

Abstract
A multi-component radical addition strategy enables difunctionalization of alkenes with heteroarenes and a variety of radical precursors, including N3, P(O)R2, and CF3. This unified approach for coupling diverse classes of electrophilic radicals and heteroarenes to vinyl ethers allows for direct, vicinal C-C as well as C-N, C-P, and C-Rf bond formation.
Graphical Abstract
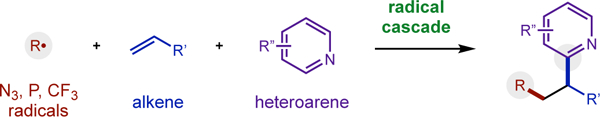
A unified strategy enables multi-component radical addition cascades to couple alkenes, heteroarenes, and various radicals, including N3, P(O)R2, and CF3.
The unique reactivity and selectivity accessible via radical addition to alkenes has significantly enabled the synthesis of valuable medicines and materials.1 Specifically, in the realm of drug discovery, certain privileged elements and architectures (e.g. N, P, F, heteroarenes) are frequently incorporated into a medicinal candidate to improve its pharmacological properties.2 Among the most common radical reactions used to introduce heteroarenes in this arena is the Minisci reaction.3,4 This two-component reaction (Fig 1a) is best suited for coupling nucleophilic radicals (e.g. α-oxy) to electrophilic heteroarenes (e.g. pyridine, quinoline). Conversely, the polarity-mismatched5 coupling of radicals and heteroarenes that are both electron-deficient is typically disfavored – with rare exceptions.6 However, Minisci also demonstrated that a polarity-reversing strategy could bring together three components in a radical cascade (Fig 1b).7 This underutilized approach chemoselectively combines an in situ generated electrophilic radical with an electron-rich alkene in the presence of an electron-deficient heteroarene. The resulting nucleophilic radical then combines with the protonated heteroarene to afford a difunctionalized product after subsequent oxidation. In a pioneering example, Minisci generated α-carbonyl radicals from acetone using Ag/S2O8 to initiate this cascade.7 Later, refluxing peroxides were employed to enable the perfluoroalkyl radical (•Rf) variant.8
Fig. 1.
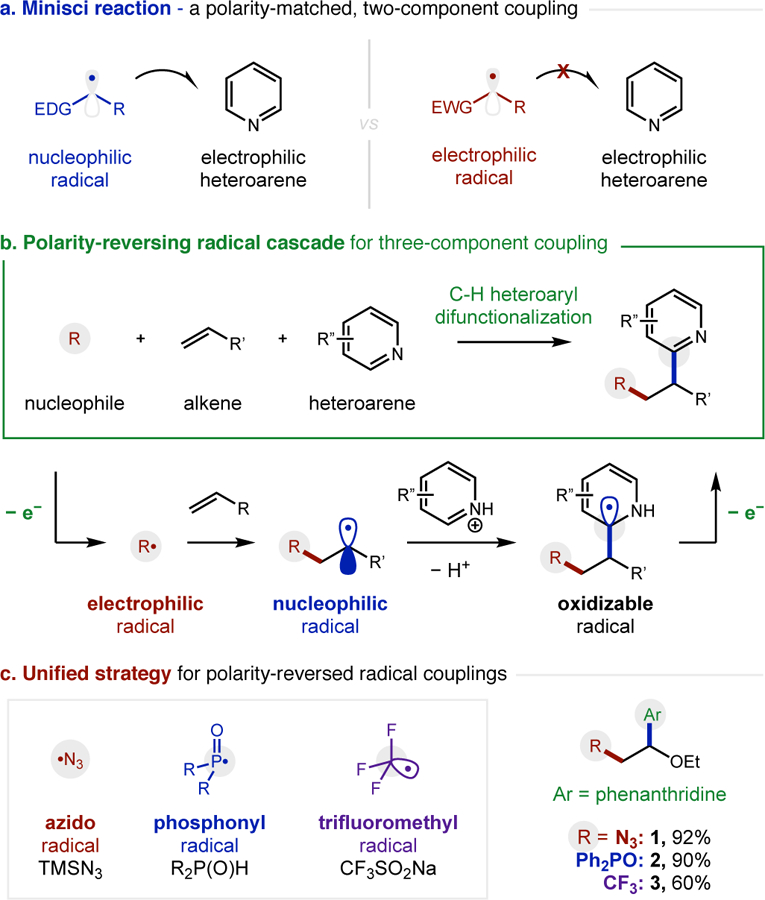
Design of a unified strategy for polarity-reversal cascades
Recent efforts have focused on extending this approach to incorporate other radical partners – and to do so under milder conditions that are more suitable to medicinal applications. For example, Barriault developed a bimetallic, gold-catalyst to incorporate α-carbonyl radicals (•REWG).9 Herzon and Baran reported hydropyridylations (•H) with Co and Fe complexes.10 Liu developed an azido variant (•N3) with iodanes, and Chu reported a photocatalytic version shortly thereafter.11 Other photolytic methods for incorporating trifluoromethyl (•CF3) and phosphonyl radicals (•P(O)R2) have been developed by Hong, Matsunaga, and us.12 However, in looking at the divergent conditions required to perform each of these specific reactions, we questioned if a single, unified strategy could be developed to enable the incorporation of multiple classes of electrophilic radicals within this cascade. The advantage of such an approach would be its utility as a robust synthetic tool to rapidly access complex molecules with vicinal substituted functionality – especially for applications in medicinal chemistry.
Towards our goal of a unified strategy, we sought to employ hypervalent iodanes, which are robust precursors for numerous electrophilic radicals (Fig 1c).13 In particular, we hypothesized azido, phosphonyl, and trifluoromethyl radicals could all be accessed by iodane-scission reactions, as shown in Fig 2. In all three cases, a neutral or anionic reagent combines with a λ3-iodane either directly or otherwise to form an electrophilic radical. Azido radicals A are formed via direct ligand displacement of PhI(OAc)2 by TMSN3, followed by homolysis of the weak N-I bond.14 Conversely, we proposed that phosphonyl radicals B may be formed indirectly by H• abstraction of the weak P-H in Ph2P(O)H by •N3.14,15 Finally, trifluoromethyl radicals C may be generated by ligand displacement of PhI(OTFA)2 with Langlois’ reagent NaSO2CF3.16
Fig. 2.
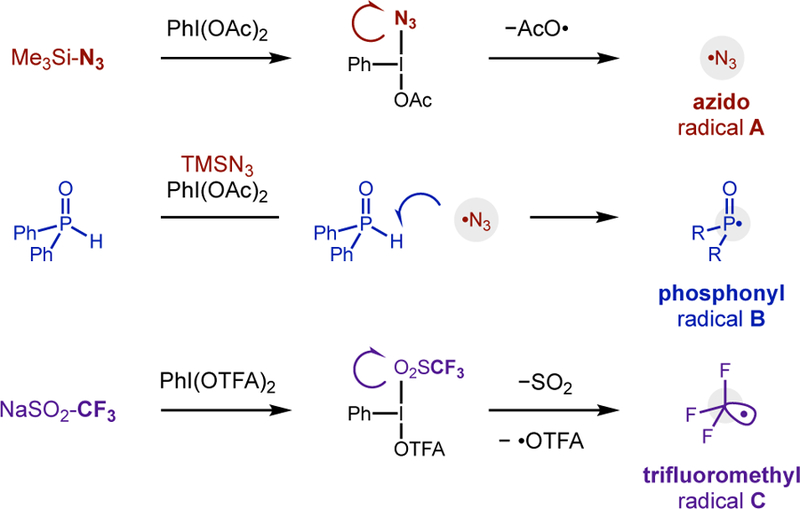
Diverse electrophilic radicals from hypervalent iodanes
To our delight, upon subjecting these three, distinct radicalprecursors (TMSN3, Ph2P(O)H, and NaSO2CF3) to an electron-rich alkene (ethyl vinyl ether) and heteroarene (phenanthridine) in the presence of PhI(OAc)2 or PhI(OTFA)2, all three classes of cascade reactions were accessible (1–3; 60–92%). As shown in Fig 3, the development of these three reactions were largely enabled by the identity of the oxidant, as well as its rate of addition. For example, in the azido variant, an initial addition of PhI(OAc)2 affords only 20% yield (entry 1), likely due to rapid formation of •N3 in too high of concentration. Alternatively, slow addition via syringe pump allows for efficient formation of the azido product 1 (entry 2, 92% yield). This strongly beneficial effect of slow addition was also seen for other heteroarene traps (see SI).
Fig. 3.
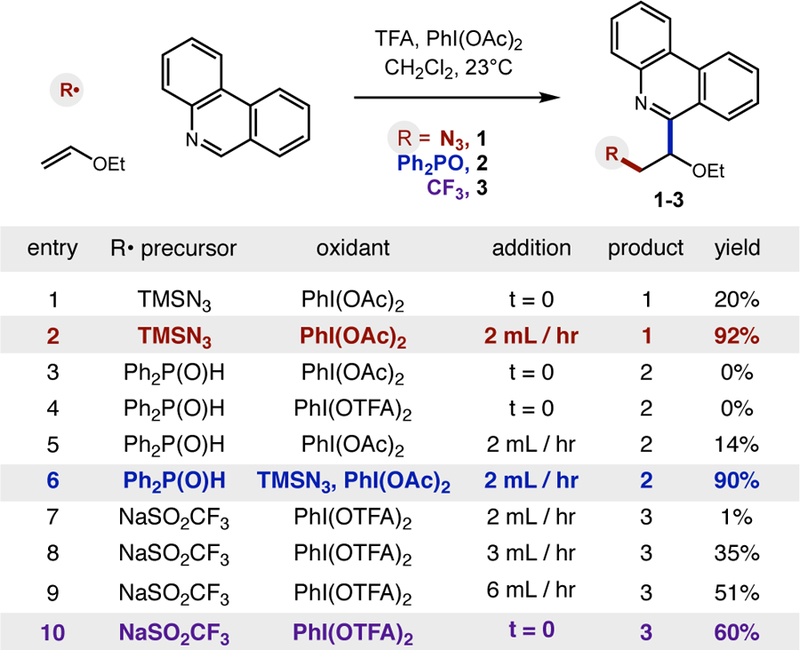
Development of three radical cascade reaction classes a
a N3 : Heteroarene (0.1 mmol), TFA (1 eq), TMSN3 (2 eq), alkene (4 eq), PhI(OAc)2 (2 eq), CH2Cl2 (0.05 M), 23 °C, 30 min. P: Heteroarene (0.1 mmol), TFA (2 eq), TMSN3 (1 eq), phosphine oxide (6 eq), alkene (2 eq), PhI(OAc)2 (2 equiv), CH2Cl2 (0.05 M), 23 °C, 30 min. CF3: Heteroarene (0.1 mmol), TFA (1 eq), NaSO2CF3 (2 eq), alkene (2 eq), PhI(OTFA)2 (2 eq), MeCN (0.1 M), 23 °C, 45 min. Yields determined by NMR.
Next, we investigated the phosphonyl cascade reaction. As expected, direct displacement of both PhI(OAc)2 and PhI(OTFA)2 by diphenyl phosphine oxide is inefficient, with up to 14% yield observed only in the case of slow oxidant addition (entries 3–5). However, addition of TMSN3 as an H-atom transfer mediator,14 enables efficient formation of phosphonyl product 2 (entry 6, 90% yield).
Finally, we investigated the trifluoromethyl radical-mediated variant of this cascade. Knowing that NaSO2CF3 does not efficiently displace the ligands of PhI(OAc)2 at room temp, we used the more reactive oxidant, PhI(OTFA)2. Interestingly, in the event of slow oxidant addition, slow rates of both iodane substitution and ensuing formation of •CF3 resulted in low reactivity (entries 7–9). Thus, for the CF3 cascade reaction, we found that addition of the reagent at t = 0 is optimal, affording trifluoromethyl product 3 (entry 10, 60% yield).
With a unified, iodane-mediated strategy in hand for heteroaryl difunctionalization of alkenes by a polarity-reversing radical cascade, we wished to investigate the generality of these three classes of reactions. In the azido three-component coupling (Fig. 4), we were pleased to find the phenanthridine-based protocol could be extended to include several other electronically diverse heteroarenes. For example, both isoquinolines (4–10) and quinolines (11–12), bearing various functional groups, including ketones, esters, amides, and halides, were tolerated. Additionally, in place of ethyl vinyl ether, 2,3-dihydrofuran could be used as the alkene partner to afford azido furan 13 as a single diastereomer (> 20:1 d.r.). Unfortunately, non-ethereal alkenes were inefficient coupling partners – providing diminished reactivity in all three radical cascades.17
Fig. 4.
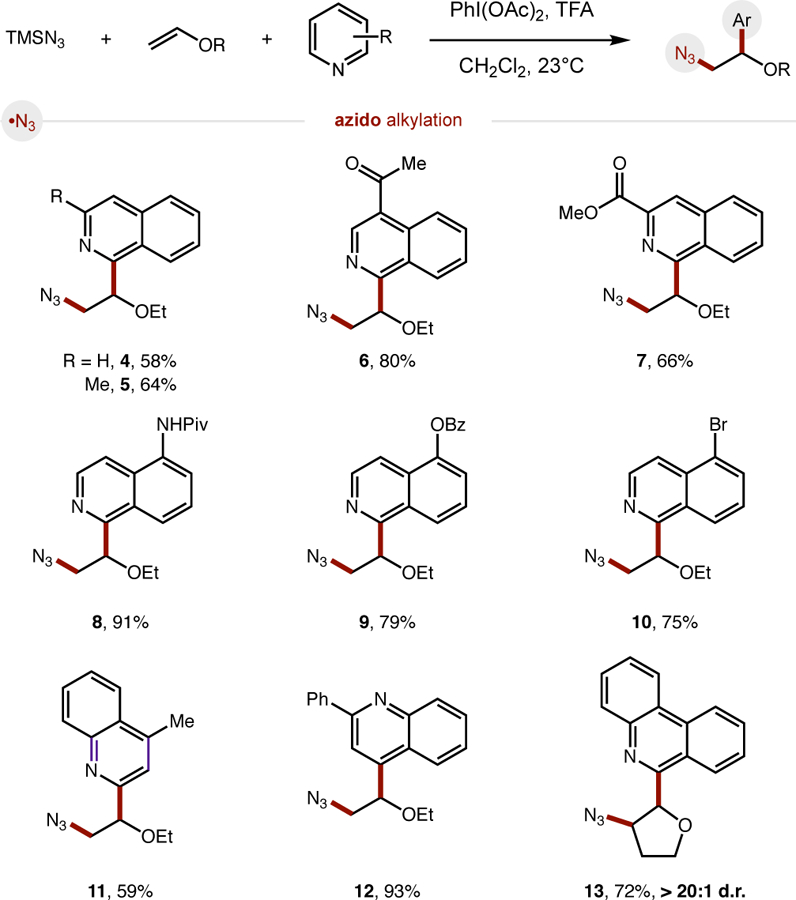
Azido three-component coupling
Due to the importance of phosphorus motifs in materials and medicines, we then turned our attention to exploring the scope of the phosphonyl three-component coupling (Fig. 5). As in the case of the azide variant, the incorporation of diphenyl phosphine oxide through phosphinylalkylation of heteroarenes was efficient and synthetically generalizable. Similarly, both isoquinolines (14–20, 23) and quinolines (21–22) were tolerated. Synthetic utility was again demonstrated by the incorporation of various functional groups, including ketones, esters, amides, and halides. Notably, aryl bromide 20, which may undergo deleterious halide abstraction by phosphonyl radicals, was tolerated. And as before, both the 2- and 4-positions of quinoline are susceptible to efficient addition when the other position is blocked with a substituent. To our delight, furan derivative 23 was also accessed from the cyclic precursor in high diastereoselectivity (> 20:1 d.r.).
Fig. 5.
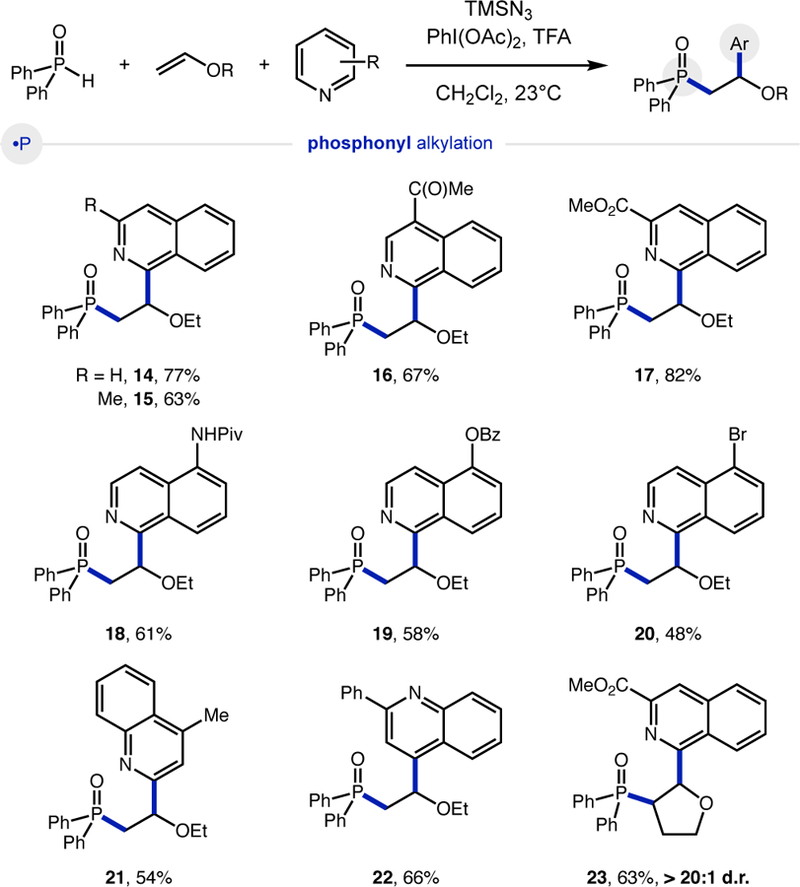
Phosphonyl three-component coupling
Finally, given the ubiquity of trifluoromethyl groups in medicine, we also sought to explore the generality of the trifluoromethyl three-component coupling (Fig. 6). Despite the use of the more reactive oxidant, PhI(OTFA)2, the reaction scope appears similarly general to the other two cascade reaction classes. To test this reaction in more complex and challenging cases, ketones and halides were included in the heteroarene component at varying positions without issue (24–27). Similarly, a cyclic alkene was employed – affording a single diastereomer of the fluoroalkylated heteroarene (> 20:1 d.r.).
Fig. 6.
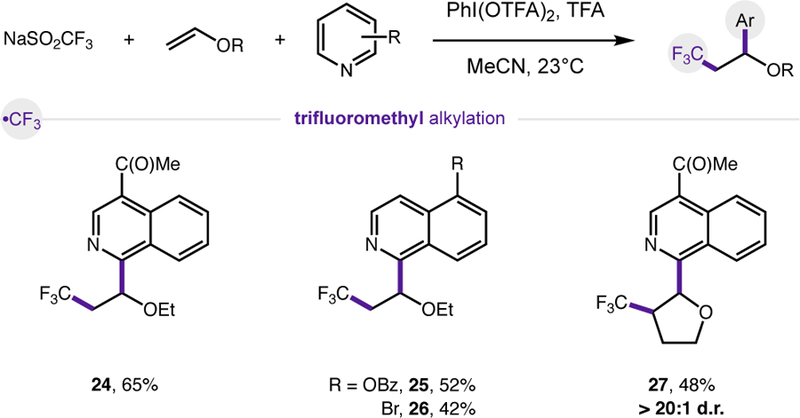
Trifluoromethyl three-component coupling
In conclusion, we have demonstrated the development of a unified, modular strategy for three-component couplings to access alkyl heteroarenes in which three contiguous carbons contain biologically valuable substitution (N, P, Rf, O, pyridine). The synthetically facile approach employs hypervalent iodine reagents to generate azido, phosphonyl, and trifluoromethyl radicals, which chemo- and regio-selectively combine with enol ethers and heteroarenes. The mildness of the platform permits wide functional group tolerance and enables the synthesis of diverse classes of heteroarenes with medicinal relevance.
Supplementary Material
Acknowledgments
We thank the National Institutes of Health (NIH R35 GM119812), National Science Foundation (NSF CAREER 1654656), and the United States Air Force (J.Q.B.) for financial support.
Footnotes
Electronic Supplementary Information (ESI) available: Procedures, Characterization, and Spectroscopic data of all new compounds. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare
References
- 1.Hart DJ, Science, 1984, 223, 883–887; M. Yan, J. C. Lo, J. T. Edwards and P. S. Baran, J. Am. Chem. Soc., 2016, 138, 12692–12714. [DOI] [PubMed] [Google Scholar]
- 2.Smith BR, Eastman CM and Njardarson JT, J. Med. Chem, 2014, 57, 9764–9773; E. Vitaku, D. T. Smith and J. T. Njardarson, J. Med. Chem., 2014, 57, 10257–10274. [DOI] [PubMed] [Google Scholar]
- 3.Duncton MAJ, Med. Chem. Commun, 2011, 2, 1135–1161; 10.1002/anie.201900977. [DOI] [Google Scholar]
- 4.Recent examples of the Minisci reaction: Devari S and Shah BA, Chem. Commun, 2016, 52, 1490–1493; M. C. Quattrini, S. Fujii, K. Yamada, T. Fukuyama, D. Ravelli, M. Fagnoni and I. Ryu, Chem. Commun., 2017, 53, 2335–2338; L. Zhang, G. Zhang, Y. Li, S. Wang and A. Lei, Chem. Commun., 2018, 54, 5744–5747; A. H. Jatoi, G. G. Pawar, F. Robert and Y. Landais, Chem. Commun., 2019, 55, 466–469. [DOI] [PubMed] [Google Scholar]
- 5.Roberts BP, Chem. Soc. Rev, 1999, 28, 25–35; F. De Vleeschouwer, V. Van Speybroeck, M. Waroquier, P. Geerlings and F. De Proft, Org. Lett., 2007, 9, 2721–2724. [Google Scholar]
- 6.Nagib DA and MacMillan DWC, Nature, 2011, 480, 224–228; Y. Ji, T. Brueckl, R. D. Baxter, Y. Fujiwara, I. B. Seiple, S. Su, D. G. Blackmond and P. S. Baran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14411–14415; Y. Fujiwara, J. A. Dixon, F. O’hara, E. D. Funder, D. D. Dixon, R. A. Rodriguez, R. D. Baxter, B. Herlé, N. Sach, M. R. Collins, Y. Ishihara and P. S. Baran, Nature, 2012, 492, 95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Citterio A, Gentile A and Minisci F, Tetrahedron Lett, 1982, 23, 5587–5590. [Google Scholar]
- 8.18 Antonietti F, Mele A, Minisci F, Punta C, Recupero F and Fontana F, J. Fluor. Chem, 2004, 125, 205–211. [Google Scholar]
- 9.McCallum T and Barriault L, Chem. Sci, 2016, 7, 4754–4758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma X and Herzon SB, J. Am. Chem. Soc, 2016, 138, 8718–8721; J. C. Lo, D. Kim, C.-M. Pan, J. T. Edwards, Y. Yabe, J. Gui, T. Qin, S. Gutiérrez, J. Giacoboni, M. W. Smith, P. L. Holland and P. S. Baran, J. Am. Chem. Soc., 2017, 139, 2484–2503; X. Ma, H. Dang, J. A. Rose, P. Rablen and S. B. Herzon, J. Am. Chem. Soc., 2017, 139, 5998–6007. [DOI] [PubMed] [Google Scholar]
- 11.Liu Z and Liu Z-Q, Org. Lett, 2017, 19, 5649–5652; J. Chen, S. Zhu, J. Qin and L. Chu, Chem. Commun., 2019, 55, 2336–2339. [DOI] [PubMed] [Google Scholar]
- 12.He Y-T, Kang D, Kim I and Hong S, Green Chem, 2018, 20, 5209–5214; J. Q. Buquoi, J. M. Lear, X. Gu, and D. A. Nagib. ACS Catal., 2019, DOI: 10.1021/acscatal.9b01580 ; Y. Kumagai, N. Murakami, F. Kamiyama, R. Tanaka, T. Yoshino, M. Kojima, and S. Matsunaga, Org. Lett., 2019, DOI: 10.1021/acscatal.9b0158010.1021/acs.orglett.9b01015; Y. Kumagai, N. Murakami, F. Kamiyama, R. Tanaka, T. Yoshino, M. Kojima, and S. Matsunaga, Org. Lett., 2019, DOI: 10.1021/acs.orglett.9b01015 . [Google Scholar]
- 13.Togo H and Katohgi M, Synlett, 2001, 2001, 0565–0581; A. Yoshimura and V. V. Zhdankin, Chem. Rev., 2016, 116, 3328–3435. [Google Scholar]
- 14.Fontana F, Minisci F, Yong MY and Lihua Z, Tetrahedron Lett, 1993, 34, 2517–2520; K. Matcha and A. P. Antonchick, Angew. Chem. Int. Ed., 2013, 52, 2082–2086; A. P. Antonchick and L. Burgmann, Angew. Chem. Int. Ed., 2013, 52, 3267–3271; K. Matcha, R. Narayan and A. P. Antonchick, Angew. Chem. Int. Ed., 2013, 52, 7985–7989. [Google Scholar]
- 15.Williams RH and Hamilton LA, J. Am. Chem. Soc, 1952, 74, 5418–5420; S. Deprèle and J.-L. Montchamp, J. Org. Chem., 2001, 66, 6745–6755. [Google Scholar]
- 16.Yang Y-D, Iwamoto K, Tokunaga E and Shibata N, Chem. Commun, 2013, 49, 5510–5512. L. Shi, X. Yang, Y. Wang, H. Yang and H. Fu, Adv. Synth. Catal., 2014, 356, 1021–1028. [DOI] [PubMed] [Google Scholar]
- 17. Non-ethereal alkenes afford the following yields for each reaction: N3 (53%), P (35%), and CF3 (10%). A likely explanation is the decreased nucleophilicity of the intermediate, non-ketyl, alkyl radical is less suited to heteroarene addition than to competing pathways (e.g. oxidation or H-atom transfer).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.