

Abstract
Background:
Attention deficit-hyperactivity disorder (ADHD) is common in fetal alcohol spectrum disorders (FASD), but also in patients without prenatal alcohol exposure (PAE). Many patients diagnosed with idiopathic ADHD may actually have ADHD and covert PAE, a treatment-relevant distinction.
Methods:
We compared proton magnetic resonance spectroscopic imaging (MRSI; N=44) and diffusion tensor imaging (DTI; N=46) of the anterior corona radiata (ACR)-- a key fiber tract in models of ADHD-- at 1.5 T in children with ADHD with PAE (ADHD+PAE), children with ADHD without PAE (ADHD-PAE), children without ADHD with PAE (non-ADHD_PAE), and children with neither ADHD nor PAE (non-ADHD-PAE, i.e., typically developing controls). Levels of choline-compounds (Cho) were the main MRSI endpoint, given interest in dietary choline for FASD; the main DTI endpoint was fractional anisotropy (FA), as ACR FA may reflect ADHD-relevant executive control functions.
Results:
For ACR Cho, there was an ADHD-by-PAE interaction (p=0.038) whereby ACR Cho was 26.7% lower in ADHD+PAE than in ADHD-PAE children (p<0.0005) but there was no significant ACR Cho difference between non-ADHD+PAE and non-ADHD-PAE children. Voxelwise false-discovery rate (FDR)-corrected analysis of DTI revealed significantly (q≤0.0101–0.05) lower FA in ACR for subjects with PAE (ADHD+PAE or non-ADHD+PAE) than for subjects without PAE (ADHD-PAE or non-ADHD-PAE). There was no significant effect of ADHD on FA. Thus, in overlapping samples, effects of PAE on Cho and FA were observed in the same white-matter tract.
Conclusions:
These findings point to tract-focal, white-matter pathology possibly specific for ADHD+PAE subjects. Low Cho may derive from abnormal choline metabolism; low FA suggests suboptimal white-matter integrity in PAE. More advanced MRSI and DTI—and neurocognitive assessments— may better distinguish ADHD+PAE from ADHD-PAE, helping identify covert cases of FASD.
Keywords: fetal alcohol spectrum disorder, attention deficit hyperactivity disorder, magnetic resonance spectroscopy, diffusion tensor imaging, anterior corona radiata
1|. INTRODUCTION
Fetal alcohol spectrum disorders (FASD) affect up to 5% of children in the United States (May et al., 2018), representing a significant public health concern. Neurobehavioral deficits such as hyperactivity, impulsivity, distractibility, inattention, and impaired executive functions are among the disabling effects of prenatal alcohol exposure (PAE; Kable et al.,2016; Mattson and Riley, 1998; Rasmussen, 2005; Schonfeld et al., 2009; Streissguth & O’Malley, 2000). Accordingly, a diagnosis of ADHD is very common in children with PAE (Mattson et al., 2013l O’Connor & Paley, 2009). ADHD in the presence of PAE (ADHD+PAE) may represent a distinct subtype with earlier onset and different clinical presentation (Coles et al., 1997; Mattson et al., 2013). The differential diagnosis of ADHD+PAE versus ADHD without PAE (ADHD-PAE), however, is often challenging: the PAE key criterion of maternal drinking during pregnancy often cannot be established as many children with PAE are adopted or in foster care with the birth mother unavailable (Chernoff et al., 1994); and many other mothers are reluctant to admit to drinking while pregnant (CDC, 2004). Also, many children with PAE lack the characteristic (but not essential) facial stigmata of the condition (Mattson & Riley 2011; Mattson et al., 2013). Thus, occult PAE is well represented in clinical ADHD populations (Coles, 2001). Further, stimulants are frequently used for these children (O’Malley & Nanson, 2002), although the few published studies (Doig et al., 2008; Oesterheld et al., 1998; Snyder et al. 1997) suggest that stimulants are only partially effective in children with PAE, with some effect upon hyperactivity but not upon inattention – a prominent feature of ADHD+PAE (Peadon & Elliott, 2010). Clearly, there is a clinical need to understand better the pathophysiology underlying ADHD+PAE, to improve our ability to distinguish ADHD+PAE from ADHD-PAE children in order to avoid ineffective therapies, and to establish markers for development of improved therapies.
Alongside more refined neuropsychological testing, which indicates lower executive function in ADHD+PAE versus ADHD-PAE (Mattson et al., 2013), the field looks to neuroimaging for objective methods to distinguish the two conditions. Preclinical data (for review see Isayama et al., 2009), for example, indicate impaired brain white-matter development and integrity after PAE resulting, perhaps, from apoptosis in subcortical cholinergic nuclei. In vivo human investigations of PAE using magnetic resonance (MR)-based modalities potentially substantiate these preclinical findings. For example, in an investigation using proton magnetic resonance spectroscopy (MRS), Astley et al. (2009) demonstrated below-normal levels of choline-compounds (Cho) in children with FASD in white matter of the corona radiata, proximal to the anterior corona radiata (ACR) portion of the tract. As reported among Astley et al.’s secondary findings, Cho also decreased with increasing amount of PAE across subjects. Gonçalves et al. (2009) found a below-normal ratio of Cho to creatine+phosphocreatine (“Cho/Cr+PCr”) in an MRS acquisition volume (“voxel”) containing the internal capsule-- the ventral extension of the corona radiata—as well as the putamen and caudate. Thus, there are indications of a possible role of ACR Cho in FASD.
Human MRS findings of abnormal Cho are consistent with rodent models revealing teratogenic effects of PAE on developing central cholinergic systems (Lauder and Schambra, 1999; Schambra et al., 1990; Sulik & Johnston, 1982,1983; Sulik, 1984), including depletion of the acetylcholine (ACh)-synthesizing enzyme choline acetyl-transferase (ChAT), ACh being critical for memory, cognition, and mood functions (Drachman, 1977). Astley et al. (1995; 2009) discuss Cho metabolism in detail with respect to PAE. The MRS Cho signal is an amalgam of the signals for phosphocholine (PC), glycerophosphocholine (GPC), and (a small amount) of choline proper (Ross & Sachdev, 2004). PC and GPC are chief precursors to longer-chain cell membrane phospholipids such as phosphatidylcholine (PtdCho) and sphingomyelin. Importantly, once incorporated into a membrane, PC and GPC are no longer MRS-visible (Tunggal et al., 1990). Thus, the magnitude of the MRS Cho signal is proportional to the free concentrations of these compounds. The Cho peak is, thus, frequently interpreted as being dominated by lipid monomers being mobilized for membrane synthesis or being released upon membrane breakdown (Gujar et al., 2005). Choline, on the other hand, is a precursor both to PC and GPC and (in a rate-limiting fashion) to the neurotransmitter ACh (Freeman & Jenkins, 1976). Wurtman et al. (1985) hypothesized that neuronal membranes undergo “autocannibalism”, as needed, to supply choline for ACh production. Hence, elevated Cho may represent accelerated membrane breakdown for this or other purposes, while diminished Cho may reflect choline loss beyond the ability of membrane breakdown to compensate. Cho levels are higher in neuroglia than in neurons and, hence, in white than in gray matter. Myelin production requires abundant PC, hence, it is perhaps unsurprising that white matter in volumetric MRI studies is more impacted than gray matter by PAE (Riikonen et al., 1999; Sowell et al., 2001,2002 Meanwhile, Zeisel (2011) points out that ethanol may increase metabolic demand for choline and Astley et al. (2009) explain that ACh released from growing axons regulates growth, differentiation, and plasticity of developing CNS neurons. PAE-induced reduction in choline may, therefore, via ACh, lead to complex alterations of brain development, including, perhaps, irregular white-matter tract formation. Addition of extracellular choline, on the other hand, protects membranes from drops in PtdCho and other phospholipids (Ulus et al., 1989). In the rat, neonatal and perinatal dietary supplementation with choline affords some apparent protection against the effects of ethanol exposure (Thomas et al., 2000,2004,2007,2009; Thomas & Tran, 2011; Wagner and Hunt, 2008; Balaraman et al., 2017). Recent results of a long-anticipated human randomized controlled trial of dietary choline for FASD affirm that the treatment is well tolerated and that it yields at least a limited cognitive benefit (Wozniak et al., 2013a; 2015). Thus, there are ample grounds for suspecting abnormal choline neurometabolism in PAE.
Studies using another MR modality, diffusion tensor imaging (DTI), have measured below-normal fractional anisotropy (FA)—a possible sign of diminished white-matter integrity-- in children with PAE in several brain tracts, including the corona radiata and specifically ACR (Fryer et al., 2009) and internal capsule (Sowell et al., 2008). DTI investigations in typically developing individuals have linked FA in the ACR to the mental function of executive control (Fan et al., 2002). Specifically, greater fiber integrity in the ACR is associated with greater executive control and superior conflict resolution (Ge et al., 2013; Niogi, 2008,2010; Yin et al., 2013).
The foregoing findings motivated us to target the ACR in a reanalysis of a neuroimaging dataset we had previously acquired for other purposes (partial results in Hamilton et al., 2008; Luders et al., 2009; Tafazoli et al., 2013) in a pediatric cohort containing subjects with ADHD+PAE, ADHD-PAE, and typically developing controls. As it happens, magnetic resonance spectroscopic imaging (MRSI) Cho and/or DTI FA values in bilateral ACR could be extracted for a majority of subjects from this dataset. We undertook this reanalysis because there are relatively few extant MRS studies of FASD (Astley et al., 1995,2009; Cortese et al., 2006; Fagerlund et al., 2006; O’Leary-Moore et al., 2008; du Plessis et al., 2014; Hess et al., 2014; Howells et al., 2016), and which typically do not account for ADHD. There are many MRS and DTI studies of ADHD (reviewed in Chen et al., 2016; Perlov et al., 2009; O’Neill et al., 2013b), but, these studies rarely account for PAE. We therefore pursued further post-processing and analysis of these data as described below to test the specific hypotheses that ACR Cho and FA are lower in subjects with than in subjects without PAE. We further hypothesized that these endpoints help distinguish ADHD+PAE from ADHD-PAE.
2|. MATERIALS AND METHODS
2.1|. Subjects: Overall Cohort
Data were reanalyzed from an earlier neuroimaging study of ADHD. Partial results were reported previously (Hamilton et al., 2008; Luders et al., 2009; Tafazoli et al., 2013). This was a study of 62 African-American and Hispanic children and adolescents from underserved communities in Southern California, including subjects with ADHD and typically developing controls without psychiatric afflictions. This cohort was chosen because—unlike most neuroimaging ADHD research—the assessment protocol explicitly included inquiry into possible prenatal exposure to ethanol, nicotine, and/or illegal drugs of abuse. Since neuroimaging studies of ADHD that even screen subjects for PAE are rare, we felt obligated and privileged to mine these prior data for possible insights into brain sequelae of PAE.
ADHD and control subjects (aged 7–17 years) were recruited from clinics, schools, and health organizations in the South Los Angeles area. After complete description of the study, written informed assent from the subject and consent from the subject’s parent or guardian were obtained. Experimental protocols were approved by the UCLA Institutional Review Board according to the US Federal Policy for the Protection of Human Subjects. Inclusion for ADHD subjects required meeting DSM-IV criteria for ADHD by parental interview using the NIMH Diagnostic Interview Schedule for Children, Version IV (NIMH DISC-IV; Schaffer et al., 2000). Further criteria included obtaining a score over 1.5 SD from the mean on the parent-rated and/or teacher-rated Inattentive and Hyperactive-Impulsive subscales of the SNAP-IV (Swanson, 1995). The ultimate diagnosis, however, was established by clinical interview and subsequent case consensus with the senior clinicians on the project that included a clinical psychologist, a neuropsychologist, and a psychiatrist. Subjects with the co-occurrence of a known genetic syndrome associated with ADHD including fragile X, tuberous sclerosis, generalized resistance to thyroid hormone, and those taking nonstimulant psychotropic medication were excluded. Medication was restricted to stimulant agents commonly used to treat ADHD (methylphenidates, amphetamines) and one subject was taking atomoxetine.
Control subjects were required to be free from any current or lifetime history of major Axis I mental disorder as assessed by DISC interview. Additional exclusions were presence of serious medical or neurologic illness (in particular closed head injury), first-degree relative with history of any disruptive behavior disorder (including ADHD), antisocial personality disorder, schizophrenia, or bipolar disorder. Also excluded from both ADHD and control groups were subjects with weight or height smaller than the fifth or larger than the 95th percentile. As growth restriction is a major symptom of PAE, this criterion unfortunately likely excluded many FASD subjects, biasing the present investigation towards more mild presentations of FASD.
Full-scale intelligence quotient (FSIQ) in ADHD and control subjects was assessed using the Block Design and Vocabulary subtests of the Wechsler Intelligence Scale for Children, 3rd edition (WISC-III) (Wechsler, 1991). A FSIQ cutoff of 60 was used in order to capture low IQ as a possible symptom of PAE. Subjects with FSIQ between 60 and 80 were evenly distributed between the major subject groups in the present reanalysis and these groups did not differ significantly in FSIQ (Tables 1–2).
TABLE 1.
MRSI Cho Sample Demographics by ADHD and Prenatal Alcohol Exposure Subgroup (N=44)
| Subgroup(N) | ||||||
|---|---|---|---|---|---|---|
| +ADHD | +ADHD | +ADHD | -ADHD | -ADHD | -ADHD | |
| +PAE(10) | -PAE(13) | ?PAE(4) | +PAE(7) | -PAE(8) | ?PAE(2) | |
| Sex | 1 F/9 M | 2 F/11 M | 2 F/2 M | 4 F/3 M | 6 F/2 M | 2 F/0 M |
| Age, years | 12.4±1.9 (7.9–14.2) |
11.9±2.6 (8.8–16.1) |
12.6±0.8 (11.4–13.2) |
12.4±2.4 (10.0–16.5) |
12.9±2.3 (10.3–16.9) |
12.0±0.3 (11.8–12.3) |
| FSIQ | 87.6±17.1 (65–123) |
92.7±13.2 (68–117) |
78.8±17.8 (62–97) |
93.0±18.7 (65–120) |
92.9±16.2 (62–109) |
106.0±0.0 (106–106) |
| Ethnicity | 7 AA/3 H | 9 AA/4 H | 3 AA/1 H | 4 AA/3 H | 5 AA/5 H | 1 AA/1 H |
| Handedness | 1 L/9 R | 1 L/12 R | 0 L/4 R | 0 L/7 R | 2 L/6 R | 1 L/1 R |
| ADHD subtype | 4 I/6 C | 6 I/6 C/1 NOS | 3 C/1 NOS | -- | -- | -- |
| Current medication, N | 1 MPH 1 Amp 1 Atmx |
1 MPH | 1 Amp | -- | -- | -- |
|
Past medication |
2 MPH 1 Amp |
1 MPH | -- | -- | -- | -- |
| Comorbidities | 7 ODD 1 CD 1 Disrupt 1 Tics |
7 ODD 2 SAD 1 SepAnx 1 Enuresis |
2 ODD 1 CD 1 Tric 1 Phob 1 Dysthm |
-- | -- | -- |
| Cumulative PAE, Drinks | 720±920 (13–2488) |
-- | -- | 316±482 (39–1382) |
-- | -- |
| Prenatal nicotine, N | 5 Y/4 N/1 ? | 0 Y/13 N | 1 Y/3 ? | 2 Y/5 N | 0 Y/8 N | 2 ? |
| Prenatal other drugs, N | 7 Y/3 N 5 Cocaine 3 Marijuana 2 Amp 1 Opioids |
0 Y/13 N | 4 ? | 1 Y/6 N 1 Cocaine |
0 Y/8 N | 2 ? |
+ADHD = attention deficit hyperactivity disorder (ADHD) diagnosis; -ADHD = no ADHD diagnosis; +PAE = prenatal alcohol exposure; -PAE = no PAE; ?PAE = unknown if any PAE; FSIQ = full-scale intelligence quotient (Wechsler 1991); AA = African-American; H = Hispanic; I = ADHD Inattentive subtype; C = ADHD Combined subtype; NOS = ADHD not otherwise specified; MPH = methylphenidate; Amp = amphetamines; Atmx = atomoxetine; ODD = oppositional defiant disorder; CD = conduct disorder; Disrupt = disruptive behavior disorder NOS; Tics = tic disorder; SAD = social anxiety disorder; SepAnx = separation anxiety disorder; Tric = trichotillomania; Phob = specific phobias; Dysthm = dysthymia. Values are numbers of subjects or subgroup-means±standard deviations (ranges)
TABLE 2.
DTI FA Sample Demographics by ADHD and Prenatal Alcohol Exposure Subgroup (N=46)
| Subgroup(N) | ||||||
|---|---|---|---|---|---|---|
| +ADHD | +ADHD | +ADHD | -ADHD | -ADHD | -ADHD | |
| +PAE(9) | -PAE(11) | ?PAE(4) | +PAE(7) | -PAE(9) | ?PAE(6) | |
| Sex | 1 F/8 M | 2 F/9 M | 3 F/1 M | 4 F/3 M | 7 F/2 M | 3 F/3 M |
| Age, years | 12.4±2.1 (7.9–14.2) |
11.7±2.4 (8.8–16.1) |
11.8±1.4 (10.0–13.2) |
12.4±2.4 (10.0–16.5) |
13.1±2.2 (10.3–16.9) |
11.4±2.5 (8.3–15.0) |
| FSIQ | 88.1±18.0 (65–123) |
92.7±14.1 (68–117) |
81.0±20.3 (62–100) |
93.0±18.7 (65–120) |
92.0±15.4 (62–109) |
95.5±9.0 (85–106) |
| Ethnicity | 6 AA/3 H | 7 AA/4 H | 3 AA/1 H | 4 AA/3 H | 6 AA/3 H | 4 AA/2 H |
| Handedness | 1 L/8 R | 1 L/10 R | 1 L/3 R | 0 L/7 R | 2 L/7 R | 1 L/4 R |
| ADHD subtype | 3 I/6 C | 5 I/5 C/1 NOS | 2 C/2 NOS | -- | -- | -- |
| Current medication, N | 1 MPH 1 Amp 1 Atmx |
-- | 1 Amp | -- | -- | -- |
|
Past medication |
1 MPH 1 Amp |
1 MPH | -- | -- | -- | -- |
| Comorbidities | 6 ODD 1 CD 1 Disrupt 1 Tics |
7 ODD 1 SAD 1 SepAnx 1 Enuresis 1 Phob 1 Dysthm |
2 ODD 1 CD 1 Tric 1 Phob 1 Dysthm |
-- | -- | -- |
| Cumulative PAE, Drinks | 794±985 (13–2488) |
-- | -- | 316±482 (39–1382) |
-- | -- |
| Prenatal nicotine, N | 5 Y/3 N/1 ? | 0 Y/11 N | 1 Y/3 ? | 2 Y/5 N | 0 Y/9 N | 6 ? |
| Prenatal other drugs, N | 7 Y/2 N 4 Cocaine 3 Marijuana 2 Amp 1 Opioids |
0 Y/11 N | 4 ? | 1 Y/6 N 1 Cocaine |
0 Y/9 N | 6 ? |
+ADHD = attention deficit hyperactivity disorder (ADHD) diagnosis; -ADHD = no ADHD diagnosis; +PAE = prenatal alcohol exposure; -PAE = no PAE; ?PAE = unknown if any PAE; FSIQ = full-scale intelligence quotient (Wechsler 1991); AA = African-American; H = Hispanic; I = ADHD Inattentive subtype; C = ADHD Combined subtype; NOS = ADHD not otherwise specified; MPH = methylphenidate; Amp = amphetamines; Atmx = atomoxetine; ODD = oppositional defiant disorder; CD = conduct disorder; Disrupt = disruptive behavior disorder NOS; Tics = tic disorder; SAD = social anxiety disorder; SepAnx = separation anxiety disorder; Tric = trichotillomania; Phob = specific phobias; Dysthm = dysthymia. Values are numbers of subjects or subgroup-means±standard deviations (ranges)
Within this overall cohort, those subjects were identified who both had had MRSI acquired and whose data passed quality control (see below), in particular subjects who had usable values of MRSI Cho in the ACR (“MRSI Cho sample”). Similarly, within the overall cohort, those subjects were identified who both had had DTI acquired and whose data passed quality control, in particular those who had usable values of DTI FA in the ACR (“DTI FA sample”).
2.2|. Subjects: MRSI Cho Sample
This sample comprised 44 subjects (Table 1). It included 17 girls and 27 boys, consistent with the higher prevalence of ADHD in males. Group-mean age was 12.3±2.1 years (range: 7.9–16.9 years). Mean FSIQ was 91.0±15.9 (62–123). A diagnosis of ADHD was found in 27 subjects and 17 were diagnosed not to have ADHD. Five subjects were on psychotropic medication for ADHD at time of study (Table 1). Multiple ADHD subjects had one or more psychiatric comorbidities (Table 1). Some degree of PAE was identified in 17 subjects, while PAE was denied in 21 subjects. For 6 subjects, PAE status was unknown. Frequency of drinking by mothers during pregnancy ranged from 1 drink/week to 9 drinks/day. Some subjects had prenatal exposure to nicotine and/or to illegal drugs of abuse (Table 1).
2.3|. Subjects: DTI FA Sample
This sample comprised 46 subjects (Table 2), including 20 girls and 26 boys. Mean age was 12.2±2.2 years (7.9–16.9 years). Mean FSIQ was 91.1±15.5 (62–123). A diagnosis of ADHD was made in 24 subjects and 22 were diagnosed not to have ADHD. Four subjects were on psychotropic medication for ADHD (Table 2). Again, multiple psychiatric comorbidities were scattered across the ADHD subsample (Table 2). Some degree of PAE was identified in 16 subjects, while PAE was denied for 20 subjects, and PAE was unknown for 6 subjects. Frequencies of drinking during pregnancy were similar to those of the MRSI Cho sample. Some subjects had prenatal exposure to nicotine and/or illegal drugs-of-abuse (Table 2).
2.4|. Subjects: Overlap Between MRSI and DTI FA Samples
There was ample subject overlap between the two samples. A total of 40 subjects were present in both. Classifying by ADHD clinical status, these included 23 subjects with and 17 subjects without ADHD; classifying by PAE, they included 16 subjects with PAE, 19 with PAE denied, and 5 with PAE unknown. There were 6 subjects in the DTI FA sample who were not in the MRSI Cho sample and 4 subjects in the MRSI Cho sample who were not in the DTI FA sample. Thus, overlap was appreciable between the total samples (80%) and the major clinical categories (75–95%).
2.5|. Magnetic Resonance Acquisition and Post-Processing
MRSI and DTI were acquired in the same scanning session in a 1.5-T Siemens Sonata at the UCLA Ahmanson-Lovelace Brain Mapping Center using a standard quadrature headcoil. Subjects on psychotropics withheld medication for at least 24 hours before MR scanning. Considering this, the fact that hyperactivity and inattention are cardinal symptoms of ADHD, and comorbid oppositional defiant disorder in some subjects, compliance with scanning procedures was much better than expected, greatly facilitated by allowing each child to watch a video of his or her choice during scanning and other techniques. Of the original 62-subject sample, MR data from 5 subjects were excluded due to undue head motion. To enable prescription of subsequent MRSI and DTI pulse-sequences and for offline tissue-segmentation into gray-matter, white-matter, and CSF subvolumes (Shattuck et al., 2001) two sagittal high-resolution (voxels 1.3×0.9 ×1.2 mm3) T1-weighted spoiled gradient-echo (SPGR) whole-brain volumes were acquired with repetition-time (TR) of 24 ms and echo-time (TE) of 12.6 ms (flip angle 22°). Separate acquisition of these two scans diminished the likelihood of subject movement during scanning; offline, the two scans were co-registered to each other and averaged to yield higher signal-to-noise ratio. SPGR volumes were corrected for intensity inhomogeneities (Shattuck et al., 2001) and manually skull-stripped for post-processing with MRSI and DTI data. SPGR scans were reviewed by a pediatric neuroradiologist and subjects with clinically relevant structural findings (e.g., congenital, traumatic, vascular, neoplastic, or infectious lesions of the brain or head visible on MRI) were liable for exclusion from the study. No subjects were excluded for that reason.
MRSI was acquired and post-processed as in our prior work (Bejjani et al., 2012; Joe et al., 2018; O’Neill et al., 2012,2013a). Water-suppressed MRSI was acquired from a 2D-slab using a point-resolved spectroscopy (PRESS) sequence (TR/TE=1500/30 ms, 4 excitations). The slab (Fig. 1) was an axial-oblique (parallel to the genu-splenium plane) 9-mm thick 16×16 planar array of 11×11 mm2 voxels, including a central 4×4 subarray (“PRESS excitation volume”) from which usable data were obtained, one MR spectrum per voxel. The excitation volume had its posterior face abutting the rostrum of the corpus callosum about which it was positioned symmetrically dorsoventrally. Mesiolaterally, the excitation volume straddled the longitudinal fissure symmetrically. As such, bilaterally, mesial and posterior portions of the slab sampled the pregenual anterior cingulate cortex (pACC), mesial and anterior portions sampled mesial superior frontal cortex, and lateral portions sampled the anterior corona radiata (ACR), the target region of the present reanalysis. An identical, non-water-suppressed MRSI sequence (1 excitation) was acquired in immediate succession.
FIGURE 1.
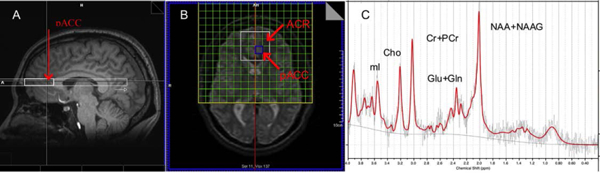
Positioning of 1H magnetic resonance spectroscopic imaging (MRSI) and sample spectrum. (A) sagittal T1-weighted MRI of the human brain showing cross-section of full MRSI slab (light white) and point-resolved spectroscopy (PRESS) excitation volume (heavy white) from which usable spectra are acquired. The posterior face of the PRESS volume abuts the genu corpus callosum about which the slab is placed symmetrically. Posterior portions of the volume sample bilateral pregenual cingulate cortex (pACC; red arrow). (B) axial-oblique (genu-splenium parallel) T2-weighted MRI displaying slab in-plane. The slab is a 9-mm thick 16×16 rectangular array (green mesh in yellow frame) of 11×11 mm2 acquisition volumes (“voxels”), each of which yields an MR spectrum. The excitation volume constitutes the central 4×4 subarray (heavy white). As mentioned, mesial and posterior portions of this subarray sample bilateral pACC; lateral portions sample bilateral anterior corona radiata (ACR), the target of the present study. (C) Sample LCModel PRESS spectrum from pACC showing major peaks for choline-compounds (Cho), as well as N-acetyl compounds (NAA+NAAG), glutamate+glutamine (Glu+Gln), creatine+phosphocreatine (Cr+PCr), and myo-inositol (mI). The jagged gray trace denotes the raw data, red the fit data, and smooth gray the spectral baseline.
Spectra were fit automatically offline using LCModel (Provencher, 2001). In addition to the target Cho, the fit included the metabolites N-acetyl-aspartate+N-acetyl-aspartyl-glutamate (NAA+NAAG), glutamate+glutamine (Glu+Gln), creatine+phosphocreatine (Cr+PCr), and myo-inositol (mI), as well as lipids and macromolecules. Our MRSI Voxel Picker (MVP) program (Seese et al., 2011) was used for co-processing of MRI and MRSI data. MVP reconstructed each subject’s whole-brain SPGR, gray-matter, white-matter, and CSF volumes into the space of the MRSI slab. MVP then returned volume percentages of gray matter, white matter, and CSF, as well as CSF-corrected metabolite levels, for each voxel in the slab. Separately for left and right pACC, values were averaged together for all voxels containing ≥65% gray matter, a signal-to-noise ratio ≥3, and a full width at half maximum (FWHM) ≤0.1 ppm. Also, only metabolite peaks satisfying the LCModel criterion of ≤20% standard deviation were included in the average. The same was done for left and right ACR, except the tissue-content criterion was ≥75% white matter. MVP implemented these quality-control criteria automatically. Additionally, with the help of the MVP guided user interface, all voxels passing these criteria were manually inspected by raters blind to diagnosis. Voxels that showed significant artifact (e.g., contaminating signals from extracranial lipids) were eliminated from analysis. Metabolite levels were normed to the unsuppressed water signal and expressed in Institutional Units (IU).
DTI was acquired and, in part, post-processed as in our prior work (Clark et al., 2012; Hamilton et al., 2008; Hudkins et al., 2012; Tobias et al., 2010; Ringman et al., 2007). The following post-processing was applied to the whole-brain DTI acquisition to prepare for subsequent analysis within the bilateral ACR subvolume. Whole-brain echo-planar DTI was performed in 6 noncollinear directions (b=1000 s/mm2) with one non-diffusion-weighted acquisition (b=0). The DTI pulse sequence used dual bipolar diffusion gradient pulses and a double spin-echo to suppress eddy currents. Fifty 3-mm slices (3×3 mm2 in-plane resolution) oriented parallel to the anterior-posterior commissure plane were acquired for each direction, and the entire pulse-sequence was repeated four times (1 average each). Offline 6-parameter rigid registration between these successive repetitions was used to correct for minor head movements. The diffusion tensor, fractional anisotropy (FA), and mean diffusivity (MD) were calculated within each voxel (Basser et al., 1994) using software developed at the UCLA Laboratory of NeuroImaging. FA volumes were skull-stripped using rigid body registered masks from the SPGR. The left and right ACR volume-of-interest (VOI) masks from the JHU-ICBM-DTI-81 white-matter labels atlas (Mori et al., 2008; http://fsl.fmrib.ox.ac.uk/fsl/fslwiki/Atlases) were registered to each subject’s FA map using Automated Image Registration with a 12-parameter affine registration followed by a second order (30-parameter) through fourth order (105-parameter) nonlinear warp (Woods et al., 1998). DTI data were then further processed using tract-based spatial statistics (TBSS; Smith et al., 2006) in the Functional MRI of the Brain (FMRIB) Diffusion Toolbox (Behrens et al., 2003) of FMRIB’s Software Library (FSL) version 4.1.3 (http://www.fmrib.ox.ac.uk/fsl; Smith et al.,2004). Thereby, all subjects’ FA maps were aligned to the most representative subject and thence into MNI152 space. An average FA image was generated and thinned to create a white-matter skeleton representing the centers of all white-matter tracts common to all participants. This FA skeleton was then thresholded to FA ≥0.3 to include the major white-matter pathways but to avoid smaller tracts. Voxelwise cross-subject statistics were then performed on this skeletonized data using the Randomise (v2.1) permutation testing software in FSL. Analysis-of-variance (ANOVA) was performed on the measure FA with main effects for the between-subjects factors “ADHD” (2 levels: Yes, No) and “PAE” (2 levels: Yes, No) and the ADHD-by-PAE interaction as the model. Statistical parametric maps were corrected for multiple comparisons using False Discovery Rate (FDR; Benjamini & Hochberg, 1995) applied to the bilateral ACR search space. Following analysis of FA, MD volumes were projected onto the skeleton using the same nonlinear warping parameters calculated for FA and a similar analysis was performed.
2.6|. Statistical Analyses
The statistical strategy focused on testing the a priori hypotheses that ACR Cho and FA would be lower in subjects with PAE than in subjects without PAE and that these endpoints could distinguish ADHD+PAE from ADHD-PAE. As there were no strong grounds for anticipating laterally asymmetric findings, neuroimaging endpoints were examined for the average of the left and the right ACR. In cases where a significant effect was found for the left-right average, we did, however, follow-up to determine whether the effect also existed for left ACR and right ACR separately. Other analyses ancillary to the main goals included ruling-out alternative explanations or evaluating the specificity of the effects to Cho vs. other neurometabolites, to ACR white matter vs. neighboring gray matter of the pACC, or to FA vs. another DTI index, MD.
Within the MRSI Cho sample, the subsample with ADHD and the subsample without ADHD were compared for possible confounding effects of sex, age, FSIQ, cumulative PAE (total drinks during pregnancy), and volume percent gray matter and white matter in the ACR and pACC MRSI voxels (both averaged across left and right hemispheres). Volume percent CSF was not tested as all metabolite levels were already corrected for CSF content. Except for sex (using the χ2-test), all these comparisons made use of independent T-tests. Analogous procedures compared these variables for the subsample with PAE and the subsample without PAE (except for cumulative PAE which was 0 for all non-PAE subjects). The first and second study hypotheses of diminished Cho in PAE and particularly in ADHD+PAE versus ADHD-PAE subjects were tested with an omnibus 2-way analysis-of-covariance (ANCOVA) with Cho in ACR (average of left and right) as measure testing for a main effect of ADHD, a main effect of PAE, and the ADHD-by-PAE interaction. Sex, age, FSIQ, cumulative PAE, and volume percent gray and white matter represented potential covariates, depending on whether they differed significantly between the subsamples. In the event of a significant main effect of ADHD or PAE, ANCOVA was followed by protected post-hoc tests on the overall group for effects of ADHD, respectively, PAE. In the event of a significant ADHD-by-PAE interaction, ANCOVA was followed by protected post-hoc tests among the four subgroups, in particular comparing ADHD+PAE to ADHD-PAE and non-ADHD+PAE to non-ADHD-PAE.
In the event of a significant main effect or interaction, effects of ADHD and/or PAE on Cho levels were examined within the overall group (for main effects) or subgroups (for an interaction) in left and right ACR separately to identify any hemispheric laterality of the effect. The specificity of the putative effects to Cho was tested by comparing (mean left+right) ACR levels of the other major neurometabolites NAA+NAAG, Glu+Gln, Cr+PCr, and mI. The ACR being a white-matter tract, the specificity of the putative effects on Cho to white matter was tested by comparing the overall group or subgroups for Cho in the neighboring gray matter of (left+right) pACC. All these comparisons used independent T-tests. Finally, a receiver operating characteristic (ROC) curve was constructed quantifying sensitivity and selectivity of ACR Cho to predict PAE within subjects in the sample. Criterion for statistical significance was p<0.05, uncorrected given the a priori hypotheses.
Within the DTI FA sample, analogous procedures were followed. The subsample with ADHD and the subsample without ADHD were compared for sex, age, FSIQ, and cumulative PAE. The same variables (except cumulative PAE) were compared for the subsample with PAE and the subsample without PAE. Further statistical analyses were integrated into DTI post-processing as described above. In particular, the principal study FA hypothesis (that ACR FA is lower in subjects with PAE) was tested by preparing statistical parametric maps (SPMs) of a 2-way ANOVA with FA as measure showing DTI voxels within left and right ACR where there was a significant main effect of ADHD, main effect of PAE, or ADHD-by-PAE interaction. These SPMs were examined for possible hemispheric lateral asymmetry. The specificity of effects for the FA metric was tested by performing a similar analysis for the DTI MD index. Voxel-based statistics were corrected for multiple comparisons using FDR within the subvolume of the bilateral ACR. Criterion for statistical significance was a q-value, i.e., an FDR-adjusted p-value, of <0.05.
3|. RESULTS
3.1|. Potential Confounds: Sex, Age, FSIQ, Cumulative PAE, and Voxel Tissue Content
Within the MRSI Cho sample, there were 5 girls and 22 boys with ADHD and 12 girls and 5 boys without ADHD (reflecting the well-known higher prevalence of ADHD in boys). This difference in sex was significant (χ2(1,44) = 11.9, p=0.001). Therefore, sex was used as a covariate in comparisons of ADHD subjects vs. non-ADHD subjects. Age (ADHD: 12.2±2.1 years; non-ADHD: 12.6±2.1 years; t(34.4)=0.6, p=0.525), FSIQ (88.7±15.6 versus 94.5±16.2; t(33.0)=1.2, p=0.255), cumulative PAE (721±920 drinks versus 316±482 drinks; t(9.1)=1.0, p=0.329), volume percent pACC gray matter (75.8±3.1% versus 75.9±3.5%; t(29.6)=0.1, p=0.959), volume percent pACC white matter (16.7±4.0% versus 16.2±4.3%; t(30.6)=−0.4, p=0.709) did not differ significantly between the ADHD subsample and the non-ADHD subsample (subsample means ± standard deviations in parentheses). Volume percent ACR gray matter (13.2±4.0% versus 9.7±4.3%; t(30.4)=−2.6, p=0.013) was slightly higher in the ADHD subsample, while volume percent ACR white matter (85.9±4.1% versus 89.5±4.2%; t(31.8)=2.7, p=0.01) was a few percent lower. Therefore, volume percent ACR white matter was included as a covariate in comparisons of ADHD subjects vs. non-ADHD subjects involving MRSI metabolites in the ACR. (Since volume percent gray matter and volume percent white matter in any MRSI voxel are linearly dependent under present conditions, it is only necessary to covary for one of the two.) In the PAE subsample, there were 5 girls and 12 boys, while in the non-PAE subsample there were 8 girls and 13 boys. This difference in sex distribution was not significant (χ2(1,38) = 0.3, p=0.575). Age (PAE: 12.4±2.0 years; non-PAE: 12.3±2.4 years; t(36.0)=−0.1, p=0.913), FSIQ (89.8±17.4 versus 92.8±14.0; t(30.5)=0.6, p=0.577), volume percent ACR gray matter (12.6±5.5% versus 10.8±3.8%; t(23.7)=−−1.0, p=0.304), volume percent ACR white matter (86.3±4.2% versus 88.3±4.2%; t(26.7)=−1.2, p=0.233), volume percent pACC gray matter (74.9±4.0% versus 76.2±2.6%; t(29.6)=1.0, p=0.306), volume percent pACC white matter (17.7±4.4% versus 16.1±3.9%; t(28.4)=−1.1, p=0.275) did not differ significantly between the PAE subsample and the non-PAE subsample.
Within the DTI FA sample, there were 6 girls and 18 boys with ADHD and 14 girls and 8 boys without ADHD (χ2(1,46) = 7.0, p=0.008), a significant difference. Age (ADHD: 12.0±2.1 years; non-ADHD: 12.4±2.3 years; t(42.2)=0.6, p=0.542), FSIQ (89.0±16.5 versus 93.3±14.5; t(43.9)=0.9, p=0.360), cumulative PAE (795±984 drinks versus 316±482 drinks; t(7.0)=−1.1, p=0.314). In the PAE subsample, there were 5 girls and 11 boys, while in the non-PAE subsample there were 9 girls and 11 boys. This difference in sex distribution was not significant (χ2(1,36) = 0.7, p=0.400). Age (12.4±2.1 years versus 12.4±2.3 years; t(33.5)=−0.02, p=0.969) and FSIQ (90.2±17.9 versus 92.4±14.3; t(28.4)=0.4, p=0.698) did not differ significantly between the PAE subsample and the non-PAE subsample.
3.2|. Group Differences in ACR Cho Involving ADHD and PAE
Within the MRSI Cho sample, two-way ANCOVA on the measure ACR Cho for main effect of ADHD, main effect of PAE, and the ADHD-by-PAE interaction covarying for sex and volume per cent white matter in the ACR MRSI voxel, yielded no significant effect of ADHD (F(1,33)=0.6, p=0.446), a trend-level effect of PAE (F(1,33)=3.2, p=0.087 trend), and a significant ADHD-by-PAE interaction (F(1,33)=4.8, p=0.038). Given the significant interaction, we proceeded to examine the factors underlying the interaction through separate post-hoc protected T-tests involving the four subject subgroups ADHD+PAE, ADHD-PAE, non-ADHD+PAE, and non-ADHD-PAE. Within the MRSI Cho sample, the ADHD+PAE subgroup (N=10) had 26.7% lower ACR Cho than the ADHD-PAE subgroup (N=18), a significant difference (t(16.9)=−4.3, p<0.0005; Fig. 2A). The non-ADHD+PAE subgroup (N=10) had 3.5% lower ACR Cho than the non-ADHD-PAE subgroup (N=8), but this difference was not significant (t(10.9)=0.3, p=0.741; Fig. 2B). Thus, the Cho deficit appeared specific to subjects who had both ADHD and PAE. Note further that all but 1 ADHD+PAE subject had a Cho level below the mean of the non-ADHD-PAE subjects (typically developing controls; Fig. 2A).
FIGURE 2.
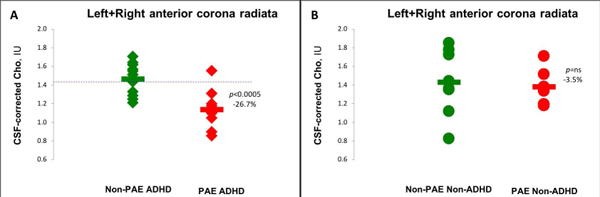
Interaction of effects of ADHD and prenatal alcohol exposure (PAE) on CSF-corrected levels of choline-containing compounds (Cho) obtained with MRSI in anterior corona radiata (ACR) and ability of these levels to distinguish ADHD+PAE from ADHD-PAE. Children with ADHD are marked with diamonds and children without ADHD are marked with circles; children with PAE appear in red, children without PAE appear in green. Horizontal bars mark group means. (A) Comparing subjects with (N=10) and subjects without (N=13) PAE within the ADHD subsample, the former exhibited 26.7% lower Cho (p<0.0005). The blue dashed line represents the mean value for N=8 non-ADHD-PAE (i.e., healthy control) children. Note that Cho is below this control mean for all but one ADHD+PAE child. (B) Comparing subjects with (N=10) and subjects without (N=8) PAE within the non-ADHD subsample, the former exhibited only 3.5% lower Cho, a non-significant difference (p=0.741). Thus, the ACR Cho deficit appears specific to children with both PAE and ADHD.
3.3|. Hemispheric Laterality of ACR Cho Deficit in ADHD+PAE
Mean Cho in left ACR was 20.0% lower (t(8.5)=3.4, p=0.012) in ADHD+PAE subjects than in ADHD-PAE subjects within the MRSI Cho sample. In right ACR, Cho was 28.6% lower (t(12.9)=4.5, p=0.001) in ADHD+PAE subjects than in ADHD-PAE subjects. Thus, the Cho deficit in ACR in ADHD+PAE was present in both cerebral hemispheres.
3.4|. Specificity of ACR Neurometabolite Subgroup Differences to Cho
Comparing ADHD+PAE to ADHD-PAE subjects within the MRSI Cho sample for mean levels of NAA+NAAG (t(16.6)=0.9, p=0.396), Glu+Gln (t(12.4)=1.1, p=0.283), Cr+PCr (t(12.2)=0.9, p=0.405), and mI (t(11.3)=0.2, p=0.839), there were no significant differences between the subsamples (Table 3). Thus, Cho was the only neurometabolite identified showing a difference between ADHD+PAE and ADHD-PAE in the ACR.
TABLE 3.
MRSI Cho Sample Voxel Tissue Composition and Neurometabolite Levels
| Subgroup(N) | ||||||
|---|---|---|---|---|---|---|
| +ADHD | +ADHD | +ADHD | -ADHD | -ADHD | -ADHD | |
| +PAE(10) | -PAE(13) | ?PAE(4) | +PAE(7) | -PAE(8) | ?PAE(2) | |
| Left+Right anterior corona radiata (ACR) | ||||||
| Gray matter, vol% | 13.8±5.6 (1.6–19.0) |
13.1±2.4 (10.2–16.2) |
13.8±2.1 (11.3–16.2) |
10.7±5.4 (4.7–20.3) |
8.4±3.5 (2.8–13.8) |
11.8±4.5 (8.6–15.0) |
| White matter, vol% | 85.2±5.5 (80.8–98.4) |
86.8±2.6 (83.2–86.8) |
86.0±2.3 (83.2–88.6) |
88.0±4.7 (79.7–92.4) |
91.1±3.6 (85.7–97.2) |
88.0±4.8 (84.6–91.4) |
| CSF, vol% | 1.0±1.4 (0–3.3) |
0.1±0.3 (0–2.8) |
0.2±0.3 (0–0.6) |
1.3±1.2 (0–2.9) |
0.6±1.0 (0–2.8) |
0.2±0.3 (0–0.4) |
| Cho, IU | 1.1±0.2 (0.9–1.6) |
1.5±0.2 (1.2–1.7) |
1.3±0.1 (1.2–1.5) |
1.4±0.2 (1.2–1.7) |
1.4±0.4 (0.9–1.9) |
1.4±0.3 (0.8–1.9) |
| NAA+NAAG, IU | 5.8±1.4 (4.0–8.2) |
6.3±1.2 (4.6–8.8) |
6.5±0.6 (5.9–7.3) |
6.3±0.3 (5.8–6.6) |
6.2±1.2 (3.8–7.8) |
6.3±0.8 (3.8–7.8) |
| Glu+Gln, IU | 9.4±2.1 (6.5–12.5) |
10.3±1.6 (8.2–13.8) |
9.1±0.9 (8.1–10.0) |
9.6±0.9 (8.7–11.1) |
9.2±2.2 (4.5–11.7) |
9.6±1.7 (4.5–11.7) |
| Cr+PCr, IU | 3.3±1.1 (1.4–4.9) |
3.7±0.6 (2.7–4.7) |
4.0±0.4 (3.6–4.6) |
3.8±0.5 (3.2–4.4) |
3.6±0.9 (1.7–4.4) |
3.7±0.7 (1.7–4.4) |
| mI, IU | 3.6±0.9 (2.2–4.7) |
3.7±0.7 (2.9–5.0) |
3.8±0.4 (3.4–4.4) |
3.3±0.4 (3.0–3.9) |
3.4±1.0 (1.6–5.0) |
3.4±0.8 (1.6–5.0) |
| Left+Right pregenual Anterior Cingulate Cortex (pACC) | ||||||
| Gray matter, vol% | 75.7±3.5 (70.3–81.7) |
76.5±2.7 (74.4–80.2) |
76.0±2.8 (74.4–80.2) |
73.9±4.7 (65.4–77.2) |
76.5±1.8 (72.9–78.3) |
79.4±1.4 (78.4–80.4) |
| White matter, vol% | 16.5±4.8 (10.2–25.8) |
14.6±3.7 (11.3–18.8) |
15.4±3.8 (12.0–18.8) |
19.5±3.2 (16.1–25.4) |
14.6±4.0 (9.8–20.3) |
12.6±2.5 (10.9–14.4) |
| CSF, vol% | 7.5±3.2 (1.4–11.7) |
8.8±2.9 (6.7–13.5) |
8.6±3.3 (6.7–13.5) |
6.6±2.4 (3.9–9.3) |
8.3±3.2 (1.9–11.7) |
8.0±1.0 (7.3–8.7) |
| Cho, IU | 1.3±0.2 (1.0–1.6) |
1.3±0.2 (0–3.3) |
1.2±0.2 (1.0–1.3) |
1.3±0.2 (1.1–1.6) |
1.3±0.2 (1.0–1.6) |
1.3±0.2 (1.0–1.7) |
In two-way ANCOVA, no significant main effect of ADHD (F(1,33)=0.6, p=0.446), trend-level effect of PAE (F(1,33)=3.2, p=0.087 trend), and significant ADHD-by-PAE interaction (F(1,33)=4.8, p=0.038), representing 26.7% lower ACR Cho in ADHD+PAE than in ADHD-PAE (post-hoc t(16.9)=−4.3, p<0.0005) but no significant difference between -ADHD+PAE and -ADHD-PAE (3.5%; t(10.9)=0.3, p=0.741).
+ADHD = attention deficit hyperactivity disorder (ADHD) diagnosis; -ADHD = no ADHD diagnosis; +PAE = prenatal alcohol exposure; -PAE = no PAE; ?PAE = unknown if any PAE; vol% = percent by volume of tissue type in MRSI voxel; CSF = cerebrospinal fluid; Cho = choline-containing compounds; NAA+NAAG = N-acetyl-aspartate+N-acetyl-aspartyl-glutamate; Glu+Gln = glutamate+glutamine; Cr+PCr – creatine+phosphocreatine; mI = myo-inositol; IU = Institutional Units. Values are numbers of subjects or subgroup-means±standard deviations (ranges)
3.5|. Specificity of ACR Cho Deficit in ADHD+PAE to White Matter
The MRSI voxels in the ACR in the present investigation—like most voxels in most MRS clinical studies-- exhibited some degree of “partial-voluming”, i.e., inclusion of more than one tissue-type (e.g., gray matter and white matter) in the same volume. For mixed-tissue voxels, one must ask whether a given observed effect is due to gray matter, to white matter, or to both. The high white-matter content (≥75%) of ACR voxels in our study implies that below-normal Cho in PAE is a white-matter effect. As an additional check on this proposition, however, for the MRSI Cho sample, we compared the ADHD+PAE subsample to the ADHD-PAE subsample for Cho levels in the pACC, a neighboring structure to the ACR that would have been a major source of any contaminating gray matter. pACC Cho did not differ significantly between the ADHD+PAE subsample and the ADHD-PAE subsample (t(16.5)=−0.2, p=0.848), supporting the inference that the Cho deficit in ADHD+PAE it is indeed a white-matter effect.
3.6|. Receiver Operating Characteristic (ROC) Curve For Effect of PAE on ACR Cho
For the MRSI Cho sample, an ROC curve (Fig. 3) was prepared displaying the ability of low ACR Cho to predict positive PAE status. Contributing to the curve were 17 subjects with PAE and 21 without PAE. Low ACR Cho significantly predicted positive PAE status with an area under the curve (AUC) of 0.76±0.08 (p=0.007). AUC-values in the range 0.7–0.8 are considered “fair” predictors.
FIGURE 3.
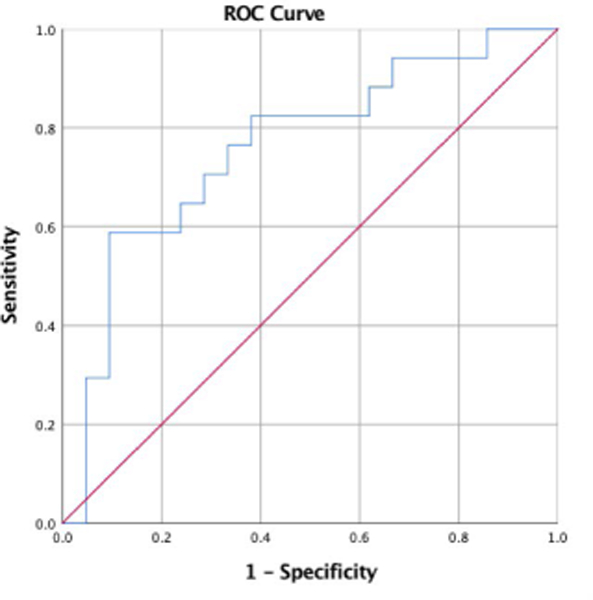
Receiver operating characteristic (ROC) curve for the ability of low ACR Cho to predict positive status for prenatal alcohol exposure (PAE) for 17 subjects with PAE and 21 without PAE. Low ACR Cho significantly predicted positive PAE status with an area under the curve (AUC) of 0.76±0.08 (p=0.007). This implies a “fair” ability of ACR Cho to predict PAE status.
3.7|. Effects of ADHD and PAE on ACR DTI FA
Within the DTI FA sample, FDR-corrected two-way ANOVA on the measure ACR FA for main effect of ADHD, main effect of PAE, and the ADHD-by-PAE interaction, yielded no voxels with a significant effect of ADHD (all voxels q>0.10) or a significant ADHD-by-PAE interaction (all voxels q>0.10). There were, however, clusters of voxels in both left and right ACR showing significantly lower FA in PAE subjects than in non-PAE subjects (q=0.0102–0.05; Fig. 4). When a similar two-way ANOVA was run on the DTI MD measure, no significant voxels resulted for effects of ADHD, effects of PAE, or the ADHDX PAE interaction (all voxels q>0.10). Thus, the effect of PAE was specific to the FA index.
FIGURE 4.
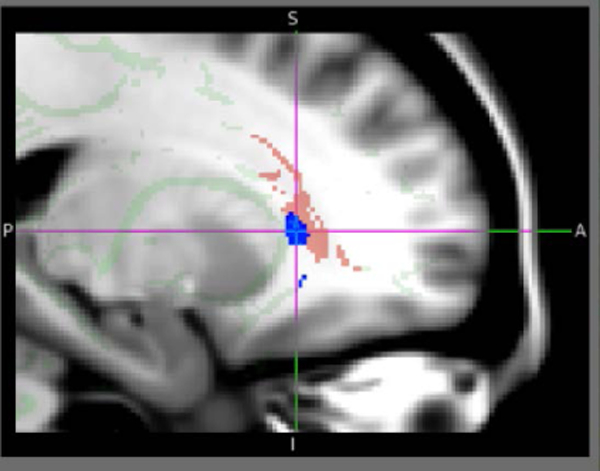
Left parasagittal section of the MNI152 atlas of the human brain with superposed tract-based spatial statistics (TBSS) diffusion tensor imaging (DTI) skeleton of the major white-matter tracts (light green) and mask of the anterior corona radiata (ACR) target tract of this investigation (pink). In blue is a superposed statistical parametric map (SPM) indicating portions of the ACR where significantly (q=0.0102–0.05, FDR-corrected) lower DTI fractional anisotropy (FA) was seen for subjects with prenatal alcohol exposure (PAE) than for subjects without PAE. A similar cluster of voxels with significantly lower FA in PAE subjects was seen in right ACR. This finding may imply lower white-matter integrity in PAE. This is approximately the region where lower Cho was observed (bilaterally) in an overlapping ADHD+PAE sample (Fig. 2A). Thus, ACR data evince two putative neuroimaging indices of focal white-matter pathology in PAE: MRS Cho and DTI FA.
4|. DISCUSSION
4.1|. Summary of Major Findings
ADHD is very common in children with prenatal alcohol exposure (PAE), but also occurs frequently in children without PAE. This targeted reanalysis of MRSI and DTI data previously acquired in a study of pediatric ADHD (partially reported on in Hamilton et al., 2008; Luders et al., 2009; Tafazoli et al., 2013) tested for neuroimaging signs of PAE in the anterior corona radiata (ACR) that might distinguish ADHD+PAE from ADHD-PAE. Prior literature pointed to the ACR as a possible site of PAE-associated MRS and DTI abnormalities and as a white-matter tract that helps mediate executive functions that are disturbed in ADHD. There were two major findings: 1) An ADHD-by-PAE interaction whereby levels of MRSI Cho in bilateral ACR were lower in ADHD+PAE than in ADHD-PAE children; 2) Again in bilateral ACR, DTI FA was lower in children with PAE than in children without PAE, no such effect was observed for children with ADHD vs. children without ADHD. Taken together, these findings indicate possible tract-focal, white-matter pathology that may be specific for the PAE etiology of ADHD.
4.2|. ADHD-by-PAE Interaction: Lower ACR Cho in ADHD+PAE versus ADHD-PAE
The first major finding was an ADHD-by-PAE interaction whereby ACR Cho was specifically lower in children with ADHD+PAE than in children with ADHD-PAE. There was no significant difference in Cho between non-ADHD+PAE and non-ADHD-PAE children. Hence, a Cho deficit was notable only for the subgroup of children with ADHD who had been exposed to prenatal ethanol. The finding was present bilaterally, perhaps to be expected from exposure to a systemic toxin like ethanol. The finding was specific to Cho and was not present for any of the other canonical 1H MRS neurometabolites. Low Cho was also specific to white matter, i.e., the effect was not present in neighboring gray matter. This finding approximately reproduces the 7–12% white-matter Cho deficit found in children with FASD (without regard to presence or absence of ADHD) by Astley et al. (2009). Astley et al. (2009) failed to find effects of FASD on NAA+NAAG or Cr+PCr in white matter; we failed to find effects of PAE on NAA+NAAG, Glu+Gln, Cr+PCr, or mI. One difference between the studies is that the Astley et al. voxel was more dorsal and caudal representing mixed superior/anterior corona radiata, while the present more ventrorostral voxel contained more purely anterior corona radiata (ACR). Also, the Astley et al. single-voxel finding was assayed in the right cerebral hemisphere only, while we report independent findings in left and right ACR. Another difference was that the Cho deficit in Astley et al. was observed for subjects with fetal alcohol syndrome (FAS) or partial FAS (pFAS) only and was not present for subjects with less severe forms of FASD. Our subjects were evaluated for presence and amount of PAE only, but were not formally classified into the various diagnostic subcategories of FASD. Therefore, we cannot say whether or not our white-matter Cho data support the distinctions between different subtypes of FASD drawn by Astley et al. (2009). In a simian PAE model, the same investigators (Astley et al., 1995) measured elevated Cho/Cr+PCr in PAE relative to non-PAE macaques in a voxel containing internal capsule, other proximal white matter, thalamus, and “basal ganglia” (although the most severely impaired monkey had low Cho/Cr+PCr). Gonçalves et al. (2009) did not observe any effects of PAE on Cho or other metabolites in a left “frontal-lobe” voxel centered on ACR. This voxel, however, contained large amounts of neighboring gray matter, which may have diluted any putative Cho effect in white matter. These authors did, however, measure diminished Cho/Cr+PCr in PAE relative to control subjects in a voxel containing internal capsule, caudate, and putamen, somewhat close to the Astley et al. (1995) monkey voxel. du Plessis et al. (2014) measured below-normal Cho in the cerebellum of children with PAE (note that their voxel also contained a good deal of white matter). Our present results, combined with this prior literature, suggest that PAE-associated Cho abnormalities (depressions or elevations) occur in various parts of the corona radiata, the contiguous internal capsule, and various subcortical nuclei. Thereby, in addition to FASD subtype, it is prudent to consider MRS voxel tissue composition. Present results point to higher Cho levels in white matter than in gray matter except in the ADHD+PAE subgroup (Table 3).
As indicated by the significant ADHD-by-PAE interaction in the absence of significant main effects of ADHD and PAE, the Cho deficit was only significant for subjects who had both ADHD and PAE. To explain this, one might consider that PAE, in principle, can induce abnormalities anywhere in the brain leading to a variety of symptoms. In a given subject, the ACR may or may not be affected. Low Cho may mark those cases of PAE in which the ACR is sufficiently compromised to produce symptoms of ADHD. Normal Cho may mark cases where the ACR has remained functionally intact enough to avoid the emergence of ADHD.
Using neuropsychological profiling (e.g., Boseck et al., 2015; Mattson et al., 2013), some progress has been made in discriminating ADHD+PAE from ADHD-PAE. The present Cho findings suggest that neuroimaging may contribute to such efforts. There are multiple reports of elevated Cho in ADHD (Colla et al., 2005; Courvoisie et al., 2004; Ferreira et al., 2009; Jin et al., 2001; Perlov et al., 2007; reviewed in O’Neill et al., 2013b; Perlov et al., 2009). The meta-analysis of Perlov et al. (2009) concludes that regional elevation of Cho is the most common MRS sign of ADHD. Thus, one might interpret the present finding as an elevation of Cho in ADHD+PAE that is somehow absent in ADHD-PAE. The foregoing findings of high Cho in ADHD, however, are typically restricted to MRS voxels with majority cortical or subcortical gray-matter, rather than white-matter, content. Also, as seen in Fig. 2A, ACR Cho levels in ADHD-PAE are comparable to those of healthy controls in our study. Thus, we interpret the present result as a decrement in Cho in ADHD+PAE, rather than an increment in Cho in ADHD-PAE. In any case, the present result suggests that there exist alternative neurophysiological routes towards the production of ADHD symptoms. In our Cingulocentric Model of ADHD (O’Neill et al., 2013b), which draws on Carlsson’s Dual Accelerator-Brake Model (Carlsson, 2000a,b) and Middleton (2009), dysfunction of an action-impulsive behaviors cortico-subcortical reentrant circuit responsible for production and selection of voluntary skeletal movements leads to symptoms of ADHD. This circuit includes the ACR as a principal pathway connecting cingulate cortex (pACC and the immediately caudal anterior middle cingulate cortex—aMCC) to striatum and is modulated by dopaminergic input from the ventral tegmental area. One might propose that, while ADHD-PAE (especially familial ADHD) arises from inadequate dopaminergic modulation of this circuit because of suboptimally active cortical dopamine receptors or transporters in aMCC and/or pACC, ADHD+PAE stems from improper signal transmission within the circuit due to PAE-associated maldevelopment of the ACR.
4.3|. FA Decrement in ACR in PAE
The second major finding was that ACR FA was lower bilaterally in subjects with PAE. Low FA was not due to the presence of ADHD. The finding was specific to FA and was not present for the DTI MD index. Several prior DTI studies of FASD (for review see Wozniak & Muetzel, 2011) measured below-normal FA in various white-matter tracts (Fan et al., 2016; Li et al., 2009; Lebel et al., 2008; Sowell et al., 2008; Wozniak et al., 2009), with a few reporting elevated MD (Fan et al., 2016; Wozniak et al., 2006). Present findings agree with Fryer et al. (2009) who found diminished FA in FASD in ACR, a tract not examined in all of the foregoing studies. Also note that, with the exception of O’Conaill et al. (2015), we are aware of no prior DTI study of FASD that factored the effects of ADHD into the analysis, ADHD having very high incidence in FASD. Similar to our study, O’Conaill et al. (2015) found no effects of ADHD on their DTI endpoints (FA and MD in the inferior longitudinal fasciculus). There have been many DTI studies of ADHD (reviewed in Chen et al., 2016; van Ewijk et al., 2012). Some of these report below-normal (Kobel et al., 2010; Pavuluri et al., 2009) or above-normal (Davenport et al., 2010) FA in ACR. None of these studies screened subjects for PAE, as is widely the case for DTI (or MRS) studies of ADHD (Wozniak & Muetzel, 2011). There were no effects of ADHD in the ACR in our investigation of a more recent sample using more advanced DTI (Lawrence et al., 2013) which had screened subjects for PAE. The present study of the ACR, which accounted for both ADHD and PAE found effects of PAE only.
Low FA may imply reduced axonal integrity (Alexander et al., 2007), reduced myelination (Alexander et al., 2007; Assaf et al., 2008), reduced homogeneity of fiber orientation (Alexander et al., 2007), or increased proportion of “crossing fibers” in the tract. Low axonal integrity and/or myelination would be consistent with our above finding of low Cho in the same tract in subjects with PAE of a partly overlapping sample. Low FA and low Cho would, hence, represent dual in vivo imaging indices of regional white-matter pathology in PAE. Lower homogeneity of fiber orientation and/or higher proportion of crossing-fibers, on the other hand, might result from PAE-influenced dysregulation of axonal growth and migration. Wozniak et al. (2011,2013b) have detected abnormal blood-oxygen-level dependent (BOLD) functional connectivity in FASD that may be linked to aberrant organization of underlying white-matter tracts. Future investigations with advanced DTI, relaxometry, and other techniques taking account of both PAE and ADHD may be able to confirm and characterize this putative PAE-induced FA deficit in the ACR.
4.4|. Limitations and Conclusion
Subjects with PAE were not formally diagnosed with a FASD but included any subject for whom the mother endorsed any amount of PAE across the course of pregnancy. As typical of clinical PAE samples, some mothers of subjects with PAE also consumed other teratogens, e.g., nicotine or cocaine, during pregnancy. Hence, exposure to these other substances may have influenced results. As polysubstance abuse is more the rule than the exception in FASD research, recruitment of samples with monosubstance exposure is frequently a challenge in the field. Another limitation the present study shares with FASD research generally is that assessment of PAE status (and of the amount of PAE) usually depends on information provided by the birth mother. Many birth mothers are understandably reluctant to admit to consuming alcohol during pregnancy, some may have unreliable memories, others (e.g., in the common case of foster children) may be difficult to locate. Thus, our samples contain 6–10 subjects of unknown PAE status and one can rarely be absolutely sure that a negative PAE status is accurate. Our sample screened out children with growth deficiencies which likely excluded many moderate-to-severe cases of FASD. Accordingly, results may be more representative of less severe and covert cases of PAE. Several subjects suffered from psychiatric comorbidities as is typical of PAE (O’Connor and Paley, 2009; Weyrauch et al., 2017) and ADHD (Jensen et al., 1997) samples. A few subjects were taking psychotropic medication at time of study, although they were required to abstain from medication on day of scan. MRSI and (elementary) DTI were acquired at 1.5 T using a quadrature coil. Preferable alternatives nowadays would include acquisition at 3 T with a multichannel phased-array headcoil. More sophisticated DTI using 64 or more directions and other advancements would also be preferred. In particular, newer techniques provide for superior handling of crossing fibers, which are prevalent in the ACR and can obscure reductions in FA (Chao et al., 2009). These weaknesses are counterbalanced by strengths of our study. As stated, it is rare for neuroimaging studies of ADHD to assess PAE at all and it is rare for neuroimaging studies of FASD to analyze effects of ADHD explicitly, both of which were done for the present study. MRSI was acquired at relatively high spatial resolution (1.1 cc) and metabolite levels were expressed relative to water rather than to Cr+PCr. The tissue-composition of each MRSI voxel was calculated and incorporated into the analyses. This calculation did not account for the point-spread function of MRSI, but, gray- and white-matter are still estimated to be quite high, given the tight packing of these tissues in pediatric brains. Bearing these limitations in mind, the present study profiles the ACR as a white-matter tract that may be particularly vulnerable to PAE; neuroimaging endpoints measured in the ACR may figure in the development of objective assays to distinguish ADHD+PAE from ADHD-PAE.
ACKNOWLEDGEMENTS
This research was supported by NIAAA R01AA025066 (Levitt/O’Connor), NIMH R01MH081864 (O’Neill/Piacentini), NCRR P41RR13642, and NCMHD/NIA/NIDA/NIMH P01MH063357 (Strickland).
Funding information
Supported by NIAAA R01AA025066 (Levitt/O’Connor), NIMH R01MH081864 (O’Neill/Piacentini), NCRR P41RR13642, and NCMHD/NIA/NIDA/NIMH P01MH063357 (Strickland).
Footnotes
CONFLICT OF INTEREST
There is some overlap in content in the introduction and methods sections and in the limitations and discussion to other publications we have authored. My coauthors and I do not have any conflicts of interest or financial disclosures to report related to this publication and APA ethical standards were followed in the conduct of the study.
REFERENCES
- Alexander AL, Lee JE, Lazar M, Field AS (2007). Diffusion tensor imaging of the brain. Neurotherapeutics, 4, 316–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Assaf Y, Pasternak O (2008). Diffusion tensor imaging (DTI)-based white matter mapping in brain research: a review. Journal of Molecular Neuroscience, 34, 51–61. [DOI] [PubMed] [Google Scholar]
- Astley SJ, Richards T, Aylward EH, Olson HC, Kerns K, Brooks A, Coggins TE, Davies J, Dorn S, Gendler B, Jirikowik T, Kraegel P, Maravilla K (2009). Magnetic resonance spectroscopy outcomes from a comprehensive magnetic resonance study of children with fetal alcohol spectrum disorders. Magnetic Resonance Imaging, 27, 760–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astley SJ, Weinberger E, Shaw D, Richards T, Clarren SK (1995). Magnetic resonance imaging and spectroscopy in fetal ethanol exposed Macaca nemestrina. Neurotoxicology and Teratology, 17, 523–530. [DOI] [PubMed] [Google Scholar]
- Balaraman S, Idrus NM, Miranda RC, Thomas JD (2017). Postnatal choline supplementation selectively attenuates hippocampal microRNA alterations associated with developmental alcohol exposure. Alcohol 60, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basser PJ, Mattiello J, LeBihan D (1994). MR diffusion tensor spectroscopy and imaging. Biophysics Journal, 66, 259–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens TE, Woolrich MW, Jenkinson M, Johansen-Berg H, Nunes RG, Clare S, Matthews PM, Brady JM, Smith SM (2003). Characterization and propagation of uncertainty in diffusionweighted MR imaging. Magnetic Resonance in Medicine 50, 1077–1088. [DOI] [PubMed] [Google Scholar]
- Bejjani A, O’Neill J, Kim JA, Frew AJ, Yee VW, Ly R, Kitchen C, Salamon N, McCracken JT, Toga AW, Alger JR, Levitt JG (2012). Elevated glutamatergic compounds in pregenual anterior cingulate in pediatric autism spectrum disorder demonstrated by 1H MRS and 1H MRSI. PLoS ONE 7(7). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, & Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Ser B, 57, 289–300. [Google Scholar]
- Boseck JJ, Davis AS, Cassady JC, Finch WH, Gelder BC (2015). Cognitive and adaptive skill profile differences in children with attention-deficit hyperactivity disorder with and without comorbid fetal alcohol spectrum disorder. Applied Neuropsychology: Child, 4(4), 230–236. [DOI] [PubMed] [Google Scholar]
- Carlsson ML (2000a). On the role of cortical glutamate in obsessive-compulsive disorder and attention deficit hyperactivity disorder, two phenomenologically antithetical conditions. Acta Psychiatrica Scandinavica, 102, 401–413. [DOI] [PubMed] [Google Scholar]
- Carlsson ML (2000b). On the role of prefrontal cortex glutamate for the antithetical phenomenology of obsessive-compulsive disorder and attention deficit hyperactivity disorder. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 25, 5–26. [DOI] [PubMed] [Google Scholar]
- CDC, Centers for Disease Control and Prevention. (2004). Fetal alcohol syndrome: Guidelines for referral and diagnosis Washington, DC; National Center on Birth Defects and Developmental Disabilities. Department of Health and Human Services. [Google Scholar]
- Chao T-C, Chou M-C, Yang P, Chung H-W, Wu M-T (2009). Effects of interpolation methods in spatial normalization of diffusion tensor imaging data on group comparison of fractional anisotropy. Magnetic Resonance Imaging 27, 681–690. [DOI] [PubMed] [Google Scholar]
- Chen L, Hu X, Ouyang L, He N, Liao Y, Liu Q, Zhou M, Wu M, Huang X, Gong Q (2016). A systematic review and meta-analysis of tract-based spatial statistics studies regarding attention-deficit/hyperactivity disorder. Neuroscience and Biobehavioral Reviews, 68, 838–847. [DOI] [PubMed] [Google Scholar]
- Chernoff R, Combs-Orme T, Risley-Curtiss C, Heisler A (1994). Assessing the health status of children entering foster care. Pediatrics 93, 594–601. [PubMed] [Google Scholar]
- Clark K, Narr K, O’Neill J, Levitt J, Siddarth P, Phillips O, Toga A, Caplan R (2012). White matter integrity, language, and childhood onset schizophrenia. Schizophrenia Research 138(2–3), 150–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coles CD, Platzman KA, Raskind-Hood CL, Brown RT, Falek A, Smith IE (1997). A comparison of children affected by prenatal alcohol exposure and attention deficit, hyperactivity disorder. Alcoholism: Clinical and Experimental Research 21, 150–61. [PubMed] [Google Scholar]
- Coles CD (2001). Fetal alcohol exposure and attention: moving beyond ADHD. Alcohol Research & Health, 25(3), 199–203. [PMC free article] [PubMed] [Google Scholar]
- Colla M, Ende G, Alm B, Deuschle M, Heuser I, Kronenberg G (2008). Cognitive MR spectroscopy of anterior cingulate cortex in ADHD: Elevated choline signal correlates with slowed hit reaction times. Journal of Psychiatric Research, 42, 587–595. [DOI] [PubMed] [Google Scholar]
- Cortese BM, Moore GJ, Bailey BA, Jacobson SW, Delaney-Black V, Hannigan JH (2006). Magnetic resonance and spectroscopic imaging in prenatal alcohol-exposed children: preliminary findings in the caudate nucleus. Neurotoxicology and Teratology, 28, 597–606. [DOI] [PubMed] [Google Scholar]
- Courvoisie H, Hooper SR, Fine C, Kwock L. Castillo M (2004). Functioning and neuropsychological correlates in children with ADHD-H: Preliminary findings. Journal of Neuropsychiatry and Clinical Neurosciences, 16, 63–69. [DOI] [PubMed] [Google Scholar]
- Davenport ND, Karatekin C, White T, Lim KO (2010). Differential fractional anisotropy abnormalities in adolescents with ADHD or schizophrenia. Psychiatry Research, 181, 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doig J, McLennan JD, Gibbard WB (2008). Medication effects on symptoms of attention-deficit/hyperactivity disorder in children with fetal alcohol spectrum disorder. Journal of Child and Adolescent Psychopharmacology, 18, 365–371. [DOI] [PubMed] [Google Scholar]
- Drachman DA (1977). Memory and cognitive functions in man: does the cholinergic system have a specific role? Neurology, 27, 783–790. [DOI] [PubMed] [Google Scholar]
- du Plessis L, Jacobson JL, Jacobson SW, Hess AT, van der Kouwe A, Avison MJ, Molteno CD, Stanton ME, Stanley JA, Peterson BS, Meintjes EM (2014). An in vivo (1)H magnetic resonance spectroscopy study of the deep cerebellar nuclei in children with fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 38(5), 1330–1338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fagerlund A, Heikkinen S, Autti-Rämö I, Korkman M, Timonen M, Kuusi T, Riley EP, Lundbom N (2006). Brain metabolic alterations in adolescents and young adults with fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 30, 2097–2104. [DOI] [PubMed] [Google Scholar]
- Fan J, Jacobson SW, Taylor PA, Molteno CD, Dodge NC, Stanton ME, Jacobson JL, Meintjes EM (2016). White matter deficits mediate effects of prenatal alcohol exposure on cognitive development in childhood. Human Brain Mapping, 37, 2943–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, McCandliss BD, Sommer T, Raz A, Posner MI (2002). Testing the efficiency and independence of attentional networks. Journal of Cognitive Neuroscience, 14, 340–347. [DOI] [PubMed] [Google Scholar]
- Ferreira PEMS, Palmini A, Bau CHD, Grevet EH, Hoefel JR, Rohde LA, Anés M, Ferreira EE, Belmonte-de-Abreu P (2009). Differentiating attention-deficit/hyperactivity disorder inattentive and combined types: a 1H-magnetic resonance spectroscopy study of fronto-striato-thalamic regions. Journal of Neural Transmission, 116, 623–629. [DOI] [PubMed] [Google Scholar]
- Freeman JJ, Jenden DJ (1976). The source of choline for acetylcholine synthesis in brain. Life Sciences, 19, 949–962. [DOI] [PubMed] [Google Scholar]
- Fryer SL, Schweinsburg BC, Bjorkquist OA, Frank LR, Mattson SN, Spadoni AD, Riley EP (2009). Characterization of white matter microstructure in fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 33, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge H, Yin X, Xu J, Tang Y, Han Y, Xu W, Pang Z, Meng H, Liu S (2013). Fiber pathways of attention subnetworks revealed with tract-based spatial statistics (TBSS) and probabilistic tractography. PLoS One, 8, e78831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonçalves R.deC.F., Vasconcelos MM, Faleiros LO, Cruz LC Jr., Domingues RC, Brito AR, Werner J Jr., Herdy GV (2009). Proton magnetic resonance spectroscopy in children with fetal alcohol spectrum disorders. Arquivos de Neuro-Psiquiatria 67, 254–61. [DOI] [PubMed] [Google Scholar]
- Gujar SK, Maheshwari S, Bjorkman-Burtscher I, Sundgren PC (2005). Magnetic resonance spectroscopy. Journal of Neuroophthalmology, 25, 217–226. [DOI] [PubMed] [Google Scholar]
- Hamilton LS, Levitt JG, O’Neill J, Alger JR, Luders E, Phillips OR, Caplan R, Toga AW, McCracken JT, Narr KL (2008). Reduced white matter integrity in attention-deficit hyperactivity disorder. Neuroreport 19(17), 1705–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hess AT, Jacobson SW, Jacobson JL, Molteno CD, van der Kouwe AJW, Meintjes EM (2014). A comparison of spectral quality in magnetic resonance spectroscopy data acquired with and without a novel EPI-navigated PRESS sequence in school-aged children with fetal alcohol spectrum disorders. Metabolic Brain Disease, 29(2), 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howells FM, Donald KA, Roos A, Woods RP, Zar HJ, Narr KL, Stein DJ (2016). Reduced glutamate in white matter of male neonates exposed to alcohol in utero: a 1H-magnetic resonance spectroscopy study. Metabolic Brain Disease, 31, 1105–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudkins M, O’Neill J, Tobias MC, Bartzokis G, London ED (2012) Cigarette smoking and white matter microstructure. Psychopharmacology, 221(2), 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isayama RN, Leite PE, Lima JP, Uziel D, Yamasaki EN (2009). Impact of ethanol on the developing GABAergic system. The Anatomical Record 292, 1922–19839. [DOI] [PubMed] [Google Scholar]
- Jensen PS, Martin D, Cantwell DP. (1997). Comorbidity in ADHD: implications for research, practice and DSM-V. Journal of the American Academy of Child and Adolescent Psychiatry, 36, 1065–1079. [DOI] [PubMed] [Google Scholar]
- Jin Z, Zang YF, Zeng YW, Zhang L, Wang YF (2001). Striatal neuronal loss or dysfunction and choline rise in children with attention-deficit hyperactivity disorder: a 1H-magnetic resonance spectroscopy study. Neuroscience Letters, 315, 45–48. [DOI] [PubMed] [Google Scholar]
- Joe E, Medina LD, Ringman JM, O’Neill J (2018). 1H MRS spectroscopy in preclinical autosomal dominant Alzheimer disease. Brain Imaging and Behavior (in press). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable JA, O’Connor MJ, Carmichael Olson H, Paley B, Mattson SN, Anderson SM, & Riley EP (2016). Neurobehavioral Disorder Associated with Prenatal Alcohol Exposure (ND-PAE); Proposed DSM-5 Diagnosis. Child Psychiatry and Human Development, 47(2), 335–346. doi 10:1007/s10578-015-0566-7. [DOI] [PubMed] [Google Scholar]
- Kobel M, Bechtel N, Specht K, Klarhofer M, Weber P, Scheffler K, Opwis K, Penner IK, (2010). Structural and functional imaging approaches in attention deficit/hyperactivity disorder: does the temporal lobe play a key role? Psychiatry Research, 183, 230–236. [DOI] [PubMed] [Google Scholar]
- Lauder JM, & Schambra UB (1999). Morphogenetic roles of acetylcholine. Environmental Health Perspectives, 107, 65–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence KE, Levitt JG, Loo SK, Ly R, Yee V, O’Neill J, Alger J, Narr KL (2013). White matter microstructure in attention-deficit/hyperactivity disorder subjects and their siblings. Journal of the American Academy of Child and Adolescent Psychiatry 52, 431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebel C, Rasmussen C, Wyper K, Walker L, Andrew G, Yager J, Beaulieu C (2008). Brain diffusion abnormalities in children with fetal alcohol spectrum disorder. Alcoholism: Clinical and Experimental Research 32(10), 1732–1740. [DOI] [PubMed] [Google Scholar]
- Li L, Coles CD, Lynch ME, Hu X (2009). Voxelwise and skeleton-based region of interest analysis of fetal alcohol syndrome and fetal alcohol spectrum disorders in young adults. Human Brain Mapping, 30, 3265–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luders E, Narr KL, Hamilton LS, Phillips OR, Thompson PM, Valle JS, Del’Homme M, Strickland T, McCracken JT, Toga AW, Levitt JG (2009). Decreased callosal thickness in attention-deficit/hyperactivity disorder. Biological Psychiatry 65, 84–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, & Riley EP (1998). A review of the neurobehavioral deficits in children with fetal alcohol syndrome or prenatal exposure to alcohol. Alcoholism: Clinical and Experimental Research, 22(2), 279–294. [DOI] [PubMed] [Google Scholar]
- Mattson SN, Roesch SC, Glass L, Deweese BN, Coles CD, Kable JA, May PA, Kalberg WO, Sowell ER, Adnams CM, Jones KL, Riley EP, CIFASD. (2013). Further development of a neurobehavioral profile of fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 37, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson SN, & Riley EP (2011). The quest for a neurobehavioral profile of heavy prenatal alcohol exposure. Alcohol Research and Health, 34, 51–55. [PMC free article] [PubMed] [Google Scholar]
- May PA, Baete A, Russo J, Elliott AJ, Blankenship J, Kalberg WO, Buckley D, Brooks M, Hasken J, Abdul-Rahman O, Adam MP, Robinson LK, Manning M, Hoyme HE (2014). Prevalence and characteristics of fetal alcohol spectrum disorders. Pediatrics 134(5), 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- May PA, Chambers CD, Kalberg WO, Zellner J, Feldman H Buckley MA, Kopald D Hoyme HE (2018). Prevalence of Fetal Alcohol Spectrum Disorders in 4 US Communities, Journal of the American Medical Association, 319(5), 474–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Middleton FA (2009). The contribution of anterior cingulate-basal ganglia circuitry to complex behavior and psychiatric disorders In: Vogt BA (ed.) Cingulate neurobiology and disease. New York, Oxford University Press. [Google Scholar]
- Mori S, Oishi K, Jiang H, Jiang L, Li X, Akhter K, Hua K, Faria AV, Mahmood A, Woods R, Toga A,W, Pike GB, Neto PR, Evans A, Zhang J, Huang H, Miller MI, van Zijl P, Mazziotta J (2008). Stereotaxic white matter atlas based on diffusion tensor imaging in an ICBM template. Neuroimage 40, 570–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niogi S, Mukherjee P, Ghajar J, McCandliss BD (2010). Individual differences in distinct components of attention are linked to anatomical variations in distinct white matter tracts. Frontiers in Neuroanatomy, 4, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niogi S, Mukherjee P, Ghajar J, Johnson CE, Kolster R, Lee H, Suh M, Zimmerman RD, Manley GT, McCandliss BD (2008). Structural dissociation of attentional control and memory in adults with and without mild traumatic brain injury. Brain, 131, 3209–3221. [DOI] [PubMed] [Google Scholar]
- O’Connor MJ, & Paley B (2009). Psychiatric conditions associated with prenatal alcohol exposure. Developmental Disabilities Research Review 15, 225–234. [DOI] [PubMed] [Google Scholar]
- O’Leary-Moore SK, McMechan AP, Galloway MP, Hannigan JH (2008). Neonatal alcohol-induced region-dependent changes in rat brain neurochemistry measured by high-resolution magnetic resonance spectroscopy. Alcoholism: Clinical and Experimental Research 32(10), 1697–1707. [DOI] [PubMed] [Google Scholar]
- Oesterheld JR, Kofoed L, Tervo R, Fogas B, Wilson A, Fiechtner H (1998). Effectiveness of methylphenidate in Native American children with fetal alcohol syndrome and attention deficit/hyperactivity disorder: a controlled pilot study. Journal of Child and Adolescent Psychopharmacology 8(1), 39–48. [DOI] [PubMed] [Google Scholar]
- O’Conaill CR, Malisza KL, Buss JL, Bolster RB, Clancy C, Dreessen de Gervai P, Chudley AE, Longstaffe S (2015). Visual search for feature conjunctions: an fMRI study comparing alcohol-related neurodevelopmental disorder (ARND) to ADHD. Journal of Neurodevelopmental Disorders, 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Malley KD, & Nanson J (2002). Clinical implications of a link between fetal alcohol spectrum disorder and attention-deficit hyperactivity disorder. The Canadian Journal of Psychiatry 47, 349–354. [DOI] [PubMed] [Google Scholar]
- O’Neill J, Gorbis E, Feusner JD, Yip JC, Chang S, Maidment KM, Levitt JG, Salamon N, Saxena S. Effects of intensive cognitive-behavioral therapy on cingulate neurochemistry in obsessive-compulsive disorder. Journal of Psychiatry Research, 47(4),494–504 (2013a). [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill J, Levitt JG, Alger JR (2013b). Magnetic resonance spectroscopy studies of attention deficit hyperactivity disorder In: Blüml S, Panigrahy A (eds.) MR spectroscopy of pediatric brain disorders, pp. 229–276. NY, Springer. [Google Scholar]
- O’Neill J, Piacentini JC, Chang S, Levitt JG, Rozenman M, Bergman L, Salamon N, Alger JR, McCracken JT MRSI correlates of cognitive-behavioral therapy in pediatric obsessive-compulsive disorder. Progress in Neuro-Psychopharmacology and Biological Psychiatry, 36(1),161–168 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavuluri MN,. Yang S, Kamineni K, Passarotti AM, Srinivasan G, Harral EM, Sweeney JA, Zhou XJ (2009). Diffusion tensor imaging study of white matter fiber tracts in pediatric bipolar disorder and attentiondeficit/ hyperactivity disorder. Biological Psychiatry, 65, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peadon E, & Elliott EJ (2010). Distinguishing between attention-deficit hyperactivity and fetal alcohol spectrum disorders in children: clinical guidelines. Journal of Neuropsychiatric Disease and Treatment, 6, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perlov E, Philipsen A, Matthies S, Drieling T, Maier S, Bubl E, Hesslinger B, chert M, Henning J, Ebert D, Tebartz van Elst L (2009). Spectroscopic findings in attention-deficit/hyperactivity disorder: Review and meta-analysis. World Journal of Biological Psychiatry, 10(4), 355–365. [DOI] [PubMed] [Google Scholar]
- Perlov E, Philipsen A, Hesslinger B, Büchert M, Ahrendts J, Feige B, Bubl E, Henning J, Ebert D, Tebartz van Elst L (2007). Reduced cingulate glutamate/glutamine-to-creatine ratios in adult patients with attention deficit/hyperactivity disorder – A magnet resonance spectroscopy study. Journal of Psychiatric Research, 41, 934–941. [DOI] [PubMed] [Google Scholar]
- Provencher S,W (2001). Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR in Biomedicine, 14, 260–264. [DOI] [PubMed] [Google Scholar]
- Rasmussen C (2005). Executive functioning and working memory in fetal alcohol spectrum disorder. Alcoholism: Clinical and Experimental Research 29, 1359–1367. [DOI] [PubMed] [Google Scholar]
- Riikonen R, Salonen I, Partanen K, Verho S (1999). Brain perfusion SPECT and MRI in foetal alcohol syndrome. Developmental Medicine and Child Neurology, 41, 652–659. [DOI] [PubMed] [Google Scholar]
- Ringman J, O’Neill J, Geschwind D, Medina L, Schaffer B, Varpetian A, Apostolova L, Tseng P, Fitten J, Ortiz F, Cummings J, Bartzokis G (2007). Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain 130, 1767–1776. [DOI] [PubMed] [Google Scholar]
- Ross AJ, Sachdev PS (2004). Magnetic resonance spectroscopy in cognitive research. Brain Research: Brain Research Reviews 44, 83–102. [DOI] [PubMed] [Google Scholar]
- Schambra UB, Lauder JM, Perusz P, Sulik KK (1990). Development of neurotransmitter systems in the mouse embryo following acute ethanol exposure: a histological and immunocytochemical study. International Journal of Developmental Neuroscience, 8, 507–522. [DOI] [PubMed] [Google Scholar]
- Shattuck DW, Sandor-Leahy SR, Schaper KA, Rottenberg DA, Leahy RM (2001). Magnetic resonance image tissue classification using a partial volume model. NeuroImage, 13, 856–876. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Woolrich MW, Beckmann CF, Behrens TE, Johansen-Berg H, Bannister PR, De Luca M, Drobnjak I, Flitney DE, Niazy RK, Saunders J, Vickers J, Zhang Y, De Stefano N, Brady JM, Matthews PM (2004). Advances in functional and structural MR image analysis and implementation as FSL. Neuroimage 23(Suppl 1), S208–S219. [DOI] [PubMed] [Google Scholar]
- Smith SM, Jenkinson M, Johansen-Berg H, Rueckert D, Nichols TE, Mackay CE, Watkins KE, Ciccarelli O, Cader MZ, Matthews PM, Behrens TE (2006). Tract-based spatial statistics: Voxelwise analysis of multi-subject diffusion data. Neuroimage 31, 1487–1505. [DOI] [PubMed] [Google Scholar]
- Seese RR, O’Neill J, Hudkins M, Siddarth P, Levitt J, Tseng B, Wu KN, Caplan R (2011). Proton magnetic resonance spectroscopy and thought disorder in childhood schizophrenia. Schizophrenia Research, 133, 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer D, Fisher P, Lucas CP, Dulcan MK, Schwab-Stone ME (2000). NIMH Diagnostic Interview Schedule for Children Version IV (NIMH DISC-IV): description, differences from previous versions, and reliability of some common diagnoses. Journal of the American Academy of Child and Adolescent Psychiatry 39, 28–38. [DOI] [PubMed] [Google Scholar]
- Schonfeld AM, O’Connor MJ, Paley B, Frankel F (2009). Behavioral Regulation as a predictor of response to Children’s Friendship Training in Children with Fetal Alcohol Spectrum Disorders, Clinical Neuropsychology 23(3), 428–445. [DOI] [PubMed] [Google Scholar]
- Snyder J, Nanson J, Snyder R, Block G (1997). A study of stimulant medication in children with FAS In: Streissguth A, Kanter J (eds.) Overcoming and preventing secondary disabilities in fetal alcohol syndrome and fetal alcohol effects, pp. 64–77. Seattle, University of Washington Press. [Google Scholar]
- Sowell ER, Johnson A, Kan E, Lu LH, Van Horn JD, Toga AW, O’Connor MJ, Bookheimer SY (2008). Mapping white matter integrity and neurobehavioral correlates in children with fetal alcohol spectrum disorders. Journal of Neuroscience, 28, 1313–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Mattson SN, Tessner KD, Jernigan TL, Riley EP, Toga AW (2001). Voxel-based morphometric analyses of the brain in children and adolescents prenatally exposed to alcohol. Neuroreport, 12, 515–523. [DOI] [PubMed] [Google Scholar]
- Sowell ER, Thompson PM, Peterson BS, Mattson SN, Welcome SE, Henkenius AL, et al. (2002). Mapping cortical gray matter asymmetry patterns in adolescents with heavy prenatal alcohol exposure. Neuroimage 17, 1807–1903. [DOI] [PubMed] [Google Scholar]
- Streissguth AP, O’Malley K (2000). Neuropsychiatric implications and long-term consequences of fetal alcohol spectrum disorders. Seminars in Clinical Neuropsychiatry 5(3), 177–190. [DOI] [PubMed] [Google Scholar]
- Sulik KK (1984). Critical periods for alcohol teratogenesis in mice with special reference to the gastrulation stage of embyrogenesis, pp. 124–141. Mechanisms of alcohol damage in utero. London, Pitman Ciba Foundation Symposium. [DOI] [PubMed] [Google Scholar]
- Sulik KK, & Johnston MC (1982). Embryonic origin of holoprosencephaly: interrelationship of the developing brain and face. Scanning Electron Microscopy, 1, 309–322. [PubMed] [Google Scholar]
- Sulik KK, & Johnston MC (1983). Sequence of developmental alterations following acute ethanol exposure in mice craniofacial features of the fetal alcohol syndrome. Americam Journal of Anatomy, 166, 257–269. [DOI] [PubMed] [Google Scholar]
- Swanson JM (1995). SNAP-IV Scale. Irvine: University of California Child Development Center. [Google Scholar]
- Tafazoli S, O’Neill J, Beijani A, Salamon N, McCracken JT, Alger JR, Levitt JG (2013). 1H MRSI of middle frontal gyrus in pediatric ADHD. Journal of Psychiatry Research 47(4), 505–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Abou EJ, Dominguez HD (2009). Prenatal choline supplementation mitigates the adverse effects of prenatal alcohol exposure on development in rats. Neurotoxicology and Teratology, 31(5), 303–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Biane JS, O’Bryan KA, O’Neill TM, Dominguez HD (2007). Choline supplementation following third-trimester-equivalent alcohol exposure attenuates behavioral alterations in rats. Behavioral Neuroscience, 121(1), 120–130. [DOI] [PubMed] [Google Scholar]
- Thomas JD, La Fiette MH, Quinn VR, Riley EP (2000). Neonatal choline supplementation ameliorates the effects of prenatal alcohol exposure on a discrimination learning task in rats. Neurotoxicology and Teratology 22(5), 703–711. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Garrison M, O’Neill TM (2004). Perinatal choline supplementation attenuates behavioral alterations associated with neonatal alcohol exposure in rats. Neurotoxicology and Teratology 26, 35–45. [DOI] [PubMed] [Google Scholar]
- Thomas JD, Tran TD (2011). Choline supplementation mitigates trace, but not delay, eyeblink conditioning deficits in rats exposed to alcohol during development. Hippocampus 22(3), 619–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobias MC, O’Neill J, Hudkins M, Bartzokis G, Dean A,C, London ED (2010). White-matter abnormalities in brain during early abstinence from methamphetamine abuse. Psychopharmacology, 209(1),13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tunggal B, Hofmann K Stoffel W (1990). In vivo “C nuclear magnetic resonance investigations of choline metabolism in rabbit brain. Magnetic Resonance in Medicine, 13, 90–102. [DOI] [PubMed] [Google Scholar]
- Ulus I, Wurtman R, Mauron C, Blusztajn J (1989). Choline increases acetylcholine release and protects against the stimulation-induced decrease in phosphatide levels within membranes of rat corpus striatum. Brain Research, 484, 217–227. [DOI] [PubMed] [Google Scholar]
- van Ewijk H, Heslenfeld DJ, Zwiers MP, Buitelaar JK, Oosterlaan J (2012). Diffusion tensor imaging in attention deficit/hyperactivity disorder: a systematic review and meta-analysis. Neuroscience and Biobehavioral Reviews, 36, 1093–1106. [DOI] [PubMed] [Google Scholar]
- Wagner AF, Hunt PS (2006). Impaired trace fear conditioning following neonatal ethanol: reversal by choline. Behavioral Neuroscience, 120(2), 482–87. [DOI] [PubMed] [Google Scholar]
- Wechsler D (1991). Wechsler Intelligence Scale for Children —3rd ed. San Antonio, Psychological Corporation. [Google Scholar]
- Weyrauch D, Schwartz M, Hart B, Klug MG, Burd L (2017). Comorbid mental disorders in fetal alcohol spectrum disorders: A systematic review. Journal of Developmental and Behavioral Pediatrics, 38, 283–291. [DOI] [PubMed] [Google Scholar]
- Woods RP, Grafton ST, Watson JD, Sicotte NL, Mazziotta JC (1998). Automated image registration: II. Intersubject validation of linear and nonlinear models. Journal of Computer Assisted Tomography, 22, 153–165. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Fuglestad AJ, Eckerle JK, Fink BA, Hoecker HL, Boys CJ, Radke JP, Kroupina MG, Miller NC, Brearley AM, Zeisel SH, Georgieff MK (2015). Choline supplementation in children with fetal alcohol spectrum disorders: a randomized, double-blind, placebo-controlled trial. American Journal of Clinical Nutrition 102, 1113–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Fuglestad AJ, Eckerle JK, Kroupina MG, Miller NC, Boys CJ, Brearley AM, Fink BA, Hoecker HL, Zeisel SH, Georgieff MK (2013a). Choline supplementation in children with fetal alcohol spectrum disorders has high feasibility and tolerability. Nutrition Research 33, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Bell CJ, Muetzel RL, Hoecker HL, Boys CJ, Lim KO. (2013b). Global functional connectivity abnormalities in children with fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 37(5), 745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Chang P-N, Muetzel RL, Caros L, Lim KO. (2006). Diffusion tensor Imaging in children with fetal alcohol spectrum disorders. Alcoholism: Clinical and Experimental Research 30(10), 1799–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, Mueller BA, Muetzel RL, Bell CJ, Hoecker HL, Nelson ML, Chang P-N, Lim KO. (2011). Inter-Hemispheric functional connectivity disruption in children with prenatal alcohol exposure. Alcoholism: Clinical and Experimental Research 35(5), 849–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak JR, & Muetzel RL (2011). What does diffusion tensor imaging reveal about the brain and cognition in fetal alcohol spectrum disorders? Neuropsychology Reviews, 21, 133–147. [DOI] [PubMed] [Google Scholar]
- Wozniak JR, Muetzel RL, Mueller BA, McGee CL, Freerks MA, Ward EE, Nelson ML, Chang P-N, Lim KO. (2009). Microstructural corpus callosum anomalies in children with prenatal alcohol exposure: An extension of previous diffusion tensor imaging findings. Alcoholism: Clinical and Experimental Research 33(10), 1825–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wurtman RJ, Bluszajn JK, Maire JC (1985). “Autocannibalism” of choline-containing membrane phospholipids in the pathogenesis of Alzheimer’s disease—a hypothesis. Neurochemistry International, 7, 369–372. [DOI] [PubMed] [Google Scholar]
- Yin X, Han Y, Ge H, Xu W, Huang R, Zhang D, Xu J, Fan L, Pang Z, Liu S (2013). Inferior frontal white matter asymmetry correlates with executive control of attention. Human Brain Mapping, 34(4), 796–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeisel SH (2011). What choline metabolism can tell us about the underlying mechanisms of fetal alcohol spectrum disorders. Molecular Neurobiology 44, 185–191. [DOI] [PMC free article] [PubMed] [Google Scholar]