Dependent Retention of Red Blood Cells in Clots Is Mediated by Fibrin α-Chain Crosslinking. Blood 2015, 126, 1940–1948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Hetényi C; Van Der Spoel D Blind Docking of Drug-Sized Compounds to Proteins with up to a Thousand Residues. FEBS Lett. 2006, 580, 1447–1450. [DOI] [PubMed] [Google Scholar]
- (30).Hardes K; Hammamy MZ; Steinmetzer T Synthesis and Characterization of Novel Fluorogenic Substrates of Coagulation Factor XIII-A. Anal. Biochem 2013, 442, 223–230. [DOI] [PubMed] [Google Scholar]
- (31).Abraham MJ; Murtola T; Schulz R; Páall S; Smith JC; Hess B; Lindah E GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar]
- (32).Van Der Spoel D; Lindahl E; Hess B; Groenhof G; Mark AE; Berendsen HJC GROMACS: Fast, Flexible, and Free. J. Comput. Chem 2005, 26, 1701–1718. [DOI] [PubMed] [Google Scholar]
- (33).Berendsen HJC; van der Spoel D; van Drunen R GROMACS: A Message-Passing Parallel Molecular Dynamics Implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- (34).Hess B; Kutzner C; Van Der Spoel D; Lindahl E GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput 2008, 4, 435–447. [DOI] [PubMed] [Google Scholar]
- (35).Jorgensen WL; Chandrasekhar J; Madura JD; Impey RW; Klein ML Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys 1983, 79, 926. [Google Scholar]
- (36).Lindorff-Larsen K; Piana S; Palmo K; Maragakis P; Klepeis JL; Dror RO; Shaw DE Improved Side-Chain Torsion Potentials for the Amber Ff99SB Protein Force Field. Proteins Struct. Funct. Bioinforma 2010, 78, 1950–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Bussi G; Donadio D; Parrinello M Canonical Sampling through Velocity-Rescaling. J. Chem. Phys 2007, 126, 014101. [DOI] [PubMed] [Google Scholar]
- (38).Parrinello M; Rahman A Polymorphic Transitions in Single Crystals: A New Molecular Dynamics Method. J. Appl. Phys 1981, 52, 7182–7190. [Google Scholar]
- (39).Nosé S; Klein ML Constant Pressure Molecular Dynamics for Molecular Systems. Mol Phys 1983, 50, 1055–1076. [Google Scholar]
- (40).Hess B; Bekker H; Berendsen HJC; Fraaije JGEM LINCS: A Linear Constraint Solver for Molecular Simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- (41).Essmann U; Perera L; Berkowitz ML; Darden T; Lee H; Pedersen LG A Smooth Particle Mesh Ewald Method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- (42).Darden T; York D; Pedersen L Particle Mesh Ewald: An N .log( N ) Method for Ewald Sums in Large Systems. J. Chem. Phys 1993, 98, 10089–10092. [Google Scholar]
- (43).Humphrey W; Dalke A; Schulten K VMD: Visual Molecular Dynamics. J. Mol. Graph 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (44).Krieger E; Vriend G YASARA View - Molecular Graphics for All Devices - from Smartphones to Workstations. Bioinformatics 2014, 30, 2981–2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Trott O; Olson AJ AutoDock Vina. J. Comput. Chem 2010, 31, 445–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Novoa EM; De Pouplana LR; Barril X; Orozco M Ensemble Docking from Homology Models. J. Chem. Theory Comput 2010, 6, 2547–2557. [DOI] [PubMed] [Google Scholar]
- (47).Peherstorfer S; Brewitz HH; Paul George AA; Wißbrock A; Adam JM; Schmitt L; Imhof D Insights into Mechanism and Functional Consequences of Heme Binding to Hemolysin-Activating Lysine Acyltransferase HlyC from Escherichia Coli. Biochim. Biophys. Acta - Gen. Subj 2018, 1862, 1964–1972. [DOI] [PubMed] [Google Scholar]
- (48).Choi Y; Deane CM FREAD Revisited: Accurate Loop Structure Prediction Using a Database Search Algorithm. Proteins Struct. Funct. Bioinforma 2010, 78, 1431–1440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.