

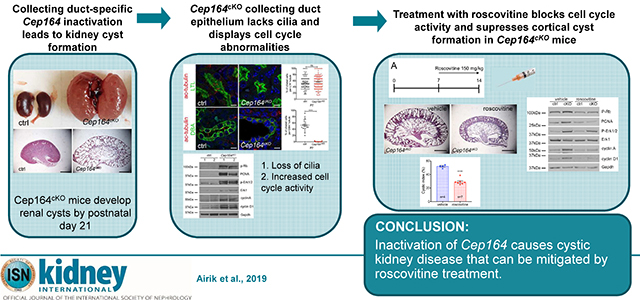
Abstract
Nephronophthisis is an autosomal recessive kidney disease with high genetic heterogeneity. Understanding the functions of the individual genes contributing to this disease is critical for delineating the pathomechanisms of this disorder. Here, we investigated kidney function of a novel gene associated with nephronophthisis, CEP164, coding a centriolar distal appendage protein, using a Cep164 knockout mouse model. Collecting duct-specific deletion of Cep164 abolished primary cilia from the collecting duct epithelium and led to rapid postnatal cyst growth in the kidneys. Cell cycle and biochemical studies revealed that tubular hyperproliferation is the primary mechanism that drives cystogenesis in the kidneys of these mice. Administration of roscovitine, a cell cycle inhibitor, blocked cyst growth in the cortical collecting ducts and preserved kidney parenchyma in Cep164 knockout mice. Thus, our findings provide evidence that therapeutic modulation of cell cycle activity can be an effective approach to prevent cyst progression in the kidney.
Keywords: CEP164, NPHP, PKD, cilia, centrosome
Graphical Abstract
INTRODUCTION
Renal ciliopathies are genetic disorders that present a spectrum of kidney phenotypes ranging from small degenerative kidneys with small cysts, such as in nephronophthisis (NPHP)1 to grossly enlarged kidneys with highly proliferative cysts, such as in autosomal dominant or autosomal recessive polycystic kidney disease.2, 3 Regardless of the underlying genetic involvement, the disease mechanisms of renal ciliopathies have been attributed to the function of the primary cilium, a cell surface organelle to which cystoproteins localize.4
The primary cilium is a non-motile, microtubule-based hair-like projection on the apical surface of ciliated cells. It consists of microtubular axoneme which is surrounded by a ciliary membrane and is rooted in the basal body, a modified mother centriole, that is anchored to the cell membrane by distal appendages.5, 6 Besides mediating membrane attachment, centriolar distal appendages are essential for initiating the early steps in ciliogenesis by binding to endosomal distal appendage vesicles which aggregate to form a single ciliary vesicle that fuses with the cell membrane.7, 8 In addition, distal appendages are recruitment sites for intraflagellar transport (IFT) complex proteins,9 which facilitate the growth of axonemal microtubules and transport of ciliary components in and out of the cilium.10 To date, 5 proteins have been identified as forming the core of the distal appendages – CEP83, CEP89, SCLT1, CEP164, and FBF1, defined by their recruitment to the mother centriole distal end prior to cilia formation.11 Loss of function of any of the distal appendage proteins invariably leads to absence of cilia in cell culture models,9, 11–14 underlining the critical requirement of distal appendages for cilia formation. Knockout mouse models have been described for Sclt1 and Cep164, both of which present ciliogenesis defects – Sclt1−/− mice develop pleiotropic ciliopathy phenotypes, including postnatal cystic kidneys,15 whereas Foxj1-Cre controlled deletion of Cep164 leads to absence of multiciliated cells in the lung, brain and testis.16
Although, Cep164 was the first distal appendage protein to be identified by utilizing an RNA interference screen in cell culture,14 and mutations in CEP164 were associated with pleiotropic congenital abnormalities, including nephronophthisis in humans,17 CEP164 role in the kidney has not been previously studied. In order to address the role of Cep164 in kidney development and homeostasis we generated a conditional Cep164 knockout mouse model. Collecting duct-specific deletion of Cep164 by using the Hoxb7-Cre transgene18 abolished cilia formation and led to rapid postnatal cyst growth in Cep164 knockout mice. Our biochemical analyses show that a blockade of cell cycle activity with roscovitine suppresses the formation of cortical collecting duct cysts in Cep164cKO mouse, identifying cell cycle dysregulation as the primary mechanism of cyst growth in early postnatal kidneys.
RESULTS
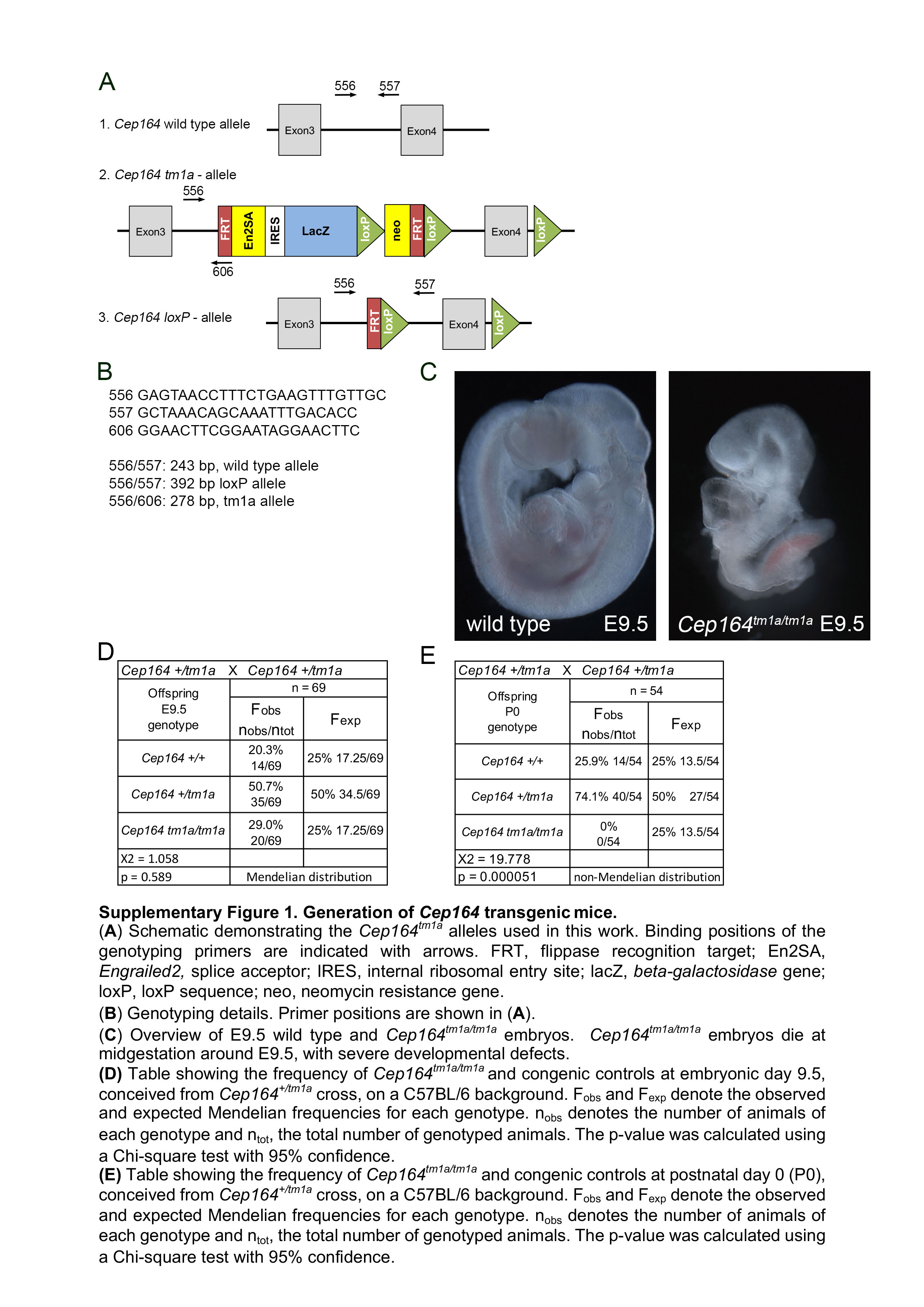
Cep164 knockout mice were generated using targeted Cep164tm1a embryonic stem cells from the European Conditional Mouse Mutagenesis Program (EUCOMM)19. Allele-specific primers were used to genotype the mice (Supplementary Figure S1A and S1B). Since homozygous Cep164tm1a/tm1a animals died around midgestation before the kidneys develop (Supplementary Figure S1C – S1E) we crossed Cep164tm1a mice with the FLPo deleter mice20 and generated a conditional Cep164loxP/loxP mouse line. To study Cep164 kidney function we deleted Cep164 from the ureteric bud-derived collecting duct epithelium using the Hoxb7-Cre strain.18 The resulting Hoxb7Cre+;Cep164loxP/loxP mice are hereafter referred to as mutant or Cep164cKO mice, and Hoxb7Cre−;Cep164loxP/loxP mice are referred to as control mice in all of the assays described in this paper.
Cep164cKO mice were born at Mendelian ratio with no visible abnormalities until up to 3-weeks of age when they became lethargic. Male and female mice were affected equally, and there were no differences in the phenotype among different age-matched mutants. Necropsy of postnatal day (P) 21 mice revealed that the mutant mice had severely enlarged kidneys compared to control mice (Figure 1A). Quantification of kidney weight to body weight (KW/BW) ratio in mice at P7, P14 and P21, showed no differences between mutant and control mice at P7. However, the KW/BW ratio was increased by 3-fold in mutants at P14 (****p<0.0001), and 7-fold at P21 (****p<0.0001) (Figure 1B). Despite of the rapid cyst growth between P7 and P14, measurements of blood urea nitrogen were normal until P14 in the mutant mice. However, a precipitous decline in kidney function occurred by P21 (Figure 1C), underlying the subsequent rapid lethality in Cep164cKO mice (Figure 1D).
Figure 1. Deletion of Cep164 from collecting duct epithelium leads to postnatal cystogenesis.

(A) Overview of P21 kidneys. Cep164cKO mice develop polycystic kidney disease by P21.
(B) Quantification of kidney weight to body weight ratio of Cep164cKO mice at postnatal days 7, 14 and 21. P7 ctrl 0.75 ± 0.04, n=4 vs. Cep164cKO 0.77 ± 0.03, n=5, ns; P14 ctrl 0.82 ± 0.03, n=6 vs. Cep164cKO 3.09 ± 0.18, n=4, ****p<0.0001; P21 difference ctrl 0.74 ± 0.02, n=5 vs. Cep164cKO 7.2 ± 0.53, n=8, ****p<0.0001.
(C) Blood urea nitrogen levels are significantly increased in Cep164cKO animals by P21 and indicate kidney failure. BUN values are comparable in control and Cep164cKO mice at P2 and P14. At P21 the BUN levels in Cep164cKO mice indicated kidney failure (ctrl 14.31 ± 1.09, n=7 vs. Cep164cKO 72.87 ± 9.69, n=5, ****p<0.0001).
(D) Mantel-Cox logrank test shows a statistically significant difference between control and Cep164cKO mouse survival (**p<0.001). Median survival time of Cep164cKO mice is 25 days (n = 10).
(E) P7 kidney sections stained with antibodies against acetylated α-tubulin to mark primary cilia (red) and Lotus tetragonolobus lectin (LTL) to mark proximal tubules (green). Both, control and Cep164cKO mice have primary cilia in the proximal tubular epithelium. DAPI nuclei stained in blue. Scale bar 7.5 μm. There is no change in the fraction of ciliated cells in the proximal tubule of control vs. Cep164cKO kidneys at P7; ctrl (53.87 % ± 2.00, n=105 tubules, n=699 cells) vs. Cep164cKO (56.55 % ± 1.89, n=131 tubules, n=707 cells), p=0.3336.
(F) P7 kidney sections stained with antibodies against acetylated α-tubulin to mark primary cilia (red) and Dolichos biflorus agglutinin (DBA) lectin to mark collecting ducts (green). Primary cilia are visible in the lumen of the collecting ducts in control kidneys, while no cilia are present in the Cep164cKO collecting duct epithelium. DAPI nuclei stained in blue. Scale bar 7.5 μm. Cep164cKO kidneys lack primary cilia in the collecting duct at P7; ctrl (52.95 % ± 3.58, n=23 tubules, n=292 cells) vs. Cep164cKO (0.36 % ± 0.20, n=25 tubules, n=643 cells), ****p<0.0001.
(G) There is no change in the fraction of ciliated cells in the proximal tubule of control vs. Cep164cKO kidneys at P14; ctrl (49.64 % ± 1.811, n=130 tubules, n=909 cells) vs. Cep164cKO (49.39 % ± 1.617, n=133 tubules, n=887 cells), p=0.9180.
(H) Cep164cKO kidneys lack primary cilia in the collecting duct at P14; ctrl (53.77 % ± 3.96, n=24 tubules, n=266 cells) vs. Cep164cKO (0.24 % ± 0.11, n=37 tubules, n=562 cells), ****p<0.0001.
(I–P) Comparison of hematoxylin and eosin-stained mid-sagittal sections from control (I, K, M, O) and Cep164cKO kidneys (J, L, M, P) at various postnatal stages, reveals the formation of collecting duct dilations in the medullary region of the mutant kidneys at P7 (K, L). By P14 the medullary cysts have greatly expanded in size, and cystogenesis has occurred in the cortical collecting ducts (M, N). By P21, corticomedullary border and kidney parenchyma has been destroyed by the expanding cysts in Cep164cKO kidneys (O, P). Scale bar, 1 mm (I, J), 2 mm (K – P).
(Q) Quantification of the cystic area (%) in Cep164cKO kidneys at postnatal stages P7, P14, P21. There is a sudden increase in the cystic index from P7 to P14, coinciding with the appearance of cortical collecting duct cysts.
To confirm that Cep164 expression was abolished from the collecting duct epithelial cells in Cep164cKO mice we co-stained kidneys with antibodies against γ-tubulin (centrosomes), Cep164 (centrosomal distal appendages), and LTL or DBA, which label the proximal tubules and collecting ducts, respectively. Cep164 expression was readily detected at the centrosomes in the proximal tubular cells of control and Cep164cKO kidneys, and in the collecting duct epithelial cells of control kidneys (Supplementary Figure S2A and S2B). In contrast, no Cep164 staining was observed at the centrosomes in the collecting duct epithelium of Cep164cKO kidneys (Supplementary Figure S2B), demonstrating that deletion of Cep164 was specific to the collecting duct cells.
Since Cep164 is essential for the growth of primary cilia in cell culture,8, 14, 17 we next examined whether ciliogenesis is affected in Cep164cKO kidneys. Primary cilia were present at similar levels in the proximal tubules of control and Cep164cKO kidneys at P7 and P14 (Figure 1E,G). In contrast, no cilia were observed in the collecting duct lining epithelium of the mutant kidneys at P7 or P14 (Figure 1F,H), demonstrating that Cep164 is required for renal ciliogenesis.
To characterize the onset and progression of kidney cyst growth in Cep164cKO mice in detail, we sectioned mutant kidneys at P2, P7, P14 and P21, and analyzed for structural changes. The first tubular dilations in Cep164cKO medullary collecting ducts appeared at P2 (Figure 1I,J). Within the first postnatal week the cysts expanded in size, but remained largely restricted to the kidney medullary region (Figure 1K,L). In contrast, by P14 both medullary and cortical areas were filled with radially oriented, cystic distensions of collecting ducts in Cep164cKO kidneys (Figure 1M,N), which further increased in diameter by P21 (Figure 1O,P). Quantification of cystic indices at P7, P14 and P21, showed that kidney cyst growth accelerated between P7 and P14, coinciding with the cyst formation in cortical collecting ducts (Figure 1Q).
Early postnatal kidney cystogenesis is a common feature of cilia-lacking mouse models – Kif3a, Ift20,21, 22 and has been attributed to tubular hyperproliferation. To characterize the role of cell proliferation in Cep164cKO kidneys and to measure the rates of normal cell proliferation in postnatal collecting ducts we administered EdU to P2, P7, P14 and P21 days old mice, and collected the kidneys 2 hours later for analysis. Representative images from P14 and P21 kidneys (Supplementary Figure S3A – S3D) show that there are only a few EdU-positive proliferating cells in control kidneys compared to Cep164cKO kidneys. Quantification of the EdU-positive cells in DBA-positive cortical collecting ducts revealed that cell proliferation rates are similar between Cep164cKO and control kidneys at P2 (Supplementary Figure S3E), but thereafter decline in the control kidneys, while high proliferation rates are maintained in the mutant kidneys (Supplementary Figure S3E). To further characterize the cell cycle abnormalities on molecular level we performed a western blot analysis against various markers of cell cycle activity, including P-Rb, PCNA, P-Erk1/2, cyclinD1 and cyclin A, which all showed increased expression levels in Cep164cKO kidneys (Supplementary Figure S3F). We also examined the levels of apoptosis by TUNEL assay. Apoptotic nuclei were not seen in P14 kidneys, but we did observe a noticeable number of apoptotic figures in P21 mutant kidneys (Supplementary Figure S3G,H).
Together, our data demonstrate that cystic growth in Cep164cKO kidneys is primarily driven by collecting duct epithelial cell hyperproliferation. In contrast to models of PKD, such as jck and cpk mice23, apoptosis is not a prominent feature of the cystic epithelium in Cep164cKO kidneys at early stages, but appears later, possibly as a physiologic response to tubular cell injury.
Since our histological and molecular analysis indicated that hyperproliferation is the main driver of renal cyst growth in Cep164cKO mice, we next tested whether treatment with roscovitine – a cyclin-dependent kinase (Cdk) inhibitor, would attenuate cyst growth in Cep164cKO mice. We administered roscovitine (150 mg/kg) daily for one week starting at P7 (Figure 2A). Mice in the control arm received a vehicle only injection. Kidneys were collected at P14 and analyzed for morphological and histological changes. At gross morphology kidneys from the roscovitine treated Cep164cKO mice were smaller than those of vehicle-treated mice (Figure 2B–D), indicating that blockage of cell cycle inhibited cyst formation. In addition, the kidney weight to body weight ratio was improved in roscovitin-treated Cep164cKO mice (Figure 2D). At the same time there did not appear to be any adverse effect of roscovitine to control kidneys. Histologic analysis on control and Cep164cKO kidneys (Figure 2E–H) confirmed the beneficial effect of roscovitine in repressing the cystic phenotype in the mutant animals, particularly in the cortical region of the kidneys, where cyst formation ensued after P7 (Figure 2H and 1F). Quantification of the cystic area on the histologic sections showed that roscovitin led to 50% reduction in the cystic index in mutant mice (Figure 2I). To confirm that roscovitine specifically inhibited cell cycle activity we performed a western blot analysis against P-Rb (Ser807/811), PCNA, P-Erk1/2, cyclinD1 and cyclin A on P14 kidneys (Figure 2J). Our data shows that roscovitine effectively suppressed Rb phosphorylation and decreased cyclin A and cyclin D levels in the mutant kidneys (Figure 2J and K).
Figure 2. Roscovitine slows cyst growth in Cep164cKO kidneys.

(A) Schematic of timeline of roscovitin injections and kidney analysis.
(B) Overview of kidney morphology of vehicle treated control (ctrl) and Cep164cKO kidneys at P14. Scale bar 5mm.
(C) Overview of kidney morphology of roscovitine (150 mg/kg) treated control (ctrl) and Cep164cKO kidneys at P14. Scale bar 5mm.
(D) Roscovitine treatment reduces the kidney weight to body weight ratio in P14 Cep164cKO mice. Vehicle 2.76 ± 0.10, n=5, vs. roscovitine 1.79 ± 0.15, n=6, ***p<0.001.
(E – H) Comparison of hematoxylin and eosin-stained mid-sagittal sections of vehicle treated (E,G) and roscovitine-treated kidneys (F,H) demonstrate a significant reduction in the size of the cortical collecting duct cysts in roscovitine-treated P14 Cep164cKO kidneys.
(I) Cystic index is significantly reduced in P14 Cep164cKO kidneys after roscovitin treatment. Vehicle 52.25 % ± 1.15 %, n=4 vs. roscovitine 28.92 % ± 1.68 %, n=7, **** p<0.0001.
(J) Roscovitine treatment blocks cell cycle activity in Cep164cKO kidneys. Western blotting against markers of cell cycle activity pRb, PCNA, pERK1/2, cyclin A and cyclin D1 are significantly decreased in roscovitine-treated Cep164cKO kidneys at P14 compared to vehicle-treated kidneys. Erk1 and Gapdh are used as loading controls. Phospho-proteins were compared with total protein or GAPDH as loading control for densitometric analysis which is presented graphically in i, ii, iii and iv. i) pRb ctrl vs cKOveh (1 ± 0.2, n=3 vs. 3.6 ± 0.7, n=3), cKOveh vs. cKOros (3.4 ± 0.7, n=3 vs. 0.9 ± 0.0, n=3, *p<0.05); ii) cyclinA ctrl vs. cKOveh (1 ± 0.1, n=3 vs. 2.6 ± 0.1, n=3, **p<0.01), cKOveh vs. cKOros (2.6 ± 0.1, n=3 vs. 1.3 ± 0.1, n=3, **p<0.01); iii) pErk1 ctrl vs. cKOveh (1 ± 0.1, n=3 vs. 4.8 ± 0.6, n=3, **p<0.01), cKOveh vs. cKOros (4.8 ± 0.6, n=3 vs. 2.8 ± 0.2, n=3, *p<0.05); iv) cyclinD1 ctrl vs. cKOveh (1 ± 0.2, n=3 vs. 7.8 ± 0.9, n=3, **p<0.01), cKOveh vs. cKOros (7.8 ± 0.9, n=3 vs. 2.7 ± 0.4, n=3, **p<0.01). Data from 3 individual animals in a given group were used for densitometric analysis.
Together, our data shows that treatment with roscovitine improves the kidney morphology in Cep164cKO mice by suppressing cell cycle activity.
DISCUSSION
The data presented here demonstrate for the first time the role of Cep164 in kidney development and collecting duct pathogenesis. We show that Cep164 is essential for primary cilia formation in the kidney tubular epithelium. Similar to other mouse models with a ciliogenesis defect – Kif3a21 and Ift2022, Cep164cKO kidneys undergo rapid cyst formation shortly after birth. Our biochemical analysis and therapeutic rescue experiments established that tubular cell hyperproliferation was the primary driver of tubular cyst formation in the mutant mice. The role of cell cycle dysregulation in kidney cyst growth has been previously demonstrated in orthologous mouse models of PKD, such as jck, cpk, pkd1, pkd2 mice.23, 24 However, it was recently suggested that abnormal ciliary signaling, and not cilia disruption, is the main driver of kidney cyst growth in PKD.25 The newly generated Cep164cKO mouse model which lacks cilia (and ciliary signaling) in the collecting duct-lining epithelial cells, provides an excellent in vivo system to distinguish between these two possibilities. Using roscovitine, a therapeutic drug, which blocks cyclin dependent kinases at multiple cell cycle phases,26 we showed that epithelial cell hyperproliferation during early postnatal kidney growth is the major component of kidney cystogenesis. Sustained suppression of cell proliferation by roscovitine preserved much of the normal histology in the cortex of Cep164cKO kidneys. Why roscovitine failed to inhibit cyst growth in the medullary collecting ducts remains unclear. Given that the morphogenesis of different collecting duct segments is differentially regulated by local cues and cellular factors during postnatal kidney growth27–29 it is likely that roscovitine does not target all of these mechanisms. Also, it is plausible that the roscovitin dosing schedule used here (every 24 hours) does not provide sustained inhibition to the hyperproliferation of the medullary collecting duct cells that have endogenously high proliferative capacity during normal kidney growth27 or that timing of roscovitine treatment was too late, as the first medullary cysts were present already at P7. Subsequent studies will be needed to address these questions. Together, this proof-of-principle study provides further evidence that therapeutics targeting cell cycle are feasible and beneficial modalities against PKD.
SHORT METHODS
Mouse Breeding and Maintenance
Targeted Cep164 embryonic stem cell clone EPD0342–5-B01 was obtained from the Knockout Mouse Project (KOMP) Repository. Chimeric mice were prepared by blastocyst microinjection and bred with C57BL/6J mice to obtain germline transmission. Genotyping primers are shown on Figure S1. Cep164 wild type littermates were used as controls for mutant mice. FLPo deleter mice (stock #011065) were obtained from The Jackson Laboratory and Hoxb7-Cre mice from Dr. Carlton M. Bates. For timed matings, noon on the day a plug was found was designated as E0.5. The person who genotyped the embryos or new born animals was not involved in collecting the material. The experimental protocol was reviewed and approved by the Animal Care Committee of the University of Pittsburgh.
Roscovitine administration
Roscovitine (R-1234) was purchased from LC Laboratories (Boston, Massachusetts), and dissolved in DMSO at 200 mg/ml. For injections roscovitine solutions were prepared at concentrations of 150 mg/kg in 30% 2-hydroxypropyl-β-cyclodextrin in water. 30% 2-hydroxypropyl-β-cyclodextrin was used as a vehicle control. Injection solutions were made fresh every day.
Histological Analyses
Tissues were fixed in 4% (w/v) paraformaldehyde in PBS at 4°C. All tissues were then dehydrated through a methanol series and embedded in paraffin. Sections were taken at 5 μm. Hematoxylin and eosin staining followed standard protocols. Phenotyping in mouse embryos was performed blinded. More than 5 regions were selected at random and analyzed using Adobe Photoshop.
Biochemical Analyses
On the day of euthanasia, blood was immediately collected, and serum levels of BUN were measured using a Urea Assay Kit (K024-H1, Arbor Assays, Michigan).
Immunohistochemistry and antibodies
E10.5 embryos were fixed in 4% (w/v) paraformaldehyde (PFA) in PBS at 4°C. Embryos were then immersed in 15% and 30% sucrose and embedded in Tissue Freezing Medium (Triangle Biomedical Sciences, Inc.). Sections were taken at 8 μm. For immunostaining sections were blocked in 10% donkey serum/1% BSA and permeabilized in 0.1% Tween-20. Primary antibodies are listed in Supplementary Table 1. Secondary antibodies were: donkey anti-mouse Alexa Fluor 488, donkey anti-mouse Alexa Fluor 594, donkey anti-rabbit Alexa Fluor 488, donkey anti-rabbit Alexa Fluor 594 (Molecular Probes), goat anti-chicken Alexa Fluor 488 (Molecular Probes), and donkey anti-rabbit Alexa Fluor 647 (Molecular Probes). Samples were mounted in ProlongGold (Molecular Probes) and images captured using a Leica DM2500 optical microscope or Leica TSC 5SP X confocal microscope (Leica Microsystems). EdU staining on paraffin embedded kidney sections was conducted using Click-iT™ EdU imaging kit (Invitrogen, Carlsbad, CA) according to the manufacturer’s protocol. Animals were injected EdU at 25 mg/kg and euthanized 2 hours later for analysis. A least 3 individual kidneys were analyzed per genotype per age group. From each kidney 10 images from the cortex were taken at 200x magnification, and the total number of EdU+ nuclei and DAPI+ nuclei in the DBA+ collecting ducts were counted for calculating the EdU ratio.
Western Blotting
Kidney tissues were lysed in RIPA Lysis Buffer (Pierce, Rockford, IL) and homogenized with a douncer. Cleared tissue lysates were produced by centrifugation of the resulting samples at 16,0003g for 30 minutes at 4°C. Gel electrophoresis of tissue lysates was performed using the NuPAGE System (Invitrogen). Samples were resolved on 4%–12% Bis-Tris gels in 3-(N-morpholino) propanesulfonic acid buffer and transferred to a nitrocellulose membrane that was then probed for the protein of interest using antibodies diluted in Tris-buffered saline containing 5% milk or 5% BSA and 0.1% Tween-20 (Sigma-Aldrich). Densitometric analysis of western blot bands was performed using ImageJ (Version 2.0.0, NIH). For each blot, the relative density (calculated by dividing the percent value for each sample by the percent value for the standard) of the target protein in each lane is divided by the ratio of density of the loading control. This will give the normalized density to the loading control. Vehicle control was taken to represent 1, the expression ratios of other samples were normalized against the vehicle control, and data blotted as bar graphs with SEM.
Statistics
χ2 test was used to test whether the distribution of mouse genotypes corresponds to Mendelian ratio. Mantel-Cox log-rank test was used to analyze mouse survival. t-test was used to compare data between two groups. Significance was determined at P < 0.05 and represented by * to denote P<0.05, ** P<0.01, *** P<0.001, **** P<0.0001. All experiments were carried out three times or more (n≥3). Data were analyzed using Prism 6 software (GraphPad Software, San Diego, CA) and are given as the mean ± SEM.
Study approval
The procedures for all animal experiments were reviewed and approved by the IACUC of the University of Pittsburgh.
Data availability
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author on request.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
TRANSLATIONAL STATEMENT.
Animal models of human cystic kidney diseases are highly valuable as preclinical models for testing compounds to counter the pathology of these genetic conditions. Our results here demonstrate that treatment with roscovitine, a cyclin-dependent kinase inhibitor effectively blocks kidney cyst growth in a mouse model with collecting duct-specific deletion of primary cilia. Together, this proof-of-principle study provides further evidence that therapeutics targeting cell cycle are feasible and beneficial modalities against cystic kidney disease.
ACKNOWLEDGEMENTS
This research was supported by grants from the National Institutes of Health to R.A. (DK099434, P30DK079307) and F.H. (DK068306). F.H. is a William E. Harmon Professor of Pediatrics.
Footnotes
DISCLOSURE
F.H. is a cofounder of Goldfinch-Bio. The other authors have no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Hildebrandt F, Benzing T, Katsanis N: Ciliopathies. The New England journal of medicine, 364: 1533–1543, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Igarashi P, Somlo S: Genetics and pathogenesis of polycystic kidney disease. Journal of the American Society of Nephrology : JASN, 13: 2384–2398, 2002. [DOI] [PubMed] [Google Scholar]
- 3.Ward CJ, Hogan MC, Rossetti S, Walker D, Sneddon T, Wang X, Kubly V, Cunningham JM, Bacallao R, Ishibashi M, Milliner DS, Torres VE, Harris PC: The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat Genet, 30: 259–269, 2002. [DOI] [PubMed] [Google Scholar]
- 4.Yokoyama T: Ciliary subcompartments and cysto-proteins. Anat Sci Int, 92: 207–214, 2017. [DOI] [PubMed] [Google Scholar]
- 5.Sorokin SP: Centriole formation and ciliogenesis. Aspen Emphysema Conf, 11: 213–216, 1968. [PubMed] [Google Scholar]
- 6.Ishikawa H, Kubo A, Tsukita S, Tsukita S: Odf2-deficient mother centrioles lack distal/subdistal appendages and the ability to generate primary cilia. Nature cell biology, 7: 517–524, 2005. [DOI] [PubMed] [Google Scholar]
- 7.Lu Q, Insinna C, Ott C, Stauffer J, Pintado PA, Rahajeng J, Baxa U, Walia V, Cuenca A, Hwang YS, Daar IO, Lopes S, Lippincott-Schwartz J, Jackson PK, Caplan S, Westlake CJ: Early steps in primary cilium assembly require EHD1/EHD3-dependent ciliary vesicle formation. Nature cell biology, 17: 228–240, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schmidt KN, Kuhns S, Neuner A, Hub B, Zentgraf H, Pereira G: Cep164 mediates vesicular docking to the mother centriole during early steps of ciliogenesis. The Journal of Cell Biology, 199: 1083–1101, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wei Q, Xu Q, Zhang Y, Li Y, Zhang Q, Hu Z, Harris PC, Torres VE, Ling K, Hu J: Transition fibre protein FBF1 is required for the ciliary entry of assembled intraflagellar transport complexes. Nature communications, 4: 2750, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deane JA, Cole DG, Seeley ES, Diener DR, Rosenbaum JL: Localization of intraflagellar transport protein IFT52 identifies basal body transitional fibers as the docking site for IFT particles. Current biology : CB, 11: 1586–1590, 2001. [DOI] [PubMed] [Google Scholar]
- 11.Tanos BE, Yang HJ, Soni R, Wang WJ, Macaluso FP, Asara JM, Tsou MF: Centriole distal appendages promote membrane docking, leading to cilia initiation. Genes & development, 27: 163–168, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Joo K, Kim CG, Lee MS, Moon HY, Lee SH, Kim MJ, Kweon HS, Park WY, Kim CH, Gleeson JG, Kim J: CCDC41 is required for ciliary vesicle docking to the mother centriole. Proc Natl Acad Sci U S A, 110: 5987–5992, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sillibourne JE, Hurbain I, Grand-Perret T, Goud B, Tran P, Bornens M: Primary ciliogenesis requires the distal appendage component Cep123. Biology open, 2: 535–545, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Graser S, Stierhof YD, Lavoie SB, Gassner OS, Lamla S, Le Clech M, Nigg EA: Cep164, a novel centriole appendage protein required for primary cilium formation. J Cell Biol, 179: 321–330, 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li J, Lu D, Liu H, Williams BO, Overbeek PA, Lee B, Zheng L, Yang T: Sclt1 deficiency causes cystic kidney by activating ERK and STAT3 signaling. Hum Mol Genet, 26: 2949–2960, 2017. [DOI] [PubMed] [Google Scholar]
- 16.Siller SS, Sharma H, Li S, Yang J, Zhang Y, Holtzman MJ, Winuthayanon W, Colognato H, Holdener BC, Li FQ, Takemaru KI: Conditional knockout mice for the distal appendage protein CEP164 reveal its essential roles in airway multiciliated cell differentiation. PLoS genetics, 13: e1007128, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chaki M, Airik R, Ghosh AK, Giles RH, Chen R, Slaats GG, Wang H, Hurd TW, Zhou W, Cluckey A, Gee HY, Ramaswami G, Hong CJ, Hamilton BA, Cervenka I, Ganji RS, Bryja V, Arts HH, van Reeuwijk J, Oud MM, Letteboer SJ, Roepman R, Husson H, Ibraghimov-Beskrovnaya O, Yasunaga T, Walz G, Eley L, Sayer JA, Schermer B, Liebau MC, Benzing T, Le Corre S, Drummond I, Janssen S, Allen SJ, Natarajan S, O’Toole JF, Attanasio M, Saunier S, Antignac C, Koenekoop RK, Ren H, Lopez I, Nayir A, Stoetzel C, Dollfus H, Massoudi R, Gleeson JG, Andreoli SP, Doherty DG, Lindstrad A, Golzio C, Katsanis N, Pape L, Abboud EB, Al-Rajhi AA, Lewis RA, Omran H, Lee EY, Wang S, Sekiguchi JM, Saunders R, Johnson CA, Garner E, Vanselow K, Andersen JS, Shlomai J, Nurnberg G, Nurnberg P, Levy S, Smogorzewska A, Otto EA, Hildebrandt F: Exome Capture Reveals ZNF423 and CEP164 Mutations, Linking Renal Ciliopathies to DNA Damage Response Signaling. Cell, 150: 533–548, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao H, Kegg H, Grady S, Truong HT, Robinson ML, Baum M, Bates CM: Role of fibroblast growth factor receptors 1 and 2 in the ureteric bud. Dev Biol, 276: 403–415, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V, Mujica AO, Thomas M, Harrow J, Cox T, Jackson D, Severin J, Biggs P, Fu J, Nefedov M, de Jong PJ, Stewart AF, Bradley A: A conditional knockout resource for the genome-wide study of mouse gene function. Nature, 474: 337–342, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu Y, Wang C, Sun H, LeRoith D, Yakar S: High-efficient FLPo deleter mice in C57BL/6J background. PloS one, 4: e8054, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lin F, Hiesberger T, Cordes K, Sinclair AM, Goldstein LS, Somlo S, Igarashi P: Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc Natl Acad Sci U S A, 100: 5286–5291, 2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jonassen JA, San Agustin J, Follit JA, Pazour GJ: Deletion of IFT20 in the mouse kidney causes misorientation of the mitotic spindle and cystic kidney disease. J Cell Biol, 183: 377–384, 2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bukanov NO, Smith LA, Klinger KW, Ledbetter SR, Ibraghimov-Beskrovnaya O: Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature, 444: 949–952, 2006. [DOI] [PubMed] [Google Scholar]
- 24.Lantinga-van Leeuwen IS, Leonhard WN, van der Wal A, Breuning MH, de Heer E, Peters DJ: Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum Mol Genet, 16: 3188–3196, 2007. [DOI] [PubMed] [Google Scholar]
- 25.Ma M, Tian X, Igarashi P, Pazour GJ, Somlo S: Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat Genet, 45: 1004–1012, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Meijer L, Borgne A, Mulner O, Chong JP, Blow JJ, Inagaki N, Inagaki M, Delcros JG, Moulinoux JP: Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem, 243: 527–536, 1997. [DOI] [PubMed] [Google Scholar]
- 27.Marquez MG, Cabrera I, Serrano DJ, Sterin-Speziale N: Cell proliferation and morphometric changes in the rat kidney during postnatal development. Anat Embryol (Berl), 205: 431–440, 2002. [DOI] [PubMed] [Google Scholar]
- 28.Li J, Ariunbold U, Suhaimi N, Sunn N, Guo J, McMahon JA, McMahon AP, Little M: Collecting duct-derived cells display mesenchymal stem cell properties and retain selective in vitro and in vivo epithelial capacity. Journal of the American Society of Nephrology : JASN, 26: 81–94, 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guaytima EDV, Brandan YR, Favale NO, Santacreu BJ, Sterin-Speziale NB, Marquez MG: Bradykinin mediates the association of collecting duct cells to form migratory colonies, through B2 receptor activation. J Cell Physiol, 233: 6173–6195, 2018. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all other data supporting the findings of this study are available within the article and its Supplementary Information files, or from the corresponding author on request.