

Graphic Abstract
Introduction
A wide range of compounds have been identified that can potentiate antibiotic activity. Several studies have examined the structure-activity of various NorA efflux pump inhibitors for their utility against Staphylococcus aureus.1–3 There are also several reviews that have highlighted the significant affect that such agents can have on lowering the minimal inhibitory concentrations (MICs) observed for clinical antibiotics against otherwise resistant Gram (−) bacteria.4–15 Among the agents that can potentiate antibiotic activity are those that can increase the permeability of the bacterial membrane or inhibit bacterial efflux pumps. PAβN (Figure 1) is one of the more extensively studied potentiators of antibiotic activity. It has been shown to affect membrane permeability in a concentration-dependent manner as well as inhibit the efflux pumps of certain bacteria.16 Structural factors that influence the effectiveness of PAβN and related compounds have been investigated.17–23
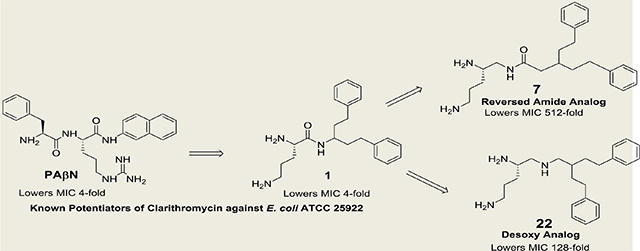
Figure 1.

PAβN, 1 and its reversed amide homologues 2–7.
Studies from our laboratory have indicated that amides, such as 1 (Figure 1), derived from 3-amino-1,5-diphenylpentane can potentiate the antibiotic activity of clarithromycin against Escherichia coli.24 In the present study, the impact on activity of reversing the amide moiety within this series of compounds was investigated. In general, the reversed amides exhibited enhanced potency as potentiators of antibiotic activity against E. coli. Desoxy derivatives of three of the reversed amide derivatives were also synthesized and evaluated for their ability to potentiate antibiotic activity.
Chemistry
The reversed amide derivative of 1, together with several of its structurally-related homologues 2–7 were synthesized. These compounds are listed in Table 1. These compounds were prepared by reaction of N,N’-diprotected triaminopentane derivatives with the requisite carboxylic acid intermediates.
Table 1.
Impact of PAβN and 1–9 on the Activity of Clarithromycin against E. coli ATCC 25922.
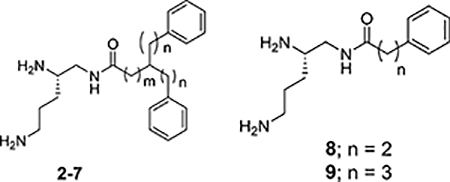 | ||||
|---|---|---|---|---|
| Compound* | m | n | MIC of Clarithromycin (μg/ml) | Fold Reduction in MIC |
| None | - | - | 64 | - |
| 1 | 16 | 4 | ||
| 2 | 0 | 0 | 32 | 2 |
| 3 | 0 | 1 | 64 | 1 |
| 4 | 0 | 2 | 4 | 16 |
| 5 | 1 | 0 | 64 | 1 |
| 6 | 1 | 1 | 4 | 16 |
| 7 | 1 | 2 | 0.125 | 512 |
| 8 | - | 2 | 64 | 1 |
| 9 | - | 3 | 64 | 1 |
| PAβN | - | - | 16 | 4 |
When present, all compounds were evaluated at 12.5 μg/ml, a concentration at least four-fold lower than the intrinsic MIC of each compound (given in Table S1).
The general method used for the preparation of the N,N’-diprotected triaminopentane derivatives is outlined in Scheme 1.
Scheme 1.

General method used for the preparation of N,N’-diprotected triaminopentane derivatives. Reagents and conditions: a) i) N-methylmorpholine, isobutylchloroformate, DME, ii) NaBH4, DME/H2O; b) phthalimide, DIAD, PPh3, THF; c) hydrazine, methanol.
The methods used for the synthesis of the various carboxamides, 2–7, is outlined in Scheme 2.
Scheme 2.

General method for forming the amide derivatives 2–7 using N’,N”-diprotected 2,5-diaminopentylamine and the appropriate acid. Reagents and conditions: a) EDC, HOBT, 2,6-lutidine; b) one step deprotection (20% Pd(OH)2/C, H2, ethanol) or a 2-step deprotection (i)TFA, DCM; ii) 20% Pd(OH)2/C, H2, ethanol).
2,2-Diphenylacetic acid, 2-benzyl-3-phenylpropanoic acid and 3,3-diphenylpropanoic acid were commercially available. The synthesis of 2-phenethyl-4-phenylbutanoic acid was accomplished as previously described. Using a Wittig-Horner reaction, 1,3-diphenyl-2-propanone and 1,5-diphenyl-3-pentanone were converted to 3-benzyl-4-phenylbutanoic acid and 3-phenethyl-5-phenylpentanoic acid by formation of their intermediate ester derivatives followed by subsequent hydrolysis.
The 1-phenylpropanamide and 1-phenylbutyramide derivatives, 8 and 9, were similarly prepared as outlined in Scheme 3 using commercially available phenylpropionic and phenylbutyric acids.
Scheme 3.

Reagents and conditions: a) appropriate carboxylic acid, EDC, HOBT, 2,6-lutidine, DMF (82% for 8) (40% for 9); b) 20% Pd(OH)2/C, H2, ethanol (100% for 8 and 9),
Using 3-carboxy-1,5-diphenylpentane and varied di-protected pentane triamines, a series of isomeric N-(diaminopentyl) 2-phenethyl-4-phenylbutanamides, 10–12, related to 4 were prepared. The general method for the preparation of each of these 2-phenethyl-4-phenylbutanamide derivatives are outlined in Schemes 4.
Scheme 4.

Reagents and conditions: a) i) N-methylmorpholine, isobutylchloroformate, DME, ii) NaBH4, DME/H2O (91%); b) phthalimide, DIAD, PPh3, THF (73%); c) hydrazine, methanol (33%); d) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine, DMF (50%); e) TFA/DCM (86%); f) 20% Pd(OH)2/C, H2, ethanol (78%)
Homologues of 4 were also selected for synthesis and evaluation. These include homologues wherein the alkyl diamine linkage was shortened by one methylene unit (13) and two methylene units (14), requiring the synthesis of di-protected butane and propane triamine derivatives. Using these N’,N”-di-protected triamines, the N-diaminopropane and N-diaminobutane amide derivatives, 13 and 14, were synthesized as illustrated in Scheme 7.
Scheme 7.

Reagents and conditions: a) i) N-methylmorpholine, isobutylchloroformate, DME, ii) NaBH4, DME/H2O (48% for 13) (96% for 14); b) phthalimide, DIAD, PPh3, THF (41% for 13) (54% for 14); c) hydrazine, methanol (61.5% for 13) (44% for 14); d) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine, DMF (86% for 13) (86% for 14); e) 20% Pd(OH)2/C, H2, ethanol (69% for 13) TFA/DCM (79% for 14); f) 20% Pd(OH)2/C, H2, ethanol (81% for 14)
Several structurally-modified derivatives of 4 were also synthesized. These include N-methyl and the N,N-dimethyl derivatives, 15 and 16, respectively. The method used for the preparation of these derivatives is outlined in Scheme 8. We also prepared the hydrazino and the guanidine analogs, 17 and 18 of compound 4 as outlined in Scheme 9.
Scheme 8,

Methods used for the preparation of 15 and 16. Reagents and conditions: a) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine (86%); b) TFA, CH2Cl2 (82%); c) (i) benzaldehyde, ethanol, 3A molecular sieves, NaBH3CN, (ii) 37% formaldehyde, NaBH3CN (for 15) (28%); 20% MeOH/DCM, 37% formaldehyde, sodium triacetoxyborohydride (for 16) (47%); d) 20% Pd(OH)2/C, H2, ethanol (100% for 15), (91% for 16)
Scheme 9,

Methods used for the preparation of 17. Reagents and conditions a) EDC, HOBT, dibenzylamine, DIPEA, 54%; b) LiOH, THF/methanol/water (3:2:2), 95%; c) (i) t-butyl carbazate, EDC, (ii) TFA, DCM, 64%; d) (i) borane-THF, (ii) Boc2O, DCM, 18%; e) (i) 20% Pd(OH)2/C, (ii) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine, 51%; f) TFA, CH2Cl2, 88%;
Both N-(5-amino-2-(methylamino)pentyl)-2-phenethyl-4-phenylbutanamide, 19 and N-(5-amino-2-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide, 20, were synthesized as outlined in Scheme 11.
Scheme 11.

Methods used for the preparation of 19 and 20. Reagents and conditions: a) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine (45%); b) TFA, CH2Cl2 (78%); c) (i) benzaldehyde, ethanol, 3A molecular sieves, NaBH3CN, (ii) 37% formaldehyde, NaBH3CN (for 19) (31%); 20% MeOH/DCM, 37% formaldehyde, sodium triacetoxyborohydride (for 20) (79%); d) (i) 20% Pd(OH)2/C, H2, ethanol, Boc2O, (ii) TFA, DCM (61% for 19); (100% for 20)
The effect of modifying the secondary amide moiety to a tertiary amide was examined by the synthesis and evaluation of N-(2,5-diaminopentyl)-N-methyl-2-phenethyl-4-phenylbutanamide, 21, which was prepared as outlined in Scheme 12.
Scheme 12:

Methods used for the preparation of 21. Reagents and conditions: a) HOBT, EDC, DIPEA (82%); b) TFA, DCM (100%); c) (i) BH3·THF, THF, (ii) Boc2O, DCM (39%); d) 20% Pd(OH)2/C, H2, ethanol e) 2-phenethyl-4-phenylbutanoic acid, PyBrOP, Et3N, DCM (78%); f) TFA, DCM (100%)
Results and Discussion
The relative enhancement in the observed potency of clarithromycin against E. coli ATCC 25922 in the presence of the varied acyl bis-alkylphenyl derivatives of 1,2,5-triaminopentane (2-7) is provided in Table 1.
For this series of compounds, a greater reduction in clarithromycin MIC is seen when the acyl moiety is attached directly to a carbon atom with bis-(2-phenyl)ethyl groups (4 > 2 and 3) and even more so when the acyl moiety is attached via a methylene linker (7 >> 5 and 6). In this regard, 7 represents the most potent potentiator elucidated from the present study with a 128-fold greater potency than PAβN. The acyl mono-alkylphenyl derivatives of 1,2,5-triaminopentane (8 and 9) had no effect on the activity of clarithromycin.
Compound 10 (the R-enantiomer of 4) was also prepared and exhibited similar potentiation of clarithromycin activity (see Table 2). Several compounds derived from 2-phenethyl-4-phenylbutanoic acid were also synthesized and evaluated for their potentiation activity. The relative activities for amide derivatives formed from two isomeric triaminopentanes (11 and 12) and two isomeric trimaminobutanes (13 and 14) are listed in Table 2. While 11 had similar activity to 4, compound 12 was slightly more potent and 13 was significantly less potent as potentiators of clarithromycin activity.
Table 2.
Impact of 10–14 and 22–24 on the Activity of Clarithromycin against E. coli ATCC 25922.
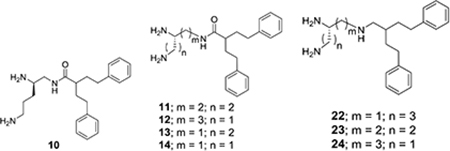 | ||||
|---|---|---|---|---|
| Compound* | m | n | MIC of Clarithromycin (μg/ml) | Fold Reduction in MIC |
| 10 | 4 | 16 | ||
| 11 | 2 | 2 | 4 | 16 |
| 12 | 3 | 1 | 2 | 32 |
| 13 | 1 | 2 | 32 | 2 |
| 14 | 1 | 1 | 4 | 16 |
| 22a | 1 | 3 | 0.5 | 128 |
| 23b | 2 | 2 | 1 | 64 |
| 24c | 3 | 1 | 2 | 32 |
All compounds were present at 12.5 μg/ml, a concentration at least four-fold lower than the intrinsic MIC of each compound (given Table S1).
Desoxy derivative of 4.
Desoxy derivative of 11
Desoxy derivative of 12.
Methylation of either of the primary amines of 4, as in either 15 and 16 or in 19 and 20, resulted in derivatives with decreased activity (see Table 3). While the hydrazine derivative 17 was inactive, the guanidine derivative 18 did retain similar activity to 4. Conversion of 4 to its N-methyl tertiary amide, 21, resulted in a loss of activity.
Table 3.
Impact of 15–21 on the Activity of Clarithromycin against E. coli ATCC 25922.
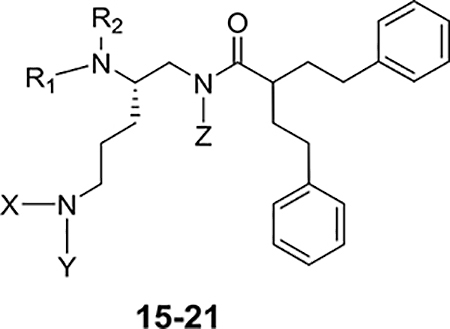 | |||||||
|---|---|---|---|---|---|---|---|
| Compound* | X | Y | Z | R1 | R2 | MIC of Clarithromycin (μg/ml) | Fold Reduction in MIC |
| 15 | CH3 | H | H | H | H | 32 | 2 |
| 16 | CH3 | CH3 | H | H | H | 32 | 2 |
| 17 | NH2 | H | H | H | H | 61 | 1 |
| 18 | CNH(NH2) | H | H | H | H | 16 | 4 |
| 19 | H | H | H | CH3 | H | 64 | 1 |
| 20 | H | H | H | CH3 | CH3 | 32 | 2 |
| 21 | H | H | CH3 | H | H | 64 | 1 |
All compounds were present at 12.5 μg/ml, a concentration at least four-fold lower than the intrinsic MIC of each compound (given in Table S1).
One of the more dramatic effects on activity from structure modification was seen for 22, 23, and 24, which represent the desoxy derivatives of 4, 11 and 12, respectively (see Table 2). All of these triamine derivatives were significantly more active as potentiators than their analogous amides. Conversion of the amide 4 to the triamine 22 provided one of the more potent potentiators in this study with a 32-fold greater potency than PAβN.
The two most potent compounds (7 and 22) at potentiating the antibacterial activity of clarithromycin were also evaluated for their ability to potentiate the activity of four additional antibiotics (minocycline, novobiocin, clindamycin, and azithromycin) against E. coli ATCC 25922. Significantly, both 7 and 22 were effective at potentiating the activities of each of the four additional antibiotics (see Table 4), with 7 inducing MIC reductions in the range of 8- to 128-fold and 22 inducing MIC reductions in the range of 4- to 32-fold. Both compounds potentiated novobiocin activity the most and minocycline activity the least. Aerobic and many anaerobic Gram-negative bacteria are resistant to clindamycin, a spectrum of activity consistent with the high intrinsic MIC of this antibiotic (128 μg/ml) against E. coli (Table 4). Interestingly, both 7 and 22 reduced the MIC of clindamycin by 16-fold (from 128 to 8 μg/ml). This observation suggests that potentiating compounds like 7 and 22 offer the potential for expanding the spectrum of clindamycin activity to include a broader array of Gram-negative bacterial species.
Table 4.
Impact of 7 and 22 on the Activities of the Antibiotics Minocycline, Novobiocin, Clindamycin, and Azithromycin against E. coli ATCC 25922
| Antibiotic | Intrinsic MIC (μg/ml) | In the Presence of 7 @ 12.5 μg/ml | In the Presence of 22 @ 12.5 μg/ml | ||
|---|---|---|---|---|---|
| MIC (μg/ml) | Fold Reduction in MIC | MIC (μg/ml) | Fold Reduction in MIC | ||
| Minocycline | 1 | 0.125 | 8 | 0.25 | 4 |
| Novobiocin | 64 | 0.5 | 128 | 2 | 32 |
| Clindamycin | 128 | 8 | 16 | 8 | 16 |
| Azithromycin | 8 | 0.125 | 64 | 0.5 | 16 |
Additional cell-based studies were conducted on 7 and 22 with the aim of understanding the mechanism underlying the actions of these compounds as antibiotic potentiators. Initial studies were focused on probing the potential of 7 and 22 to inhibit the activity of efflux pumps in E. coli ATCC 25922, which include the AcrAB-TolC efflux pump, using a cell-based fluorescence assay of efflux, with Hoechst 33342 (H33342), a known substrate of Gram-negative bacterial efflux pumps, as the readout fluorophore.25,26 When bound to chromosomal DNA inside the cell, H33342 fluoresces brightly, with unbound dye outside the bacterial cell exhibiting very weak fluorescence. Efflux of H33342 from inside to outside the cell is therefore associated with a decrease in fluorescence. In the efflux assay, efflux pump activity of the E. coli cells is initially repressed by depletion of ATP using carbonyl cyanide 3-chlorophenylhydrazone (CCCP). In the absence of ATP, H33342 accumulates inside the cell, resulting in increased DNA binding and thus increased fluorescence. Efflux pump activity in the cells is then assayed in real-time by reenergizing the pumps through the addition of glucose. With the addition of glucose, pump-mediated efflux of H33342 outside the cell induces a time-dependent decrease in fluorescence. No such fluorescence decrease occurs in the absence of added glucose (Figure 2). Examination of the impact that increasing concentrations of 7 and 22 have on the fluorescence decrease stimulated by the addition of glucose reveals that both compounds inhibit pump-mediated efflux of H33342 to a similar degree, with the extent of this inhibition being dependent on the concentration of the test compound (Figure 2).
Figure 2.

Assay for inhibition of the efflux of the fluorophore Hoechst 33342 (H33342) by 7 (A) or 22 (B) in E. coli ATCC 25922. All reactions were initiated by the addition of 50 mM glucose, except where indicated. H33342 was used at a concentration of 10 μM. Both compounds were used at concentrations ranging from 1/32X to ¼X MIC (1.56 to 12.5 μg/ml for 7 and 3.125 to 25 μg/ml for 22). The intrinsic MICs of 7 and 22 against E. coli ATCC 25922 are 50 and 100 μg/ml, respectively. Compound vehicle was DMSO.
In addition to being assessed for their abilities to inhibit efflux pump activity, 7 and 22 were also evaluated for their potential to permeabilize the outer and inner cell membranes of E. coli ATCC 25922 cells. A cell-based fluorescence assay using N-phenyl-1-naphthylamine (NPN) as the readout fluorophore27 was used to assess 7 and 22 for their potential to permeabilize the outer cell membrane. In the absence of a permeabilizer, NPN cannot penetrate the outer cell membrane, which contains the lipopolysaccharide (LPS) layer. Permeabilizers of the outer membrane allow NPN to interact with hydrophobic lipids in the outer as well as inner cell membranes, whereupon the dye undergoes a marked increase in fluorescence emission.27 As shown in Figures 3A and B, both 7 and 22 significantly increase NPN fluorescence in a concentration dependent manner, indicating that both compounds effectively permeabilize the outer cell membrane. Note that NPN has been shown to be substrate of the AcrAB-TolC efflux pump in E. coli.13 Thus, the efflux pump inhibiting activities associated with 7 and 22, as described above, may also be contributing factors to the increase in NPN fluorescence induced by these compounds.
Figure 3.

Membrane permeabilization assays of 7 and 22 in E. coli ATCC 25922. Both test compounds were used at concentrations ranging from 1/32X to ¼X MIC (1.56 to 12.5 μg/ml for 7 and 3.125 to 25 μg/ml for 22). The intrinsic MICs of 7 and 22 against E. coli ATCC 25922 are 50 and 100 μg/ml, respectively. The compound vehicle was DMSO. (A,B) Impact of E. coli treatment with 7 (A) or 22 (B) on the intensity of the fluorophore N-phenyl-1-naphthylamine (NPN) at a concentration of 10 μM. (C,D) Impact of treatment with 7 (C) or 22 (D) on the percentage of E. coli cells stained with the fluorophore propidium iodide (PI) at a concentration of 50 μM.
We further characterized the membrane permeabilizing activities of 7 and 22 using a fluorescence-activated cell sorting (FACS) assay and propidium iodide (PI) as the readout fluorophore.28 In the absence of a permeabilizer, PI cannot cross the outer or inner cell membranes to enter the cell. However, a compound that permeabilizes both membranes enables PI to enter the cell and interact with the chromosomal DNA, whereupon the dye undergoes a significant increase in fluorescence emission.28 The FACS assay determines the percentage of cells that exhibit increased PI fluorescence. The results of this assay (shown in Figures 3C and D) reveal that 7 and 22 permeabilize not only the outer cell membrane, but the inner cell membrane as well. Furthermore, 7 appears to be a more potent permeabilizer of the inner membrane than 22. It is interesting to note that the compounds with the more potent activity as antibiotic potentiators are also the compounds with the greater intrinsic antibacterial activity (compare Tables 1–4 with Table S1). This general correlation is consistent with membrane permeabilization being an important contributor to the antibiotic potentiating activities of the compounds.
Another possible mechanism for potentiation of antibiotic activity is through disruption of the proton gradient and potential across the inner cell membrane. We examined 7 and 22 for this possibility in E. coli ATCC 25922 cells using a FACS assay and 3,3′-diethyloxacarbocyanine iodide (DiOC2) as the readout fluorophore.29,30 In healthy bacterial cells that maintain a membrane potential, DiOC2 accumulates inside the cell at high concentrations, causing the dye to self-associate and exhibit red fluorescence emission. However, when the potential across the membrane is disrupted, the intracellular concentration and thus self-association of DiOC2 is significantly reduced, whereupon its fluorescence emission shifts from red to green wavelengths.30 This shift is manifested by a corresponding decrease in the ratio of red to green fluorescence (λred/λgreen). For comparative purposes, we used CCCP, a prototypical disruptor of membrane potential, as a positive control in this assay. The results of this assays are shown in Figure S1. In marked contrast to CCCP, neither 7 nor 22 disrupted the membrane potential to any significant degree (Figure S1). In fact, 22 appeared to increase rather than decrease this potential.
Conclusions
These studies indicate that the reversed analogs of 1, in general, were significantly more active in potentiating the antimicrobial of clarithromycin against E. coli ATCC 25922. Major differences were observed in the ability of the structurally-related diamino N-acyl arylalkylamides 2–9 to potentiate the antibacterial of clarithromycin. The absence of a bis-arylalkyl moiety as in 8 and 9 was associated with a loss in activity. Relative to 4, mono- or dimethylation of either of its primary amines (as in 15,16, 19, or 21) significantly reduced its ability to potentiate the antibacterial activity of clarithromycin. In the case of 7, the N1-1,2,5-triaminopentyl amide of 3,3,-dibenzylpropanoic acid, potency was at least 32-fold greater than 2–6. Compound 7 proved to be the most active analog, lowering the MIC of clarithromycin by 512-fold and having 128-fold greater potency than PAβN.
The desoxy derivatives 22 and 23 were 8-fold and 4-fold more potent than their carboxamides 4 and 11, respectively. Compound 22 was among the more potent potentiators evaluated in this study with a 128-fold decrease in the MIC when evaluated together with clarithromycin. These data indicate that such alkyl triamines can also exhibit significant activity.
Compounds 7 and 22 were effective at potentiating the activity of not only clarithromycin, but also of other antibiotics, including minocycline, novobiocin, clindamycin, and azithromycin. The collective results of cell-based studies suggest that the antibiotic potentiating activities of 7 and 22 reflect contributions from both membrane permeabilization and efflux pump inhibition, but not from disruption of the proton gradient and potential across the membrane.
Experimental
Chemistry: General Methods
All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates with UV254 (Sorbent Technologies), visualizing with ultraviolet light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO using hexane, ethyl acetate, dichloromethane, and methanol. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were done in CDCl3, Methanol-d4, and DMSO-d6 and recorded on a Bruker Avance III (400 MHz) Multinuclear NMR Spectrometer. Data is expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), m (multiplet), and bs (broad singlet), and coupling constants (J) are reported in Hertz. Melting points were determined using Mel-temp II apparatus and are uncorrected. Analytical HPLC was performed on a Shimadzu LC-20AT Prominence liquid chromatograph using a 150 × 4.6 mm Princeton SPHER-100 RP C18 1000A 5 u column using 0% water for 2 minutes and a 0–100% water/methanol gradient over a 5 minute period and 5 minutes at 100% methanol at a 2.0 ml/minute flow rate monitoring uv absorbance at 254 and 296 nm. Using this method of analysis, the purity of all compounds used in bioassays was determined to be ≥ 95%. HRMS experiments were conducted by Washington University Resource for Biomedical and Bioorganic Mass Spectrometry Department of Chemistry.
(S)-N-(2,5-Diaminopentyl)-2,2-diphenylacetamide (2)
Benzyl t-butyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate
To a solution of (S)-5-(((benzyloxy)carbonyl)amino)-2-((t-butoxycarbonyl)amino)-pentanoic acid (5.0 g, 13.6 mmol) in DME (25 mL) at −15 °C were successively added a solution of N-methyl morpholine (1.7 mL, 15.4 mmol) and isobutyl chloroformate (1.8 mL, 13.65 mmol). The reaction was stirred at 15 °C to −10 °C for 15 minutes. The precipitated N-methyl morpholine HCl was removed by filtration and washed with DME (10 mL), the combine filtrates were chilled to −15 °C in an ice-salt bath. Then a solution of sodium borohydride (1.55 g, 40.95 mmol) in water (10 mL) was added in one portion at −15 °C. This reaction mixture was stirred at this temperature for 10 minutes. The reaction was quenched by the addition of saturated aq. NH4Cl and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate. The solution was then filtered and concentrated under reduced pressure the purified on an ISCO chromatograph (0–70% ethyl acetate/hexane) to give the product as a colorless oil (3.81g, 79%); 1H NMR (CDCl3) (400 MHz) δ 7.34 (s, 5H), 5.29 (brs, 1H), 5.07 (s, 2H), 5.04 (brs,1H), 3.55 (m, 3H), 3.18 (m, 2H), 1.53 (m, 4H), 1.43 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.6, 156.3, 136.6, 128.4, 128.0, 79.4, 66.5, 64.9, 52.1, 40.8, 28.6, 28.4, 26.4.
Benzyl t-butyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(S)-dicarbamate
Triphenylphosphine (3.26 g 12.44 mmol) and phthalimide (1.83 g, 12.44 mmol) were added to a flask containing dry THF (15 mL). Benzyl t-butyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate (3.65g, 10.4 mmol) was added and the flask was cooled to 0°C. DIAD (2.45 mL, 12.4 mmol) was added dropwise and reaction was allowed to stir for 30 minutes at 0 °C and then overnight at room temperature. The mixture was concentrated under reduced pressure and the residue purified using an ISCO chromatograph with silica (0–70% ethyl acetate/hexane) to give product as a pale yellow solid. (3.9 g, 79%); MP 131–133 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.85 (m, 2H), 7.71 (m, 2H), 7.37 ( m, 5H), 5.10 (s, 2H), 4.99 (brs, 1H), 4.70 (d, 1H, J = 8), 3.98 (m, 1H), 3.70 (m, 2H), 3.24(m, 2H), 1.60 (m, 4H), 1.24 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 168.5, 156.4, 155.7, 136.6, 133.9, 132.0, 128.4, 128.0, 123.3, 79.2, 66.5, 49.7, 42.2, 40.7, 30.0, 28.0, 26.4
Benzyl t-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate
Benzyl t-butyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(S)-dicarbamate (3.92 g, 8.1 mmol) was dissolved in methanol (30 mL) and hydrazine monohydrate (0.8 mL, 16.3 mmol) was added. The reaction mixture was then refluxed for 2 hours and cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was concentrated under reduced pressure and the remaining solid purified using an ISCO chromatograph (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a yellow oil (500 mg, 18%); 1H NMR ( CDCl3) ( 400 MHz) δ 7.32 (m, 5H), 5.28 (m, 1H), 5.08 (s, 2H), 4.85 (d, 1H, J = 8), 3.50 (m, 1H), 3.19 (m, 2H), 2.71 (m, 1H), 2.60 (m, 1H), 1.51 (m, 4H), 1.43 (s, 9H); 13C NMR (CDCl3) ( 100 MHz) δ 156.5, 156.1, 136.6, 128.4, 128.0, 79.1, 66.5, 52.6, 45.9, 40.8, 30.0, 28.4, 26.5.
Benzyl t-butyl (5-(2,2-diphenylacetamido)pentane-1,4-diyl)(S)-dicarbamate
2,2-Diphenylacetic acid (63 mg, 0.29 mmol) was dissolved in dry DMF (5 mL) and EDC (113 mg, 0.59 mmol) and HOBt (80 mg, 0.59 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl t-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (95 mg, 0.27 mmol) was added, followed by 2,6-lutidine (0.09 mL, 0.81 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated under reduced pressure and purified using an ISCO chromatograph with silica (0 – 10% MeOH/dichloromethane) to give a white flaky solid (110 mg, 75%); MP 158–160 °C; 1H NMR (400 MHz) (CDCl3) δ 7.31 (m, 16H), 6.49 ( brs, 1H), 5.12 (brs, 2H), 5.10 (s,2H), 4.89 (m, 2H), 3.61 (m, 1H), 3.27 (m, 2H), 3.15 (m, 2H), 1.50 (m,4H), 1.43 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 172.7, 156.5, 156.3, 139.4, 136.6, 128.8, 128.6, 128.5, 128.0, 127.2, 79.50, 66.5, 58.9, 50.7, 44.3, 40.6, 30.0, 23.4, 26.2
Benzyl (S)-(4-amino-5-(2,2-diphenylacetamido)pentyl)carbamate
Benzyl t-butyl (5-(2,2-diphenylacetamido)pentane-1,4-diyl)(S)-dicarbamate (100 mg, 0.18 mmol) was dissolved in dichloromethane ( 3mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2 mL) was added and reaction stirred at that temperature for 2 hours. The reaction mixture was dissolved in saturated NaHCO3 and the organic layer separated. The combined organic layers were dried over sodium sulfate and solvent removed under reduced pressure to give product as a colorless oil (76 mg, 94%); 1H NMR (400 MHz) (CDCl3) δ 7.18 (m, 15H), 6.66 (brs, 1H), 5.11 (brs, 1H), 4.97 (s, 2H), 4.82 (s, 1H), 3.17 (m, 3H), 2.98 (m, 3H), 2.70 (brs, 1H), 1.21 (m, 5H); 13C NMR (100 MHz) (CDCl3) δ 172.8, 156.6, 139.47, 139.44, 136.6, 128.84, 128.81, 128.7, 128.5, 128.1, 128.0, 127.2, 66.6, 58.7, 50.9, 44.6, 40.7, 31.4, 26.1
(S)-N-(2,5-Diaminopentyl)-2,2-diphenylacetamide
Benzyl (S)-(4-amino-5-(2,2-diphenylacetamido)pentyl)carbamate (35 mg, 0.08 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg) was added. The reaction mixture was then purged and stirred under hydrogen atmosphere overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried on a vacuum pump to give product as a colorless oil. (20 mg, 83%); 1H NMR (400 MHz) (CDCl3) δ 7.21 (m, 10H), 6.64 (m, 1H), 4.87 (s, 1H), 3.25 (m, 1H), 2.95 (m, 1H), 2.84 (brs, 4H), 2.70 (m, 1H), 2.57 (m, 2H), 1.38 (m,4H); 13C NMR (100 MHz) (CDCl3) δ 172.5, 139.6, 139.5, 128.9, 128.8, 128.6, 127.2, 58.8, 50.8, 45.5, 41.2, 32.7, 28.5; HRMS (ESI) Calculated for C19H25N3O (M+H)+ 312.2070, found 312.2065
(S)-2-Benzyl-N-(2,5-diaminopentyl)-3-phenylpropanamide (3)
Benzyl t-butyl (5-(2-benzyl-3-phenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate
2-Benzyl-3-phenylpropanoic acid (68 mg, 0.29 mmol) was dissolved in dry DMF (5 mL) and EDC (109 mg, 0.57 mmol) and HOBt (77 mg, 0.57 mmol) were added and the reaction stirred at room temperature for 5minutes. Benzyl t-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (91 mg, 0.26 mmol) was added followed by 2,6-lutidine (0.09 mL, 0.78 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated under reduced pressure and purified using an ISCO chromatograph with silica (0°C; 1H NMR (400 MHz) (CDCl–10% MeOH/dichloromethane) to give a white flaky solid, (119 mg, 80%); MP 132–134 °C; 1H NMR (400 MHz) (CDCl3) δ 7.26 (m, 15H), 5.59 (brs, 1H), 5.11 (s, 3H), 4.46 (d, 1H, J = 8), 3.32 (m, 1H), 3.05 (m, 6H), 2.80 (m, 2H), 2.61 (m, 1H), 1.44 (s, 9H), 1.41 (m, 2H), 1.07 (m, 2H); 13C NMR (400 MHz) (CDCl3) δ 174.8, 156.5, 155.8, 139.69, 139.62, 136.6, 128.9, 128.5, 128.49, 128.41, 128.0, 126.4, 126.3, 79.3, 66.5, 52.3, 50.5, 42.9, 40.6, 38.9, 38.6, 29.1, 28.4, 26.1
Benzyl (S)-(4-amino-5-(2-benzyl-3-phenylpropanamido)pentyl)carbamate
Benzyl t-butyl (5-(2-benzyl-3-phenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate (81 mg, 0.14 mmol) was dissolved in dichloromethane (3 mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2mL) was added and reaction stirred at that temperature for 2hrs. The reaction mixture was dissolved in saturated NaHCO3 and the organic layer separated. The combined organic layers were dried over sodium sulfate and solvent removed under reduced pressure to give product as a colorless oil. (62 mg, 93%); 1H NMR (400 MHz) (CDCl3) δ 7.17 (m, 15H), 6.19 (brs, 1H), 5.12 (brs, 1H), 4.98 (s, 2H), 4.85 (brs, 2H), 2.80 (m, 10H), 1.32 (m, 2H), 1.05 (m, 2H); 13C NMR (400 MHz) (CDCl3) δ 175.0, 156.7, 139.58, 139.50, 136.4, 129.0, 128.9, 128.5, 128.49, 128.47, 128.1, 128.0, 126.46, 126.42, 66.7, 52.0, 51.0, 43.1, 40.4, 38.9, 38.8, 29.6, 25.8
(S)-2-Benzyl-N-(2,5-diaminopentyl)-3-phenylpropanamide
Benzyl (S)-(4-amino-5-(2-benzyl-3-phenylpropanamido)pentyl)carbamate (42 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg) was added. The reaction mixture was then purged and stirred under hydrogen atmosphere for overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried on a vacuum pump to give product as a colorless oil. (26 mg, 86%); 1H NMR (CDCl3) (400 MHz) δ 7.15 (m, 10H), 6.02 (m, 1H), 3.81 (brs, 4H), 3.03 −2.37 (m, 10H), 1.46 (m, 1H), 1.36 (m, 1H), 0.92 (m,1H), 0.80 (m, 1H); 13C NMR (CDCl3) (100 MHz) δ 174.5, 139.7, 139.6, 129.0, 128.9, 128.45, 128.44, 126.3, 52.2, 50.5, 44.8, 40.6, 39.1, 38.8, 32.0, 29.7, 27.5; HRMS (ESI) Calculated for C21H29N3O (M+H)+ 340.2383, found 340.2380
(S)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide (4)
Diethyl 2,2-diphenethylmalonate
A 60% dispersion of sodium hydride (1.25 g, 31.25 mmol) was added to a solution of diethyl malonate (1.90 mL, 12.5 mmol) in DMF (20 mL). The mixture was stirred at room temperature for 15 minutes. Then 2-bromoethyl) benzene (7 mL, 52.5 mmol) was added and the reaction mixture was warmed to 50 °C and stirred for 4 hours. The reaction was then allowed to reach room temperature, diluted with brine and extracted with diethyl ether. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The concentrate was purified using an ISCO chromatograph with silica using 0–5% ethyl acetate/ hexane to give a colorless oil (1.06 g, 23%); 1H NMR (400 MHz) (CDCl3) δ 7.42 (m, 4H), 7.34 (m, 6H), 4.34 (q, 4H), 2.72 (m, 4H), 2.47 (m, 4H), 1.41 (t, 6H, J = 4); 13C NMR (100 MHz) (CDCl3) δ 171.3, 141.5, 128.6, 128.5, 126.2, 61.2, 57.6, 34.9,30.9, 14.3
2-(Ethoxycarbonyl)-2-phenethyl-4-phenylbutanoic acid
Diethyl 2,2-diphenethylmalonate (1g, 2.9 mmol) was dissolved in 95% ethanol (25 mL) and water (7 mL) and KOH (178 mg, 3.2 mmol) was added. The mixture was refluxed for 4 hours. The ethanol was removed under reduced pressure and water was added. The mixture was washed with ether and the aqueous solution was acidified with conc. HCl at 0 °C. The mixture was extracted with ether and the combined ethereal layers washed with water and dried over anhydrous sodium sulfate to give a colorless oil (520 mg, 53%); 1H NMR (400 MHz) (MeOD) δ 7.21 ( m, 10H), 4.17 (q, 2H), 2.53 (m, 4H), 2.24 (m, 4H), 1.25 (t, 3H, J = 8); 13C NMR (100 MHz) (MeOD) δ 173.2, 172.7, 142.9, 142.7, 129.6, 129.59, 129.53, 129.47, 129.45, 127.2, 127.1, 62.5, 62.4, 58.97, 58.94, 36.0, 35.9, 31.9, 31.8, 14.64, 14.60
Ethyl 2-phenethyl-4-phenylbutanoate
A solution of 2-(ethoxycarbonyl)-2-phenethyl-4-phenylbutanoic acid (520 mg, 1.53 mmol) in pyridine/water solution (14 mL) (6:1) was heated to reflux for 72 hours. The excess solvent was evaporated under reduced pressure and the residue acidified to pH = 2 with 1M HCl. The mixture was extracted with ethyl acetate and the combined organic fractions were washed with brine and dried over sodium sulfate. It was then filtered and evaporated and purified on ISCO chromatograph with silica (0–50% ethyl acetate/ hexane) to give product as a colorless oil (228 mg, 50%); 1H NMR (400 MHz) (CDCl3) δ 7.10 (m, 10H), 4.05 (q, 2H), 2.48 (m, 4H), 2.33 (m, 1H), 1.89 (m, 2H), 1.68 (m, 2H), 1.19 (t, 3H, J = 4); 13C NMR (100 MHz) (CDCl3) δ 175.8, 141.7, 128.5, 128.49, 128.46, 126.0, 60.3, 44.8, 34.2, 33.6, 14.0
2-Phenethyl-4-phenylbutanoic acid
A mixture of ethyl 2-phenethyl-4-phenylbutanoate (200 mg, 0.44 mmol) and KOH (98 mg, 1.76 mmol) in ethanol/ water (3 mL: 2 mL) was heated at 70 °C for 20 hours. The mixture was cooled to room temperature under reduced pressure and residue was extracted with ethyl acetate. The combined extracts were washed with brine and dried over sodium sulfate. This was then filtered and evaporated under reduced pressure to give product was a colorless oil; (117 mg, 100%); 1H NMR (400 MHz) (CDCl3) δ 10.74 (brs, 1H), 7.13 (m, 10H), 2.55 (m, 4H), 2.38 (m, 1H), 1.93 (m, 2H), 1.74 (m, 2H); 13C NMR (100 MHz) (CDCl3) δ 182.5, 141.4, 128.4, 126.0, 44.5, 33.8, 33.5
Dibenzyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate
To a solution of (S)-2,5-bis(((benzyloxy)carbonyl)amino)pentanoic acid (1000 mg, 2.5 mmol) in DME (10 mL) at −15 °C were successively added a solution of N-methyl morpholine (310 μL, 2.82 mmol) and isobutyl chloroformate (320 μL, 2.5 mmol). The reaction was stirred at −15 °C to −10 °C for 15 minutes. The precipitated N-methyl morpholine HCl was removed by filtration and washed with DME (10 mL), the combine filtrates were chilled to −15 °C in an ice-salt bath. Then a solution of sodium borohydride (283 mg, 7.5 mmol) in water (4 mL) was added in one portion at −15 °C. This reaction mixture was stirred at this temperature for 10 minutes. The reaction was quenched by the addition of saturated aq. NH4Cl and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate. The solution was then filtered and concentrated under reduced pressure, purified on column (0–70% ethyl acetate/ hexane) to give product as a white powder (508 mg, 52%); MP 128–129 °C; 1H NMR (CDCl3) (400 MHz) δ 7.34 ( m, 10H), 5.07 (m, 6H), 3.69 (m, 3H), 3.22 (m, 2H), 1.54 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 156.6, 156.5, 136.5, 136.3, 128.54, 128.52, 128.2, 128.1, 66.8, 66.7, 65.1, 52.8, 40.7, 28.5, 26.5
Dibenzyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(S)-dicarbamate
Triphenylphosphine (325 mg, 1.24 mmol) and phthalimide (182 mg, 1.24 mmol) were added to a flask containing dry THF (5 mL). Dibenzyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate (400 mg, 1.03 mmol) was added and the flask was cooled to 0 °C. DIAD (250 mg, 1.24 mmol) was added dropwise and reaction allowed to stir for 30 minutes at 0 °C and then overnight at room temperature. The mixture was concentrated under reduced pressure and residue purified using an ISCO chromatograph with silica (0–70% ethyl acetate/hexane) to give product as a white solid. (491 mg, 92%); MP 99–101 °C; 1H NMR (CDCl3) (400 MHz) δ 7.83 (m, 2H), 7.72 (m, 2H), 7.32 (m, 10H), 5.22 (brs,1H), 5.09 (s, 2H), 5.04 (brs,1H), 4.97 (s, 2H), 4.03 (m, 1H) 3.76 (m, 2H), 3.24 (m, 2H), 1.57 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 168.5, 156.4, 156.2, 136.6, 136.5, 134.0, 132.1, 123.0, 131.9, 131.8, 128.6, 128.5, 128.4, 128.3, 128.0, 127.9, 127.8, 123.4, 66.6, 66.5, 50.7, 41.7, 40.6, 30.0, 26.3.
Dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate
Dibenzyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(S)-dicarbamate (400 mg, 0.78 mmol) formed was dissolved in methanol (20 mL) and hydrazine monohydrate (80 μL, 1.55 mmol) was added. The reaction mixture was then refluxed for 2 hours and cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was concentrated under reduced pressure and the remaining solid purified using an ISCO chromatograph with silica (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a white powder. (206 mg, 68%); MP 113–115 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.36 (m, 10H), 5.29 (brs, 1H), 5.22 (brs, 1H), 5.09 (s, 4H), 3.60 ( m, 1H), 3.19 (m, 2H), 2.70 (m, 2H), 1.70 (s, 2H), 1.46 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 156.6, 136.6, 136.5, 128.53, 128.51, 128.1, 128.0, 66.6, 66.5, 53.0, 45.6, 40.7, 29.7. 26.5
Dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (86.3 mg, 0.28 mmol) was dissolved in dry dichloromethane (5 mL) and oxalyl chloride (48 μL, 0.55 mmol) was added followed by a catalytic amount of DMF (2 drops). The reaction was stirred at room temperature for 1 hour. The solvent was then evaporated and residue was pumped dry. The residue was redissolved in dichloromethane (5 mL), then dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (87 mg, 0.23 mmol) and triethylamine (60 μL, 0.40 mmol) was added. The reaction was allowed to stir at room temperature overnight. The reaction mixture was then dissolved in saturated sodium bicarbonate and extracted with dichloromethane. The combined organic layers were then dried over sodium sulfate and purified using an ISCO chromatograph (0–10% MeOH/dichloromethane) to give product as a white solid. (106.4 mg, 73%); MP 171–173 °C; 1H NMR (400 MHz) (CDCl3) δ 7.32 (m, 21H), 5.06 (m, 3H), 4.85 (d, 1H, J = 12), 3.71 (m, 1H), 3.43 (m, 1H), 3.19 (m, 3H), 2. 54 (m, 4H), 2.10 (m, 1H), 1.95 (m, 2H), 1.52 (m, 6H); 13C NMR (100 MHz) (CDCl3) δ 176.30, 156.8, 156.5, 141.6, 136.6, 136.2, 128.7, 128.5, 128.46. 128.43, 128.3, 128.0, 125.9, 125.7, 66.8, 66.4, 53.4, 51.8, 46.5, 43.6, 40.6, 34.4, 34.3, 33.6, 33.5, 30.0, 26.3.
(S)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
A gel like suspension of dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate, (75 mg, 0.12 mmol), 20% Pd(OH)2 /C (20 mg) and ethanol (10 mL) was purged and stirred under a hydrogen atmosphere overnight. The catalyst was then filtered and washed with 20% MeOH/dichloromethane. The solution was then concentrated and purified on ISCO chromatograph (0–20% MeOH/dichloromethane) to give the product as a colorless oil. (19.5 mg, 45%); 1H NMR of triflate salt (400 MHz) (MeOD) δ 7.22 (m, 10H), 3.46 (m, 2H), 2.97 (m, 2H), 2.61 (m, 4H), 2.40 (m, 1H), 1.96 (m, 2H), 1.83 (m, 6H); 13C NMR (100 MHz) (MeOD) δ 180.1, 143.0, 129.4, 129.3, 126.9, 52.7, 47.5, 42.0, 40.1, 35.6, 35.5, 34.7, 28.5, 24.4; HRMS (ESI) Calculated for C23H34N3O (M+H)+ 368.2696, found 368.2687
(S)-N-(2,5-Diaminopentyl)-3,3-diphenylpropanamide (5)
Benzyl t-butyl (5-(3,3-diphenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate
3,3-Diphenylpropanoic acid (61 mg, 0.27 mmol) was dissolved in dry DMF (5mL) and EDC (105 mg, 0.55 mmol) and HOBt (74 mg, 0.55 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl t-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (86 mg, 0.25 mmol) was added followed by 2,6-lutidine (0.09 mL, 0.75 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated under reduced pressure and purified using an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a whitish yellow flaky solid. (101 mg, 77%); MP 102–104 °C; 1H NMR (400 MHz) (CDCl3) δ 7.29 (m, 16H), 6.17 (brs, 1H), 5.12 (s, 3H), 4. 57 (t, 2H, J = 8), 3.44 (m, 1H), 3.13 (m, 3H), 2.92 (m, 3H), 1.46 (s, 9H), 1.24 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 171.8, 156.6, 156.1, 143.6, 136.6, 128.6, 128.5, 128.1, 128.0, 127.7, 126.5, 79.5, 66.6, 50.5, 47.4, 43.5, 43.1, 40.6, 29.2, 28.4, 26.2.
Benzyl (S)-(4-amino-5-(3,3-diphenylpropanamido)pentyl)carbamate
Benzyl t-butyl (5-(3,3-diphenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate (74 mg, 0.13 mmol) was dissolved in dichloromethane (3 mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2mL) was added and reaction stirred at that temperature for 2 hours. The reaction mixture was dissolved in saturated NaHCO3 and the organic layer separated. The combined organic layers were dried over sodium sulfate and solvent removed under reduced pressure to give the product as a colorless oil. (56 mg, 94%); 1H NMR (400 MHz) (CDCl3) δ 7.34 (brs, 1H), 7.15 (m, 15H), 5,20 (m, 1H), 4.94 (s, 2H), 4.42 (t, 1H, J = 8), 3.16 (m, 2H), 2.86 (m, 5H), 1.25 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 173.1, 157.0, 143.4, 143.3, 136.3, 128.63. 128.61, 128.5, 128.2, 127.8, 127.75, 127.70, 126.6, 66.8, 51.80, 47.4, 42.3, 41.0, 40.1, 27.3, 25.4
(S)-N-(2,5-Diaminopentyl)-3,3-diphenylpropanamide
Benzyl (S)-(4-amino-5-(3,3-diphenylpropanamido)pentyl)carbamate (40 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried under vacuum to give product as a colorless oil. (31 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.13 (m, 11H), 4.43 (m, 1H), 3.25 – 3.09 (m, 2H), 2.89 (m, 3H), 2.70 (m, 2H), 1.53 (m, 2H), 1.23 (m, 2H); 13C NMR (400 MHz) (MeOD) δ 175.2, 145.08, 145.04, 129.6, 128.9, 128.8, 127.6, 52.2, 43.8, 43.1, 42.9, 40.3, 29.2, 24.7; HRMS (ESI) Calculated for C20H27N3O (M+H)+ 326.2227, found 326.2221
(S)-3-Benzyl-N-(2,5-diaminopentyl)-4-phenylbutanamide (6)
Ethyl 3-benzyl-4-phenylbut-2-enoate
To a round bottom flask containing 60% dispersion NaH (571 mg, 14.3 mmol) and anhydrous THF (20 mL) at 0 °C was added triethylphosphonoacetate (3.1 mL, 15.7 mmol) dropwise. The reaction mixture was naturally warmed to room temperature followed by a dropwise addition of 1,3-diphenyl acetone (1.9 mL, 9.5 mmol). The reaction mixture was stirred for 12 hours and then poured in water and extracted with dichloromethane. The combined organic layer was washed with brine and dried over sodium sulfate, filtered and concentrated under reduced pressure. This was purified on an ISCO chromatograph with silica to give the product as a colorless oil. (880 mg, 33%); 1H NMR (400 MHz) (CDCl3) δ 7.29 (m, 10H), 5.84 (s, 1H), 4.27 (q, 2H), 4.09 (s, 2H), 3.42 (s, 2H), 1.36 (t, 3H, J = 8), 13C NMR (100 MHz) (CDCl3) δ 166.5, 159.8, 138.8, 137.7, 129.4, 129.1, 128.6, 128.5, 126.7, 126.4, 118.4, 59.9, 43.4, 36.8, 14.3.
Ethyl 3-benzyl-4-phenylbutanoate
Ethyl 3-benzyl-4-phenylbut-2-enoate (777 mg, 2.77 mmol) was dissolved in ethanol (20 mL) and 10% Pd/C (280 mg) was added. The mixture was purged and stirred overnight under hydrogen atmosphere. The reaction was then filtered to remove catalyst and solvent removed under reduced pressure. The residue was purified on an ISCO chromatograph with silica gel (0–10% ethyl acetate/ hexane) to give the product as a colorless oil. (698 mg, 89%); 1H NMR (400 MHz) (CDCl3) δ 7.31 (m, 4H), 7.22 (m, 6H), 4.08 (q, 2H), 2.62 (m, 5H), 2.25 (d, 2H, J = 8), 1.25 (t, 3H, J = 8); 13C NMR (100 MHz) (CDCl3) δ172.9, 140.0, 129.3, 128.3, 126.1, 60.2, 40.1, 39.0, 37.9, 14.2.
3-Benzyl-4-phenylbutanoic acid
A mixture of ethyl 3-benzyl-4-phenylbutanoate (260 mg, 0.92 mmol) and KOH (206, 3.68 mmol) in ethanol/water (3:2) (5mL) was heated at 70 °C for 4 hours. The mixture was cooled to room temperature and acidified to pH = 2 with 1 M HCl. The solvent was evaporated under reduced pressure and residue extracted with ethyl acetate. The combined extracts were washed with brine, dried over Na2SO4, filtered and evaporated under reduced pressure to give the product as a colorless oil (176 mg, 75%); 1H NMR (400 MHz) (CDCl3) δ 11.73 (brs, 1H), 7.10 (m, 10H), 2.47 (m, 5H), 2.15 (d, 2H, J = 8); 13C NMR (100 MHz) (CDCl3) δ 180.0, 139.9, 129.3, 128.4, 128.3, 126.3, 40.1, 38.9, 37.6.
Dibenzyl (5-(3-benzyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
3-Benzyl-4-phenylbutanoic acid (119 mg, 0.47 mmol) was dissolved in DMF (5mL). EDC (179 mg, 0.94 mmol) and HOBt (126 mg, 0.94 mmol) were added and reaction stirred at room temperature for 5 minutes. Dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (150 mg, 0.39 mmol) was added followed by 2,6-lutidine (0.18 mL, 1.55 mmol) and reaction mixture stirred at room temperature overnight. The mixture was then diluted with ethyl acetate, washed with water, 1 M HCl, saturated NaHCO3, water and brine, dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure and purified on an ISCO chromatograph with silica (0–10% methanol/dichloromethane) to give product as a flaky yellow solid, (176 mg, 60%); MP 134–136 °C; 1H NMR (CDCl3) (400 MHz) δ 7.11 (m, 20H), 6.11 (s, 1H), 5.38 (d, 1H, J = 8), 5.08 (s, 1H), 4.87 (m, 4H), 3.54 (s, 1H), 3.09 (m, 4H), 2.45 (m, 5H), 1.92 (m, 2H), 1.35 (m, 4H); 13C NMR (400 MHz) (CDCl3) δ 173.2, 156.9, 156.6, 140.2, 140.1, 136.6, 136.4, 129.3, 128.53, 128.51, 128.3, 128.1, 128.06, 128.00, 126.0, 66.7, 66.6, 51.6, 43.8, 40.6, 40.03, 39.9, 38.9, 29.8, 26.3.
(S)-3-Benzyl-N-(2,5-diaminopentyl)-4-phenylbutanamide
Dibenzyl (5-(3-benzyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (75 mg, 0.12 mmol) was dissolved in ethanol (12 mL) and 20% Pd(OH)2/C (40 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere overnight, then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The filtrate was concentrated under reduced pressure to give the product as a brown colored oil. (31 mg, 73%); 1H NMR (400 MHz) (CDCl3) δ 7.13 (m, 11H), 3.07 (m, 1H), 2.93 (m, 1H), 2.57 (m, 8H), 2.04 (d, 2H, J = 4), 1.43 (m, 3H), 1.20 (m, 1H); 13C NMR (400 MHz) (CDCl3) δ 175.7, 141.6, 130.3, 129.4, 127.1, 51.9, 46.5, 41.8, 41.1, 40.5, 32.9, 28.4; HRMS (ESI) Calculated for C22H31N3O (M+H)+ 354.2540, found 354.2530
(S)-N-(2,5-Diaminopentyl)-3-phenethyl-5-phenylpentanamide (7)
Ethyl 3-phenethyl-5-phenylpent-2-enoate
To a round bottom flask containing 60% dispersion NaH (125 mg, 5.2 mmol) and anhydrous THF (10 mL) at 0 °C was added triethylphosphonoacetate (1.1 mL, 5.78 mmol) dropwise. The reaction mixture was allowed to warm to room temperature. This this reaction mixture was added dropwise 1,5-diphenylpentan-3-one, 32, (820 mg, 3.5 mmol). The reaction mixture was stirred for 12 hours and then poured into water and extracted with dichloromethane. The combined organic layer was washed with brine and dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified on an ISCO chromatograph to give product as a colorless oil. (270 mg, 25%); 1H NMR (400 MHz) (CDCl3) δ 7.16 (m, 6H), 7.06 (m, 4H), 5.61 (s, 1H), 4.05 (q, 2H), 2.82 (m, 2H), 2.66 (m, 4H), 2.30 (m, 2H), 1.16 (t, 3H, J = 8): 13C NMR (100 MHz) (CDCl3) δ 166.3, 162.3, 142.0, 141.8, 141.1, 128.8, 128.5, 128.4, 128.3, 128.2, 128.0, 126.4, 126.2, 126.0, 116.3, 59.6, 58.4, 44.5, 40.5, 35.1, 34.7, 34.1, 33.2, 29.9, 29.8, 26.6, 26.3, 18.5, 14.4, 14.2.
Ethyl 3-phenethyl-5-phenylpentanoate
Ethyl 3-phenethyl-5-phenylpent-2-enoate (270 mg, 0.88 mmol) was dissolved in ethanol (10 mL) and 10% Pd/C (100 mg) was added. The mixture was purged and stirred overnight under a hydrogen atmosphere. The reaction was then filtered to remove catalyst and solvent removed in under reduced pressure. The residue was purified on an ISCO chromatograph with silica gel (0–10% ethyl acetate/hexane) to give the product as a colorless oil, (188 mg, 69%); 1H NMR (CDCl3) (400 MHz) δ 7.11 (m, 10H), 4.03 (m, 2H), 2.52 (m, 4H), 2,27 (m, 2H), 1.90 (m, 1H), 1.59 (m, 4H), 1.14 (m, 3H); 13C NMR (100 MHz) (CDCl3) δ173.3, 173.1, 142.6, 142,4, 142.1, 128.5, 128.4, 128.2, 126.1, 125.8, 125.5, 60.3, 44.5, 39.2, 39.0, 37.9, 35.8, 35.1, 34.6, 34.1, 33.5, 33.1, 33.0, 30.9, 29.8, 26.8, 26.5, 14.3
3-Phenethyl-5-phenylpentanoic acid
A mixture of ethyl 3-phenethyl-5-phenylpentanoate (180 mg, 0.58 mmol) and KOH (130, 2.31 mmol) in ethanol/water (3:2) (5mL) was heated at 70 °C for 4 hours. The mixture was cooled to room temperature and acidified to pH = 2 with 1 M HCl. The solvent was evaporated under reduced pressure and residue extracted with ethyl acetate. The combined extracts were washed with brine, dried over Na2SO4, filtered and evaporated under reduced pressure to give the product as a colorless oil (121 mg, 74%); 1H NMR (CDCl3) (400 MHz) δ 11.54 (brs, 1H), 7.15 (m, 10H), 2.43 (m, 6H), 1.93 (m, 1H), 1.67 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 179.9, 142.2, 128.5, 128.4, 125.9, 38.7, 35.7, 34.5, 33.0
Dibenzyl (5-(3-phenethyl-5-phenylpentanamido)pentane-1,4-diyl)(S)-dicarbamate
Ethyl 3-phenethyl-5-phenylpentanoate (90 mg, 0.32 mmol) was dissolved in DMF (5 mL). EDC (122 mg, 0.64 mmol) and HOBt (86 mg, 0.64 mmol) were added and the reaction stirred at room temperature for 5 minutes. Dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (123 mg, 0.32 mmol) was added followed by 2,6-lutidine (0.12 mL, 0.96 mmol) and reaction mixture stirred at room temperature overnight. The mixture was then diluted with ethyl acetate, washed with water, 1M HCl, saturated NaHCO3, water and brine. The mixture was then dried over sodium sulfate and filtered. The filtrate was then concentrated under reduced pressure and the residue purified on an ISCO chromatograph using silica (0–10% methanol/dichloromethane) to give the product as a flaky white solid. (98 mg, 48%); 1H NMR (CDCl3) (400 MHz) δ 7.27 (m, 10H), 6.11 (s, 1H), 5.16 (m, 6H), 3.69 (s, 1H), 3.25 (m, 4H), 2.62 (m, 3H), 1.99 (m, 3H), 1.51 (m, 9H); 13C NMR (100 MHz) (CDCl3) δ 173.2, 156.9, 156.6, 142.4, 136.6, 136.4, 128.5, 128.4, 128.3, 128.1, 128.07, 128.03, 125.8, 66.7, 66.6, 51.7, 43.9, 41.3, 40.6, 35.5, 34.8, 32.9, 29.7, 26.3.
(S)-N-(2,5-Diaminopentyl)-3-phenethyl-5-phenylpentanamide
Dibenzyl (5-(3-phenethyl-5-phenylpentanamido)pentane-1,4-diyl)(S)-dicarbamate (60 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (30 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere overnight. The catalyst was filtered and washed with 20% MeOH/dichloromethane. The filtrate was concentrated under reduced pressure to give the product as a brown colored oil. (31 mg, 73%); 1H NMR (MeOD) (400 MHz) (trifluoroacetic acid salt) δ 7.11 (m, 11H), 3.24 (m, 2H), 2.89 (m, 3H), 2.53 (t, 4H, J = 8), 2.25 (d, 2H, J = 4), 1.89 (m, 1H), 1.62 (m, 8H); 13C NMR (100 MHz) (MeOD) δ 177.2, 143.7, 129.4, 129.3, 126.8, 52.8, 52.7, 47.6, 44.0, 41.7, 40.1, 36.8, 35.9, 33.8, 30.7, 28.4; HRMS (ESI) Calculated for C24H36N3O (M+H)+ 382.2853, found 382.2846
(S)-N-(2,5-Diaminopentyl)-3-phenylpropanamide (8)
Dibenzyl (5-(3-phenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate
3-Phenylpropionic acid (41 mg, 0.28 mmol) was dissolved in DMF (5 mL), EDC ( 105 mg, 0.55 mmol) and HOBT (74 mg, 0.55 mmol) were added and reaction was stirred at room temperature for 5 minutes under nitrogen. Dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (97 mg, 0.25 mmol) was added followed by 2,6-lutidine (90 μL, 0.75 mmol). The reaction mixture was then stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layers were dried over sodium sulfate, concentrated and purified on an ISCO chromatograph (0–10% MeOH/dichloromethane) to give product as a white solid. (106 mg, 82%); MP 159–161 °C; 1H NMR (400 MHz) (CDCl3) δ 7.22 (m, 12H), 7.09 (m, 3H), 5.91 (brs, 1H), 5.00 (s, 2H), 4.99 (s, 2H), 4.95 (brs, 1H), 4.88 (brs, 1H), 3.55 (m, 1H), 3.13 (m, 4H), 2.82 (m, 2H), 2.34 (m, 2H), 1.72 (m, 1H), 1.44 (m, 2H), 1.30 (m, 2H); 13C NMR (100 MHz) (CDCl3) δ 173.0, 156.8, 156.5, 140.7, 136.5, 136.4, 128.5, 128.3, 128.2. 128.1, 128.0, 126.2, 66.7, 66.6, 51.5, 43.7, 40.5, 38.2, 31.6, 29.5, 26.3
(S)-N-(2,5-Diaminopentyl)-3-phenylpropanamide
Dibenzyl (5-(3-phenylpropanamido)pentane-1,4-diyl)(S)-dicarbamate (85 mg, 0.16 mmol) was dissolved in ethanol (15 mL) and 20% Pd(OH)2/C (20mg) was added. Reaction was purged and stirred under hydrogen atmosphere for overnight. The catalyst was filtered and washed with 20% methanol/dichloromethane. The filtrate was concentrated under reduced pressure and dried under vacuum to obtain the product as a yellow oil. (41 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.12 (m, 5H), 3.08 (m, 1H), 2.92 (m, 1H), 2.82 (t, 2H, J = 8), 2.56 (m, 3H), 2.41 (t, 2H, J = 8), 1.40 (m, 2H), 1.26 (m, 1H), 1.12 (m, 1H); 13C NMR (100 MHz) (MeOD) δ 142.1, 129.5, 129.4, 127.2, 51.8, 46.5, 42.3, 38.9, 32.9, 32.8, 30.7, 29.5; HRMS (ESI) Calculated for C14H24N3O (M+H)+ 250.1914, found 250.1914
(S)-N-(2,5-Diaminopentyl)-4-phenylbutanamide (9)
Dibenzyl (5-(4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
4-Phenylbutanoic acid (47 mg, 0.29 mmol) was dissolved in dry DMF (5mL) and EDC (109 mg, 0.57 mmol) and HOBt (76 mg, 0.57 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl t-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (100 mg, 0.26 mmol) was added followed by 2,6-lutidine (0.09 mL, 0.75 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. Filtrate was then concentrated and purified on an ISCO chromatograph (0–10% MeOH/dichloromethane) to give a white solid. (55 mg, 40%); MP 143–145 °C; 1H NMR (100 MHz) (CDCl3) δ 7.26 (m, 16H), 6.13 (brs, 1H), 5.10 (s, 2H), 5.09 (s, 1H), 5.07 (brs, 1H), 3.72 (m, 1H), 3.27 (m, 4H), 2.63 (t, 2H, J = 8), 2.14 (m, 2H), 1.96 (m, 2H), 1.52 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 173.6, 156.5, 141.4, 136.5, 136.3, 128.5, 128.4, 128.3, 128.1, 128.08, 128.04, 125.9, 66.8, 66.6, 51.7, 43.9, 40.5, 35.7, 35.1, 29.8, 27.0, 26.4
(S)-N-(2,5-Diaminopentyl)-4-phenylbutanamide
Dibenzyl (5-(4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (57 mg, 0.11 mmol) was dissolved in ethanol (10mL) and 20% Pd(OH)2/C (35 mg) was added. The reaction mixture was then purged and stirred under hydrogen atmosphere overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried under vacuum to give the product as a pale yellow oil, (26 mg, 100%); 1H NMR (MeOD) (400 MHz) δ 7.10 (m, 6H), 3.14 (m, 1H), 2.98 (m, 1H), 2.61 (m, 5H), 2.13 (m, 2H), 1.81 (m, 2H), 1.45 (m, 3H), 1.20 (m, 1H); 13C NMR (MeOD) (100 MHz) δ 176.3, 142.9, 129.5, 129.4, 127.0, 51.9, 46.4, 41.9, 36.5, 36.3, 32.9, 28.7, 28.6; HRMS (ESI) Calculated for C15H25N3O (M+H)+ 264.2070, found 264.2072
(R)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide (10)
Benzyl t-butyl (5-hydroxypentane-1,4-diyl)(R)-dicarbamate
To a solution of (R)-2-(((benzyloxy)carbonyl)amino)-5-((t-butoxycarbonyl)amino)-pentanoic acid (1000 mg, 2.73 mmol) in DME (10 mL) at −15 °C were successively added a solution of N-methyl morpholine (0.34 mL, 3.08 mmol) and isobutyl chloroformate (0.35 mL, 2.73 mmol). The reaction was stirred at −15 °C to −10 °C for 15 minutes. The precipitated N-methyl morpholine HCl was removed by filtration and washed with DME (10 mL), the combine filtrates were chilled to −15 °C in an ice-salt bath. Then a solution of sodium borohydride (310 mg, 8.19 mmol) in water (4 mL) was added in one portion at −15 °C. This reaction mixture was stirred at this temperature for 10 minutes. The reaction was quenched by the addition of saturated aq. NH4Cl and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate. The solution was then filtered and concentrated under reduced pressure, purified on column (0–70% ethyl acetate/hexane) to give product as a colorless oil (855 mg, 91%); 1H NMR (CDCl3) ( 400 MHz) δ 7.28 (s, 5H), 5.46 (brs, 1H), 4.98 (s, 2H), 4.84 (brs, 1H), 4.03 (m, 1H), 3.60 (m, 2H), 2.99 (m, 2H), 1.42 (m, 4H), 1.36 (s, 9H); 13C NMR (CDCl3) ( 400 MHz) δ 156.6, 156.1, 136.4, 128.5, 128.1, 128.0, 79.3, 66.8, 65.0, 62.7, 52.9, 52.4, 40.3, 29.8, 28.4, 26.7, 26.0.
Benzyl t-butyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl) (R)-dicarbamate
Triphenylphosphine (652 mg, 2.49 mmol) and phthalimide (366 mg, 2.49 mmol) were added to a flask containing dry THF (5 mL). Benzyl t-butyl (5-hydroxypentane-1,4-diyl)(R)-dicarbamate (730 mg, 2.27 mmol) was added and the flask was cooled to 0 °C. DIAD (503 mg, 2.49 mmol) was added dropwise and reaction was allowed to stir for 30 minutes at 0 °C and overnight at room temperature. The mixture was concentrated under reduced pressure and the residue purified on an ISCO chromatograph with silica (0–70% ethyl acetate/hexane) to give product as a yellow solid. (736 mg, 73%); MP 74–76 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.82 (m, 2H), 7.71 (m, 2H), 7.27 ( m, 5H), 5.22 (brs, 1H), 4.95 (s, 2H), 4.70 (brs, 1H), 4.02 (m, 1H) 3.75 (m, 2H), 3.14 (m, 2H), 1.55 (m, 4H), 1.44 (s, 9H); 13C NMR ( CDCl3) ( 100 MHz) δ 168.4, 156.3, 156.0, 136.6, 133.9, 131.8, 128.4, 128.3, 127.8, 127.7, 123.3, 78.9, 66.3, 50.7, 41.9, 40.2, 29.9, 28.4, 26.4
Benzyl t-butyl (5-aminopentane-1,4-diyl)(R)-dicarbamate
Benzyl t-butyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(R)-dicarbamate (700 mg, 1.45 mmol) formed was dissolved in methanol (15 mL) and hydrazine monohydrate (0.14 mL, 2.90 mmol) was added. The reaction mixture was then refluxed for 2 hours and then cooled to room temperature. The precipitate formed was filtered and methanol used to wash the solid. The filtrate was concentrated under reduced pressure and the residue purified using an ISCO chromatograph with silica (0–10% methanol/dichloromethane + 1% NH4OH) to give the desired compound as a yellow oil, (166 mg, 33%); 1H NMR (CDCl3) ( 400 MHz) δ 7.25 (m, 5H), 5.41 (d, 1H, J = 8), 5.00 ( s, 2H), 4.76 (brs, 1H); 3.53 (m, 1H), 3.02 ( m, 2H), 2.61 (m, 2H), 1.40 (m, 4H), 1.36 (s, 9H); 13C NMR (CDCl3) ( 100 MHz) δ 156.6, 156.0, 136.6, 128.4, 128.1, 128.0, 78.9, 66.6, 53.2, 50.3, 45.7, 40.2, 29.7, 28.4, 26.6.
Benzyl t-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(R)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (75.5 mg, 0.28 mmol) was dissolved in dry DMF (5 mL) and EDC (109 mg, 0.57 mmol) and HOBt (77 mg, 0.57 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl t-butyl (5-aminopentane-1,4-diyl)(R)-dicarbamate (90 mg, 0.26 mmol) was added followed by 2,6-lutidine (90 μL, 0.78 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. Filtrate was then concentrated and the residue purified using an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a tan colored solid, (78 mg, 50%); MP 148–150 °C; 1H NMR (400 MHz) δ 7.11 (m, 15H), 6.09 (s, 1H), 5.30 (d, 1H, J = 8), 4.98 (d, 1H, J = 12), 4.78 (d, 1H, J = 12), 4.63 (s, 1H), 3.67 (m, 1H), 3.34 (m, 1H), 3.19 (m, 1H), 3.03 (m, 2H), 2.46 (m, 4H), 1.93 (m, 3H), 1.66 (m,2H), 1.45 (m, 4H), 1.35 (s, 9H); 13C NMR δ 176.3, 156.9, 156.1, 141.66, 141.64, 136.2, 128.45, 128.43, 128.3, 128.0, 126.1, 125.9, 79.2, 66.8, 51.8, 46.6, 43.7, 40.1, 34.49, 34.4, 34.2, 33.9, 33.6, 30.1, 28.4, 26.5
Benzyl (R)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate
Benzyl t-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(R)-dicarbamate (71 mg, 0.12 mmol) was dissolved in dichloromethane ( 3mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid was added and reaction stirred at that temperature for 2 hours. The reaction mixture was dissolved in saturated NaHCO3 and the organic layer separated. The combined organic layers were dried over sodium sulfate and solvent removed under reduced pressure to give product as a yellow oil (51 mg, 86%); 1H NMR (MeOD) (400 MHz) δ 7.09 (m, 16H), 4.81 (m, 1H), 3.69 (m, 1H), 3.11 (m, 2H), 2.70 (m, 2H), 2.43 (m, 4H), 2.17 (m, 1H), 1.79 (m, 2H), 1.62 (m, 6H); 13C NMR δ (MeOD) (100 MHz) 178.7, 158.8, 143.2, 138.0, 129.4, 128,9, 128.8, 126.9, 67.5, 52.2, 47.7, 44.3, 41.3, 36.0, 34.8, 34.7, 31.1, 27.4
(R)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
Benzyl (R)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate (45.3 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg) was added. The reaction mixture was then purged and stirred under hydrogen atmosphere for overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was concentrated under reduced pressure and dried under vacuum to give product was a colorless oil. (26.7 mg, 78%); 1H NMR (400 MHz) (MeOD) δ 7.10 (m, 11H), 3.11 (m, 2H), 2.70 (m, 3H), 2.46 (m, 4H), 2.21 (m 1H), 1.80 (m, 2H), 1.59 (m, 4H), 1.18 (m, 2H); 13C NMR (100 MHz) (MeOD) δ 181.2, 145.6, 131.9, 131.8, 129.5, 54.5, 50.9, 50.2, 49.1, 44.0, 38.4, 37.3, 35.4, 30.1, 24.9; HRMS (ESI) Calculated for C23H33N3O (M+H)+ 368.2696, found 368.2684
(S)-N-(3,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide (11)
Dibenzyl (5-diazo-4-oxopentane-1,3-diyl)(S)-dicarbamate
(S)-2,4-Bis(((benzyloxy)carbonyl)amino)butanoic acid (1000 mg, 2.59 mmol) was dissolved in THF ( 15 mL) and triethylamine (0.38 mL, 2.72 mmol) and reaction mixture cooled to −15 °C. Ethyl chloroformate (0.26 mL, 1.05 mmol) was added and the mixture was stirred for 30 minutes at −5 °C and the precipitate formed was filtered off. Acetonitrile (dry) (10 mL) and trimethylsilyldiazomethane (2.0 M solution in hexane) (2.6 mL, 5.18 mmol) were added to the filtrate and the mixture stirred at +4 °C for 24 hours. Diethyl ether was added to the mixture and extracted with 10% HCl, sat NaHCO3 and brine. The organic layer was dried over sodium sulfate and solvents evaporated to obtain the crude diazoketone. This was purified on an ISCO chromatograph (0–70%) ethyl acetate/hexane to obtain pure product as a yellow colored oil. (578 mg, 54%); 1 H NMR (400 MHz) (CDCl3) δ 7.37 (m, 10H), 5.72 (brs, 1H), 5.37 (brs, 1H), 5.30 (brs, 1H), 5.03 (s, 4H), 4.21(brs, 1H), 3.48 (m, 1H), 3.09 (m, 1H), 2.00 (m, 1H), 1.74 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 192.8, 156.6, 156.4, 136.5, 136.1, 128.5, 128.2, 128.1, 128.0, 67.2, 66.7, 55.3, 54.2, 37.1, 33.1
Methyl (S)-3,5-bis(((benzyloxy)carbonyl)amino)pentanoate
The diazoketone, 57, (560 mg, 1.36 mmol) was suspended in methanol and a solution of silver benzoate (62 mg, 0.27 mmol) in triethylamine ( 0.8 mL) was gradually added while the mixture was sonicated in an ultrasound bath until completion of the reaction (30 minutes). The methanol was removed unde reduced pressure and the residue dissolved in ethyl acetate and extracted with sat. NaHCO3, 5% HCl and brine and dried over sodium sulfate, concentrated and purified using anISCO chromatograph (0–50% ethyl acetate/ hexane) to give product as a colorless oil. (312 mg, 55%); 1H NMR (CDCl3) (400 MHz) δ 7.33 (m, 10H), 5.70 (m, 2H), 5.10 (s, 4H), 4.06 (m, 1H), 3.63 (s, 3H), 3.41 (m, 1H), 3.03 (m, 1H), 2.54 (d, 2H, J = 4), 1.68 (m, 2H); 13C NMR (100 MHz) (CDCl3) 171.8, 156.5, 156.4, 136.7, 136.5, 128.5, 128.4, 128.2, 128.1, 128.0, 66.7, 66.5, 51.7, 45.6, 38.9, 37.6, 37.1, 34.6
Dibenzyl (5-hydroxypentane-1,3-diyl)(S)-dicarbamate
Methyl (S)-3,5-bis(((benzyloxy)carbonyl)amino)pentanoate was dissolved in ethanol/THF (10:1) (11 mL) under nitrogen. Lithium borohydride was added and reaction stirred at room temperature for overnight. The reaction mixture was dissolved in water and extracted with ethyl acetate and purified on an ISCO chromatograph (0–60% ethyl acetate/hexane) to give product as a colorless oil (216 mg, 78%); 1H NMR (MeOD) (400 MHz) δ 7.35 (m, 12H), 5.10 (s, 4H), 3.80 (s, 1H), 3.22 (m, 4H), 1.69 (m, 4H); 13C NMR (MeOD) (100 MHz) δ 158.94, 158.82, 138.4, 129.5, 129.0, 128.85, 128.8, 67.47. 59.8, 47.4, 39.0, 38.9, 36.4
Dibenzyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,3-diyl)(S)-dicarbamate
Triphenylphosphine (175 mg, 0.67 mmol) and phthalimide (99 mg, 0.67 mmol) were added to a flask containing dry THF (5 mL). Dibenzyl (5-hydroxypentane-1,3-diyl)(S)-dicarbamate (217 mg, 0.56 mmol) was added and the flask was cooled to 0 °C. DIAD (0.13 mL, 0.67 mmol) was added dropwise and the reaction was allowed to stir for 30 minutes at 0 °C and overnight at room temperature. The mixture was concentrated under reduced pressure and the residue purified on an ISCO chromatograph with silica (0 –70% ethyl acetate/hexane) to give product as a white solid. (151 mg, 53%); MP 103–105 °C; 1H NMR (400 MHz) (CDCl3) δ 7.79 (m, 2H), 7.66 (m, 2H), 7.22 (m,10H), 5.61 (m, 1H), 5.33( d, 1H, J= 12), 5.08 (m, 4H0, 3.74(m, 3H), 4.43 (m, 1H), 3.02 (m, 1H), 1.83 (m, 3H), 1.51 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 168.2, 156.6 156.5, 136.7, 136.4, 133.9, 132.0, 128.5, 128.4, 128.1, 128.0, 127.9, 123.2, 66.7, 66.5, 47.0, 37.4, 35.1. 33.7,
Dibenzyl (5-aminopentane-1,3-diyl)(R)-dicarbamate
Dibenzyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,3-diyl)(S)-dicarbamate (120 mg, 0.23 mmol) was dissolved in methanol (10 mL) and hydrazine monohydrate (0.02 mL, 0.47 mmol) was added. The reaction mixture was then refluxed for 2 hours and cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was concentrated under reduced pressure and the remaining solid purified on an ISCO chromatograph with silica (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a colorless oil. (80 mg, 90%); 1H NMR (CDCl3) (400 MHz) δ 7.30 (m, 10H). 5.05 (s, 4H), 3.68 (m, 1H), 3.19 (m, 2H), 2.62 (t, 2H, J = 8 ), 1.64 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 161.4, 161, 3, 141.0, 140.9, 132.0, 131.5, 131.3, 131.2, 69.9, 50.31, 41.9. 41.7, 41.3, 39.0
Dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,3-diyl)(R)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (57.4 mg, 0.21 mmol) was dissolved in dry DMF (5mL) and EDC (80 mg, 0.42 mmol) and HOBt (57 mg, 0.42 mmol) were added and the reaction stirred at room temperature for 5 minutes. Dibenzyl (5-aminopentane-1,3-diyl)(R)-dicarbamate (75 mg, 0.19 mmol) was added followed by 2,6-lutidine (0.07 mL, 0.57 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated and purified with an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a white solid. (90 mg, 75%); MP 152–154 °C; 1H NMR( CDCl3) (400 MHz) δ 7.26 ( m, 20H), 6.21 (brs, 1H), 5.32 (brs, 1H), 5.06 (m, 5H), 3.75 (m, 1H), 3.63 (m, 1H), 3.39 (m, 1H), 3.06 ( m, 2H), 2.58 (m 4H), 2.07 (m. 3H), 1.75 (m, 4H), 1.56 (m, 2H); 13C NMR (100 MHz) (CDCl3) δ 175.4, 157.0, 156.5, 141.7, 136.6, 136.3, 128.6, 128.5, 128.4, 128.2, 128.1, 128.0, 125.9, 66.9, 66.6, 60.4, 46.77, 46.6, 37.7, 36.0, 35.6, 34.5
(S)-N-(3,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
Dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,3-diyl)(R)-dicarbamate (65.5 mg, 0.103 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C ( 30 mg ) was added. Reaction mixture was purged and stirred under a hydrogen atmosphere. The catalyst was removed by filtration and washed residue with 20% methanol/ dichloromethane to give product as a colorless oil. (36.1 mg, 97%); 1H NMR (400 MHz) (CDCl3) δ 7.10 ( m, 10H), 2.42 ( m, 1H), 3.21 (m, 2H), 2.63 (m, 2H), 2.45 (m, 4H), 2.16 (m, 1H), 1.80 (m, 2H), 1.56 (m, 6H); 13C NMR (100 MHz) (CDCl3) δ 178.2, 143.1, 129.4, 129.3, 127.0, 126.9, 68.3, 54.3, 47.6, 45.9, 40.5, 39.5, 38.4, 37.5, 37.1, 36.6, 35.9, 34.8, 32.8; HRMS (ESI) Calculated for C23H33N3O (M+H)+ 368.2696, found 368.2698
(S)-N-(4,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide (12)
Methyl (S)-2-(((benzyloxy)carbonyl)amino)-5-((tert-butoxycarbonyl)amino)-pentanoate
Cbz ornithine (Boc) acid (1 g, 2.73 mmol) was dissolved in DMF (5 mL) and K2CO3 (452.6 mg, 3.26 mmol). The reaction was cooled to 0 °C and methyl iodide (775 mg, 5.46 mmol) was added. The reaction was allowed to warm to room temperature and stirred at the temperature overnight. Then the reaction mixture was washed with saturated sodium bicarbonate solution and extracted with ethyl acetate. The combined organic layers were washed with brine, dried over sodium sulfate, concentrated and purified on an ISCO chromatograph (0–60% ethyl acetate/ hexane) to give product as a colorless oil. (761 mg, 73%); 1H NMR δ 7.19 ( s, 5H), 6.06 ( d, 1H, J = 8), 5.12 ( brs, 1H), 4.94 (s, 2H), 4.17 (m, 1H), 3.55 (s, 3H), 2.94 (m, 2H), 1.69 (m, 1H), 1.55 (m, 1H), 1.40 (m, 2H), 1.27 (s, 9H); 13C NMR δ 172.7, 156.0, 155.9, 136.3, 128.2, 128.1, 127.9, 127.8, 78.6, 67.2, 66.5, 53.7, 39.8, 29.2, 28.2, 25.9
Benzyl tert-butyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate
To a solution of methyl (S)-2-(((benzyloxy)carbonyl)amino)-5-((tert-butoxycarbonyl)-amino)pentanoate, (431 mg, 1.13 mmol) in THF (5mL)/ethanol (1 mL) was added LiBH4 (32 mg, 1.47 mmol) at 0 °C. The mixture was stirred at that temperature for 30 minutes and warmed to room temperature and stirred overnight. The reaction mixture was poured into water and extracted with ethyl acetate. The combine organic layers were washed with brine and dried over sodium sulfate and concentrated. It was purified on an ISCO chromatograph (0–70% ethyl acetate/ hexane) to give product as a colorless oil. (385 mg, 97%); 1H NMR (CDCl3) ( 400 MHz) δ 7.28 ( m, 5H), 5.02 (s, 3H), 3.60 (m, 4H), 3.04 (m, 2H), 1.47 (m, 4H), 1.36 (m, 9H); 13C NMR (CDCl3) ( 100 MHz)δ 156.6, 156.1, 136.4, 128.5, 128.1, 128.0, 79.3, 66.8, 65.0, 62.7, 52.9, 52.4, 40.3, 29.8, 28.4, 26.7, 26.0
Benzyl tert-butyl (5-(1,3-dioxoisoindolin-2-yl)pentane-1,4-diyl)(S)-dicarbamate
Triphenylphosphine (325 mg, 1.24 mmol) and phthalimide (182 mg, 1.24 mmol) were added to a flask containing dry THF (5 mL). Dibenzyl (5-hydroxypentane-1,4-diyl)(S)-dicarbamate (400 mg, 1.03 mmol) was added and the flask was cooled to 0 °C. DIAD (250 mg, 1.24 mmol) was added dropwise and the reaction was allowed to stir for 30 minutes at 0 °C and overnight at room temperature. The mixture was concentrated under reduced pressure and the residue purified on an ISCO chromatograph (0–70% ethyl acetate/hexane) to give product as a white solid. (340 mg, 69%); MP 74–76 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.82 (m, 2H), 7.71 (m, 2H), 7.27 ( m, 5H), 5.18 (brs, 1H), 4.96 (s, 2H), 4.67 (brs, 1H), 4.02 (m, 1H) 3.75 (m, 2H), 3.14 (m, 2H), 1.60 (m, 4H), 1.44 (s, 9H); 13C δ 168.4, 156.3, 156.0, 136.6, 133.9, 131.8, 128.4, 128.3, 127.8, 127.7, 123.3, 78.9, 66.3, 60.3, 50.7, 41.9, 40.2, 29.9, 28.4, 26.4
Benzyl tert-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate
The phthalimide (340 mg, 0.71 mmol) formed was dissolved in methanol (20 mL) and hydrazine monohydrate (0.07 mL, 1.41 mmol) was added. The reaction mixture was then refluxed for 2 hours and then cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was rotavapped and the remaining solid purified on an ISCO chromatograph (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a colorless oil. ( 164 mg, 66%); 1H NMR ( CDCl3) ( 400 MHz) δ 7.25 (m, 5H), 5.41 (d, 1H, J = 8), 5.00 ( s, 2H), 4.84 (brs, 1H); 3.50 (m, 1H), 3.01 ( m, 2H), 2.61 (m, 2H), 1.40 (m, 4H), 1.36 (s, 9H); 13C NMR (CDCl3) (100 MHz) δ 156.6, 156.0, 136.6, 128.4, 128.1, 128.0, 78.9, 66.6, 53.2, 45.7, 40.2, 29.7, 28.4, 26.6, 25.0, 24.9
Dibenzyl tert-butyl pentane-1,2,5-triyl(S)-tricarbamate
Benzyl tert-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (162.2 mg, 0.46 mmol) was dissolved in dichloromethane (5 mL) under nitrogen atmosphere and triethylamine (.08 mL, 0.55 mmol) was added. CbzCl (0.08 mL, 0.55 mmol) was added and the reaction stirred at room temperature until the reaction was completed as indicated by TLC. The dichloromethane was removed under reduced pressure and the residue purified on an ISCO chromatograph to give product as a white solid. (126 mg, 57%); MP 125–126 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.34 (m, 10), 5.36 (brs, 1H), 5.23 (d, IH, J = 4), 5.09 ( s, 4H), 4.72 (m, 1H), 3.72 (m, 1H), 3.21 (m, 4H), 1.45 (m, 4H), 1.42 (s, 9H); 13C NMR ( CDCl3) ( 100 MHz) δ 157.0, 156.5, 156.1, 136.4, 128.5, 128.1, 128.07, 128.0, 79.1, 66.8, 66.7, 51.7, 45.0, 40.1, 29.5, 28.4, 26.4
Dibenzyl (5-aminopentane-1,2-diyl)(S)-dicarbamate
Dibenzyl tert-butyl pentane-1,2,5-triyl(S)-tricarbamate (189 mg, 0.39 mmol) was dissolved in dichloromethane (3mL) and the reaction mixture cooled to 0 °C under nitrogen. Trifluoroacetic acid (2 mL) was added and the reaction stirred at 0 °C for 3 hours. Upon completion of the reaction, the reaction was quenched with saturated solution of NaHCO3 and extracted with dichloromethane. The organic layer was concentrated under reduced pressure to give product as a yellow solid (101.7 mg, 67%); MP 85–87 °C; 1H NMR ( CDCl3) ( 400 MHz) δ 7.17 (m, 10H), 5.51 (m, 1H), 4.90 (m, 4H), 3.54 (m, 4H), 3.05 (m, 2H), 2.84 (m, 2H), 1.43 (m, 4H); 13 C δ 135.7, 128.6, 128.3, 127.7, 67.4, 51.2, 44.6, 39.9, 28.6, 23.4
Dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,2-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (46 mg, 0.17 mmol) was dissolved in dry DMF (5 mL) and EDC (63 mg, 0.33 mmol) and HOBt (44 mg, 0.33 mmol) were added and the reaction stirred at room temperature for 5 minutes. Dibenzyl (5-aminopentane-1,2-diyl)(S)-dicarbamate (59 mg, 0.15 mmol) was added followed by 2,6-lutidine (0.05 mL, 0.45 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated under reduced pressure and the residue purified using an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a white solid. (63 mg, 67%); MP 145–147 °C; 1H NMR (400 MHz) δ 7.25 (m, 20H), 5.77 (brs, 1H), 5.32 (m, 2H), 5.09 (s, 2H), 5.08 (s, 2H), 3.77 (m, 1H), 3.32 (m, 4H), 2.60 (m, 4H), 2.03 (m, 3H), 1.78 (m, 2H), 1.58 (m, 4H)
(S)-N-(4,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
Dibenzyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,2-diyl)(R)-dicarbamate (58 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (30 mg) was added. The reaction mixture was then purged and stirred under hydrogen atmosphere for overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried under vacuum to give the product as a colorless oil. (14 mg, 42%); 1H NMR (MeOD) (400 MHz) δ 7.09 (m, 11H), 3.14 (m, 2H), 2.66 (m, 1H), 2.45 (m, 6H), 2.13 (m, 1H), 1.79 (m, 2H), 1.50 (m, 6H); 13C NMR (100 MHz) (MeOD) δ 178.2, 143.1, 129.4, 129.3, 126.9, 53.3, 47.6, 47.5, 40.2, 35.9, 34.8, 32.8, 27.1; HRMS (ESI) Calculated for C23H33N3O (M+H)+ 368.2696, found 368.2687
(S)-N-(2,3-Diaminopropyl)-2-phenethyl-4-phenylbutanamide (13)
Benzyl t-butyl (3-hydroxypropane-1,2-diyl)(S)-dicarbamate
To a solution of (S)-2-(((benzyloxy)carbonyl)amino)-3-((t-butoxycarbonyl)amino)-propanoic acid (900 mg, 2.66 mmol) in DME (10 mL) at −15 °C were successively added a solution of N-methyl morpholine (0.33 mL, 3 mmol) and isobutyl chloroformate (0.35 mL, 2.66 mmol). The reaction was stirred at −15 °C to −10 °C for 15 minutes. The precipitated N-methyl morpholine HCl was removed by filtration and washed with DME (10 mL), the combined filtrates were chilled to −15 °C in an ice-salt bath. Then a solution of sodium borohydride (301 mg, 7.98 mmol) in water (5 mL) was added in one portion at −15 °C. This reaction mixture was stirred at this temperature for 10 minutes. The reaction was quenched by the addition of saturated aq. NH4Cl and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate. The solution was then filtered and concentrated under reduced pressure, purified on a ISCO chromatograph (0–70% ethyl acetate/ hexane) to give product as a white powder (675 mg, 78%); MP 117–120 °C; 1H NMR (400 MHz) (MeOD) δ 7.34 (m, 5H), 5.09 (s, 2H), 3.73 (m, 1H), 3.24 (m, 4H), 1.44 (s, 9H); 13C NMR (100 MHz) (MeOD) δ 158.6, 138.3, 129.5, 129.0, 128.9, 80.3, 67.5, 63.0, 54.6, 42.1, 28.8.
Benzyl t-butyl (3-(1,3-dioxoisoindolin-2-yl)propane-1,2-diyl)(S)-dicarbamate
Triphenylphosphine (709 mg, 2.71 mmol) and phthalimide (398 mg, 2.71 mmol) were added to a flask containing dry THF (6 mL). Benzyl t-butyl (3-hydroxypropane-1,2-diyl)(S)-dicarbamate (730 mg, 2.26 mmol) was added and the flask was cooled to 0 °C. DIAD (548 mg, 2.71 mmol) was added dropwise and the reaction was allowed to stir for 30 minutes at 0 °C and overnight at room temperature. The mixture was concentrated under reduced pressure and residue purified on an ISCO chromatograph with silica (0 – 70% ethyl acetate/hexane) to give the product as a white solid. (556 mg, 55%); MP 92–95 °C; 1H NMR (400 MHz) (CDCl3) δ 7.83 (m, 2H), 7.71 (m, 2H), 7.28 (m, 5H), 5.70 (m, 1H), 5.26 (brs, 1H), 5.02 (s, 2H), 4.06 (m, 1H), 3.84 (m, 2H), 3.31 (m, 2H), 1.44 (s, 9H), 13C NMR (400 MHz) (CDCl3) δ 168.5, 156.6, 156.3, 136.4, 134.1, 131.8, 128.3, 127.9, 123.4, 79.7, 66.6, 51.6, 41.9, 39.2, 28.3, 21.9
Benzyl t-butyl (3-aminopropane-1,2-diyl)(S)-dicarbamate
The benzyl t-butyl (3-(1,3-dioxoisoindolin-2-yl)propane-1,2-diyl)(S)-dicarbamate (450 mg, 0.99 mmol) formed was dissolved in methanol (10 mL) and hydrazine monohydrate (0.1 mL, 1.98 mmol) was added. The reaction mixture was then refluxed for 2 hours and cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was concentrated under reduced pressure and the remaining solid purified on an ISCO chromatograph with silica (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a colorless oil. (140 mg, 44%); 1H NMR (400 MHz) (MeOD) δ 7.27 (m, 5H), 6.37 (s, 1H), 5.87 (s, 1H), 5.02 (s, 2H), 3.94 (s, 4H), 3.60 (m, 1H), 3.12 (m, 2H), 2.70 (m, 2H), 1.36 (s, 9H); 13C NMR (100 MHz) (MeOD) δ 158.9, 129.4, 129.0, 128.9, 80.3, 67.6, 54.7, 43.4, 2.5, 28.7
Benzyl t-butyl (3-(2-phenethyl-4-phenylbutanamido)propane-1,2-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (100 mg, 0.37 mmol) was dissolved in dry DMF (5 mL) and EDC (139 mg, 0.72 mmol) and HOBt (98 mg, 0.72 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl t-butyl (3-aminopropane-1,2-diyl)(S)-dicarbamate (109 mg, 0.33 mmol) was added followed by 2,6-lutidine (0.11 mL, 0.99 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated and purified using an ISCO chromatograph with silica (0 – 10% MeOH/dichloromethane) to give a white solid. (126 mg, 67%); MP 177–179 °C; 1H NMR (400 MHz) (CDCl3) δ 7.14 (m, 15 H), 6.60 (brs, 1H), 6.06 (brs, 1H), 5.18 (m, 1H), 4.97 (m, 2H), 3.56 (m, 2H), 3.21 (m, 3H), 2.50 (m, 4H), 2.11 (m, 1H), 1.92 (m, 2H), 1.69 (m, 2H), 1.37 (m, 9H); 13C NMR (100 MHz) (CDCl3) δ 177.1, 157.3, 156.6, 141.6, 141.5, 136.4, 128.5, 128.4, 128.3, 128.1, 128.0, 125.9, 80.0, 66.7, 53.6, 46.6, 40.8, 40.0, 34.7, 34.4, 33.7, 33.6, 28.3, 28.2
Benzyl (S)-(1-amino-3-(2-phenethyl-4-phenylbutanamido)propan-2-yl)carbamate
Benzyl t-butyl (3-(2-phenethyl-4-phenylbutanamido)propane-1,2-diyl)(R)-dicarbamate (114 mg, 0.20 mmol) was dissolved in dichloromethane ( 3 mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2 mL) was added and reaction stirred at that temperature for 2 hours. The reaction mixture was dissolved in saturated NaHCO3 and the organic layer separated. The combined organic layers were dried over sodium sulfate and solvent removed under reduced pressure to give the product as a white flaky powder. (74.5 mg, 79%); MP 112–115 °C; 1H NMR (400 MHz) (CDCl3) δ 7.12 (m, 15H), 6.16 (m, 1H), 5.66 (m, 1H), 4.89 (m, 2H), 3.60 (m, 1H), 3.33 (m, 2H), 2.71 (m, 2H), 2.47 (m, 4H), 1.93 (m, 3H), 1.65 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 176.3, 156.8, 141.5, 136.2, 128.5, 128.4, 128.3, 128.1, 128.0, 125.9, 66.8, 52.9, 46.5, 43.0, 41.3, 34.4, 33.6, 33.5
(S)-N-(2,3-Diaminopropyl)-2-phenethyl-4-phenylbutanamide
Benzyl (S)-(1-amino-3-(2-phenethyl-4-phenylbutanamido)propan-2-yl)carbamate (51 mg, 0.11 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent from the filtrate was concentrated under reduced pressure under to give product as a yellow oil. (29.6 mg, 81%); 1H NMR (400 MHz) (MeOD) δ 7.20 (m, 11H), 3.27 (m, 2H), 2.88 (m, 1H), 2.73 (m, 1H), 2.58 (m, 4H), 2.32 (m, 2H), 1.92 (m, 2H), 1.78 (m, 2H); 13C NMR (100 MHz) (MeOD) δ 178.8, 143.0, 129.4, 129.3, 126.9, 53.8, 47.6, 45.8, 43.9, 35.8, 34.8; HRMS (ESI) Calculated for C21H29N3O (M+H)+ 340.2383, found 340.2377
(S)-N-(2,4-Diaminobutyl)-2-phenethyl-4-phenylbutanamide) (14)
Dibenzyl (4-hydroxybutane-1,3-diyl)(S)-dicarbamate
To a solution of (S)-2,4-bis(((benzyloxy)carbonyl)amino)butanoic acid (1000 mg, 2.77 mmol) in DME (10 mL) at −15 °C were successively added N-methyl morpholine (340 μL, 3.13 mmol) and isobutyl chloroformate (360 μL, 2.77 mmol). The reaction was stirred at −15 °C to −10 °C for 15 minutes. The precipitated N-methyl morpholine HCl was removed by filtration and washed with DME (10 mL) and the combine filtrates were chilled to −15 °C in an ice-salt bath. Then a solution of sodium borohydride (378 mg, 8.31 mmol) in water (5 mL) was added in one portion at −15 °C. This reaction mixture was stirred at this temperature for 10 minutes. The reaction was quenched by the addition of saturated aq. NH4Cl and the resulting mixture was extracted with ethyl acetate. The organic layer was washed with brine and dried over sodium sulfate. The solution was then filtered and concentrated under reduced pressure, purified on column (0–70% ethyl acetate/ hexane) to give product as a white powder (491 mg, 48%); MP 90–92 °C; 1H NMR (400 MHz) (CDCl3) δ 7.33 (s, 10H), 5.72 (s, 1H), 5.63 (d, 1H, J = 8), 5.08 (s, 4H), 3.48 (m, 4H), 3.02 (m, 1H), 1.71 (m, 1H), 1.57 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 157.0, 156.7, 136.5,136.3, 128.55, 128.50, 128.1, 128.07, 128.02, 66,8, 66.6, 64.6, 50.4, 37.7, 31.7
Dibenzyl (4-(1,3-dioxoisoindolin-2-yl)butane-1,3-diyl)(S)-dicarbamate
Triphenylphosphine (365 mg, 1.39 mmol) and phthalimide (204 mg, 1.39 mmol) were added to a flask containing dry THF (6 mL). Dibenzyl (4-hydroxybutane-1,3-diyl)(S)-dicarbamate (432 mg, 1.39 mmol) was added and the flask was cooled to 0 °C. DIAD (281 mg, 1.39 mmol) was added dropwise and the reaction was allowed to stir for 30 minutes at 0 °C and then overnight at room temperature. The mixture was concentrated under reduced pressure and the residue purified on an ISCO chromatograph with silica (0–70% ethyl acetate/hexane) to give product as a white solid. (237 mg, 41%); MP 152–154 °C; 1H NMR (400 MHz) (CDCl3) δ 7.83 (m, 2H), 7.70 (m, 2H), 7.36 (m, 10H), 5.61 (brs, 1H), 5.46 (d, 1H, J = 8), 5.10 (m, 4H), 4.12 (m, 1H), 3.78 (m, 2H), 3.51 (m, 1H), 3.08 (m, 1H), 1.83 (m, 1H), 1.54 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 168.5, 156.7, 156.5, 136.7, 136.4, 134.1, 131.7, 128.46, 128.41, 128.0, 127.9, 127.7, 123.4, 66.6, 66.5, 53.4, 48.8, 41.8, 37.4, 33.2.
Dibenzyl (4-aminobutane-1,3-diyl)(S)-dicarbamate
Dibenzyl (4-(1,3-dioxoisoindolin-2-yl)butane-1,3-diyl)(S)-dicarbamate (170 mg, 0.34 mmol) was dissolved in methanol (5 mL) and hydrazine monohydrate (0.03 mL, 0.68 mmol) was added. The reaction mixture was then refluxed for 2 hours and cooled to room temperature. The precipitate formed was filtered and methanol used to wash the filtrate. The filtrate was concentrated under reduced pressure and the remaining solid purified on an ISCO chromatograph with silica (0–10% methanol/dichloromethane + 1% NH4OH) to give product as a colorless oil. (77 mg, 61%); 1H NMR (400 MHz) (CDCl3) δ 7.34 (m, 10H), 5.77 (brs, 1H), 5.56 (d, 1H, J = 8), 5.09 (m, 4H), 3.69 (m, 1H), 3.44 (m, 1H), 3.02 (m, 1H), 2.74 (m, 2H), 2.26 (s, 2H), 1.68 (m, 1H), 1.47 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 157.0, 156.5, 136.7, 136.4, 128.5, 128.4, 128.1, 128.0, 66.8, 66.5, 50.5, 45.5, 37.6, 33.0
Dibenzyl (4-(2-phenethyl-4-phenylbutanamido)butane-1,3-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (55.6 mg, 0.20 mmol) was dissolved in dry DMF (5mL) and EDC (79 mg, 0.41 mmol) and HOBt (56 mg, 0.41 mmol) were added and the reaction stirred at room temperature for 5 minutes. Dibenzyl (4-aminobutane-1,3-diyl)(S)-dicarbamate (70 mg, 0.19 mmol) was added followed by 2,6-lutidine (0.07 mL, 0.56 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated and purified with an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a white solid. (100 mg, 86%); MP 187–189 °C; 1H NMR (400 MHz) (CDCl3) δ 7.13 (m, 20H), 5.89 (m, 1H), 5.61 (m, 1H), 5.54 (m, 1H), 4,98 (m, 3H), 4.76 (m, 1H), 3.68 (m, 1H), 3.38 (m, 2H), 3.14 (m, 2H), 2.93 (m, 1H), 2.43 (m, 4H), 1.87 (m, 3H), 1.64 (m, 3H), 1.42 (m, 1H); 13C NMR (100 MHz) (CDCl3) δ 176.7, 157.2, 156.6, 141.54, 141.50, 136.6, 136.2, 128.5, 128.4, 128.3, 128.07, 128.00, 126.0, 125.9, 66.8, 66.6, 50.1, 46.4, 43.3, 37.4, 34.38, 37.35, 33.6, 33.5.
(S)-N-(2,4-Diaminobutyl)-2-phenethyl-4-phenylbutanamide
Dibenzyl (4-(2-phenethyl-4-phenylbutanamido)butane-1,3-diyl)(S)-dicarbamate (54 mg, 0.09 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (30 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere for overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried under vacuum to give product was a colorless oil. (21.5 mg, 69%); 1H NMR ( MeOD) (400 MHz) δ 7.13 (m, 10H), 6.49 (brs, 1H), 3.30 (m, 1H), 2.92 (m, 2H), 2.49 (m, 6H); 1.97 (m, 3H), 1.54 (m, 4H); 13C NMR (100 MHz) (MeOD) δ 175.6, 141.7, 128.4, 128.3, 125.9, 49.9, 46.5, 45.5, 38.9, 37.6, 34.4, 33; HRMS (ESI) Calculated for C22H31N3O (M+H)+ 354.2540, found 354.2532
(S)-N-(2-Amino-5-(methylamino)pentyl)-2-phenethyl-4-phenylbutanamide (15)
Benzyl tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (204 mg, 0.76 mmol) was dissolved in dry DMF (5mL) and EDC (290 mg, 1.52 mmol) and HOBt (204 mg, 1.52 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl tert-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (295 mg, 0.83 mmol) was added followed by 2,6-lutidine (0.29 mL, 2.5 mmol). Reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated and purified with an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a white solid. (100 mg, 86%); MP 160–162 °C; 1H NMR (CDCl3) (400 MHz) (CDCl3) δ 7.12 (m, 15H), 5.94 (brs, 1H), 5.17 (brs, 1H), 4.99 (d, 1H, J = 12), 4.80 (d, 1H, J = 12), 4.57 (brs, 1H), 3.66 (s, 1H), 3.34 (m, 1H), 3.20 (m, 1H), 3.04 (m, 1H), 2.47 (m, 4H), 1.93 (m, 3H), 1.67 (m, 2H), 1.46 (m, 4H), 1.36 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.2, 156.8, 156.1, 141.64, 141.63, 136.2, 128.5, 128.4, 128.3, 128.0, 125.9, 79.2, 66.8, 51.9, 46.6, 43.7, 40.1, 34.4, 34.3, 33.6, 33.5, 30.1, 28.4, 26.5
Benzyl (S)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate
Benzyl (S)-5-((tert-butoxycarbonyl)amino)-2-((2-phenethyl-4-phenylbutanamido)-methyl)pentanoate (145 mg, 0.24 mmol) was dissolved in dichloromethane (3 mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2 mL) was added and reaction stirred at 0 °C for 2 hours. The solvents were rotavapped on completion of the reaction and residue dissolved in saturated sodium bicarbonate. Crude product was extracted with dichloromethane and purified on an ISCO chromatograph (0–10% methanol/ dichloromethane with 1% NH4OH) to give product as a pale yellow solid. (98 mg, 82%); MP 122–123 °C; 1H NMR (400 MHz) (CDCl3) δ 7.23 (m, 15 H), 6.26 (brs, 1H), 5.63 ( d, 1H, J = 8), 5.10 (d, 1H, J = 12). 4.91 (d, 1H, J = 12), 3.75 ( m, 1H), 3.39 (m, 2H), 2.60 (m, 6H), 2.10 (m, 1H), 1.98 (m, 2H), 1.76 (m, 2H), 1.54 (m, 4H), 1.41 (s, 2H); 13C NMR (100 MHz) (CDCl3) δ176.1, 157.0, 141.6, 136.3, 128.5, 128.4, 128.3, 128.0, 125.9, 66.7, 51.8, 46.6, 43.9, 41.7, 34.4, 34.3, 33.6, 33.5, 30.3, 29.3
Benzyl (S)-(5-(benzyl(methyl)amino)-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate
To a stirred solution of benzyl (S)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate, (81 mg, 0.16 mmol) in ethanol (10 mL) in 3Å molecular sieves at room temperature was added benzaldehyde (20 μL, 0.20 mmol). This was stirred for 30 minutes, then sodium cyanoborohydride (13 mg, 0.20 mmol) was added and reaction stirred under nitrogen overnight. Then 37% formaldehyde (30 μL, 0.33 mmol) was added, followed by sodium cyanoborohydride (14 mg, 0.23 mmol). Reaction was stirred at room temperature for 1 hour. The reaction mixture was filtered to remove the sieves and washed residue with methanol. The filtrate was concentrated under reduced pressure, the residue redissolved in ethyl acetate and quenched with saturated ammonium chloride solution. The organic layer was washed with saturated NaHCO3 solution and brine, dried over sodium sulfate. It was purified on an ISCO chromatograph (0–10% MeOH/dichloromethane) to give product as a colorless oil. (27 mg, 28%); 1H NMR (400 MHz) (CDCl3) δ 7.24 (m, 20H), 6.34 (brs, 1H), 6.04 (brs, 1H), 5.10 (m, 1H), 4.91 (m, 1H), 3.73 (m, 1H), 3.56 (s, 2H), 3.37 (m, 2H), 2.55 (m, 6H), 2.22 (s, 3H), 1.99 (m, 2H), 1.66 (m, 6H); 13C NMR (100 MHz) (CDCl3) δ 176.2, 157.2, 141.7, 136.3, 129.3, 128.8, 128.5, 128.4, 128.3, 128.2, 128.0, 127.9, 127.5, 126.9, 125.9, 66.7, 62.1, 56.6, 51.6, 46.7, 44.3, 41.6, 34.4, 33.6, 30.6, 23.0
tert-Butyl (S)-(4-((tert-butoxycarbonyl)amino)-5-(2-phenethyl-4-phenyl-butanamido)-pentyl)(methyl)carbamate
Benzyl (S)-(5-(benzyl(methyl)amino)-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)-carbamate (25.5 mg, 0.042 mmol) was dissolved in ethanol (5 mL). Boc2O (22.9 mg, 0.11 mmol) and Pd(OH)2/C (20 mg) were added and the reaction mixture stirred under hydrogen for overnight. The reaction mixture was then filtered to remove catalyst. Filtrate was concentrated and purified on an ISCO chromatograph to give product as a colorless oil (12.7 mg, 52%); 1H NMR (400 MHz) (CDCl3) δ 7.23 (m, 10H), 3.72 (m, 1H), 3.35 (m, 2H), 3.24 (m, 2H), 2.84 (s, 3H), 2.64 (m, 2H), 2.55 (m, 2H), 2.12 (m 1H), 2.00 (m, 2H), 1.79 (m, 3H), 1.62 (m, 3H), 1.47(s, 9H), 1.39 (m, 9H); 13C NMR (100 MHz) (CDCl3) δ 175.9, 156.5, 141.7, 128.4, 125.9, 79.6, 79.3, 46.6, 44.3, 34.4, 34.1, 33.7, 33.6, 28.4, 28.3
(S)-N-(2-Amino-5-(methylamino)pentyl)-2-phenethyl-4-phenylbutanamide
tert-Butyl (S)-(4-((tert-butoxycarbonyl)amino)-5-(2-phenethyl-4-phenylbutanamido)-pentyl)(methyl)carbamate, 155, (8.8 mg, 0.015 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) was added. The reaction mixture was stirred at room temperature for 2 hours. Then, the solvents were removed under reduced pressure and the residue dried under vacuum to obtain product as a colorless oil. (8.7 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.21 (m, 11H), 3.45 (m, 2H), 3.01 (m, 2H), 3.71 (s, 3H), 2.61 (m, 4H), 2.38 (m, 1H), 1.83 (m, 9H); 13C NMR (100 MHz) (MeOD) δ 180.1, 142.9, 129.4, 129.3, 127.0, 52.7, 47.5, 42.0, 35.6, 35.5, 34.8, 34.7, 33.5, 28.5, 22.9; HRMS (ESI) Calculated for C24H35N30 (M+H)+ 382.2853, found 382.2846
(S)-N-(2-Amino-5-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide (16)
Benzyl (S)-(5-(dimethylamino)-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate
Benzyl (S)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate (82 mg, 0.16 mmol) was dissolved in 20% methanol/dichloromethane (10 mL) and a solution of 37% formaldehyde (0.5 mL) was added and allowed to stir for 10 minutes. Sodium triacetoxyborohydride (345 mg, 1.6 mmol) was added and reaction was stirred at room temperature overnight. The reaction mixture was dissolved in saturated sodium bicarbonate solution and extracted with dichloromethane. It was purified on an ISCO chromatograph (0–10 MeOH/dichloromethane + 1 NH4OH) to give product as a colorless oil (39.9 mg, 47%); 1H NMR (400 MHz) (CDCl3) δ 7.12 (m, 15H), 6.23 (m, 2H), 5.01 (d, 1H, J = 12), 4.83 (d, 1H, J = 12 ) 3.63 (m, 1H), 3.30 (d, 2H, J = 4), 2.48 (m, 4H), 2.21 (s, 2H0, 2.12 (s, 6H), 2.02 (m, 1H), 1.89 (m, 2H), 1.66 (m, 2H), 1.50 (m, 4H); 13C NMR (400 MHz) (CDCl3) δ 175.9, 157.3, 141.7, 136.4, 128.4, 128.3, 128.0, 127.9, 125.8, 66.6, 59.0, 51.59, 46.8, 45.1, 44.3, 34.5, 34.4, 33.7, 33.6, 30.7, 23.5.
(S)-N-(2-Amino-5-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide
Benzyl (S)-(5-(dimethylamino)-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)-carbamate (40 mg, 0.075 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (20 mg)was added. Reaction mixture was purged and stirred at room temperature under hydrogen. On completion, reaction was filtered and residue washed with 20% MeOH/dichloromethane. The filtrate was concentrated under reduced pressure and dried under vacuum to give product as a yellow oil. (30mg, 91%); 1H NMR (400 MHz) (MeOD) 7.16 (m, 11H), 3.19 (m, 2H), 2.85 (m, 1H), 2.54 (m, 3H), 2.28 (m, 10H), 1.87 (m, 2H), 1.63 (m, 6H); 13C NMR (400 MHz) (MeOD) δ 181.2, 145.6, 131.9, 131.8, 129.4, 63.0, 54.6, 50.2, 48.7, 47.5, 38.4, 38.3, 37.3, 35.8, 27.0; HRMS (ESI) Calculated for C25H37N3O (M+H)+ 396.3009, found 396.2999
(S)-N-(2-Amino-5-hydrazinylpentyl)-2-phenethyl-4-phenylbutanamide (17)
Benzyl (S)-4-((tert-butoxycarbonyl)amino)-5-(dibenzylamino)-5-oxopentanoate
(S)-5-(Benzyloxy)-2-((tert-butoxycarbonyl)amino)-5-oxopentanoic acid ( 3000 mg, 8.9 mmol) was dissolved in dichloromethane (50 mL) at room temperature under nitrogen. EDC (1875 mg, 9.8 mmol) and HOBt (1321 mg, 9.8 mmol) were added and reaction stirred at the same temperature for 5 minutes. Dibenzylamine (1.8 mL, 9.8 mmol) was added, followed by DIPEA ( 2.3 mL, 13.7 mmol). Reaction was then stirred overnight until completion of the reaction. Then, the organic layer was washed with water, saturated NaHCO3, 1 M HCl, water and brine. The organic layer was dried over sodium sulfate and the solvent removed under reduced pressure to give a residue which was purified by ISCO chromatograph (0 – 70 % ethyl acetate/hexane) to obtain product as a colorless oil (2.5 g, 54%); 1H NMR (400 MHz) (CDCl3) δ 7.29 (m, 15H), 5.48 (brs, 1H), 5.12 (s, 2H), 4.80 (m, 3H), 4.49 (d, 1H, J = 16), 4.34 (d, 1H, J = 16), 2.50 (m, 2H), 2.12 (m, 1H), 1.87 (m, 1H), 1.47 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 172.7, 155.6, 136.7, 136.0, 135.8, 128.9, 128.7, 128.5, 128.2, 128.1, 127.8, 127.5, 127.1, 79.7, 66.4, 49.7, 47.9, 29.8, 28.7, 28.3
(S)-4-((tert-Butoxycarbonyl)amino)-5-(dibenzylamino)-5-oxopentanoic acid
Benzyl (S)-4-((tert-butoxycarbonyl)amino)-5-(dibenzylamino)-5-oxopentanoate (2.4 g, 4.73 mmol) was dissolved in THF/methanol/water (3:2:2) (40 mL) at room temperature over nitrogen. Lithium hydroxide (1.9 g, 47.3 mmol) was added and the reaction was stirred for 6 hours until the completion of the reaction. Then the resulting mixture was acidified to pH = 3 with 2N HCl. The aqueous layer was washed with ethyl acetate twice. The combined organic phase was dried over sodium sulfate and the solvent removed under reduced pressure to give product as a colorless oil. (1.9 g, 95%); 1H NMR (400 MHz) (MeOD) δ 7.28 (m, 10H), 4.86 (brs, 1H), 4.78 (m, 2H), 4.57 (m, 2H), 4.35 (d, 1H, J = 12), 2.37 (t, 2H, J = 8), 2.01 (m, 1H), 1.85 (m, 1H), 1.45 (s, 9H); 13C NMR (100 MHz) (MeOD) δ 176.4, 175.0, 157.9, 142.7, 138.1, 137.8, 129.9, 129.7, 129.4, 128.9, 128.8, 128.5, 128.3, 128.0, 80.6, 65.2, 51.5, 51.2, 30.7, 28.7, 28.6
(S)-2-Amino-N,N-dibenzyl-5-hydrazinyl-5-oxopentanamide
To a solution of (S)-4-((tert-butoxycarbonyl)amino)-5-(dibenzylamino)-5-oxopentanoic acid ( 1.8 g, 4.23 mmol) in dichloromethane ( 24 mL) was added EDC ( 890 mg, 4.65 mmol), then tertbutylcarbazate ( 1.1 g, 8.47 mmol) and the reaction mixture was stirred at room temperature overnight. Then the organic layer was washed with saturated NaHCO3 and brine. The organic layer was then dried over sodium sulfate and the solvent removed under reduced pressure. The residue was purified by ISCO chromatograph (0 – 50 % ethyl acetate/hexane) to give the product as a colorless oil. This oil was dissolved in DCM (5 mL) and stirred at 0 °C under nitrogen. Trifluoroacetic acid (5 mL) was also added and the reaction stirred at that temperature for 1 hour, then room temperature for another hour. On completion of the reaction, the solvents were removed under reduced pressure and triturated with methanol to obtain product as a colorless oil. (910 mg, 64%); 1H NMR (400 MHz) (MeOD) δ 7.32 (m, 10H), 4.62 (m, 1H), 4.48 (d, 1H, J = 20), 4.28 (d, 1H, J = 12), 4.00 (s, 1H), 2.59 (m, 2H), 2.29 (m, 1H), 2.15 (m, 1H); 13C NMR (100 MHz) (MeOD) δ 172.5, 170.4, 137.6, 136.8, 130.1, 129.8, 129.3, 129.1, 128.8, 128.2, 51.7, 51.0, 49.8, 49.7, 28.6, 27.0
Di-tert-butyl (S)-1-(4-((tert-butoxycarbonyl)amino)-5-(dibenzylamino)pentyl)hydrazine-1,2-dicarboxylate
(S)-2-Amino-N,N-dibenzyl-5-hydrazinyl-5-oxopentanamide (900 mg, 2.0 mmol) was dissolved in anhydrous THF (30 mL) and BH3.THF (15 mL, 15 mmol) was added. The reaction mixture was heated at 83 °C for overnight. Then, methanol (20 mL) was added to the reaction mixture and reaction stirred at 83 °C for 2 hours. Water (8 mL) was added and the reaction cooled to room temperature. Solvents were then removed under reduced pressure and residue dissolved in DCM. The organic layer was dried over sodium sulfate and the solvent removed under reduced pressure which gave a colorless oil which was taking to the next step without any purification. The crude oil was dissolved in dichloromethane (20 mL) and Boc2O (2.5 g, 11.6 mmol) was added. Reaction was stirred at room temperature overnight. The organic layer was washed with water, dried over sodium sulfate and the solvent removed under reduced pressure. The residue was purified on ISCO chromatograph ( 0 – 40% ethyl acetate/hexane ) to give product as a colorless oil. (220 mg, 18%); 1H NMR (400 MHz) (CDCl3) δ 7.31 (m, 10H), 4.49 (m, 2H), 3.71 (m, 3H), 3.40 (m, 4H), 2.40 (d, 2H, J = 8), 1.52 (m, 4H), 1.51 (s, 9H), 1.50 (s, 9H), 1.49 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.0, 155.9, 139.3, 139.2, 128.98, 128.96, 128.2, 128.18, 128.16, 127.0, 126.9, 126.8, 81.2, 58.5, 28.4, 28.4, 28.3, 28.1
Di-tert-butyl (S)-1-(4-((tert-butoxycarbonyl)amino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-hydrazine-1,2-dicarboxylate
Di-tert-butyl (S)-1-(4-((tert-butoxycarbonyl)amino)-5-(dibenzylamino)pentyl)hydrazine-1,2-dicarboxylate (220 mg, 0.36 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (60 mg) was added and the reaction mixture purged. This mixture was stirred at room temperature overnight under hydrogen atmosphere to give product as a colorless oil. 2-phenethyl-4-phenylbutanoic acid (102 mg, 0.38 mmol) was dissolved in DMF (5 mL), then EDC (147 mg, 0.77 mmol) and HOBt (103 mg, 0.77 mmol) were added. The reaction mixture was stirred at room temperature for 5 minutes under nitrogen. Then di-tert-butyl (S)-1-(5-amino-4-((tert-butoxycarbonyl)amino)pentyl)hydrazine-1,2-dicarboxylate (150 mg, 0.35 mmol) was added, followed by 2,6-lutidine ( 0.12 mL, 1.05 mmol). The reaction mixture was stirred at room temperature overnight. On completion of the reaction, the organic layer was washed with the organic layer was washed with water, saturated NaHCO3, 1 M HCl, water and brine. The organic layer was dried over sodium sulfate and the solvent removed under reduced pressure to give a residue which was purified by ISCO chromatograph (0 – 10 % methanol/dichloromethane) to obtain product as a colorless oil. (130 mg, 51%); 1H NMR (400 MHz) (CDCl3) δ 7.23 (m, 10H), 6.60 (brs, 2H), 3.70 (m, 1H), 3.43 (m, 2H), 2.59 (m, 4H), 2.15 (m, 1H), 1.99 (m, 2H), 1.76 (m, 2H), 1.46 (m, 6H), 1.40 (s, 9H), 1.39 (s, 9H), 1.37 (m, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.1, 156.5, 156.1, 155.9, 155.4, 141.8, 141.7, 141.5, 128.3, 125.8, 125.8, 81.2, 80.4, 79.5, 64.2, 51.0, 46.8, 46.4, 44.3, 34.5, 34.3, 33.7. 33.6, 29.5, 28.4, 28.3, 28.2, 28.1, 26.4, 25.3
(S)-N-(2-amino-5-hydrazinylpentyl)-2-phenethyl-4-phenylbutanamide
Di-tert-butyl (S)-1-(4-((tert-butoxycarbonyl)amino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-hydrazine-1,2-dicarboxylate ( 90 mg, 0.13 mmol) was dissolved in dichloromethane (1 mL) and TFA (0.5 mL) was added. Reaction was stirred at room temperature for 2 hours. The solvents were removed under reduce pressure and triturated with methanol to obtain the product as a colorless oil. (66 mg, 88%); 1H NMR (400 MHz) (MeOD) δ 7.22 (m, 11H), 3.46 (m, 2H), 3.33 (m, 2H), 3.06 (t, 1H, J = 8), 2.97 (t, 1H, J = 8), 2.60 (m, 4H), 2.40 (m, 1H), 1.86 (m, 8H); 13C NMR (100 MHz) (MeOD) δ 180.1, 143.0, 129.4, 129.3, 126.9, 52.9, 51.3, 47.5, 42.1, 40.0, 35.6, 34.7, 28.7, 28.5, 24.4, 22.5; HRMS (ESI) Calculated for C23H34N4O (M+H)+ 383.2805 found 383.2811
(S)-N-(2-Amino-5-guanidinopentyl)-2-phenethyl-4-phenylbutanamide (18)
tert-Butyl (S)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate
Benzyl tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (125, mg, 0.21 mmol) was dissolved in ethanol(15 mL) and 20% Pd(OH)2/C (30 mg) was added. The reaction mixture was then purged and stirred under a hydrogen atmosphere for overnight. Then the catalyst was filtered and the residue washed with 20% MeOH/dichloromethane. The solvent of the filtrate was removed under reduced pressure and dried under vacuum to give product was a colorless oil. (84 mg, 86%); 1H NMR (400 MHz) ( CDCl3)δ 7.06 (m, 10H), 6.43 (s, 1H), 5.14 (d, 1H, J = 4), 3.54 (m, 1H), 3.19 (m, 2H), 2.46 (m, 7H), 1.94 (m, 1H), 1.84 (m, 2H), 1.62 (m, 2H), 1.40 (m, 3H), 1.24 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.0, 156.6, 141.8, 141.7, 128.3, 125.8, 79.4, 57.9, 51.1, 46.6, 44.1, 41.4, 34.4, 33.7, 33.6, 30.4, 28.9, 28.3
Tris-Boc-(S)-N-(2-amino-5-guanidinopentyl)-2-phenethyl-4-phenylbutanamide
tert-Butyl (S)-(5-amino-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate (80 mg, 0.17 mmol) was dissolved in dichloromethane (5 mL). Triethylamine (0.03 mL, 0.21 mmol) followed by 1,3-diboc-2-(trifluoromethylsulfonyl)guanidine (80 mg, 0.21 mmol) was added. The reaction was stirred at room temperature overnight under nitrogen. The reaction mixture was dissolved in saturated sodium bicarbonate and extracted with dichloromethane. The organic layer was dried over sodium sulfate and concentrated. Crude product was purified on an ISCO chromatograph (0–30% ethyl acetate hexane) to give product as a colorless oil (35.3 mg, 29%); 1H NMR (400 MHz) (CDCl3) δ 11.48 (s, 1H), 8.37 (s, 1H), 7.22 (m, 10H), 6.19 (s, 1H), 5.07 (s, 1H), 3.73 (s, 1H), 3.42 (m, 4H), 2.59 (m, 5H), 2.01 (m, 6H), 1.67 (m, 2H), 1.51 (s, 18H), 1.40 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 175.9, 163.5, 165.2, 153.3, 141.6, 128.3, 125.8, 83.1, 79.2, 51.1, 46.6, 40.4, 34.4, 33.6, 30.1, 28.33, 28.31, 28.07, 25.9.
(S)-N-(2-Amino-5-guanidinopentyl)-2-phenethyl-4-phenylbutanamide
Tris-Boc-(S)-N-(2-amino-5-guanidinopentyl)-2-phenethyl-4-phenylbutanamide (30 mg, 0.042 mmol) was dissolved in dichloromethane (2 mL) and cooled to 0 °C. Trifluoroacetic acid (1 mL) was added and reaction stirred at room temperature for 2 hours. The solvents were removed under reduced pressure and the residue dried under vacuum. The product was submitted as its trifluoroacetic acid salt. (37.6 mg, quantitative). 1H NMR (400 MHz) (MeOD) δ 7.10 (m, 11H), 3.34 (m, 2H), 3.10 (m, 3H), 2.47 (m, 4H), 2.27 (m, 1H), 1.83 (m, 2H), 1.68 (m, 6H); 13C NMR (100 MHz) (MeOD) δ 180.1, 158.7, 143.0, 129.4, 129.3, 126.9, 52.9, 47.4, 42.2, 42.0, 35.5, 34.7, 28.8, 25.6; HRMS (ESI) Calculated for C24H35N5O (M+H)+ 410.2914, found 410.2919
((S)-N-(5-Amino-2-(methylamino)pentyl)-2-phenethyl-4-phenylbutanamide (19)
Benzyl tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acid (546 mg, 2.04 mmol) was dissolved in dry DMF (5mL) and EDC (816 mg, 4.27 mmol) and HOBt (575mg, 4.27 mmol) were added and the reaction stirred at room temperature for 5 minutes. Benzyl tert-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (682 mg, 1.94 mmol) was added followed by 2,6-lutidine (0.67 mL, 3 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was then diluted with ethyl acetate and washed with water, 1M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtered. The filtrate was then concentrated and purified on an ISCO chromatograph with silica (0–10% MeOH/dichloromethane) to give a white solid. (520 mg, 45%); MP 155–157 °C; 1H NMR (CDCl3) (400 MHz) δ 7.26 (m, 15H), 6.20 (brs, 1H), 5.10 (s, 3H), 4.95 (d, 1H, J = 8), 3.70 (s, 1H), 3.29 (s, 4H), 2.59 (m, 4H), 2.11 (m, 1H), 2.01 (m, 2H), 1.78 (m, 2H), 1.55 (m, 4), 1.40 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.1, 156.5, 156.4, 141.7, 141.6,136.6, 128.5, 128.4, 128.0, 125.9, 79.6, 66.6, 51.0, 46.6, 43.9, 40.7, 34.4, 33.7, 33.6, 30.3, 28.3, 26.3
Benzyl (S)-(4-amino-5-(2-phenethyl-4-phenylbutanamido)pentyl)carbamate
Benzyl tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (107 mg, 0.18 mmol) was dissolved in dichloromethane (3 mL) and cooled to 0 °C under nitrogen. Trifluoroacetic acid (2 mL) was added and reaction stirred at 0 °C for 2 hours. The solvents were removed under reduced pressure upon completion of the reaction and residue dissolved in saturated sodium bicarbonate. Crude product was extracted with dichloromethane and purified on an ISCO chromatograph (0–10% methanol/ dichloromethane with 1% NH4OH) to give product as a colorless oil. (69 mg, 78%); 1H NMR (CDCl3) (400 MHz) δ 7.09 (m, 10H), 6.69 (s, 1H), 5.28 (s, 2H), 5.05 (s, 1H), 4.89 (s, 2H), 3.28 (m, 1H), 2.97 (s, 4H), 2.41 (m, 4H), 2.05 (m, 1H), 1.82 (m, 2H), 1.61 (m, 2H), 1.40 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 176.8, 156.8, 141.6, 136.4, 128.5, 128.4, 128.3, 128.1, 127.9, 125.9, 66.7, 51.7, 46.5, 43.1, 40.5, 34.2, 33.6, 30.0, 25.9
Benzyl (S)-(4-(benzyl(methyl)amino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-carbamate
To a stirred solution of benzyl (S)-(4-amino-5-(2-phenethyl-4-phenylbutanamido)-pentyl)carbamate, (178 mg, 0.35 mmol) in ethanol (15 mL) and 3Å molecular sieves at room temperature was added benzaldehyde (0.043 mL, 0.43 mmol) and reaction was stirred for 1 hour under nitrogen. Then NaBH3CN (26 mg, 0.425 mmol) was added and the reaction was stirred at room temperature overnight. Formaldehyde (37% aqueous solution) (0.05 ml, 0.7 mmol) was then added and reaction was stirred for 1 hour. A second batch of NaBH3CN (31 mg, 0.49 mmol) was then added and the reaction mixture stirred for 6 hours. The reaction mixture was filtered to remove the molecular sieves and the residue washed with methanol. The filtrate was concentrated under reduced pressure and the residue redissolved in ethyl acetate. The ethyl acetate layer was quenched with saturated NH4Cl solution and washed with saturated sodium bicarbonate and brine. The combined organic layers were dried over sodium sulfate, concentrated and purified on an ISCO chromatograph (0–60 % ethyl acetate/hexane). This gave product as a colorless oil (96 mg, 45%); 1H NMR (400 MHz) (CDCl3) δ 7.29 (m, 10H), 6.27 (d, 1H, J = 4), 5.14 (s, 2H), 5.04 (brs, 1H), 3.62 (m, 2H), 3.26 (m, 2H), 3.02 (m, 1H), 2.62 (m, 5H), 2.24 (s, 3H), 1.83 (m, 12H); 13C NMR (100 MHz) (CDCl3) δ 174.9, 156.5, 141.8, 141.7, 139.3, 136.6, 128.6, 128.6, 128.5, 128.43, 128.40, 128.3, 128.1, 127.2, 125.94, 125.92, 66.7, 61.1, 58.0, 46.5, 41.1, 39.5, 36.0, 34.4, 34.1, 33.6, 33.5, 27.6, 23.3
tert-Butyl (S)-(4-(methylamino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-carbamate
Benzyl (S)-(4-(benzyl(methyl)amino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-carbamate (81 mg, 0.135 mmol) was dissolved in ethanol (10 mL) and Boc2O (88 mg, 0.41 mmol) was added. The reaction mixture was purged and stirred at room temperature overnight. Then the reaction mixture was filtered to remove catalyst. The filtrate was concentrated and purified on an ISCO chromatograph to obtain product as a colorless oil (20.2 mg, 31%); 1H NMR (400 MHz) (CDCl3) δ 7.22 (m, 11H), 4.82 (brs, 1H), 3.64 (m, 1H), 3.48 (m, 1H), 3.16 (m, 2H), 3.06 (m, 1H), 2.62 (m, 7H), 2.39 (m, 1H), 2.04 (m, 2H), 1.74 (m, 6H), 1.44 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.4, 156.3, 141.7, 128.4, 128.3, 125.9, 79.6, 59.9, 46.9, 39.2, 34.6, 34.5, 33.8, 31.4, 28.4, 26.1
(S)-N-(5-Amino-2-(methylamino)pentyl)-2-phenethyl-4-phenylbutanamide
tert-Butyl (S)-(4-(methylamino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)carbamate (20 mg, 0.04 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) was added. Reaction was stirred at room temperature for 2 hours. On completion of the reaction, the solvents were removed and residue dried under vacuum to obtain product as colorless oil. (14 mg, 61%). 1H NMR (400 MHz) (MeOD) δ 7.23 (m, 11H), 3.53 (m, 2H), 3.26 (m, 1H), 2.98 (m, 2H), 2.80 (s, 3H), 2.61 (m, 4H), 2.41 (m, 1H), 1.96 (m, 8H); 13C NMR (100 MHz) (MeOD) δ 180.4, 142.9, 129.4, 129.3, 127.0, 60.0, 40.1, 39.6, 35.5, 34.8, 34.7, 31.6, 26.6, 24.4; HRMS (ESI) Calculated for C24H35N3O (M+H)+ 382.2853, found 382.2864
(S)-N-(5-amino-2-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide (20)
Benzyl (S)-(4-(dimethylamino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-carbamate
Benzyl (S)-(4-amino-5-(2-phenethyl-4-phenylbutanamido)pentyl)carbamate (53 mg, 0.11 mmol) was dissolved in 20% methanol/dichloromethane (10 mL) and a solution of 37% formaldehyde (1 mL) was added and allowed to stir for 10 minutes. Sodium triacetoxyborohydride (226 mg, 1.1 mmol) was added and reaction was stirred at room temperature overnight. The reaction mixture was dissolved in saturated sodium bicarbonate solution and extracted with dichloromethane. The organic layer was dried over sodium sulfate concentrated and purified on an ISCO chromatograph (0–10% MeOH/dichloromethane + 1 NH4OH) to give product as a colorless oil (44.3 mg, 79%); 1H NMR (400 MHz) (CDCl3) δ 7.28 (m, 15H), 6.31 (brs, 1H), 5.11 (s, 2H), 5.06 (brs, 1H), 3.61 (m, 1H), 3.24 (m, 2H), 2.97 (m, 1H), 2.65 (m, 2H), 2.56 (m, 3H), 2.28 (s, 6H), 2.07 (m, 3H), 1.77 (m, 2H), 1.58 (m, 3H); 13C NMR (100 MHz) (CDCl3) δ 175.1, 156.5, 141.8, 136.6, 128.5, 128.42, 128.40, 128.3, 128.1, 125.8, 66.6, 62.7, 46.3, 41.1, 40.1, 39.3, 34.6, 34.5, 33.7, 33.6, 27.5, 22.6
(S)-N-(5-Amino-2-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide
Step 1 : tert-Butyl (S)-(4-(dimethylamino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)-carbamate benzyl: (S)-(5-(Dimethylamino)-1-(2-phenethyl-4-phenylbutanamido)pentan-2-yl)carbamate (39 mg, 0.074 mmol) was dissolved in ethanol (5 mL), Boc2O (24 mg, 0.112 mmol) and 20% Pd(OH)2/C (20 mg) was added. Reaction mixture was purged and stirred at room temperature under hydrogen overnight. On completion of the reaction, the reaction was filtered and residue washed with 20% MeOH/dichloromethane. The filtrate was concentrated under reduced pressure and purified on an ISCO chromatograph (0–10% methanol/dichloromethane) to give product as a colorless oil (33 mg, 90%); 1H NMR (400 MHz) (CDCl3) δ 7.13 (m, 10H), 6.21 (brs, 1H), 3.51 (m, 1H), 3.05 (m, 2H), 2.85 (m, 1H), 2.49 (m, 6H), 2.81 (s, 6H), 1.95 (m, 4H), 1.66 (m, 2H), 1.51 (m, 2H), 1.37 (s, 9H);13C NMR (100 MHz) (CDCl3) δ 175.0, 156.0, 141.8, 128.4, 128.39, 128.36, 125.8, 79.1, 62.7, 46.3, 40.5, 40.1, 39.4, 34.5, 33.6, 33.3, 31.2, 28.4, 28.0, 27.7, 22.4
Step 2: (S)-N-(2-Amino-5-(dimethylamino)pentyl)-2-phenethyl-4-phenylbutanamide - tert-butyl (S)(4-(Dimethylamino)-5-(2-phenethyl-4-phenylbutanamido)pentyl)carbamate (23 mg, 0.046 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) was added. Reaction was stirred at room temperature for 2 hours under nitrogen. Then solvents were removed and dried under vacuum to give product as a trifluoroacetic acid salt which was a colorless oil. (27 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.23 (m, 11H), 3.56 (m, 1H), 3.38 (m, 1H), 3.01 (m, 2H), 2.60 (m, 4H), 2.40 (m, 1H), 1.82 (m, 4H); 13C NMR (100 MHz) (MeOD) δ 180.0, 161.9, 142.9, 129.6, 129.5, 129.4, 129.3, 127.0, 67.3, 47.3, 40.1, 38.9, 35.5, 34.8, 34.7, 25.2; HRMS (ESI) Calculated for C25H37N3O (M+H)+ 396.3009, found 396.3011
(S)-N-(2,5-diaminopentyl)-N-methyl-2-phenethyl-4-phenylbutanamide (21)
Di-tert-butyl (5-(benzyl(methyl)amino)-5-oxopentane-1,4-diyl)(S)-dicarbamate
Bis Boc-L- ornithine (1000 mg, 3 mmol) was dissolved in dichloromethane (30 mL), then EDC (631 mg, 3.3 mmol) and HOBT ( 445 mg, 3.3 mmol) was added. The reaction was stirred at room temperature under nitrogen. Then N-benzylmethyl amine (400 mg, 3.3 mmol) and DIPEA (581.6 mg, 4.5 mmol) was added. Reaction was stirred for overnight and organic layer washed with water, saturated NaHCO3, 1 M HCl, water and brine. The organic layer was dried over sodium sulfate, concentrated under reduced pressure and dried over vacuum to give product as a yellow oil. (1.08 g, 82%); 1H NMR (400 MHz) (CDCl3) δ 7.19 (m, 5H), 5.51 (m, 1H), 4.79 (brs, 1H), 4.50 (m, 3H), 3.01 (m, 2H), 2.90 (s, 3H), 1.56 (m, 4H), 1.36 (s, 9H), 1.34 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 172.3, 155.9, 155.6, 136.6, 136.1, 128.8, 128.6, 127.8, 127.7, 127.4, 126.7, 79.4, 78.9, 53.1, 51.1, 50.0, 40.1, 34.6, 30.7, 30.3, 28.4, 28.3, 28.2, 25.8
(S)-2,5-Diamino-N-benzyl-N-methylpentanamide
Di-tert-butyl (5-(benzyl(methyl)amino)-5-oxopentane-1,4-diyl)(S)-dicarbamate (524 mg, 1.2 mmol) was dissolved in dichloromethane (6 mL), then trifluoroacetic acid (2 mL) was added. Reaction was stirred at 0 °C for 2 hours. The solvents were then removed under reduced pressure and dried under vacuum to obtain product as a colorless oil. (498 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.35 (m, 5H), 4.53 (m, 2H), 3.33 (m, 1H), 2.98 (m, 5H), 1.90 (m, 4H); 13C NMR (100 MHz) (MeOD) δ 170.0, 169.9, 137.5, 137.0, 130.1, 129.8, 129.13, 129.10, 128.8, 128.0, 55.1, 53.1, 53.9, 52.3, 51.5, 51.4, 40.0, 35.0, 34.6, 29.0, 28.5, 23.8
Di-tert-butyl (5-(benzyl(methyl)amino)pentane-1,4-diyl)(S)-dicarbamate
(S)-2,5-Diamino-N-benzyl-N-methylpentanamide ( 498 mg, 1.20 mmol) was dissolved in dry THF (10 mL) and BH3.THF (1M in THF) (6 mL, 6 mmol) was added. The reaction mixture was heated at reflux overnight. Then methanol (20 mL) was added and stirred at that temperature for 2 hours and cooled to room temperature. Then water (2 mL) was added. The solvents were removed and redissolved in dichloromethane and dried over sodium sulfate to remove water. The dichloromethane was filtered and the filtrate concentrated to give a colorless oil. The oil was dissolved in dichloromethane (10 mL) and Boc2O was added. Reaction was stirred at room temperature overnight. Ethyl acetate was added to the reaction mixture and washed with brine. The organic layer was dried over sodium sulfate, concentrated and purified using an ISCO chromatograph to give product as a colorless oil. (195 mg, 39%); 1H NMR (400 MHz) (CDCl3) δ 7.26 (m, 5H), 4.77 (brs, 1H), 4.64 (brs, 1H), 3.67 (m, 1H), 3.53 (d, 1H, J = 12), 3.42 (d, 1H, J = 12), 3.10 (d, 2H, J = 4), 2.32 (m, 2H), 2.20 (s, 3H), 1.59 (m, 4H), 1.44 (s, 9H), 1.43 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 156.0, 139.0, 128.9, 128.1, 126.9, 78.8, 70.7, 62.3, 61.1, 42.6, 40.4, 30.8, 28.4, 26.4, 26.2
Di-tert-butyl (5-((1,5-diphenylpentan-3-yl)(methyl)amino)pentane-1,4-diyl)(S)-dicarbamate
Di-tert-butyl (5-(benzyl(methyl)amino)pentane-1,4-diyl)(S)-dicarbamate (180 mg, 0.42 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (50 mg) was added. The reaction mixture was purged and stirred under hydrogen atmosphere for overnight. The catalyst was filtered off and washed with 20% methanol/dichloromethane, concentrated and dried to give product (di-tert-butyl (5-(methylamino)pentane-1,4-diyl)(S)-dicarbamate) which was used in the next step without further purification. 2-Phenethyl-4-phenylbutanoic acid (94 mg, 0.35 mmol) was dissolved in dichloromethane (5 mL) and PyBrop (179 mg, 0.38 mmol) was added. Reaction was stirred at room temperature for 5 minutes. Di-tert-butyl (5-(methylamino)pentane-1,4-diyl)(S)-dicarbamate (105 mg, 0.32 mmol) was added, followed by triethylamine (0.13 mL, 0.95 mmol) and the reaction stirred at room temperature overnight under nitrogen. Then the reaction mixture was dissolved in ethyl acetate, washed with water, saturated NaHCO3, 1 M HCl, water and brine. The organic layer was dried over sodium sulfate, filtered, concentrated and purified on an ISCO chromatograph (0–10% MeOH /dichloromethane) to give product as a colorless oil. (146 mg, 78%); 1H NMR (400 MHz) (CDCl3) δ 7.18 (m, 10H), 5.14 (d, 1H, J = 8), 4.81 (brs, 1H), 3.81 (m, 2H), 3.45 (m, 1H), 3.13 (m, 2H), 2.90 (m, 1H), 2.68 (s, 3H), 2.59 (m, 4H), 2.01 (m, 2H), 1.81 (m, 2H), 1.56 (m, 4H), 1.44 (s, 9H), 1.31 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.8, 156.0, 141.7, 141.6, 128.5, 128.4, 128.38, 128.31, 125.9, 79.0, 50.6, 49.2, 40.3, 39.1, 35.2, 34.0, 33.9, 33.8, 33.4, 33.39, 33.34, 30.9, 28.4, 28.36, 28.30, 26.0
(S)-N-(2,5-Diaminopentyl)-N-methyl-2-phenethyl-4-phenylbutanamide
Di-tert-butyl (5-(N-methyl-2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (34 mg, 0.058 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) was added. Reaction was stirred under nitrogen at room temperature for 2 hours. The solvents were removed and the residue dried under vacuum pump to get product as a salt which was a light brown colored oil. (33 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.22 (m, 10H), 3.50 (m, 3H), 2.98 (t, 2H, J = 8), 2.78 (s, 3H), 2.70 (m, 1H), 2.62 (m, 4H), 1.82 (m, 8H); 13C NMR (100 MHz) (MeOD) δ 180.2, 142.8, 142.7, 129.6, 129.5, 127.1, 52.0, 51.8, 40.6, 40.2, 37.3, 34.8, 34.6, 34.3, 28.9, 24.3; HRMS (ESI) Calculated for C24H35N3O (M+H)+ 382.2853, found 382.2855
(S)-N1-(2-phenethyl-4-phenylbutyl)pentane-1,2,5-triamine (22)
Di-tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate
2-phenethyl-4-phenylbutanoic acid (158 mg, 0.59 mmol) was dissolved in DMF (5 mL) and EDC (227 mg, 1.19 mmol) and HOBT (166 mg, 1.19 mmol) were added. Reaction was stirred at room temperature for 5 minutes. Then dibenzyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (200 mg, 0.54 mmol) was added followed by 2,6 – lutidine (0.19 mL, 1.62 mmol). Reaction was stirred at room temperature overnight under nitrogen. The reaction mixture was then diluted with ethyl acetate, and washed with water, 1M HCl, saturated NaHCO3, water and brine to obtain a crude oil. This crude oil was dissolved in ethanol (15 mL), 20% Pd (OH)2/C (80 mg) and Boc2O (272 mg, 1.25 mmol) was added. The reaction mixture was stirred under hydrogen at room temperature overnight. On completion of the reaction, the mixture was filtered to remove catalyst and solvent rotavapped. The residue was purified on an ISCO chromatograph (0–10% methanol/dichloromethane) to get product as a white solid. (85 mg, 30%); MP 140–142 °C; 1H NMR (CDCl3) (400 MHz) δ 7.22 (m, 10H), 6.33 (brs, 1H), 5.02 (brs, 1H), 4.80 (m, 1H); 3.70 (m, 1H). 3.35 (m, 2H), 3.14 (m, 2H), 2.59 (m, 4H), 2.14 (m, 1H), 2.02 (m, 2H), 1.78 (m, 2H), 1.57 (m, 4H), 1.45 (s, 9H), 1.39 (m, 9H); 13C NMR (100 MHz) (CDCl3) δ 176.0, 156.5, 156.1, 141.7, 141.6, 141.4, 128.3, 125.9, 79.5, 79.1, 51.1, 46.6, 44.0, 40.2, 34.4, 33.6, 30.3, 28.4, 28.3, 26.5
(S)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
Di-tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,4-diyl)(S)-dicarbamate (80 mg, 0.14 mmol) was dissolved in dichloromethane (5 mL) and trifluoroacetic acid (2.5 mL) at 0 °C. The reaction was stirred at that temperature for 2 hours under nitrogen. The solvents were removed to obtain product as a trifluoroacetic acid salt, colorless oil. (79 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.22 (m, 10H), 3.46 (m, 2H), 2.97 (m, 2H), 2.61 (m, 4H), 2.40 (m, 1H), 1.96 (m, 2H), 1.83 (m, 6H); 13C NMR (100 MHz) (MeOD) δ 180.1, 143.0, 129.4, 129.3, 126.9, 52.7, 47.5, 42.0, 40.1, 35.6, 35.5, 34.7, 28.5, 24.4
Di-tert-butyl (5-((tert-butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)pentane-1,4-diyl)(S)-dicarbamate
(S)-N-(2,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide (79 mg, 0.14 mmol) was dissolved in dry THF (10 mL) and BH3.THF (2 mL) was added. The reaction mixture was stirred under reflux overnight under nitrogen. Methanol (10 mL) was added and the reaction was stirred at reflux for 2 hours. On completion, 2 mL of water was added and the reaction was allowed to cool to room temperature. Then the reaction was dried with sodium sulfate and filtered. The organic layer was concentrated under reduced pressure to remove solvent. The residue was dissolved in dichloromethane (5 mL) and Boc2O (92 mg, 0.42 mmol) and reaction was stirred at room temperature for 2 hours. The organic layer was concentrated under reduced pressure and the residue purified on an ISCO chromatograph (0–30% EtOAc/hexane) to give product as a colorless oil. (30 mg, 32%); 1H NMR (400 MHz) (CDCl3) δ 7.23 (m, 10H), 5.03 (brs, 1H), 4.65 (brs, 1H), 3.71 (m, 1H), 3.43 (m, 2H), 3.14 (m, 2H), 2.84 (m, 1H), 2.63 (m, 4H), 1.66 (m, 6H), 1.47 (s, 18H), 1.43 (s, 9H); 13C NMR (100 MHz) (CDCl3) δ 155.5, 128.3, 125.8, 79.9, 79.0, 70.8, 62.7, 49.7, 49.4, 40.3, 35.9, 33.1, 33.0, 32.7, 30.7, 28.43, 28.4, 26.3
(S)-N1-(2-Phenethyl-4-phenylbutyl)pentane-1,2,5-triamine
Di-tert-butyl (5-((tert-butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)pentane-1,4-diyl)(S)-dicarbamate (22 mg, 0.034 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) was added. Reaction mixture was stirred at room temperature for 2 hours. On completion of the reaction, the solvents were removed under reduced pressure. The residue was triturated with methanol three times to obtain product as a white solid. (21 mg, 100%); MP 130–13 °C; 1H NMR (400 MHz) ( MeOD) δ 7.12 (m, 10H), 3.59 (m, 1H), 3.25 (m, 1H), 3.03 (m, 2H), 2.87 (m, 2H), 2.55 (m, 4H), 1.68 (m, 10H); 13C NMR (100 MHz) ( MeOD) 142.9, 129.5, 129.4, 127.0, 54.0, 51.1, 39.9, 36.3, 33.9, 33.3, 29.4, 24.2; HRMS (ESI) Calculated for C23H35N3 (M+H)+ 354.2904, found 354.2906.
(S)-N1-(2-Phenethyl-4-phenylbutyl)pentane-1,3,5-triamine (23)
(3-Methylenepentane-1,5-diyl)dibenzene
To a suspension of methyl triphenylphosphonium bromide (2397 mg, 6.7 mmol) in THF (8 mL) at room temperature was added portion wise potassium t-butoxide (848 mg, 7.56 mmol) under nitrogen. The mixture was stirred at that temperature for 3 hours. Then a solution of 1,5-diphenylpentan-3-one (1000 mg, 4.2 mmol) in THF (2 mL) was added dropwise. The mixture was stirred overnight, then the mixture was concentrated under reduced pressure, then ice water added. The aqueous solution was extracted with ethyl acetate. The organic layer was then washed with brine and dried over sodium sulfate. The filtrate was then concentrated and purified on ISCO chromatograph (0–5% ethyl acetate/hexane) to give product as a colorless oil. (806 mg, 81%); 1H NMR (400 MHz) (CDCl3) δ 7.51 (m, 4H), 7.43 (m, 6H), 5.06 (m, 2H), 3.01 (m, 4H), 2.61 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 148.8, 142.5, 142.3, 128.9, 128.8, 128.5, 128.2, 126.4, 126.2, 126.0, 125.7, 109.9, 38.5, 38.3, 34.6
2-Phenethyl-4-phenylbutan-1-ol
To a solution of (3-methylenepentane-1,5-diyl)dibenzene (650 mg, 2.75 mmol) in THF (5 mL), cooled to 0 °C was added BH3.THF (4.1 mL, 4.1 mmol). The reaction was stirred at that temperature for 30 minutes, then warmed to room temperature and stirred for an additional 2 hours under nitrogen. Then the reaction was cooled again to 0 °C, 3 M NaOH (4.6 mL) and 30% H2O2 (1.4 mL) were added. The reaction was stirred at 0 °C for 30 minutes and heated at 60 °C for an additional hour. Solvents were removed under reduced pressure and the resulting residue diluted with water and extracted with ethyl acetate, The combined organic layers were washed with water, brine, dried over sodium sulfate, concentrated and purified on an ISCO chromatograph with 0–40% ethyl acetate/hexane) to give product as a colorless oil. (538 mg, 77%); 1H NMR (400 MHz) (CDCl3) δ 7.29 (m, 10H), 3.69 (d, 2H, J = 8), 2.71 (m, 4H, J = 8), 1.72 (m, 5H), 1.47 (brs, 1H); 13C NMR (100 MHz) (CDCl3) δ 142.5, 128.41, 128.40, 125.8, 65.3, 39.7, 33.2, 32.8
2-(2-Phenethyl-4-phenylbutyl)isoindoline-1,3-dione
Triphenylphosphine (570 mg, 2.17 mmol) and phthalimide (319 mg, 2.17 mmol) were dissolved in THF (6 mL) at 0 °C under nitrogen. Then 2-phenethyl-4-phenylbutan-1-ol (460 mg, 1.80 mmol) in THF (2 mL) and DIAD (0.43 mL, 2.17 mmol) was added dropwise to the reaction mixture. Reaction was allowed to stir for 30 minutes at 0 °C and overnight at room temperature. Then the solvents were rotavapped and residue purified on an ISCO chromatograph (0–30% ethyl acetate/hexane) to give product as a white powder. (630 mg, 91%); MP 75–76 °C; 1H NMR (400 MHz) (CDCl3) δ 7.91 (m, 2H), 7.74 (m, 2H), 7.32 (m, 10H), 3.82 (d, 2H, J = 8), 2.83 (m, 4H), 2.15 (m, 1H), 1.81 (m, 4H); 13C NMR (100 MHz) (CDCl3) δ 168.6, 142.7, 142.4, 142.1, 133.9, 132.4, 132.1, 128.9, 128.8, 128.7, 128.5, 128.4, 128.3, 128.2, 126.1, 125.9, 125.5, 123.2, 41.6, 36.9, 33.7, 32.8
2-Phenethyl-4-phenylbutan-1-amine
Hydrazine monohydrate (120 μL, 2.4 mmol) was added to a solution of 2-(2-phenethyl-4-phenylbutyl)isoindoline-1,3-dione (460 mg, 1.2 mmol) in methanol (15 mL) at room temperature. Then the reaction stirred under reflux for 2 hours. The completed reaction mixture was cooled to room temperature and the white precipitate filtered off. The residue was washed with cold methanol, the filtrate was concentrated and purified on an ISCO chromatograph (0–10% MeOH/ dichloromethane + 1% NH4OH) to give product as a colorless oil. (223 mg, 74%); 1H NMR (400 MHz) (CDCl3) δ 7.36 (m, 4H), 7.26 (m, 6H), 2.80 (d, 2H, J = 4), 2.71 (t, 4H, J = 8), 1.75 (m, 4H), 1.55 (m, 1H), 1.30 (brs, 2H); 13C NMR (100 MHz) (CDCl3) δ 142.7, 128.43, 128.41, 125.8, 45.0, 40.2, 33.4, 33.19
(S)-3,5-Bis(((benzyloxy)carbonyl)amino)pentanoic acid
Methyl (S)-3,5-bis(((benzyloxy)carbonyl)amino)pentanoate (236 mg, 0.56 mmol) was dissolved in THF/ water (12 mL) (5:1) and treated with LiOH.H2O (28 mg, 0.67 mmol) at 0 °C under nitrogen. Reaction mixture was stirred at room temperature overnight. Then 1N HCl was added and the THF removed under reduced pressure. A white solid which had precipitated was filtered. The residue was saved and the filtrate was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and solvent evaporated. The precipitated solid was added and the combined residue was dried on a vacuum pump to give product as a white solid. (150 mg, 67%); MP 110–112 °C; 1H NMR (400 MHz) (CD3OD) δ 7.24 (m, 10H), 4.96 (s, 4H), 3.92 (m, 1H); 3.13 (m, 1H), 2.99 (m, 1H), 2.40 (d, 2H, J = 4); 1.67 (m, 1H), 1.55 (m, 1H); 13C NMR (100 MHz) (CD3OD) δ 158.8, 158.4, 138.4, 129.4, 128.9, 128.8, 128.77, 128.72, 67.4, 47.5, 40.6, 38.7, 35.6
Dibenzyl (5-oxo-5-((2-phenethyl-4-phenylbutyl)amino)pentane-1,3-diyl)(S)-dicarbamate
(S)-3,5-Bis(((benzyloxy)carbonyl)amino)pentanoic acid (115 mg, 0.29 mmol) was dissolved in DMF (5 mL), EDC (110 mg, 0.58 mmol) and HOBT (78 mg, .58 mmol) were added. The reaction was stirred at room temperature for 5 minutes. Then 2-phenethyl-4-phenylbutan-1-amine (84 mg, 0.33 mmol) was added followed by 2,6-lutidine (0.11 mL, 0.99 mmol). The reaction was stirred at room temperature overnight. The reaction mixture was diluted with ethyl acetate and washed with water, saturated sodium bicarbonate, 5% HCl, water and brine. The combined organic layers were dried over sodium sulfate, concentrated and purified on an ISCO chromatograph (0–60% ethyl acetate/hexane) to give product as a white solid. (106 mg, 57%). MP 169–170 °C; 1H NMR (400 MHz) (CDCl3) δ 7.28 (m, 20H), 6.32 (brs, 1H), 5.99 (brs, 1H), 5.69 (brs, 1H), 5.08 (m, 4H), 3.98 (m, 1H), 3.41 (m, 1H), 3.29 (m, 2H), 2.99 (m, 1H), 2.57 (m, 5H), 2.29 (m, 1H), 1.66 (m, 7H); 13C NMR (100 MHz) (CDCl3) δ 171.1, 156.6, 142.2, 136.6, 136.5, 128.5, 128.4, 128.3, 128.1, 128.0, 127.9, 125.9, 66.6, 66.5, 46.0, 42.1, 40.0, 37.5, 37.2, 34.6, 33.6, 33.5, 32.9, 32.8
(S)-3,5-Diamino-N-(2-phenethyl-4-phenylbutyl)pentanamide
Dibenzyl (5-oxo-5-((2-phenethyl-4-phenylbutyl)amino)pentane-1,3-diyl)(S)-dicarbamate (90 mg, 0.14 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (40 mg) was added. The reaction mixture was purged and stirred under hydrogen atmosphere for overnight. Then the catalyst was filtered off and washed with 20% methanol/dichloromethane. The solvent was removed under reduced pressure and the residue dried under vacuum to obtain the product as a colorless oil (50.2 mg, 97%); 1H NMR (400 MHz) (CDCl3) δ 7.14 (m, 11H), 3.22 (m, 3H), 2.85 (m, 5H), 2.56 (m, 5H), 2.18 (m, 2H), 1.57 (m, 7H); 13C NMR (100 MHz) (CDCl3) δ 171.7, 171.6, 142.4, 142.3, 128.4, 128.3, 125.84, 125.8, 67.2, 53.1, 47.8, 45.3, 43.2, 43.0, 41.9, 41.6, 37.2, 37.1, 33.9, 33.8, 33.7, 33.0, 32.9, 32.6, 22.8; HRMS (ESI) Calculated for C23H34N3O (M+H)+ 368.2696, found 368.2697
(S)-N1-(2-Phenethyl-4-phenylbutyl)pentane-1,3,5-triamine
Step 1: Di-tert-butyl (5-((tert-butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)-pentane-1,3-diyl)(S)-dicarbamate - (S)-3,5-diamino-N-(2-phenethyl-4-phenylbutyl)-pentanamide (39 mg, 0.11 mmol) was dissolved in THF (10 mL) and BH3.THF (1 mL) was added. The reaction mixture was heated at reflux for overnight. Then methanol (10 mL) was added and reaction stirred at reflux for 2 hours. The reaction mixture was cooled down and water (2 mL) was added. This was then dried over sodium sulfate and filtered. The filtrate was concentrated under reduced pressure to provide the product.
Step 2: The crude product was then dissolved in dichloromethane (10 mL), Boc2O (69 mg, 0.32 mmol) was added and reaction stirred for 2 hours at room temperature under nitrogen. Workup was done by phase extraction with water and ethyl acetate. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified on an ISCO chromatograph (0–30% ethyl acetate/hexane) to give product as a colorless oil. (34 mg, 49%); 1H NMR (400 MHz) (CDCl3) δ 7.14 (m, 10H), 3.10 (m, 6H), 2.54 (m, 4H), 1.28 (m, 37H); 13C NMR (100 MHz) (CDCl3) δ 155.7, 142.5, 128.3, 125.7, 79.4, 79.2, 44.6, 43.8, 33.1, 32.9, 32.7, 29.6, 28.5, 28.5, 28.4, 28.0
Step 3; (S)-N1-(2-Phenethyl-4-phenylbutyl)pentane-1,3,5-triamine
Di-tert-butyl (5-((tert-butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)pentane-1,3-diyl)(S)-dicarbamate (10.7 mg, 0.016 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL). The reaction mixture was stirred at room temperature for 2 hours. Solvents were removed and residue was dried under vacuum to obtain product (trifluoroacetic acid salt) as a colorless oil. (9.4 mg, 91%); 1H NMR (400 MHz) (MeOD) δ 7.14 (m, 10H), 3.41 (m, 1H), 3.06 (m, 6H), 2.59 (m, 4H), 2.02 (m, 3H), 1.70 (m, 5H); 13C NMR (400 MHz) (MeOD) δ 142.9, 129.5, 129.4, 127.0, 53.1, 45.7, 44.3, 44.2, 36.3, 33.9, 33.3, 30.1, 29.8; HRMS (ESI) Calculated for C23H35N3 (M+H)+ 354.2904, found 354.2911
(S)-N5-(2-phenethyl-4-phenylbutyl)pentane-1,2,5-triamine (24)
Benzyl di-tert-butyl pentane-1,2,5-triyl(S)-tricarbamate
Benzyl tert-butyl (5-aminopentane-1,4-diyl)(S)-dicarbamate (332 mg, 0.95 mmol) was dissolved in dichloromethane (5 mL) and triethylamine (0.20 mL, 1.42 mmol) was added. Then Boc2O was added and the reaction was stirred at room temperature overnight. The reaction mixture was then washed with water and the organic layer separated. The organic layer was dried with sodium sulfate, concentrated and purified on an ISCO chromatograph (0–50% EtOAc/hexane) to give product as a yellow colored solid (252 mg, 56%); MP 75–77 °C; 1H NMR (400 MHz) (CDCl3) δ 7.33 ( m, 5H), 5.13 (brs, 1H), 5.09 (s, 2H), 4.98 (s, 1H), 4.83 (brs, 1H), 3.61 (s, 1H), 3.18 (m, 4H), 1.52 (m, 4H), 1.38 (s, 18H); 13C NMR (100 MHz) (CDCl3) δ 156.5, 156.1, 136.6, 128.4, 128.1, 128.0, 79.3, 66.5, 51.1, 44.4, 40.7, 29.9, 28.3, 26.2
Di-tert-butyl (5-aminopentane-1,2-diyl)(S)-dicarbamate
Benzyl di-tert-butyl pentane-1,2,5-triyl(S)-tricarbamate (200 mg, 0.44 mmol) was dissolved in ethanol (10 mL) and 20% Pd(OH)2/C (40 mg) and stirred under hydrogen atmosphere overnight. Then, the catalyst was filtered off and filtrate concentrated to give product as an oil (140 mg, 100%); 1H NMR (400 MHz) (CDCl3) δ 5.22 (brs, 1H), 5.13 (brs, 1H), 4.53 (m, 2H), 3.54 (m, 1H), 3.10 (m, 2H), 2.76 (m, 2H), 1.54 (m, 2H), 1.36 (s, 18H); 13C NMR (100 MHz) (CDCl3) δ 156.7, 156.4, 128.4, 127. 9, 79.22, 51.1, 44.5, 40.8, 29.9, 28.3, 27.3
Di-tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,2-diyl)(S)-dicarbamate
2-Phenethyl-4-phenylbutanoic acidwas dissolved in DMF (5 mL) and EDC (185 mg, 0.96 mmol) and HOBT (185 mg, 0.96 mmol) were added. The reaction was stirred at room temperature for 5 minutes. Then di-tert-butyl (5-aminopentane-1,2-diyl)(S)-dicarbamate (140 mg, 0.44 mmol) was added followed by 2,6-lutidine (0.15 mL, 1.32 mmol). The reaction was stirred at room temperature overnight under nitrogen. The reaction mixture was then diluted with ethyl acetate, and washed with water, 1 M HCl, saturated NaHCO3, water and brine. The organic layer was dried over sodium sulfate and filtrate was concentrated and purified on an ISCO chromatograph (0–10% methanol/dichloromethane) to obtain a solid. (85 mg, 34%); MP 135–137 °C; 1H NMR (CDCl3) (400 MHz) δ 7.23 (m, 10H), 5.96 (brs, 1H), 4.97 (brs, 2H), 3.66 (m, 1H), 3.38 (m, 1H), 3.24 (m, 3H), 2.59 (m, 4H), 2.04 (m, 3H), 1.78 (m, 2H), 1.69 (m, 4H), 1.45 (s, 9H), 1.44 (s, 9H); 13C NMR (CDCl3) (100 MHz) δ 175.2, 156.7, 156.3, 141.7, 128.4, 128.3, 125.9, 79.4, 51.2, 46.5, 44.4, 39.0, 34.3, 33.6, 30.4, 28.3, 25.9
(S)-N-(4,5-Diaminopentyl)-2-phenethyl-4-phenylbutanamide
Di-tert-butyl (5-(2-phenethyl-4-phenylbutanamido)pentane-1,2-diyl)(S)-dicarbamate (80 mg, 0.14 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL) and the reaction mixture was stirred at 0 °C for 2 hours. Then the solvents were removed under reduced pressure and dried under vacuum to obtained product as a colorless oil. (80 mg, 100%); 1H NMR (400 MHz) (MeOD) δ 7.10 (m, 10H), 3.52 (m, 1H), 3.18 (m, 4H), 2.47 (m, 4H), 2.20 (m, 1H), 1.83 (m, 2H), 1.69 (m, 6H); 13C NMR (100 MHz) (MeOD) δ 178.7, 143.1, 129.4, 129.3, 126.9, 50.5, 47.6, 42.2, 39.6, 35.8, 34.8, 29.2, 26.2
(S)-N5-(2-Phenethyl-4-phenylbutyl)pentane-1,2,5-triamine
Step 1 : di-tert-butyl (5-((tert-butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)-pentane-1,2-diyl)(S)-dicarbamate - (S)-N-(4,5-diaminopentyl)-2-phenethyl-4-phenyl-butanamide (80 mg, 0.14 mmol) was dissolved in 10 mL of THF and BH3.THF (2 mL) and the mixture was heated at 83 °C for overnight. Then methanol (10 mL) was added and the reaction mixture was stirred at that same temperature for 2 hours. Then water (1 mL) was added and the reaction cooled to room temperature. Solvents presents were then concentrated under reduced pressure and redissolved in dichloromethane, dried with sodium sulfate and organic layer filtered and solvent removed to give an oil residue.
Step 2: The residue was redissolved in dichloromethane (5 mL) and Boc2O (92 mg, 0.42 mmol) was added and reaction stirred for 2 hours. Workup was done by extraction from water with ethyl acetate. The organic layer was dried over sodium sulfate, filtered and concentrated under reduced pressure. The residue was purified on an ISCO chromatograph (0–30% ethyl acetate/hexane) to give product as a colorless oil (9 mg, 10% ); 1H NMR (400 MHz) (MeOD) δ 7.24 (m, 10H), 4.72 (m, 2H), 3.62 (brs, 1H), 3.17 (m, 6H), 2.67 (m, 4H), 1.78 (m, 1H), 1.66 (m, 8H), 1.46 (m, 27H); 13C NMR (100 MHz) (MeOD) δ 128.3, 125,7, 79.33, 57.8, 51.2, 46.8, 44.9, 33.2, 32.7, 28.4, 28.3
Step 3: (S)-N5-(2-Phenethyl-4-phenylbutyl)pentane-1,2,5-triamine - Di-tert-butyl (5-((tert-Butoxycarbonyl)(2-phenethyl-4-phenylbutyl)amino)pentane-1,2-diyl)(S)-dicarbamate (8.7 mg, 0.013 mmol) was dissolved in dichloromethane (1 mL) and trifluoroacetic acid (0.5 mL). The reaction mixture was stirred at room temperature for 2 hours. Solvents were removed and residue was dried under vacuum to obtain product (trifluoroacetic acid salt) as a white solid. (5.7 mg, 67%); MP 140–142 °C; 1H NMR (400 MHz) (MeOD) δ7.13 (m, 10H); 3.49 (m, 1H), 3.17 (m, 2H), 2.94 (m, 4H), 2.57 (m, 4H), 1.71 (m, 9H); 13C NMR (100 MHz) (MeOD) δ 142.9, 129.5, 129.4, 127.0, 52.7, 50.3, 42.0, 36.0, 33.9, 33.3, 28.8, 22.6; HRMS (ESI) Calculated for C23H36N3 (M+H)+ 354.2904, found 254.2911
MIC-based assay for potentiation of antibiotic activity against E. coli ATCC 25922
An MIC-based assay was used to evaluate the potential of all the test compounds to act as potentiators of the activity of the macrolide antibiotic clarithromycin against E. coli ATCC 25922. In addition, this assay was also used to evaluate the impact of compounds 7 and 22 on the activities of four additional antibiotics (minocycline, novobiocin, clindamycin, and azithromycin). The MIC-based assay was conducted using a protocol similar to that described previously.24 Log-phase bacteria were added to 96-well microtiter plates (at 5 × 105 colony forming units per ml) containing two-fold serial dilutions of the antibiotic in cation-adjusted Mueller-Hinton (CAMH) broth either in the absence or presence of each test compound (at a concentration of 12.5 μg/ml). Each serial dilution of antibiotic was present in duplicate. The final volume in each well was 0.1 ml, and the microtiter plates were incubated aerobically for 24 hours at 37 °C. Bacterial growth was then monitored by measuring the optical density (OD) at 600 nm using a VersaMax® plate reader (Molecular Devices, Inc., Sunnyvale, CA), with the MIC being defined as the lowest antibiotic concentration at which bacterial growth was ≥90% inhibited compared to antibiotic-free and compound-free controls. The intrinsic MIC of each test compound against E. coli ATCC 25922 was also determined using a similar assay, with the exception that the microtiter plates contained serial dilutions of test compound rather than antibiotic. Clarithromycin and minocycline hydrochloride were obtained from Sigma, while azithromycin and novobiocin (sodium salt) were obtained from Fluka. Clindamycin hydrochloride was obtained from Toku-E.
Fluorescence-based cellular assay for inhibition of pump-mediated efflux of Hoechst 33342 (H33342) in E. coli ATCC 25922
The potential of 7 and 22 to inhibit efflux pumps in E. coli ATCC 25922 cells was evaluated using a fluorescence-based assay for the pump-mediated efflux of the fluorescent dye H33342. In a similar protocol to that described previously,24–26 E. coli ATCC 25922 cells were harvested from overnight cultures by centrifugation, and the cell pellets were washed with phosphate-buffered saline (PBS) containing 1 mM MgCl2 (PBSM). After washing, the cell pellets were resuspended in PBSM to achieve a final OD at 600 nm of 1.0. The ATP required for efflux pump function was then depleted by addition of carbonyl cyanide 3-chlorophenylhydrazone (CCCP) to a final concentration of 3.13 μM, and then incubating the cells for 20 minutes at 37 °C. H33342 was then added to the cell suspension at a concentration of 10 μM, and the cells were incubated for an additional 20 minutes at 37 °C to allow for to allow for intracellular accumulation of the Hoechst dye. After the 20-minute incubation, 200 μl aliquots of the bacterial suspension were distributed into wells of a black, flat-bottom 96-well plate containing 7 or 22 at concentrations ranging from 1/32X to ¼X MIC (1.56 to 12.5 μg/ml for 7 and 3.13 to 25 μg/ml for 22), or an equivalent volume of the vehicle (DMSO) alone. A plate vortexer was used to mix the bacterial cells with the test compounds, and the plates were pre-incubated at 37 °C for 5 minutes. After the pre-incubation, efflux pump activity was initiated by reenergizing the bacterial cells with the addition of glucose to a final concentration of 50 mM. H33342 efflux was monitored using a SpectraMax® 2 fluorescent plate reader (Molecular Devices, Inc., Sunnyvale, CA) to measure the fluorescence of each well at 37 °C once per minute for 40 minutes. The excitation and emission wavelengths were set at 355 and 460 nm, respectively.
Fluorescence-based cellular assay for permeabilization of the outer cell membrane to N-phenyl-1-naphthylamine (NPN) in E. coli ATCC 25922
The fluorescent dye NPN was used to probe for the permeabilization of the outer cell membrane of E. coli ATCC 25922 cells by 7 and 22, as described previously.24,27 E. coli ATCC 25922 bacterial cultures were grown in CAMH broth to log-phase and the bacterial cells were harvested by centrifugation. The cell pellets were washed with PBS and then resuspended in PBS to achieve a final OD at 600 nm of 1.0. NPN was added to the bacterial suspension at a final concentration of 10 μM and the cells were incubated for 10 minutes at room temperature. 200 μl aliquots of the NPNcontaining bacterial suspension were then distributed into wells of a black, flat-bottom 96-well plate containing 7 or 22 at concentrations ranging from 1/32X to ¼X MIC (1.56 to 12.5 μg/ml for 7 and 3.13 to 25 μg/ml for 22), or an equivalent volume of the vehicle (DMSO) alone. For each test concentration or control well, four replicates were prepared. A plate vortexer was used to mix the bacterial cells with the test compounds and the plates were then pre-incubated at room temperature for 10 minutes in the dark. NPN fluorescence was monitored using a SpectraMax® 2 fluorescent plate reader (Molecular Devices, Inc., Sunnyvale, CA), with excitation and emission wavelengths set at 350 and 420 nm, respectively. Corrected fluorescence values associated with each given compound concentration were calculated by averaging the observed fluorescence over 10 minutes and subtracting the corresponding average fluorescence values of the vehicle-containing wells.
Fluorescence-activated cell sorting (FACS) assay for permeabilization of the outer and inner cell membranes to propidium iodide (PI) in E. coli ATCC 25922
The FACS assay used for assessing the potential of test compounds to permeabilize both the outer and inner cell membranes of E. coli ATCC 25922 bacterial cells to PI was conducted as described previously.24,28 Log-phase E. coli ATCC 25922 bacterial cells grown in CAMH broth were diluted in PBS to a concentration of 4 × 106 CFU/ml. The bacteria were aliquoted into tubes and mixed with either 7 or 22 at concentrations ranging from 1/32X to ¼X MIC (1.56 to 12.5 μg/ml for 7 and 3.13 to 25 μg/ml for 22). Control tubes to which equivalent volumes of the vehicle (DMSO) alone were also included. All test concentrations and controls were prepared in triplicate. PI was then added to all tubes at a final concentration of 50 μm, and the samples were incubated in the dark at room temperature for 1 hour. Intracellular PI fluorescence was detected by flow cytometry using a Gallios Cytometer (Beckman Coulter Inc., Indianapolis, IN). The 488 nm laser was used for excitation, with the 620/30 nm channel being used for emission. For each sample, the fluorescence of 30,000 individual bacterial cells was measured, and the percent of cells that stained positive for PI fluorescence was calculated.
FACS assay of inner membrane potential in E. coli ATCC 25922 cells using the fluorescent dye 3,3′-diethyloxacarbocyanine iodide (DiOC2)
The fluorescent dye 3,3′-diethyloxacarbocyanine iodide (DiOC2) was used to assess inner membrane potential as described previously.29,30 Log-phase E. coli ATCC 25922 cells grown in CAMH broth were diluted in PBS to a concentration of 2 × 106 CFU/ml. 1-ml aliquots of the bacterial solution were distributed into tubes and mixed with 7 or 22 at ¼X MIC (12.5 μg/ml for 7 and 25 μg/ml for 22). Control tubes to which equivalent volumes of the vehicle (DMSO) alone or 2 μg/ml CCCP were also included. The final percent DMSO in all tubes was kept constant at 0.2%. All test and control samples were prepared in triplicate. DiOC2 was then added to all tubes at a final concentration of 30 μM and the samples were incubated in the dark at room temperature for 30 min. Fluorescence was detected by flow cytometry using a Gallios Cytometer. The 488 nm laser was used for excitation, with the emission monitored at 2 different wavelengths. Green fluorescence was monitored using the 525/20 nm channel, while red florescence was monitored using the 620/30 nm channel. For each sample, the fluorescence associated with 10,000 individual bacterial cells was measured. The membrane potential was monitored by measuring the intensity ratio of the red to green fluorescence.
Supplementary Material
Table S1. Intrinsic Minimal Inhibitory Concentrations (MICs) of the test compounds and PAβN against E. coli ATCC 25922
Figure S1. Impact of test compounds 7 and 22 on the membrane potential of E. coli ATCC 25922 cells, as measured by changes in the ratio of red to green fluorescence (λred/λgreen) of 3,3′-diethyloxacarbocyanine iodide (DiOC2). Both compounds were used at a concentration of ¼ MIC (12.5 and 25 μg/ml for 7 and 22, respectively). Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), included as a positive control, was used at a concentration of 2 μg/ml. Vehicle is DMSO.
Scheme 5.

Reagents and conditions: a) i) ethyl chloroformate, Et3N, THF; ii) TMSCH2N2, ACN, 24 hr., +4 °C (54%); b) silver benzoate, Et3N, anhydrous methanol (55%); c) LiBH4, THF, ethanol (78%); d) phthalimide, DIAD, PPh3, THF (53%); e) hydrazine, methanol (90%); f) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine, DMF(75%); g) 20% Pd(OH)2/C, H2, ethanol (100%).
Scheme 6.

Reagents and conditions: a) i) N-methylmorpholine, isobutylchloroformate, DME, ii) NaBH4, DME/H2O; b) phthalimide, DIAD, PPh3, THF (92%); c) hydrazine, methanol (58%); d) CbzCl, Et3N, CH2Cl2 (65%); e) TFA, CH2Cl2 (67%); f) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine, DMF(67%); g) 20% Pd(OH)2/C, H2, ethanol (42%)
Scheme 10.

Methods used for the preparation of 18. Reagents and conditions a) 2-phenethyl-4-phenylbutanoic acid, EDC, HOBT, 2,6-lutidine; b) TFA, CH2Cl2; c) 1,3-diboc-2-(trifluoromethylsulfonyl)guanidine, Et3N, DCM (29%); d) TFA, DCM (100%)
Scheme 13.

Methods used for the preparation of 22. Reagents and conditions: a) (i) BH3·THF, THF, (ii) Boc2O, DCM (32%); b) TFA, DCM (100%)
Scheme 14.

Methods used for the preparation of 23. Reagents and conditions: a) methyltriphenylphosphonium bromide, KOtBu, THF (81%); b) (i) BH3 TFA/DCM (79%); f) 20% Pd(OH)2/C, H2, ethanol (81%)THF, (ii) NaOH, H2O2 (77%); c) phthalimide PPh3, DIAD (91%); d) hydrazine monohydrate, methanol, reflux (74%); e) LiOH, THF; f) HOBT, EDC, DIPEA; g) 20% Pd(OH)2/C, H2, ethanol (97%); h) (i) BH3·THF, THF, (ii) Boc2O, DCM (49%); i) TFA, DCM (91%)
Scheme 15.

Methods used for the preparation of 24. Reagents and conditions: a) Boc2O, Et3N, DCM (56%); b) 20% Pd(OH)2/C, H2, ethanol (100%); c) HOBT, EDC, DIPEA (34%); d) TFA, DCM; e) BH3·THF, THF (ii) Boc2O, DCM (10%); f) TFA, DCM (67%)
Acknowledgements
This research was supported in part by a research agreement between TAXIS Pharmaceuticals, Inc. and Rutgers University (to E.J.L.) and by National Institutes of Health grant RO1 AI118874 (to D.S.P.).
Footnotes
The authors declare the following competing financial interest(s): Drs. Pilch and LaVoie are co-founders of TAXIS Pharmaceuticals and therefore have a financial interest in the company.
Associated Content:
Supporting Material
Details on the synthetic methods employed for all new compounds discussed in this paper are provided. In addition, a table (Table S1) listing the intrinsic MICs of each of the test potentiators as well as a figure (Figure S1) showing the results of the assay on membrane potential are also included.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Thota N; Koul S; Reddy MV; Sangwan PL; Khan IA; Kumar A; Raja AR; Andotra SS; Qazi GN Citral derived amides as potent bacterial NorA efflux pump inhibitors. Bioorg. Med. Chem, 2008, 16, 6535–6543. [DOI] [PubMed] [Google Scholar]
- 2.Sangwan PL; Koul JL; Koul S; Reddy MV; Thota N; Khna IA; Kumar A; Kalia N; Qazi GN Piperrine analogs as potent Staphylococcus aureus NorA efflux pump inhibitors. Bioorg. Med. Chem, 2008, 16, 9847–9857. [DOI] [PubMed] [Google Scholar]
- 3.Samosorn S; Bremner JB; Ball A; Lewis K Synthesis of functionalized 2-aryl-5-nitro-1Hindoles and their activity as bacterial NorA efflux pump inhibitors. Bioorg. Med. Chem, 2005, 14, 857–865. [DOI] [PubMed] [Google Scholar]
- 4.Pagès J-M., Masi M, Barbe J Inhibitors of efflux pumps in Gram-negative bacteria. Trends Mol. Med, 2005, 11, 382–389 [DOI] [PubMed] [Google Scholar]
- 5.Kaatz GW Bacterial Efflux Pump Inhibition. Curr. Opin. Investig. Drugs 2005, 6, 191–198. [PubMed] [Google Scholar]
- 6.Opperman TJ, Nguyen ST Recent advances toward a molecular mechanism of efflux pump inhibition. Front. Microbiol 2015, 6, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schillaci D; Spanò V; Parrino B; Carbone A; Montalbano A; Barraja P; Diana P; Cirrincione G; Cascioferro SJ Pharmaceutical Approaches to Target Antibiotic Resistance Mechanisms. J. Med. Chem 2017, 60, 8268–8297. [DOI] [PubMed] [Google Scholar]
- 8.Van Bambeke F, Pagès J-M; Lee V Inhibitors of Bacterial Efflux Pumps as Adjuvants in Antibacterial Therapy and Diagnostic Tools for Detection of Resistance by Efflux. Front. AntiInfective Drug Discov 2010, 1, 138–175. [DOI] [PubMed] [Google Scholar]
- 9.Marquez B Bacterial efflux systems and efflux pump inhibitors. Biochimie. 2005, 87, 1137–1147. [DOI] [PubMed] [Google Scholar]
- 10.Worthington RJ, Melander C Combination approaches to combat multi-drug resistant bacteria. Trends Biotech. 2013, 31, 177–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gorityala BK, Guchhait G, Fernando DM, Deo S, McKenna SA, Zhanel GG, Kumar A, Schweizer F Adjuvants Based on Hybrid Antibiotics Overcome Resistance in Pseudomonas aeruginosa and Enhance Fluoroquinolone Efficacy. Angew.Chem. Int. Ed 2016, 55, 555–559 [DOI] [PubMed] [Google Scholar]
- 12.Pagès J-M, Amaral L Mechanisms of drug efflux and strategies to combat them:Challenging the efflux pump of Gram-negative bacteria. Biochim. Biophys. Acta, 2009, 1794, 826–833. [DOI] [PubMed] [Google Scholar]
- 13.Lomovskaya O; Warren MS; Lee A; Galazzo J; Fronko R; Lee M; Blais J; Cho D; Chamberland S; Renau T; Leger R; Hecker S; Watkins W; Hoshino K; Ishida H; Lee VJ; Identification and Characterization of Inhibitors of Multidrug Resistance Efflux Pumps in Pseudomonas aeruginosa: Novel Agents for Combination Therapy. Antimicrob. Agents Chemother 2001, 45, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lomovskaya O; Bostian K Practical applications and feasibility of efflux pumps inhibitors in the clinic - A vision for applied use. Biochem. Pharmacol 2006, 30, 910–918. [DOI] [PubMed] [Google Scholar]
- 15.Mahamoud A; Chevalier J; Alibert-Franco S; Kern W; Pages J Antibiotic efflux pumps in Gram-negative bacteria: the inhibitor response strategy. J. Antimicrob. Chemother 2007, 59, 1223–1229. [DOI] [PubMed] [Google Scholar]
- 16.Lamers RP, Cavallari JF, Burrows LL The efflux inhibitor phenylalanine-arginine beta- naphthylamide (PAβN) permeabilizes the outer membrane of gram-negative bacteria. PLoS One, 2013, 8, e60666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Renau T; Leger R; Flamme E; Sangalang J; She R; Yen M; Gannon C; Griffith D; Chamberland S; Lomovskaya O; Hecker S; Lee V; Ohta T; Nakayama K Inhibitors of Efflux Pumps in Pseudomonas aeruginosa Potentiate the Activity of the Fluoroquinolone Antibacterial Levofloxacin. J. Med. Chem 1999, 42, 4928–4931. [DOI] [PubMed] [Google Scholar]
- 18.Renau TE; Leger R; Yen R; She M,W.; Flamme EM; Sagalang J; Gannon CL; Chamberland S; Lomovskaya O; Lee VJ Bioorg. Med. Chem. Lett 2001, 11, 663–667. [DOI] [PubMed] [Google Scholar]
- 19.R Renau T; Leger R; Yen R; She M; Flamme E; Sangalang J; Gannon C; Chamberland S; Lomovskaya O; Lee V Peptidomimetics of Efflux Pumps Inhibitors Potentiate the Activity of Levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett 2002, 12, 763–766. [DOI] [PubMed] [Google Scholar]
- 20.Renau T; Leger R; Filonova L; Flamme E; Wang M; Yen R; Madsem D; Griffith D; Chamberland S; Dudley M; Lee V; Lomovskaya O; Watkins W; Ohta T; Nakayama K Conformationally-Restricted Analogues of Efflux Pump Inhibitors that Potentiate the Activity of Levofloxacin in Pseudomonas aeruginosa. Bioorg. Med. Chem. Lett 2003, 13, 2755–2758. [DOI] [PubMed] [Google Scholar]
- 21.Watkins W; Landaverry Y; Leger R; Litman R; Renau T; Williams N; Yen R; Zhang J; Chamberland S; Madsen D; Griffith D, Tembe V; Huie K; Dudley M; The Relationship between Physicochemical Properties, In Vitro Activity and Pharmacokinetic Profiles of Analogues of Diamine-Containing Efflux Pump Inhibitors. Bioorg. Med. Chem. Lett 2003, 13, 4241–4244. [DOI] [PubMed] [Google Scholar]
- 22.Kern W; Steinke P; Schumacher A; Schuster S; von Baum H; Bohnert J Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Escherichia coli. J. Antimicrob. Chemother 2006, 57, 339–34. [DOI] [PubMed] [Google Scholar]
- 23.Schumacher A; Steinke P; Bohnert J; Akova M; Jonas D; Kern W Effect of 1-(1-naphthylmethyl)-piperazine, a novel putative efflux pump inhibitor, on antimicrobial drug susceptibility in clinical isolates of Enterobacteriaceae other that Escherichia coli. J. Antimicrob. Chemother 2006, 57, 344–348. [DOI] [PubMed] [Google Scholar]
- 24.Blankson G; Parhi AK; Kaul M; Pilch DS; LaVoie EJ Structure-Activity Relationships of Potentiators of the Antibiotic Activity of Clarithromycin against Escherichia coli. Eur. J. Med. Chem, 2019, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coldham NG; Webber M; Woodward MJ; Piddock LJA 96-well plate fluorescence assay for assessment of cellular permeability and active efflux in Salmonella enterica serovar Typhimurium and Escherichia coli. J. Antimicrob. Chemother 2010, 65, 1655–1663. [DOI] [PubMed] [Google Scholar]
- 26.Opperman TJ; Kwasny SM; Kim HS; Nguyen ST; Houseweart C; D’Souza S; Walker GC; Peet NP; Nikaido H; Bowlin TL Characterization of a novel pyranopyridine inhibitor of the AcrAB efflux pump of Escherichia coli. Antimicrob. Agents Chemother 2014, 58, 722–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Muheim C; Gotzke H; Eriksson AU; Lindberg S; Lauritsen I; Norholm MHH; Daley DO Increasing the permeability of Escherichia coli using MAC13243. Sci. Rep 2017, 7, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Benincasa M; Pacor S; Gennaro R; Scocchi M Rapid and reliable detection of antimicrobial peptide penetration into gram-negative bacteria based on fluorescence quenching. Antimicrob. Agents Chemother 2009, 53, 3501–3504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Foss MH; Eun YJ; Grove CI; Pauw DA; Sorto NA; Rensvold JW; Pagliarini DJ; Shaw JT; Weibel DB Inhibitors of Bacterial Tubulin Target Bacterial Membranes in Vivo. MedChemComm, 2013, 4, 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Spindler EC; Hale JD; Giddings TH Jr.; Hancock RE; and Gill RT Deciphering the Mode of Action of the Synthetic Antimicrobial Peptide Bac8c. Antimicrob. Agents Chemother 2011, 55, 1706–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Intrinsic Minimal Inhibitory Concentrations (MICs) of the test compounds and PAβN against E. coli ATCC 25922
Figure S1. Impact of test compounds 7 and 22 on the membrane potential of E. coli ATCC 25922 cells, as measured by changes in the ratio of red to green fluorescence (λred/λgreen) of 3,3′-diethyloxacarbocyanine iodide (DiOC2). Both compounds were used at a concentration of ¼ MIC (12.5 and 25 μg/ml for 7 and 22, respectively). Carbonyl cyanide 3-chlorophenylhydrazone (CCCP), included as a positive control, was used at a concentration of 2 μg/ml. Vehicle is DMSO.