

Abstract
Little is known about the mechanism of amyloid assembly in immunoglobulin light chain (AL) amyloidosis, in contrast to other amyloid diseases. Early events in the aggregation pathway are especially important as these soluble species could be cytotoxic intermediates playing a critical role in the initiation of amyloid assembly. In this work, we discuss the mechanism of the early events in in vitro fibril formation of immunoglobulin light chain AL-09 and AL-12 (involved in cardiac amyloidosis) and its germline (control) protein κI O18/O8. Previous work from our laboratory showed that AL-12 adopts a canonical dimer conformation (like the germline protein), while AL-09 presents an altered dimer interface as a result of somatic mutations. Both AL-12 and AL-09 aggregate with similar rates and significantly faster than the germline protein. AL-09 is the only protein in this study that forms stable oligomeric intermediates during the early stages of the aggregation reaction with some structural rearrangements that increase the Thioflavin T fluorescence but maintain the same number of monomers in solution. The presence of the restorative mutation AL-09 H87Y changes the kinetics and the aggregation pathway compared to AL-09. The single restorative mutation AL-12 R65S slightly delayed the overall rate of aggregation as compared to AL-12. Collectively, our study provides a comprehensive analysis of species formed during amyloid nucleation in AL amyloidosis, shows a strong dependence between the altered dimer conformation and the formation of stable oligomeric intermediates, and sheds light on the structural features of amyloidogenic intermediates associated with cellular toxicity.
Graphical Abstract
INTRODUCTION
The amyloid formation reaction starts when soluble proteins sampling partially folded states (under denaturing conditions) adopt an alternative conformation that promotes self-assembly into higher order structures known as amyloid fibrils.1, 2 Amyloid fibrils are highly organized and self-assembled structures, rich in β-sheets characteristically arranged in a cross-β conformation. The formation of amyloid fibrils has been implicated in many protein aggregation diseases, collectively called amyloidosis. Although current investigations aim to elucidate the molecular mechanisms behind amyloid formation and cellular toxicity in vitro, there are a host of factors that influence the kinetics of aggregation and formation of toxic intermediates in vivo. Specifically, interaction of proteins with cell membrane and lipids can alter the rate of aggregation and formation of toxic species can increase the nucleation rate.3–8
In vitro amyloid formation is a nucleated growth polymerization reaction.9–11 This reaction has three distinct phases: 1) Lag/nucleation phase– the monomeric protein self-assembles to form a nucleus which acts as fibrillar precursors, 2) Elongation phase– characterized by rapid fibril elongation by the addition of monomeric and/or oligomeric units to the nucleus or growing end of the fibrils and 3) Stationary/plateau phase– characterized by the maturation of amyloid fibrils.11–13 None of the aforementioned phases (lag, elongation, and stationary) has a well-defined zone, as all of these phases are active during the entire aggregation pathway and are governed by the rate constants and availability of the reacting species.14 Nucleation events are particularly important in amyloid reactions as they directly influence the overall kinetics of the reaction.9, 11, 12, 15 For some amyloid precursor proteins and peptides, the lag phase also features the presence of a detectable population of metastable intermediates which can act as precursors for elongation of fibrils.16–20 Characterization of these intermediate species is especially important as many have been reported to be cytotoxic.21, 22
While the mechanisms of fibril formation in some of the amyloid associated diseases are well documented,23–25 there are other amyloid diseases where the mechanism is poorly understood. One example is immunoglobulin light chain (AL) amyloidosis, the most common form of systemic amyloidosis. AL amyloidosis is characterized by the over production of immunoglobulin light chain due to abnormal proliferation of monoclonal plasma cells. These excess light chains are secreted into circulation, misfold, and deposit as insoluble amyloid fibrils in the extracellular space of various organs, leading to impaired organ function and failure.26, 27 Fibril formation in AL amyloidosis is influenced by pH, presence of specific cofactors, and somatic mutations, affecting the protein’s thermodynamic and kinetic stability.28–30 It is, however, unclear the type and properties of amyloid intermediates involved in the initiation of amyloid assembly, the role of intermediates stability and heterogeneity in the overall aggregation process and amyloid stability in light chains.
In this work, we explored the formation of the amyloid precursors/intermediates and the role of protein concentration in the formation of these intermediates for two AL variable domain light chains, AL-09 and AL-12 belonging to the κI O18/O8 (IGKV 1–33) subgroup. Both AL-09 and AL-12 (obtained from two different patients with cardiac amyloidosis) share >90% sequence homology with the κI O18/O8 germline (control) protein. For the purpose of simplicity, throughout the manuscript we address the variable domain AL proteins tested with their alphanumeric codes only.
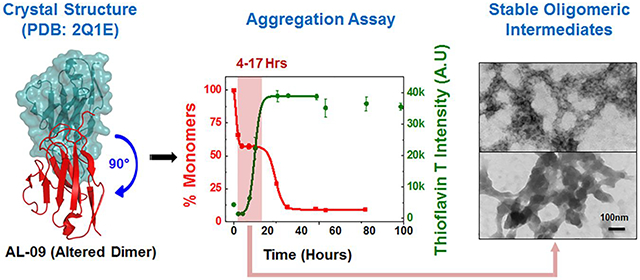
AL-12 retains the canonical dimer interface (observed in the germline structure)31 whereas AL-09 adopts an altered dimer interface where the dimers are rotated by 90° with respect to the canonical dimer of the κI O18/O8 germline protein (Figure 1-Inset).31 Previous work by our group has shown that AL-09 altered dimer interface results from the mutation Y87H favoring altered conformations which promote amyloid formation in AL-09.32 AL-09 presents the fastest amyloid formation kinetics of all κI proteins characterized in our laboratory to date.
Figure 1: Comparative analysis of aggregation rate in different light chain variable domain proteins at 20 μM.

Shown are the representative kinetic traces of different time points obtained from sedimentation assay to calculate the %monomer remaining in the reaction (■) vs ThT Assay (●) at 20 μM of (A) κI O18/O8 (PDB: 2Q20), (B) AL-09 (PDB: 2Q1E), (C) AL-09 H87Y (PDB: 3CDY), (D) AL-12 (PDB: 3DVF), (E) AL-12 R65Sa. Kinetic traces at each time point are from duplicate readings. Aggregation mixture for all the above experiments were prepared in 10 mM ABC buffer at pH 2.0. The highlighted area in salmon in the graphs represents the nucleation phase presumed for this study (based on % monomer data). The highlighted area in green in the AL-09 graph represents a stable plateau phase during which the concentration of monomers remains unchanged. Inset: Crystallographic models showing the orientation of the dimeric interface for each protein. Restorative mutation in AL-09 H87Y (C) and AL-12 R65S (E) are highlighted in yellow. Error bars represent Standard error of mean
a AL-12 R65S model was derived from the crystallographic structure of AL-12.
We used a combination of assays (sedimentation assay (SA) to determine the amount of monomer incorporated into the amyloid, the Thioflavin T binding assay (TA), dynamic light scattering (DLS), transmission electron microscopy (TEM), and size exclusion chromatography (SEC)), to qualitatively and quantitatively assess the species populated during the amyloid nucleation phase at two different concentrations. Restorative mutants AL-09 H87Y, AL-12 R65S and the control κI O18/O8 germline protein were also analyzed.
Together, our results suggest that the location of somatic mutations causing structural variation in AL light chain dimers promote formation of oligomeric intermediates varying in size and stability for the different proteins, causing them to aggregate through different pathways.
Material and Methods
Chemicals:
All chemicals and reagents used in the experiments were procured from Sigma-Aldrich unless specified otherwise. Milli Q grade water was used as a solvent to prepare different solutions.
Cloning, expression, extraction and purification of recombinant κI O18/O8, AL-09, AL-09 H87Y, AL-12 and AL12 R65S variable domain proteins:
κI OI8/O8 germline DNA (GenBank accession number EF640313) was generated by mutating AL-103 cDNA (GenBank accession number AY701640).32 DNA sequences of the mutant proteins AL-09 (GenBank accession number AF490909) and AL-12 (GenBank accession number AF490912) belonging to the κI gene family were obtained from patients exhibiting cardiac involvement.33 DNA sequence of restorative mutants AL-09 H87Y and AL-12 R65S were generated by mutating the cDNA of AL-09 and AL-12 using QuickChangeTM multisite directed mutagenesis kit (Stratagene, La Jolla, CA).34, 35 AL-09 has seven somatic mutations compared to the κI O18/O8 germline sequence, three of those mutations are non-conservative; AL-12 has eight somatic mutations out of which seven are non-conservative.28 Protein expression was performed as reported previously.28, 31, 32 Briefly, plasmids containing the recombinant DNA were transformed in Escherichia coli BL21 (DE3) gold competent cells (Stratagene, La Jolla, CA) and the cultures were grown until approximately A600nm of 0.75, at which point the cultures were induced with 0.8 mM of isopropyl-β-D-thiogalactopyranoside. After 20 h of post induction growth, bacterial cells were pelleted and re-suspended in phosphate buffered saline (PBS), and frozen at −20°C. For the following study, irrespective of whether the overexpressed proteins are found in the periplasmic space or inclusion bodies, one freeze-thaw cycle using PBS was used as the first step to extract all the proteins. AL-09 was extracted from the dialyzed resolubilized insoluble fraction while all other proteins (κIO18/O8, AL-09 H87Y, AL-12, and AL-12 R65S) were extracted from periplasmic space by breaking the cells through one freeze thaw cycle using PBS buffer at pH 7.4. All proteins were purified using size exclusion chromatography (SEC) (HiLoad 16/60 Superdex 75 Column) on an AKTA FPLC (GE Healthcare) system with 10 mM Tris-HCl at pH 7.4. Chromatographic fractions containing pure protein were verified by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and analytical SEC. After confirmation of protein purity, various chromatographic fractions were pooled together and concentrated between 200 and 300 μM, flash frozen, and stored in 1.5 mL microcentrifuge tubes at −80°C. Protein concentration was determined by UV absorbance at 280 nm using extinction coefficient calculated from amino acid sequence as follows: ε=14890 M-1 ·cm-1 for κI O18/O8 and AL-09 H87Y, and ε=13610 M-1 ·cm-1 for AL-09, AL-12, and AL-12 R65S.
CD Spectroscopy:
We used the Circular Dichroism (CD) spectropolarimeter (JASCO 810) with a 0.2 cm path-length quartz cuvette. The temperature was controlled within ±0.01°C using a Peltier system. All the protein samples (20 μM) investigated were prepared in either 10 mM Tris HCl buffer at pH 7.4 or 10 mM sodium acetate, boric acid and sodium citrate buffer (ABC buffer) at pH 2.0 (Figure S1). The thermal unfolding/refolding experiments were conducted following the ellipticity at 217 nm over a temperature range of 10–80°C as reported previously.29, 35, 36
Sample preparation for experimental assays:
Prior to the experiments, all proteins were thawed on ice and centrifuged at 643,867×g for 3 h and 20 min in a Beckman Coulter (Optima L-100 XP) ultracentrifuge. After centrifugation, the top three-fourth parts of the supernatant were gently removed and filtered through 0.2 μM low protein binding membrane filter. This step removes preformed aggregates (if any) formed during the freeze-thaw process. The filtered protein solution is additionally checked in analytical size exclusion chromatography (SEC) and dynamic light scattering (DLS) to ensure the integrity and monomeric state of the proteins before the experimental assays. The ultracentrifuged and filtered protein solution was used to prepare aggregation reaction mixtures (20μM or 220 μM protein,150 mM NaCl in 10 mM sodium acetate, boric acid and sodium citrate buffer (ABC buffer) at pH 2.0) for the different approaches: For the HPLC sedimentation assay, 1200 μL of the reaction mixture were placed in low protein binding microcentrifuge tubes and for the thioflavin T (ThT) binding assay, 800 μL of the reaction mixture were mixed with ThT (final concentration 20 μM ThT).
Our protocol differs from our previous reported experiments because the incubation of the aggregation mixture is happening in microcentrifuge tubes with 1200 μL and 800 μL compared to 260 μL of aggregation mixture in 96-well plates.29, 30, 35 After the aggregation mixture was prepared, it was kept on ice while the t=0 reading (~30 min) was determined for the different experiments. Thereafter, the aggregation mixture was moved to an incubator kept at 37°C with orbital shaking set at 300 RPM to monitor the aggregation kinetics.
For all our experimental assays, we considered two protein concentrations, 20 μM (referred to as the low concentration) and 220 μM (referred to as the high concentration). These concentrations were chosen based on the dimer dissociation constant (Kd) values for AL-09 and κI O18/O8 respectively.32 The concentrations calculated using HPLC differed slightly from these two concentrations (calculated measuring the absorbance at 280 nm). The HPLC calculated concentrations of the AL proteins used in different experimental assays are reported in table ST1. For the sake of simplicity and comparative purposes, we will address the low concentration as 20 μM and high concentration as 220 μM.
In vitro fibril formation and quantitation:
A. HPLC (High performance liquid chromatography) Sedimentation assay:
Quantification of unreacted soluble protein in the aggregation mixture was followed by periodically removing aliquots (~80 μL) from ongoing aggregation reactions followed by centrifugation at 160,000×g and injecting the supernatant (approx. 20 μL) in duplicates into an analytical reverse phase column (Agilent Zorbax-C8 2.5 × 50 mm column). Protein samples were run against a gradient of acetonitrile (0–100%) in 0.1% TFA maintained at 300 μL·min-1 flow rate in an Agilent HPLC system (1100 series). The wavelength of the UV detector was fixed at 280 nm. HPLC grade solvents (Water and Acetonitrile) used as mobile phase in the reverse phase HPLC system were obtained from Fisher Chemicals. Concentration of the unreacted soluble protein was determined from integrated A280 nm peaks using standard curves individually acquired for each protein.37
B. Thioflavin T binding assay:
ThT fluorescence from the aggregation mixture was monitored on a plate reader (Analyst AD; Molecular Devices, Sunnyvale, CA). An aliquot (~260 μL) of the fibril formation reaction (containing ThT) was placed in duplicate on a black 96 well polystyrene plate (Greiner, Monroe, NC) at specific time points (equivalent to the time points used for the sedimentation assay) to measure the fluorescence intensity. After the measurements, we placed back the reaction mixture into the microcentrifuge tube and resumed incubation at 37°C, 300 RPM in the orbital shaker till the end of the aggregation reaction. We repeat this process periodically to obtain the ThT intensity for all the time points presented in the figures. All the fluorescence measurements were done using an excitation wavelength of 440 nm and an emission wavelength of 480 nm.
For both the sedimentation and the Thioflavin T assay, we assumed the aggregation reaction had ended when the change in the minimum concentration of monomers or the Thioflavin T fluorescence intensity maximum remained constant over a period of at least 48 hrs.
C. Kinetic data analysis:
The disappearance of monomers in solution (obtained from the sedimentation assay) was plotted in parallel to the ThT emission fluorescence signal. The t50 value was obtained by fitting each independent kinetic trace to a sigmoidal function (Boltzman function in Origin software package) as previously reported.38
| Equation 1 |
where A1 is the initial fluorescence value, A2 is the final fluorescence value, x0 is the center (t50 value), dx is defined as the time constant.
Critical concentration (Cr):
Aggregation reaction mixtures were incubated for one additional day (after the plateau phase of the reaction is reached) and were analyzed for molar concentration of monomeric protein by HPLC. This concentration is the residual monomeric protein after the aggregate reaction reaches equilibrium, also defined as critical concentration (Cr). We assumed that there is complete conversion from fully folded to partially folded states under the amyloid formation reaction conditions for these calculations. Based on this assumption, the Cr we report is in fact the residual concentration of partially folded monomeric states. The inverse of Cr represents the amyloid fibril growth equilibrium constant as described previously.37, 39 The change in the Gibbs free energy (ΔG) of fibril growth was thus obtained from the equation –RT ln(1/Cr) and was subsequently used to obtain the ΔΔG (ΔGMutated protein ˗ ΔGκIO18/O8) values.
Dynamic light scattering (DLS):
DLS measurements were performed in a DynaPro molecular sizing instrument with 1 μm upper size limit for detection of hydrodynamic radius at room temperature. The viscosity and refractive index of the solvent was set for water in the software interface of the instrument before the start of the scattering measurement. An aliquot (~40 μL) was gently pipetted into the DynaPro cell (quartz micro cuvette provided with the instrument) from the ongoing aggregation reaction. The intensity autocorrelation functions obtained from the particles undergoing Brownian motion were analyzed using the Dynamics V6 software (provided with the instrument) to obtain the hydrodynamic radii and population density of specific particles.
We determined the hydrodynamic radii of the monomeric AL proteins in 10 mM Tris-HCl pH 7.4, at room temperature at different concentrations (Figure S2). We previously reported that AL proteins in Tris-HCl buffer at pH 7.4, 37°C do not form amyloid fibrils within the time frame of the experiment.30 The values shown in parentheses adjacent to the hydrodynamic radii for respective peaks in all our figures represent the fraction (in percent mass) of various species at the specified concentrations.
During the aggregation experiments, the scattering signal from particles >1 μm in general originates from the impurities in the micro cuvette and during the transfer of samples to the micro cuvette. The populations (0.0–0.1%) of these particles (>1 μm) were too low to be reliably considered as part of aggregation mixture. Additionally we excluded the DLS data pertaining to time points during elongation phase due to interference from amyloid assemblies which might lead to misinterpretation of the overall aggregation kinetics.
Analytical size exclusion chromatography:
An aliquot (~150 μL) from the ongoing aggregation reaction was filtered and injected into a 50 μL loop attached to a Superdex 200 10/300 GL analytical size exclusion column (separation range of 10,000–600,000 Da) attached to an AKTA FPLC system (GE healthcare). Phosphate buffer (0.05 M monobasic sodium phosphate, 0.05 M dibasic sodium phosphate and 0.15 M sodium chloride) at pH 6.8 at a constant flow rate of 0.40 ml·min-1 was used to elute the proteins. The wavelength of the detector in the AKTA FPLC system was fixed at 280 nm. Molecular weight and oligomeric states were estimated from elution volumes using a molecular weight calibration curve as reported earlier.29–31
Prior to beginning of each reaction, (just as we did in DLS), we injected an increasing concentration of all AL proteins in Tris-HCl buffer at pH 7.4 as controls (Figure S3). We also injected 10 mM ABC buffer in SEC column Superdex 200 10/300 to account for the baseline of the chromatographic profile (Figure S4).
The elution volume (Ve) of the monomeric AL proteins at different concentrations –with the exception of AL-09– ranged from 16.2–17.2 mL. The apparent molecular weight calculated from the protein standards (Figure S5) within this range computes to 10.4–17.2 kDa, which is consistent with the molecular weight of the monomeric AL variable domain proteins. AL-09 has a Ve= 18.5 mL (apparent molecular weight of 4.2 kDa). Such a deviation from the ideal elution properties in proteins of similar size can be observed due to a differential degree of compaction, enhanced hydrophobicity or differences in the protein’s shape.40, 41 The discrepancy observed in the elution profile of AL-09 has only been observed with the Superdex 200 matrix30 suggesting the possibility of a strong interaction between AL-09 and Superdex 200 matrix.
Transmission electron microscopy:
Aliquots (~8 μL) from the aggregation reaction at different time points were placed on a 300 mesh copper Formvar/carbon coated grid (Electron Microscopy Science, Hatfield, PA) and allowed to adsorb for 90 s. The grid was washed with Milli Q H2O, stained with freshly filtered (2% w/v) uranyl acetate for 30 s and then again washed with Milli Q H2O. During the entire process of staining and washing excess of water and stain were blotted with filter paper. Grids were analyzed on a Philips Technai T12 transmission electron microscope (FEI, Hillsboro, OR) at 80 kV. We attempted to quantitate the aspect ratio of the species during progression of our amyloid reactions but encountered technical difficulties that may have had to do with differences in sample preparation and overall heterogeneity of amyloid formation reactions.
Results
Structure and biophysical properties of the AL proteins:
The structural features of the native conformation of pure κI O18/O8 (control), the mutants (AL-09 and AL-12), and their corresponding restorative mutants (AL-09 H87Y and AL-12 R65S) were analyzed by circular dichroism spectroscopy in 10 mM Tris-HCl buffer at pH 7.4 and 10 mM ABC buffer at pH 2.0 at 4°C and 37°C (Figure S1). This was done to corroborate the folded state conformation (pH 7.4) and the structural changes prior to the aggregation studies (pH 2.0). All proteins, with the exception of AL-09, under both pH conditions at 4°C and 37°C presented far UV-CD spectra with a minimum at ~217 nm, indicative of β-sheet structure. In addition, a minimum at ~235 nm due to aromatic residues that absorb in the far UV region was observed, in agreement with previous far UV-CD reports of natively folded immunoglobulin light chain variable domains.28, 42, 43 Under the conditions of the fibril formation reactions (ABC buffer pH 2.0 at 37°C), the far UV-CD spectrum of AL-09 suggests the presence of partially folded states and a loss of secondary structure. At 80°C, all AL proteins showed an extensive loss of secondary structure at pH 2.0 and pH 7.4 (Figure S1). The thermal unfolding transitions at pH 7.4 were reversible with different levels of hysteresis, as previously reported.29, 30, 35 From the thermal denaturation experiments at pH 7.4, we calculated the following percentage of unfolded protein at 37°C: kI O18/O8= 0.0%, AL-09= 10.6%, H87Y= 0.0%, AL-12= 2.6% and AL-12 R65S= 0.7%. Each stock of AL protein at pH 7.4 was additionally investigated by dynamic light scattering and analytical size exclusion chromatography to confirm the monomeric state and the homogeneity of the protein prior to aggregation experiments.
Comparative analysis of aggregation rate in AL proteins and its mutants:
Aggregation reactions of the selected AL proteins and the κI O18/O8 control were prepared at 20 μM and 220 μM in 10 mM ABC buffer and 150 mM NaCl at pH 2.0, conditions under which all the proteins selected are able to form fibrils, as reported previously.29, 44 The only change in the reaction parameters used for this study -as compared to our previous studies- is the reaction vessel (microcentrifuge tubes in this study instead of 96-well polysterene plates). This was a necessary change due to the need for a larger reaction volume because of the multiple approaches we used to follow the amyloid formation reaction (SA, TA, DLS, TEM and SEC).
We observed faster aggregation rates for most of the proteins using microcentrifuge tubes. The most dramatic changes were observed for κI O18/O8, AL-12, and AL-12 R65S, suggesting that the rate of aggregation of these proteins is significantly influenced by the reaction volume and the air/water interface provided by the microcentrifuge tube reactions (Table 1).45–47
Table 1: Kinetic parameters of light chain variable domain proteins.
Table shows average t50 values obtained from sedimentation and Thioflavin T assays for different proteins at 20 μM and 220 μM. “n” represents the number of biological replicates used to obtain the reported average t50 values. The critical concentration (Cr) for the amyloid formation reaction was calculated at the end of the fibril formation reaction (see methods). ΔΔG values were calculated as previously reported by Williams and Wetzel.48 Experimental conditions: 10 mM ABC buffer pH 2.0, 37°C, 300 rpm.
| Proteins | Conc (μM) | t50 (Hours) | Critical Concentration [Cr] (μM) | ΔΔG [ΔGAL protein − ΔGκI O18/O8] (kcal·mol−1) | ΔΔG [ΔGALrestorative mutant − ΔGAL protein] (kcal·mol−1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Sedimentation Assay | Thioflavin T Assay | ||||||||
| Tube | Plate | ||||||||
| κI O18/O8 | 20 | 61.8 ± 9.5 (n=3) | 59.2 ±4.5 (n=3) | 266.2 ± 10.9a | 1.2 ± 0.1 | 0 | --- | ||
| 220 | 113.1 ± 10.5 (n=3) | 99.4 ± 3.3 (n=4) | --- | 3.2 ± 0.3 | 0 | --- | |||
| AL-09 | 20 | 2.4 ± 0.9 (n=3) | 27.9 ± 4.9 (n=3) | 12.1 ± 2.4 (n=3) | 54.5 ± 0.8a | 1.9 ± 0.4 | 0.29 ± 0.04 | 0 | |
| 220 | 2.2 ± 0.7 (n=3) | 34.5 ± 2.0 (n=3) | 2.1 ± 0.5 (n=3) | 40.3 ± 5.7 (n=3) | --- | 3.4 ±0.4 | 0.05 ± 0.00 | 0 | |
| AL-09 H87Y | 20 | 152.3 ± 7.6 (n=4) | 140.4 ± 18.6 (n=4) | 635 ± 26.8b | 2.4 ± 0.2 | --- | 0.13 ± 0.04 | ||
| 220 | 274.4 ± 35.9 (n=2) | 218.9 ± 12.2 (n=2) | --- | 2.8 ± 0.2 | --- | −0.12 ± 0.01 | |||
| AL-12 | 20 | 26.7 ± 5.1 (n=5) | 37.2 ± 3.2 (n=5) | 253.2 ± 4.0c; 357.0 ± 1.7a | 1.9 ± 0.3 | 0.28 ± 0.02 | 0 | ||
| 220 | 28.7 ± 6.8 (n=4) | 30.9 ± 7.7 (n=4) | --- | 3.1 ± 0.7 | −0.02 ± 0.04 | 0 | |||
| AL-12 R65S | 20 | 46.2 ± 7.5 (n=2) | 46.6 ± 1.4 (n=2) | 333.4 ± 45.4c | 3.0 ± 0.1 | --- | 0.27 ± 0.03 | ||
| 220 | 64.9 ± 7.3 (n=3) | 49.1 ± 6.8 (n=3) | --- | 3.4 ± 0.3 | --- | 0.07 ± 0.05 | |||
In addition to the aggregation kinetics parameters, the thermodynamics of fibril stability and elongation of these proteins were calculated using the residual monomer concentration (critical concentration, Cr)13, 37, 39 at 20 μM and 220 μM (Table 1). All AL proteins tested in the current study at 20 μM and 220 μM exhibit different Cr. ΔΔG values for fibril formation were calculated with respect to κI O18/O8 control. We will describe in detail the results from the 20 μM reactions in the following section.
Main observations from our 20 μM experiments show that all proteins show sigmoidal curves for TA and corresponding reciprocal curves for SA, except for AL-09 (Figure 1 A–E). For a typical amyloid nucleation-polymerization reaction, the rate of monomer depletion should correlate well with the rate of increase in ThT fluorescence enhancement. However, under some circumstances, a deviation from this correlation is observed due to the formation of structural intermediates that present a neutral ThT enhancement and/or are large enough to sediment and therefore appear as depleted monomer. The AL-09 amyloid formation reactions present a sharp decrease in monomer concentration in the first part of the reaction, followed by a stable plateau phase with the SA data, indicating the presence of stable intermediate species encompassing the same number of monomers that may rearrange, adding more binding sites to ThT. The Cr varied from 1.2 μM – 3.0 μM for different AL-proteins tested at 20 μM (Table 1; Figure S6A).
Experiments conducted at 220 μM were designed to show any possible concentration dependence in the rate of amyloid formation (Figure S7 A–E). We observed slower rate of aggregation for κI O18/O8 and AL-09 H87Y at this high monomer concentration. A similar trend was previously reported by the Fink laboratory when the aggregation rate is slower in protein concentrations tested above their respective Kd,49, 50 suggesting that the delay in the reaction corresponds to the dissociation of dimers to misfolded monomers. The Cr varied from 2.8 μM – 3.4 μM for the different AL-proteins tested at 220 μM (Table 1; Figure S6B). The Cr values are higher at 220 μM compared to the 20 μM, indicating that the reaction is slightly more efficient (final yield) at 220 μM. The ΔΔG values were calculated to compare AL proteins with κI O18/O8 and then between AL proteins and their respective restorative mutants at the two protein concentrations (Table1; Figure S6 C,D). κI O18/O8 fibrils are more stable than AL-09 and AL-12 fibrils at 20 μM, but AL-12 fibrils are more stable at 220 μM. AL-09 H87Y fibrils are less stable than AL-09 fibrils at 20 μM, but AL-09 H87Y fibrils are more stable at 220 μM. This result suggests that AL-09 H87Y fibrils are ‘different’ at the two different concentrations with more stable fibrils at 220 μM. It is possible that by having more initial monomer available in the reaction at 220 μM, the ultrastructure is different and more stable. AL-12 fibrils are more stable than AL-12 R65S at both concentrations, but to a lesser extent at 220 μM.
Aggregation kinetics in κI O18/O8:
κI O18/O8 exhibit a significant nucleation phase at 20 μM (0–26 h) and 220 μM (0–35 h) [highlighted salmon area in Figure 2A and S8A] followed by an exponential drop in monomer concentration concomitant with an increase in Thioflavin T fluorescence intensity. By the end of 120 h (20 μM) and 160 h (220 μM), κI O18/O8 is completely aggregated. The rate of aggregation determined from the t50 values was 61.8 ± 9.5 h (SA) and 59.2 ± 4.5 h (TA) at 20 μM, and 113.5 ± 10.5 h (SA) and 99.4 ± 3.3 h (TA) at 220 μM, respectively (Table 1). Early through the nucleation phase, at 20 μM and 220 μM, κI O18/O8 shows predominant monomeric species by DLS (peaks under colored square in Figure 2B and S8B). The size of the monomers, determined by their hydrodynamic radii varies from 1.9–2.7 nm, within the size range of the monomeric species (2.1–2.8 nm) observed for κI O18/O8 by DLS and SEC (Figure S2 and S3A). We observe a predominantly monomeric population during nucleation with a small fraction (0.1–0.3%) of particles measuring 25 nm – 400 nm.
Figure 2: Composite analysis of monomer concentration, species size and abundance, and aggregate morphologies for 20 μM κI O18/O8 during nucleation phase.

Shown are (A) the representative kinetic traces of the sedimentation assay (■) vs ThT fluorescence assay intensity (●). The highlighted area in salmon represents the nucleation phase (based on % monomer data). The color coded dotted lines in (A) represents the time points when DLS was performed. (B) Peaks corresponding to hydrodynamic radii computed from scattering intensity obtained from DLS. The peaks inside the colored squares represent the radius corresponding to the monomeric protein. Values presented in the parenthesis adjacent to hydrodynamic radii represent the population density in percent mass of particles. (C) Electron Micrographs of 20 μM κI O18/O8 at different time points during the course of aggregation. The color matched dotted line in the EM images compared to (A) and (B) represents the closest time point when aliquots from the reaction mixture were studied during the course of aggregation. Arrow designation: (→) Spherical oligomers, (→) Transformation of nebular mesh to fibrillar form, (→) Aggregated nebular mesh of non-fibrillar oligomers, (→) Bundles of short fibrils. Scale bar, 100 nm. Aggregation conditions: 10 mM ABC buffer at pH 2.0, 37°C, 300 rpm. Error bars in (A) represent Standard error of mean.
In order to corroborate the findings from DLS, we tested the aggregation in 220 μM κI O18/O8 by analytical size exclusion chromatography (SEC). We were unable to conduct the analytical size exclusion chromatography with the 20 μM reaction due to the low signal observed (data not shown). κI O18/O8 at 220 μM presented a single peak consistent with a monodisperse population (Figure S9A) during the nucleation phase. The apparent molecular weight calculated from the elution volume (Ve) computes to 11.7–12.4 kDa, consistent with the molecular weight of the monomeric κI O18/O8 variable domain proteins (Figure S3A), supporting our DLS observations.
We were unable to observe any higher molecular weight species that would be associated with the 25 nm – 400 nm particles observed in DLS. This is probably due to the low abundance of those species and the exclusion volume limit of our column.
TEM is biased towards larger species and while it is not quantitative, it provides information regarding size and morphology of the larger particles not observed using DLS or SEC. TEM images of κI O18/O8 show spherical oligomers (black arrows), nebular mesh of non fibrillar oligomers (red arrows) and transformation of non-fibrillar oligomers to bundles of short fibrillar forms (green arrows) during the nucleation phase (0–35 h) (Figure 2C). At the later stages of aggregation, the non-fibrillar oligomers transform into bundles of short (blue arrows) and long fibrils (purple arrows) (Figure S10 A–C). At 220 μM, κI O18/O8 mostly shows nebular mesh of non-fibrillar oligomers (red arrows) (Figure S8C) during nucleation phase, and exhibits bundles of short fibrils (blue arrows) along with long fibrillar forms (purple arrows) (Figure S10 D–F) after nucleation.
In summary, κI O18/O8 does not appear to populate any significant amount of soluble oligomeric species during the nucleation phase. In addition, κI O18/O8 undergoes significant maturation steps that happen during the stationary phase of the reaction at later time points.
Aggregation kinetics in AL-09:
There is no evidence of concentration dependence in the kinetics of amyloid formation for AL-09. The kinetics (at both concentrations) presents a fast nucleation phase with a unique biphasic transition observed by sedimentation assay. Consequently, AL-09 displays two t50 values for SA at 20 μM (2.4 ± 0.9 and 27.9 ± 4.9 h) and 220 μM (2.6 ± 0.9 and 35.0 ± 3.4 h) (Table 1). The SA data shows a sharp drop in monomer concentration followed by a stable plateau phase (highlighted green area in Figure 3A and S11A).The monomer concentration remains unaltered over a period of 4–23 h. The TA data also shows a clear biphasic transition at 220 μM, with a minor burst in fluorescence intensity (4 times the value at zero time point) followed by a plateau phase (4–12 h) and a second transition (Figure S11A). It has been reported previously that such initial burst in fluorescence intensity during TA could be due to prefibrillar oligomers rich in β–sheet content, formed early during the aggregation pathway.51, 52 The two t50 values of AL-09 at 220 μM are 2.1 ± 0.5 h and 40.3 ± 5.7 h respectively (Table 1). AL-09 aggregates four times faster than κI O18/O8 and is completely aggregated by 40 h. DLS analysis of AL-09 at 20 μM and 220 μM show two populations of intermediate species with different sizes [species–I: 5–30 nm (peaks represented under colored squares) and species–II: 100–300 nm] (Figure 3B and S11B), consistent with SA and TA. No AL-09 monomer peaks (2.1–2.6 nm, (Figure S2B) were detected in the amyloid formation reaction at either concentration. AL-09 shows a dominant population of 5.0 nm particles at 0.5 h. A similar pattern in the relative abundance of species was also noted through the plateau phase (4–17 h) (Figure 3B). This species profile is very similar in the amyloid formation reaction at 220 μM (Figure S11B). In the next phase of the reaction (6–32 h), there is a decrease of species-I associated with an increase in species-II at both concentrations tested. This phase corresponds to the plateau in monomer concentration observed by SA. SEC experiments for AL-09 at 220 μM (Figure S9B) shows intermediate species ranging in size from 63.7 kDa to 629.4 kDa within the first 9 h of the experiment, in good agreement with the DLS data. These results suggest the presence of a stable population of oligomeric intermediates that undergo conformational changes that increases their ThT fluorescence without incorporating additional monomers.
Figure 3: Composite analysis of monomer concentration, species size and abundance, and aggregate morphologies for 20 μM AL-09 during nucleation phase.

Shown are (A) the representative kinetic traces of the sedimentation assay (■) vs ThT Assay fluorescence intensity (●). The highlighted area in cyan green represents the plateau phase of the biphasic reaction (based on constant % monomeric concentration). The color coded dotted lines in (A) represents the time points when DLS was performed. (B) Peaks corresponding to hydrodynamic radii computed from scattering intensity obtained from DLS. The peaks inside the colored squares represent the radius corresponding to oligomeric intermediates. Values presented in the parenthesis adjacent to hydrodynamic radii represent the population density in percent mass of particles. (C) Electron Micrographs of 20 μM AL-09 at different time points during the course of aggregation. The color matched dotted line in the EM images compared to (A) and (B) represents closest time points when aliquots from the reaction mixture were studied during the course of aggregation. Arrow designation: (→) Spherical oligomers, (→) Aggregated nebular mesh of non-fibrillar oligomers, (→) Bundles of short fibrils. Scale bar, 100nm. Aggregation conditions: 10 mM ABC buffer at pH 2.0, 37°C, 300 rpm. Error bars in (A) represent Standard error of mean.
The TEM studies of AL-09 at 20 μM show an aggregated nebular mesh of nonfibrillar and spherical oligomers during the initial stages of aggregation (Figure 3C). Spherical oligomers (black arrows) were observed during the initial sharp decrease in the concentration of monomers (SA). During the initial plateau phase (midpoint of the reaction) in which no changes in the monomer concentration is observed but ThT fluorescence intensity continues to increase, we observe dense nebular mesh of oligomers (red arrows). At the end of the reaction when the monomer has been all incorporated into fibrils and the ThT fluorescence intensity reaches a plateau, AL-09 displays morphologies consistent with bundles of short fibrils (blue arrows) and mature long fibrils (purple arrows) (Figure S12 A–C). AL-09 reactions at 220 μM (Figure S11C and S12 D,E) presents a similar profile to the one observed at 20 μM.
Aggregation kinetics in AL-09 H87Y:
We then wanted to determine whether restorative mutant AL-09 H87Y presented the same profile of species during early events of amyloid formation compared to those of the germline protein κI O18/O8. We based this hypothesis on our previous thermodynamics and fibril formation studies of these two proteins32.
Indeed, AL-09 H87Y shows a monophasic transition at 20 μM (t50 SA: 152.3 ± 7.6 h and t50 TA: 140.4 ± 18.6 h) similar to κI O18/O8 (Figure 4A) (Table 1). Furthermore, concentration dependent studies were also similar to κI O18/O8 (Figure S13A). The overall rate of aggregation (t50 value) was observed to be 5–7 times slower than AL-09. DLS exhibited a predominant monomeric population during the nucleation phase (Figure 4B, S2C, and S13B). The SEC chromatogram is consistent with the DLS data showing a single peak representing monomeric protein as the only aggregating species at 220 μM of AL-09 H87Y during its nucleation phase (Figure S3C and S9C). In line with AL-09, the TEM of AL-09 H87Y shows a dense population of aggregated nebular mesh of non-fibrillar (red arrows) and spherical oligomers (black arrows) during the nucleation phase at both 20μM (Figure 4C) and 220 μM (Figure S13C). A codominance of nebular mesh species and short fibrillar forms (blue arrows) and mature fibrils (purple arrows) were noted at both concentrations in AL-09 H87Y after the nucleation phase (Figure S14 A–E). Although the TEM profiles of AL-09 and AL-09 H87Y at 20 μM and 220 μM show aggregating species with very similar morphologies, the sequence of their appearance is shifted for AL-09 H87Y, consistent with its delayed fibril formation kinetics.
Figure 4: Composite analysis of monomer concentration, species size and abundance, and aggregate morphologies for 20 μM AL-09 H87Y during nucleation phase.

Shown are (A) the representative kinetic traces of the sedimentation assay (■) vs ThT fluorescence assay (●). The highlighted area in salmon represents the nucleation phase (based on % monomer data). The color coded dotted lines in (A) represents the time points when DLS was performed. (B) Peaks corresponding to hydrodynamic radii computed from scattering intensity obtained from DLS. The peaks inside the colored squares represent the radius corresponding to the monomeric protein. Values presented in the parenthesis adjacent to hydrodynamic radii represent the population density in percent mass of particles. (C) Electron Micrographs of 20 μM AL-09 H87Y at different time points during the course of aggregation. The color matched dotted line in the EM images compared to (A) and (B) represents closest time points when aliquots from the reaction mixture were studied during the course of aggregation. Arrow designation: (→) Spherical oligomers, (→) Aggregated nebular mesh of non-fibrillar oligomers. Scale bar, 100 nm. Aggregation conditions: 10 mM ABC buffer at pH 2.0, 37°C, 300 rpm. Error bars in (A) represent Standard error of mean.
Aggregation kinetics in AL-12:
In our previous reports using 96-well plates, AL-12 presented the slowest fibril formation kinetics of all the proteins we have reported so far.35 We hypothesized that AL-12 would allow us to interrogate the early events of AL amyloid formation in greater detail. Interestingly, AL-12 showed the largest change in kinetics in our microcentrifuge tube assays for this study, with t50 values that resemble the fast kinetics observed at all conditions with AL-09. AL-12 aggregation reaction is characterized by a very short nucleation phase (0–13 h) (highlighted salmon area in Figure 5A and S15A) followed by a sharp drop in the concentration of monomers. The rate of aggregation observed for AL-12 showed no concentration dependence and was the second fastest aggregation reaction in this study with similar t50 values at 20 μM (SA: 26.7 ± 5.1 h and t50 TA: 37.2 ± 3.2 h) and 220 μM (SA: 28.7 ± 6.8 h and t50 TA: 30.9 ± 7.7 h) (Table 1). Unlike AL-09, aggregation kinetics in AL-12 exhibits a monophasic transition similar to κI O18/O8. In the course of the short nucleation phase (0–13 h) observed during the aggregation of AL-12 at both concentrations, a predominant population of particles ranging in size from 2.0–2.7 nm (peaks under colored square in Figure 5B and S15B) and a small fraction (0.2–0.3%) ranging from 100–400 nm was detected by DLS. The size of the predominant species observed during the short nucleation phase in AL-12 was in agreement with the size of monomeric species (1.8–2.9 nm) observed for different concentrations of AL-12 in Tris-HCl buffer at pH 7.4 (Figure S2D). The SEC chromatogram of AL-12 at 220 μM further corroborated the presence of monomeric protein during the nucleation phase (Figure S9D).
Figure 5: Composite analysis of monomer concentration, species size and abundance, aggregate morphologies for 20 μM AL-12 during nucleation phase.

Shown are (A) the representative kinetic traces of the sedimentation assay (■) vs ThT fluorescence intensity assay (●). The highlighted area in salmon represents the nucleation phase (based on % monomer data). The color coded dotted lines in (A) represents the time points when DLS was performed. (B) Peaks corresponding to hydrodynamic radii computed from scattering intensity obtained from DLS. The peaks inside the colored squares represent the radius corresponding to the monomeric protein. Values presented in the parenthesis adjacent to hydrodynamic radii represent the population density in percent mass of particles. (C) Electron Micrographs of 20 μM AL-12 at different time points during the course of aggregation. The color matched dotted line in the EM images compared to (A) and (B) represents the closest time points when aliquots from the reaction mixture were studied during the course of aggregation. Arrow designation: (→) Spherical oligomers, (→) Aggregated nebular mesh of non-fibrillar oligomers. Scale bar, 100nm. Aggregation conditions: 10 mM ABC buffer at pH 2.0, 37°C, 300 rpm. Error bars in (A) represent Standard error of mean.
The TEM images of AL-12 show a co-dominant population of spherical oligomers (black arrows) and nebulae like oligomers (red arrows) during the nucleation phase [20μM (0–13 h)] (Figure 5C). At later stages (after 30 h) short fibrils arranged in bundles (blue arrows) populate the aggregation reaction (Figure S16 A,B). The morphologies of the aggregating species at 220 μM (Figure S16 D,E) were very similar to those observed at 20 μM and differed only in the sequence of their appearance as a function of time.
Aggregation kinetics in AL-12 R65S:
In our previous report, AL-12 R65S presents delayed fibril formation kinetics with respect to AL-12.35 In this study, using the microcentrifuge tube assay, AL-12 R65S shows a delay in fibril formation with a nucleation phase twice as long as that observed for AL-12 (highlighted salmon area in Figure 6A and S17A). Similar to AL-12, the nucleation phase in AL-12 R65S is followed by a sharp drop in concentration of monomers coupled with an increase in Thioflavin T fluorescence intensity. However the rate of aggregation at 20 μM, [t50 = 46.2 ± 7.5 h (SA) and 46.6 ± 1.4 h (TA)] as compared to 220 μM, [t50 = 64.9 ± 7.3 h (SA) and 49.9 ± 6.8 h (TA)] was observed to be slightly slower by SA (Table 1). AL-12 R65S at 20 μM and 220 μM populates an abundance of monomeric species (1.9–2.5 nm) (peaks under colored square in Figure 6B and S17B), and a small fraction (0.1–0.2%) of aggregating species ranging 40–60 nm during the nucleation phase. The size of the monomeric species during nucleation was similar to the monomer size (2.6–3.2 nm) of AL-12 R65S tested at different concentrations in Tris-HCl buffer pH 7.4 (Figure S2E). The apparent molecular weight of the monomeric species (9.9 kDa) as observed from the elution properties of 220 μM AL-12 R65S during the nucleation phase (Figure S9E) is in agreement with the molecular weight of AL-12 R65S variable domain proteins tested at different concentrations in Tris-HCl buffer at pH 7.4 (Figure S3E).
Figure 6: Composite analysis of monomer concentration, species size and abundance, and aggregate morphologies for 20 μM AL-12 R65S during nucleation phase.

Shown are (A) the representative kinetic traces of the sedimentation assay (■) vs ThT fluorescence assay (●). The highlighted area in salmon represents the nucleation phase (based on % monomer data). The color coded dotted lines in (A) represents the time points when DLS was performed. (B) Peaks corresponding to hydrodynamic radii computed from scattering intensity obtained from DLS. The peaks inside the colored squares represent the radius corresponding to the monomeric protein. Values presented in the parenthesis adjacent to hydrodynamic radii represent the population density in percent mass of particles. (C) Electron Micrographs of 20 μM AL-12 R65S at different time points during the course of aggregation. The color matched dotted line in the EM images compared to (A) and (B) represents the closest time points when aliquots from the reaction mixture were studied during the course of aggregation. Arrow designation: (→) Spherical oligomers, (→) Aggregated nebular mesh of non-fibrillar oligomers. Scale bar, 100nm. Aggregation conditions: 10 mM ABC buffer at pH 2.0, 37°C, 300 rpm. Error bars in (A) represent Standard error of mean.
TEM of AL-12 R65S at 20μM during the course of aggregation shows similar morphologies as AL-12 with spherical (black arrows) and nebular-oligomers (red arrows) (Figure 6C), followed by the appearance of bundles of short fibrils (blue arrows) and clusters of mature long fibrils (purple arrows) (Figure S18 A–C). The TEM images of the aggregating species at 220μM AL-12 R65S (Figure S18 D–F) was similar to 20μM AL-12 R65S in both the chronology and the species morphology.
Overall, the comparative analysis of the aggregation kinetics of the different AL proteins in this study suggests the time for the complete conversion of monomers to mature sedimentable fibrillar forms vary significantly for the different AL proteins tested. The fibril stability varies as well, with AL-09 having the lowest stability compared to κI O18/O8. We observe concentration dependence in the stability of the fibrils with the high concentration fibril reactions showing more stable fibrils.
κI O18/O8, AL-09 H87Y, AL-12, and AL-12 R65S present a predominant population of monomers during the nucleation phase. Based on our findings, we propose that κI O18/O8, AL-09 H87Y, AL-12 and AL-12 R65S forms clusters of sparsely populated, transient intermediates during the nucleation phase of the reaction. AL-09 exhibits a stable population of oligomeric intermediates populated from 4–20 h, with no evidence of concentration dependence in the kinetics of amyloid formation. This result is consistent with AL-09’s concentration independent fibril formation behavior reported previously by our group.38 We also proposed that for mildly amyloidogenic proteins, the inverse dependence of t50 values as a function of protein concentration indicated that the amyloid formation kinetics are dominated by a nucleation-elongation mechanism.38
Discussion
In this study, we evaluated the mechanisms of aggregation of five different immunoglobulin light chain variable domain proteins. The fastest rates of aggregation were observed for both AL-09 and AL-12 as compared to κI O18/O8 and the restorative mutants AL-09 H87Y and AL-12 R65S (Figure 1 and S7, Table 1). We found that all the proteins formed transient oligomers to varying degrees during the nucleation phase. However, AL-09 populated stable oligomers (Figure 3 and S11 A,B). Notably, our comparative studies on the stability of amyloid fibrils in this study shows that AL-09 fibrils are consistently less stable than κI O18/O8 and AL-12 fibrils. Together our results demonstrate that while AL-09 presents the fastest kinetic parameters of fibril formation, the resulting fibrils are the least stable fibrils, possibly allowing these fibrils to disaggregate and populate intermediate species to a higher extent than the other proteins in this study.
To date, just a few examples of stable oligomeric intermediates in the amyloidogenic pathway have been characterized.17, 50, 53 Our results suggest that AL-09 has the ‘worst’ of both worlds for fibril pathogenicity: AL-09 aggregates fast, populates intermediates, and the reversible (disaggregation) reaction is more favorable than for other proteins. This may explain the increased cytotoxicity observed in previous studies from our laboratory.54–56
AL-09 data regarding the fibril stability is in agreement with the proposed molecular mechanism involving the participation of a stable intermediate in the fibril formation via secondary nucleation. There is extensive experimental evidence indicating that stable oligomeric intermediates are responsible for the cytotoxicity in cell cultures.18, 57, 58 We propose that the interplay between the fast populating oligomeric species and the low stability of the AL-09 fibrils results in a large pool of cytotoxic amyloidogenic intermediates, prone to recruit and aggregate soluble monomers, catalyzing the amyloid formation of AL-09 fibrils and therefore increasing its amyloidogenicity (in the most complete sense of the term). This also supports the idea that some AL proteins are better fibril nucleators while others seems to be better fibril elongators, based on specific set of mutations in key residues in the topology of the proteins.
The fast aggregation kinetics observed in AL-09 is attributed to the altered dimer interface,34 suggesting that mutations N34I and Y87H might promote formation of stable oligomeric intermediates. Our conclusion that the altered dimer in AL-09 promotes formation of stable oligomers prior to fibrils is further reinforced by the study of aggregation kinetics in the restorative mutant AL-09 H87Y. A single restorative mutation of histidine to tyrosine at position 87 changes the dimeric interface of AL-09 to the canonical form observed in κI O18/O8 germline protein.34 This change essentially shifts the pattern of aggregation from a biphasic transition observed in AL-09 to monophasic slower transition similar to κI O18/O8 (Figure 1B and A).
While it is well documented that reduced thermodynamic stability increases the amyloidogenic propensity for a protein,20, 59 thermodynamic stability alone is not enough to completely explain the amyloidogenic properties in AL proteins. Kinetic stability, kinetic traps within the folding pathway,29, 53 and changes in the structure of AL proteins34, 35 can all play a role in the overall aggregation process. To test how the structural alteration and low thermodynamic stability individually affected the rate and pathway of aggregation, we investigated the aggregation kinetics in AL-12 variable domain protein. The primary reasons for selecting AL-12 over other AL proteins is because (1) both AL-09 and AL-12 belong to κI subtype, (2) Both AL-09 and AL-12 are derived from patients with cardiac amyloidosis, (3) AL-12 has lower thermodynamic stability compared to κI O18/O8 germline protein but higher than AL-09 (κI O18/O8: Tm= 54.7 ± 0.3 °C; ΔGfolding = −6.1 ± 0.2 kcal/mol; AL-12: Tm= 46.3 ± 0.4°C; ΔGfolding = −4.4 ± 0.8 kcal/mol; AL-09: Tm= 41.1 ± 1.0°C; ΔGfolding = −3.5 ± 0.3 kcal/mol)34, 35 and (4) AL-12 retains the canonical dimeric conformation similar to κI O18/O8.
In the present study, AL-12 aggregated as fast as AL-09 (Table 1) in contrast with previous reports.35 This discrepancy might arise in part because of AL-12 sensitivity to its surface environment, favoring fibril formation in Eppendorf tubes compared to polystyrene plates. AL-12 at both concentrations presents an insignificant transient population of spherical and non-fibrillar oligomers during the nucleation phase. AL-12 R65S (Tm= 49.0 ± 0.2 °C; ΔGfolding = −4.3 ± 0.9 kcal/mol) is slightly more stable than AL-12 and aggregated more slowly with respect to AL-12, correlating with the difference in thermodynamic stability (Table 1).
The most amyloidogenic proteins AL-09, AL-12 and the restorative mutant AL-12 R65S did not present concentration dependence in their t50 values. In contrast, a slower rate of aggregation observed at high concentrations in κI O18/O8 and AL-09 H87Y may be driven by a combined effect of their dimer dissociation constant (Kd for κI O18/O8 = 217 μM; Kd for AL-09 H87Y = 200 nM)32 and their higher thermodynamic stability.
Collectively, our results show a strong dependence between the altered dimer conformation and the formation of stable oligomeric intermediates in amyloidogenic light chains. The lower Cr values at low protein concentration observed in our reactions indicate that the aggregation reaction is slightly more efficient and the fibril ultrastructure is more stable. Future studies pertaining to the structure and role of oligomeric intermediates during AL protein aggregation at physiological pH and structure of the fibrils are currently under progress in our laboratory. These studies will further improve our understanding on the catalytic properties of these intermediates and the fibrils in homologous and heterologous amyloid recruitment and amyloid co-aggregation under physiological conditions.
Since some of the biophysical techniques (DLS, SEC and TEM) used in this study have technical limitations in terms of sample preparation, measurement and data analysis, we interpreted the data in parallel with SA and TA to provide a concise picture into the early events leading to aggregation in AL proteins in vitro. While the study shows a crucial role of intermediates in the aggregation of AL proteins, caution should be maintained in interpreting and correlating the data with the in vivo studies or the clinical manifestation of the disease in the patients. Apart from oligomeric intermediates the onset and progression of AL amyloidosis can be influenced by components of extracellular matrix, apolipoproteins, physicochemical properties of the lipid membrane and pre-existing and/or parallel pathological conditions.3–5, 30, 60–62
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH R01 grants GM128253, GM071514 (MR-A), the Mayo Foundation, and the generosity of amyloidosis patients and their families. We thank our former laboratory member Marta Marin-Argany for providing AL-12 R65S protein for the present study. We also thank Michael Bergman and Dr. Matthew Auton for proof reading the article and all the members of Dr. Marina Ramirez-Alvarado laboratory for their critical comments.
Funding Sources
Funding was provided by NIH R01 GM071514 and NIH R01 GM128253 (MR-A).
ABBREVIATIONS
- AL
amyloidogenic immunoglobulin light chain
- SA
Sedimentation assay
- TA
Thioflavin T binding assay
- CD
Circular Dichroism spectroscopy
- DLS
Dynamic light scattering
- SEC
Size exclusion chromatography
- TEM
Transmission electron microscopy
- t50
time taken to complete 50% of fibril formation reaction
- ThT
thioflavin T
- ABC buffer
acetate borate citrate buffer
- HPLC
high performance liquid chromatography
- Tm
melting temperature
Footnotes
The authors declare that they have no conflicts of interest with the contents of this article.
SUPPORTING INFORMATION
Actual concentrations of AL-proteins used in the experimental assay (Table ST1), far UV-CD spectra of monomeric AL–proteins (Figure S1), hydrodynamic and elution properties of monomeric AL–proteins (Figure S2, S3), elution properties of the buffers used in the assays and protein standards by size exclusion chromatography (Figure S4, S5), critical concentration (Cr) and ΔΔG values of different light chain variable domain proteins (Figure S6), comparative analysis of SA vs TA of different AL-proteins at 220μM (Figure S7), composite analysis of monomer concentration, species size and abundance, and morphologies at 220 μM κI O18/O8 during aggregation (Figure S8, S10), elution properties of different species during the course of aggregation in AL–proteins (Figure S9), composite analysis of monomer concentration, species size and abundance, and morphologies of AL-09, AL-09 H87Y, AL-12 and AL-12 R65S at 220 μM during aggregation (Figure S11–S18)
REFERENCES
- [1].Dobson CM (2003) Protein folding and misfolding, Nature 426, 884. [DOI] [PubMed] [Google Scholar]
- [2].Stefani M (2004) Protein misfolding and aggregation: new examples in medicine and biology of the dark side of the protein world, BBA-Molecular Basis of Disease 1739, 5–25. [DOI] [PubMed] [Google Scholar]
- [3].Jo E, McLaurin J, Yip CM, St George-Hyslop P, and Fraser PE (2000) alpha-Synuclein membrane interactions and lipid specificity, J Biol Chem 275, 34328–34334. [DOI] [PubMed] [Google Scholar]
- [4].Kim SI, Yi JS, and Ko YG (2006) Amyloid beta oligomerization is induced by brain lipid rafts, J Cell Biochem 99, 878–889. [DOI] [PubMed] [Google Scholar]
- [5].Knight JD, and Miranker AD (2004) Phospholipid catalysis of diabetic amyloid assembly, J Mol Biol 341, 1175–1187. [DOI] [PubMed] [Google Scholar]
- [6].Yip CM, Darabie AA, and McLaurin J (2002) Abeta42-peptide assembly on lipid bilayers, J Mol Biol 318, 97–107. [DOI] [PubMed] [Google Scholar]
- [7].Wall JS, Ayoub FM, and O’Shea PS (1996) A study of the interactions of an immunoglobulin light chain with artificial and B-lymphocyte membranes, Front Biosci 1, a46–58. [DOI] [PubMed] [Google Scholar]
- [8].Truran S, Weissig V, Ramirez-Alvarado M, Franco DA, Burciu C, Georges J, Murarka S, Okoth WA, Schwab S, Hari P, and Migrino RQ (2014) Nanoliposomes protect against AL amyloid light chain protein-induced endothelial injury, J Liposome Res 24, 69–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Arosio P, Knowles TPJ, and Linse S (2015) On the lag phase in amyloid fibril formation, Phys. Chem. Chem. Phys 17, 7606–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Lomakin A, Teplow DB, Kirschner DA, and Benedek GB (1997) Kinetic theory of fibrillogenesis of amyloid β-protein, Proc. Natl. Acad. Sci. USA 94, 7942–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Naiki H, and Gejyo F (1999) Kinetic analysis of amyloid fibril formation, Methods Enzymol. 309, 305–318. [DOI] [PubMed] [Google Scholar]
- [12].Ferrone F (1999) Analysis of protein aggregation kinetics, Methods Enzymol. 309, 256–274. [DOI] [PubMed] [Google Scholar]
- [13].Harper JD, and Lansbury PT Jr. (1997) Models of amyloid seeding in Alzheimer’s disease and scrapie: mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins, Annu. Rev. Biochem 66, 385–407. [DOI] [PubMed] [Google Scholar]
- [14].Cohen SI, Vendruscolo M, Dobson CM, and Knowles TP (2012) From macroscopic measurements to microscopic mechanisms of protein aggregation, J. Mol. Biol 421, 160–171. [DOI] [PubMed] [Google Scholar]
- [15].Misra P, Kodali R, Chemuru S, Kar K, and Wetzel R (2016) Rapid alpha-oligomer formation mediated by the Abeta C terminus initiates an amyloid assembly pathway, Nat. Commun 7, 12419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Benilova I, Karran E, and De Strooper B (2012) The toxic Abeta oligomer and Alzheimer’s disease: an emperor in need of clothes, Nat. Neurosci 15, 349–357. [DOI] [PubMed] [Google Scholar]
- [17].Chiti F, and Dobson CM (2009) Amyloid formation by globular proteins under native conditions, Nat. Chem. Biol 5, 15–22. [DOI] [PubMed] [Google Scholar]
- [18].Khurana R, Gillespie JR, Talapatra A, Minert LJ, Ionescu-Zanetti C, Millett I, and Fink AL (2001) Partially Folded Intermediates as Critical Precursors of Light Chain Amyloid Fibrils and Amorphous Aggregates, Biochemistry-Us 40, 3525–3535. [DOI] [PubMed] [Google Scholar]
- [19].Lorenzen N, Nielsen SB, Buell AK, Kaspersen JD, Arosio P, Vad BS, Paslawski W, Christiansen G, Valnickova-Hansen Z, Andreasen M, Enghild JJ, Pedersen JS, Dobson CM, Knowles TP, and Otzen DE (2014) The role of stable alpha-synuclein oligomers in the molecular events underlying amyloid formation, J. Am. Chem. Soc 136, 3859–3868. [DOI] [PubMed] [Google Scholar]
- [20].Wall J, Schell M, Murphy C, Hrncic R, Stevens FJ, and Solomon A (1999) Thermodynamic instability of human lambda 6 light chains: correlation with fibrillogenicity, Biochemistry-Us 38, 14101–14108. [DOI] [PubMed] [Google Scholar]
- [21].Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, and Selkoe DJ (2008) Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory, Nat. Med 14, 837–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, and Selkoe DJ (2002) Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo, Nature 416, 535–539. [DOI] [PubMed] [Google Scholar]
- [23].Masters CL, and Selkoe DJ (2012) Biochemistry of amyloid beta-protein and amyloid deposits in Alzheimer disease, Cold Spring Harb. Perspect. Med 2, a006262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Fink AL (2006) The aggregation and fibrillation of alpha-synuclein, Acc. Chem. Res 39, 628–634. [DOI] [PubMed] [Google Scholar]
- [25].Stohr J, Weinmann N, Wille H, Kaimann T, Nagel-Steger L, Birkmann E, Panza G, Prusiner SB, Eigen M, and Riesner D (2008) Mechanisms of prion protein assembly into amyloid, Proc. Natl. Acad. Sci. USA 105, 2409–2414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Solomon A (1985) Light chains of human immunoglobulins, Methods Enzymol. 116, 101–121. [DOI] [PubMed] [Google Scholar]
- [27].Buxbaum J (1992) Mechanisms of disease: monoclonal immunoglobulin deposition. Amyloidosis, light chain deposition disease, and light and heavy chain deposition disease, Hematol. Oncol. Clin. North. Am 6, 323–346. [PubMed] [Google Scholar]
- [28].Sikkink LA, and Ramirez-Alvarado M (2008) Salts enhance both protein stability and amyloid formation of an immunoglobulin light chain, Biophys. Chem 135, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Blancas-Mejia LM, Tischer A, Thompson JR, Tai J, Wang L, Auton M, and Ramirez-Alvarado M (2014) Kinetic control in protein folding for light chain amyloidosis and the differential effects of somatic mutations, J. Mol. Biol 426, 347–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Blancas-Mejia LM, Hammernik J, Marin-Argany M, and Ramirez-Alvarado M (2015) Differential effects on light chain amyloid formation depend on mutations and type of glycosaminoglycans, J. Biol. Chem 290, 4953–4965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Randles EG, Thompson JR, Martin DJ, and Ramirez-Alvarado M (2009) Structural alterations within native amyloidogenic immunoglobulin light chains, J. Mol. Biol 389, 199–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Baden EM, Owen BA, Peterson FC, Volkman BF, Ramirez-Alvarado M, and Thompson JR (2008) Altered dimer interface decreases stability in an amyloidogenic protein, J. Biol. Chem 283, 15853–15860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abraham RS, Geyer SM, Price-Troska TL, Allmer C, Kyle RA, Gertz MA, and Fonseca R (2003) Immunoglobulin light chain variable (V) region genes influence clinical presentation and outcome in light chain-associated amyloidosis (AL), Blood 101, 3801–3808. [DOI] [PubMed] [Google Scholar]
- [34].Baden EM, Randles EG, Aboagye AK, Thompson JR, and Ramirez-Alvarado M (2008) Structural insights into the role of mutations in amyloidogenesis, J. Biol. Chem 283, 30950–30956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Marin-Argany M, Guell-Bosch J, Blancas-Mejia LM, Villegas S, and Ramirez-Alvarado M (2015) Mutations can cause light chains to be too stable or too unstable to form amyloid fibrils, Protein Sci. 24, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].DiCostanzo AC, Thompson JR, Peterson FC, Volkman BF, and Ramirez-Alvarado M (2012) Tyrosine residues mediate fibril formation in a dynamic light chain dimer interface, J. Biol. Chem 287, 27997–28006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].O’Nuallain B, Thakur AK, Williams AD, Bhattacharyya AM, Chen SM, Thiagarajan G, and Wetzel R (2006) Kinetics and thermodynamics of amyloid assembly using a high-performance liquid chromatography-based sedimentation assay, Amyloid, Prions, and Other Protein Aggregates, Part C: Pt. C (Methods in Enzymology) 413, 34–74. [DOI] [PubMed] [Google Scholar]
- [38].Blancas-Mejia LM, Misra P, and Ramirez-Alvarado M (2017) Differences in Protein Concentration Dependence for Nucleation and Elongation in Light Chain Amyloid Formation, Biochemistry-Us 56, 757–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].O’Nuallain B, Shivaprasad S, Kheterpal I, and Wetzel R (2005) Thermodynamics of A beta(1–40) amyloid fibril elongation, Biochemistry-Us 44, 12709–12718. [DOI] [PubMed] [Google Scholar]
- [40].Hong P, Koza S, and Bouvier ES (2012) Size-Exclusion Chromatography for the Analysis of Protein Biotherapeutics and their Aggregates, J. Liq. Chromatogr. Relat. Technol 35, 2923–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Mant CT, Parker JM, and Hodges RS (1987) Size-exclusion high-performance liquid chromatography of peptides. Requirement for peptide standards to monitor column performance and non-ideal behaviour, J. Chromatogr 397, 99–112. [DOI] [PubMed] [Google Scholar]
- [42].Albinsson B, and Norden B (1992) Excited-state properties of the indole chromophore: electronic transition moment directions from linear dichroism measurements: effect of methyl and methoxy substituents, J. Phy. Chem 96, 6204–6212. [Google Scholar]
- [43].Sreerama N, Manning MC, Powers ME, Zhang J-X, Goldenberg DP, and Woody RW (1999) Tyrosine, Phenylalanine, and Disulfide Contributions to the Circular Dichroism of Proteins: Circular Dichroism Spectra of Wild-Type and Mutant Bovine Pancreatic Trypsin Inhibitor, Biochemistry-Us 38, 10814–10822. [DOI] [PubMed] [Google Scholar]
- [44].Blancas-Mejia LM, and Ramirez-Alvarado M (2016) Recruitment of Light Chains by Homologous and Heterologous Fibrils Shows Distinctive Kinetic and Conformational Specificity, Biochemistry-Us 55, 2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Jean L, Lee Chiu F., and Vaux David J. (2012) Enrichment of Amyloidogenesis at an Air-Water Interface, Biophysical Journal 102, 1154–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Murray AN, Palhano FL, Bieschke J, and Kelly JW (2013) Surface adsorption considerations when working with amyloid fibrils in multiwell plates and Eppendorf tubes, Protein Sci. 22, 1531–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Schladitz C, Vieira EP, Hermel H, and Möhwald H (1999) Amyloid–β-Sheet Formation at the Air-Water Interface, Biophysical Journal 77, 3305–3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Williams AD, Portelius E, Kheterpal I, Guo J. t., Cook KD, Xu Y, and Wetzel R (2004) Mapping Aβ Amyloid Fibril Secondary Structure Using Scanning Proline Mutagenesis, Journal of Molecular Biology 335, 833–842. [DOI] [PubMed] [Google Scholar]
- [49].Souillac PO, Uversky VN, Millett IS, Khurana R, Doniach S, and Fink AL (2002) Elucidation of the molecular mechanism during the early events in immunoglobulin light chain amyloid fibrillation. Evidence for an off-pathway oligomer at acidic pH, J. Biol. Chem 277, 12666–12679. [DOI] [PubMed] [Google Scholar]
- [50].Qin Z, Hu D, Zhu M, and Fink AL (2007) Structural characterization of the partially folded intermediates of an immunoglobulin light chain leading to amyloid fibrillation and amorphous aggregation, Biochemistry-Us 46, 3521–3531. [DOI] [PubMed] [Google Scholar]
- [51].Hurshman AR, White JT, Powers ET, and Kelly JW (2004) Transthyretin aggregation under partially denaturing conditions is a downhill polymerization, Biochemistry-Us 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- [52].Lindgren M, Sorgjerd K, and Hammarstrom P (2005) Detection and characterization of aggregates, prefibrillar amyloidogenic oligomers, and protofibrils using fluorescence spectroscopy, Biophys J 88, 4200–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Blancas-Mejia LM, Tellez LA, del Pozo-Yauner L, Becerril B, Sanchez-Ruiz JM, and Fernandez-Velasco DA (2009) Thermodynamic and kinetic characterization of a germ line human lambda6 light-chain protein: the relation between unfolding and fibrillogenesis, J. Mol. Biol 386, 1153–1166. [DOI] [PubMed] [Google Scholar]
- [54].Lin Y, Marin-Argany M, Dick CJ, Redhage KR, Blancas-Mejia LM, Bulur P, Butler GW, Deeds MC, Madden BJ, Williams A, Wall JS, Dietz A, and Ramirez-Alvarado M (2017) Mesenchymal stromal cells protect human cardiomyocytes from amyloid fibril damage, Cytotherapy 19, 1426–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Marin-Argany M, Lin Y, Misra P, Williams A, Wall JS, Howell KG, Elsbernd LR, McClure M, and Ramirez-Alvarado M (2016) Cell Damage in Light Chain Amyloidosis: FIBRIL INTERNALIZATION, TOXICITY AND CELL-MEDIATED SEEDING, J Biol Chem 291, 19813–19825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Sikkink LA, and Ramirez-Alvarado M (2010) Cytotoxicity of amyloidogenic immunoglobulin light chains in cell culture, Cell Death Dis 1, e98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Caughey B, and Peter T. Lansbury, J. (2003) PROTOFIBRILS, PORES, FIBRILS, AND NEURODEGENERATION: Separating the Responsible Protein Aggregates from The Innocent Bystanders, Ann. Rev. Neurosci 26, 267–298. [DOI] [PubMed] [Google Scholar]
- [58].Haass C, and Selkoe DJ (2007) Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer's amyloid β-peptide, Nat. Rev. Mol. Cell Biol 8, 101. [DOI] [PubMed] [Google Scholar]
- [59].Kim Y, Wall JS, Meyer J, Murphy C, Randolph TW, Manning MC, Solomon A, and Carpenter JF (2000) Thermodynamic modulation of light chain amyloid fibril formation, J. Biol. Chem 275, 1570–1574. [DOI] [PubMed] [Google Scholar]
- [60].Guan J, Mishra S, Falk RH, and Liao R (2012) Current perspectives on cardiac amyloidosis, Am J Physiol Heart Circ Physiol 302, H544–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Obici L, Bellotti V, Mangione P, Stoppini M, Arbustini E, Verga L, Zorzoli I, Anesi E, Zanotti G, Campana C, Vigano M, and Merlini G (1999) The new apolipoprotein A-I variant leu(174) --> Ser causes hereditary cardiac amyloidosis, and the amyloid fibrils are constituted by the 93-residue N-terminal polypeptide, Am J Pathol 155, 695–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Ren R, Hong Z, Gong H, Laporte K, Skinner M, Seldin DC, Costello CE, Connors LH, and Trinkaus-Randall V (2010) Role of glycosaminoglycan sulfation in the formation of immunoglobulin light chain amyloid oligomers and fibrils, J Biol Chem 285, 37672–37682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.