

SUMMARY
Inappropriate activation of the p53 transcription factor contributes to numerous developmental syndromes characterized by distinct constellations of phenotypes. How p53 drives exquisitely specific sets of symptoms in diverse syndromes, however, remains enigmatic. Here, we deconvolute the basis of p53-driven developmental syndromes by leveraging an array of mouse strains to modulate the spatial expression pattern, temporal profile, and magnitude of p53 activation during embryogenesis. We demonstrate that inappropriate p53 activation in the neural crest, facial ectoderm, anterior heart field and endothelium induces distinct spectra of phenotypes. Moreover, altering the timing and degree of p53 hyperactivation substantially affects the phenotypic outcomes. Phenotypes are associated with p53-driven cell cycle arrest or apoptosis, depending on the cell type, with gene expression programs, rather than extent of mitochondrial priming, largely governing the specific response. Together, our findings provide a critical framework for decoding the role of p53 as a mediator of diverse developmental syndromes.
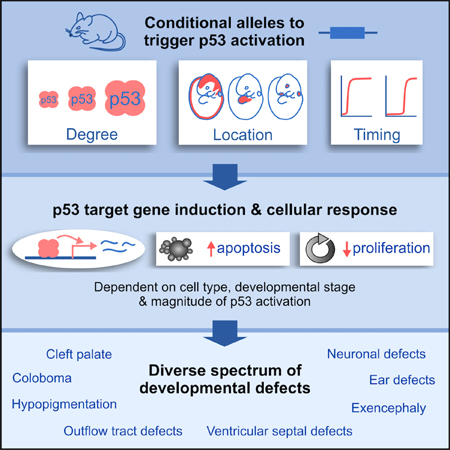
Graphical Abstract
eTOC blurb
Various developmental syndromes are associated with increased p53 activity. Bowen et al. generate a panel of mouse models to define how different spatial expression patterns, temporal profiles, and magnitudes of p53 activation during embryogenesis drive distinct spectra of developmental phenotypes.
INTRODUCTION
Developmental defects, including isolated congenital defects and those found in multi-symptom developmental syndromes, affect approximately 3% of all babies in the United States and are the leading cause of infant mortality (Egbe, 2015; Petrini et al., 2002). While the genetic or environmental causes of most isolated congenital defects are unknown (Khokha et al., 2017), inherited or de novo mutations have been identified as causative in various developmental syndromes. The cellular consequences of these mutations are varied and include alterations in ubiquitous processes such as ribosome biogenesis, DNA repair, RNA processing, centriole duplication, and epigenetic regulation (Bowen and Attardi, 2019). How these diverse causative mutations trigger very specific constellations of phenotypes has remained elusive.
Emerging evidence reveals that the p53 tumor suppressor protein is inappropriately activated in many developmental syndromes, which we refer to here as “p53-associated syndromes” (Bowen and Attardi, 2019; Van Nostrand and Attardi, 2014). p53 is a transcription factor that is normally maintained at low levels in the cell but becomes stabilized and activated in response to various types of stress (Kastenhuber and Lowe, 2017). Many of the mutations that cause developmental syndromes can trigger intracellular stress that activates p53. Accordingly, increased p53 activity is observed in clinical samples from patients with a range of developmental syndromes, and deletion of Trp53 can fully or partially rescue some or all of the developmental defects in mouse models of developmental syndromes (Figure 1A; Bowen and Attardi, 2019). Thus, a unifying theme shared by many syndromes is that their causative mutations induce stress signals that trigger p53 activation, and once active, p53 may contribute to their associated developmental defects.
Figure 1. Allelic series and Cre drivers to induce different degrees of p53 activation in specific embryonic cell types.

(A) Genetic syndromes associated with increased p53 activity. NA: not applicable, ND: not determined.
(B) p53 transactivation domain (TAD) mutants. DBD: DNA-binding domain. OD: oligomerization domain.
(C) p53 allelic series. The p5325,26,53,54 (purple) and p5325,26 (pink) proteins are stabilized due to reduced Mdm2 binding and can tetramerize with and stabilize wild-type p53, leading to increased p53 target gene induction. Target gene induction is greater in p5325,26/+ cells than p5325,26,53,54/+ cells due to the residual transactivation potential of p5325,26. In Mdm2+/− ;Mdm4+/− cells and Mdm2−/− cells, decreased negative regulation of p53 leads to increased p53 activity.
(D) Representative Immunoblot of lysates from MEFs 24hrs after AdCre or AdEmpty infection. pAB242 binds to residues 18–27 of p53 and therefore does not recognize p5325,26,53,54 or p5325,26. Note that wild-type p53 is stabilized in the presence of p5325,26,53,54 or p5325,26 to levels that are similar to what is observed in Trp53+/− MEFs after Doxorubicin (Dox) treatment. n=3.
(E) qRT-PCR analysis of p53 target gene expression (mean±SD) relative to B2m, normalized such that the mean expression in Trp53+/+ embryos is equal to 1. n=3–7 embryos per genotype. *p<0.05 when comparing to Trp53+/+ embryos.
(F) Xgal-stained E10.5 embryos carrying Cre transgenes and the Rosa26LSL-LacZ reporter. NT: neural tube. FNP: frontonasal prominence. PA: pharyngeal arches. H: heart. Te: telencephalon. OpV: optic vesicle. OP: olfactory placode. OtV: otic vesicle.
(G) Tissue-specific expression patterns of Cre transgenes. EMT: epithelial-to-mesenchymal transition. OFT: outflow tract. RV, LV: right/left ventricle. RA, LA: right/left atrium.
The notion that inappropriate p53 activation can trigger developmental defects in humans is underscored by recent findings that activating mutations in TP53 itself or hypomorphic mutations in MDM2, which encodes the main negative regulator of p53, can cause syndromes associated with certain developmental phenotypes (Figure 1A) (Lessel et al., 2017; Toki et al., 2018). Similarly, in mice, a range of developmental defects is observed in strains with activating Trp53 mutations or hypomorphic Mdm2 mutations (Bowen and Attardi, 2019). Together, analyses of both mice and humans indicate that inappropriate p53 activation, due to mutations in TP53 itself or due to mutations in p53 regulators, can drive a wide range of developmental phenotypes.
The syndromes associated with p53 hyperactivity have overlapping yet ultimately distinct sets of developmental defects, and the basis for the phenotypic differences between these syndromes is not well understood. Here, to test the hypothesis that differences in the nature of p53 activation contribute to the phenotypic diversity of these syndromes, we generate a panel of mouse models in which we modulate the magnitude, the timing, and the spatial pattern of p53 activation during embryogenesis. We thus deconvolute the threshold and spatiotemporal requirements of p53 activation needed for the manifestation of different developmental phenotypes. We further interrogate the cellular and molecular basis for how p53 promotes phenotypes in different tissues. Together, our analyses provide a critical framework for decoding the broad role of p53 in diverse developmental syndromes.
RESULTS
Establishing a platform for understanding the phenotypic diversity of p53-associated syndromes
To decipher the mechanisms by which p53 hyperactivation contributes to developmental defects in numerous human syndromes, we sought initially to establish mouse models in which we could modulate both the intensity and the spatial location of p53 activation during embryogenesis. To this end, we established a series of conditional alleles, coupled with a panel of tissue-specific Cre transgenes, to activate p53 to different degrees and in different cell compartments.
To achieve mild activation of p53, we co-expressed a p53 transcriptional mutant – p5325,26,53,54 – and wild-type p53, which had previously revealed a role for p53 in driving phenotypes characteristic of CHARGE syndrome (Van Nostrand et al., 2014). p5325,26,53,54 contains four mutations in its transactivation domains (TADs) that render it transactivation-dead, but also increase its stability due to disruption of the residues required to bind Mdm2, the main ubiquitin ligase that targets p53 for degradation (Figure 1B) (Brady et al., 2011; Mello et al., 2017). We previously showed that p5325,26,53,54 can bind to and modestly stabilize wild-type p53, leading to a mild increase in p53 target gene induction (Van Nostrand et al., 2014). Here, we used our conditional Trp53LSL−25,26,53,54 knock-in allele, which is only expressed after Cre-mediated recombination of a transcriptional stop cassette flanked by LoxP sites (LSL) (Brady et al., 2011), to express p5325,26,53,54 in a tissue-specific manner and modestly induce p53 activity (Figure 1C). We also established another system for mild p53 activation, through heterozygous Mdm2 and Mdm4 deletion, which triggers a modest increase in p53 target gene expression (Terzian et al., 2007; Zhang et al., 2017) and can be achieved using mice carrying a conditional Mdm2flox allele and an Mdm4 null allele (Grier et al., 2002; Parant et al., 2001) (Figure 1C).
To attain higher levels of p53 activity, we co-expressed another p53 mutant with alterations only in the first p53 TAD – p5325,26 – and wild-type p53 (Figure 1B,C) (Johnson et al., 2005). Although p5325,26 and p5325,26,53,54 are both more stable than wild-type p53 due to disruption of the residues required for Mdm2 binding, p5325,26 has some transactivation activity, while p5325,26,53,54 is transactivation-dead (Brady et al., 2011; Jiang et al., 2011). We thus hypothesized that p53 target gene induction would be higher in Trp5325,26/+ embryos than in Trp5325,26,53,54/+ embryos, due to the combined effects of wild-type p53 stabilization and residual p5325,26 transactivation activity. We first confirmed that p5325,26 was able to stabilize wild-type p53, by showing that wild-type p53 levels were elevated in Adenovirus Cre (AdCre)-infected Trp53LSL−25,26/+ mouse embryonic fibroblasts (MEFs) relative to AdCre-infected Trp53+/− MEFs (Figure 1D). Next, we examined p53 transcriptional activity and found that Sox2-Cre;Trp53LSL−25,26/+ embryos ubiquitously co-expressing p5325,26 and wild-type p53 displayed moderate induction of a panel of known p53 target genes, in contrast to the relatively mild induction of p53 target genes observed in Sox2-Cre;Trp53LSL−25,26,53,54/+ embryos and Mdm2+/− ;Mdm4+/− embryos (Figure 1E). These data confirm that co-expression of p5325,26 and wild-type p53 can be used to trigger higher levels of p53 activity than co-expression of p5325,26,53,54 and wild-type p53 or heterozygous Mdm2 and Mdm4 deletion.
To achieve even stronger p53 activation, we relied on conditional deletion of both alleles of Mdm2, which can induce high levels of p53 activity (Moyer et al., 2017). We confirmed that p53 accumulated to higher levels in AdCre-infected Mdm2flox/flox MEFs than in AdCre-infected Trp53LSL−25,26/+ MEFs (Figure 1D). Thus, through these approaches, we established an allelic series to achieve different levels of p53 activity, which we refer to as “mild”, “moderate” and “severe” p53 activation (Figure 1C). While it is possible that each method of activating p53 may have certain nuanced effects on p53’s functionality, our analysis of p53 target gene induction supports a model of progressively increasing p53 activity rather than driving selective gene expression programs. By coupling these conditional alleles with a panel of tissue-specific Cre transgenes (Figure 1F,G), as described below, we were poised to investigate how both the degree and spatiotemporal pattern of p53 activation contribute to the phenotypic diversity of p53-associated syndromes.
Inappropriate activation of p53 in the neural crest triggers a host of developmental defects that depend on the degree and timing of p53 activation
Craniofacial, cardiovascular and pigmentation defects are common features of p53-associated syndromes. For example, cleft palate, outer ear defects and facial bone hypoplasia are major features of Treacher Collins syndrome, heart outflow tract and ventricular septal defects (VSDs) are common in 22q11.2 deletion and CHARGE syndromes, and patches of hypopigmented hair are characteristic of Waardenburg syndrome (Corsten-Janssen et al., 2013; McDonald-McGinn et al., 2015; Vincent et al., 2016; Zaman et al., 2015). Given that neural crest cells (NCCs) give rise to melanocytes as well as components of the craniofacial skeleton and heart (Figure 1G) (Bronner and Simões-Costa, 2016), we sought to determine whether p53 activation in NCCs recapitulates the craniofacial, cardiovascular and/or pigmentation defects observed in p53-associated syndromes, and whether the degree and timing of p53 activation influences the phenotypic spectrum and severity.
To activate p53 in NCCs, we used mice carrying the Wnt1-Cre transgene (Hari et al., 2012) (Figure 1F,G). Mice with mild p53 activation in NCCs were obtained at the expected Mendelian ratios at postnatal day 21 (P21; Figure S1A, Table S1), had hypoplastic NCC-derived facial bones, displayed pigmentation defects manifesting as white belly spots, and had no overt heart defects (Figure 2A, Figure S1B–C). In contrast, moderate p53 activation in NCCs led to embryonic lethality (Table S1) and triggered a host of craniofacial and cardiovascular defects, including cleft face, short snouts and lower jaws, cleft palate, small outer ears, hypoplastic nasal passages, heart VSDs and incompletely septated heart outflow tracts, a phenotype known as persistent truncus arteriosus (PTA; Figure 2B). Even more dramatic developmental defects were observed in embryos with severe p53 activation in NCCs, which lacked most of the frontonasal region, displayed incompletely penetrant exencephaly and exhibited PTA (Figure 2C, Figure S1D). These defects could be rescued by concomitant loss of p53 (Figure S2), confirming that they are caused primarily by p53 activation rather than any potential p53-independent consequences of Mdm2 loss. Collectively, our results indicate that activating p53 to different degrees in NCCs is sufficient to trigger a broad spectrum of developmental defects involving the craniofacial skeleton, palate, outer ears, neural tube, heart outflow tract, ventricular septum and melanocytes. Thus, many of the congenital defects in p53-associated syndromes likely arise due to the sensitivity of NCCs to p53 hyperactivation.
Figure 2. Activation of p53 in the neural crest leads to diverse developmental defects that depend on the degree and timing of p53 activation.

(A) (i-ii) Wnt1-Cre;Trp53LSL−25,26,53,54/+ mice display small belly spots (arrow, 84%, n=13) and have slightly shortened snouts. (iii-v) Alizarin red (bone) staining on P21 skulls and quantification (mean±SD) of bone length (n=7 per genotype). Dotted lines outline the mesodermally-derived parietal bones (p) and the NCC-derived frontal bones (f), nasal bones (n) and mandibles (m). *p<0.05. (vi-viii) P21 Wnt1-Cre;Mdm2flox/+;Mdm4+/− mice display large white belly spots (arrow, 100%, n=9) and have short snouts.
(B) (i-iv) E12.5–13.5 Wnt1-Cre;Trp53LSL−25,26/+ embryos display cleft face (arrows i-iii, 36%, n=11) and have short snouts (iv, 100%, n=11). (v-viii) A single Wnt1-Cre;Trp53LSL−25,26/+ embryo that was viable at E15.5 (v, n=1) had cleft palate (*, vi), small outer ears (vii) and hypoplastic nasal passages (viii). (ix-x) E13.5 Wnt1-Cre;Trp53LSL−25,26/+ embryos display persistent truncus arteriosus (PTA, ix, 100%, n=6) and ventricular septal defects (VSD, x, 100%, n=3).
(C) Wnt1-Cre;Mdm2flox/flox embryos display an absence of frontonasal tissue (i, 100%, n=17), exencephaly (Ex, 47%, n=17), and persistent truncus arteriosus (PTA, ii, 100%, n=4).
(D) (i) Sox10-Cre;Trp53LSL−25,26,53,54/+ mice do not have discernable belly spots (100%, n=5). (ii) Sox10-Cre;Mdm2flox/+;Mdm4+/− mice display small white belly spots (arrow, 67%, n=3).
(E) Sox10-Cre;Trp53LSL−25,26/+ embryos display short snouts (i, 100%, n=9), small outer ears (dotted outline, ii, 100%, n=9), cleft palate (*, iii, 100%, n=4) and normally septated heart outflow tracts (iv, 100%, n=3). (F) Sox10-Cre;Mdm2flox/flox embryos display a severe loss of frontonasal tissue (i-ii, 63%, n=8) and have normally septated heart outflow tracts (iii, 100%, n=5).
Ao: aorta. PA: pulmonary artery. Scale bar: 5mm (A,D), 2mm (Bi,ii,iv,v,vii), 1mm (Ci;Ei,ii;Fi,ii), 400µm (Biii,vi,viii,x;Cii;Eiii,iv;Fiii). Sectioning plane: coronal (Biii,vi,viii;Eiii), transverse (Bx;Cii;Eiv;Fiii). See also Figure S1, Figure S2, Table S1.
To better understand the extent to which the timing of p53 activation affects NCC-based phenotypes, we used mice carrying the Sox10-Cre transgene, which is expressed in NCCs only after they start migrating, in contrast to Wnt1-Cre, which is expressed in pre-migratory NCCs (Hari et al., 2012) (Figure 1F,G). We found that the pigmentation and craniofacial phenotypes triggered by mild p53 activation in migratory NCCs (Figure 2D, Figure S1E) were less severe than those triggered by mild p53 activation in pre-migratory NCCs (Figure 2A). Similarly, moderate p53 activation in migratory NCCs (Figure 2E) was not sufficient to trigger the cleft face and PTA phenotypes induced by moderate p53 activation in pre-migratory NCCs (Figure 2Bi,ix). Furthermore, severe p53 activation in migratory NCCs (Figure 2F) was not sufficient to induce the PTA and exencephaly induced by severe p53 activation in pre-migratory NCCs (Figure 2C). As NCCs begin to express Sox10-Cre only a few hours after the onset of Wnt1-Cre expression (Hari et al., 2012), these data indicate that subtle differences in the timing of p53 activation can substantially alter the spectrum of phenotypes induced by p53. Of note, Wnt1-Cre is also expressed in the developing midbrain (Figure 1F), which likely contributes to the exencephaly in Wnt1-Cre-expressing embryos with severe p53 activation and not Sox10-Cre-expressing embryos. Together, our data suggest that even slight differences in either the timing or degree of p53 activation in NCCs could account for many of the phenotypic differences between p53-associated syndromes.
Inappropriate activation of p53 in the facial ectoderm and telencephalon induces craniofacial defects
Although p53 activation in NCCs induced craniofacial defects involving the facial skeleton, palate and outer ears (Figure 2), it did not induce other craniofacial defects observed in p53-associated syndromes, such as eye defects, inner ear defects, and choanal atresia (a blockage of the back of the nasal airway), raising the possibility that other cell types beyond NCCs contribute to p53-driven craniofacial defects. Given that the ectodermal placodes, which arise as focal thickenings on the facial ectoderm, contribute to the development of facial structures such as the eyes, inner ears and nasal airway (Steventon et al., 2014), we asked whether p53 activation in the facial ectoderm using Foxg1-Cre (Hébert and McConnell, 2000) (Figure 1F,G) could trigger craniofacial defects. Furthermore, since Foxg1-Cre is also expressed in the telencephalon, a region of the developing forebrain (Hébert and McConnell, 2000), we queried whether p53 activation in Foxg1-Cre-expressing cells could recapitulate the microcephaly observed in p53-associated syndromes.
We observed a variety of craniofacial defects in mice with p53 hyperactivation in Foxg1-Cre-expressing-cells. Mice with mild p53 activation were viable postnatally and displayed subtle craniofacial defects, including mild microphthalmia (Table S1, Figure 3A). Embryos with moderate p53 activation, which were not viable postnatally (Table S1), exhibited microcephaly due to hypoplasia of the telencephalon and displayed microphthalmia, retinal coloboma, hypoplastic nasal passages, a complete blockage of choanae and nasopharynx, and hypoplastic inner ears (Figure 3B; Figure S1F). Embryos with severe p53 activation displayed even more dramatic defects, with a near-complete absence of the frontonasal region, eyes, otic vesicle, and telencephalon, and exhibited incompletely penetrant exencephaly (Figure 3C). These data indicate that p53 activation in the facial ectoderm and telencephalon is sufficient to trigger craniofacial defects involving the eyes, inner ears, nasal airway and forebrain, with the severity of these defects highly dependent on the degree of p53 activation. Importantly, these defects overlap with those observed in human p53-associated syndromes. For example, microphthalmia is a common feature of Fanconi Anemia (Tsilou et al., 2010), retinal coloboma, choanal atresia and inner ear defects are hallmarks of CHARGE syndrome (Bergman et al., 2011), and microcephaly is characteristic of Seckel syndrome and autosomal recessive primary microcephaly (Gilmore and Walsh, 2013; Sir et al., 2011). Our data indicate further that p53 activation in the facial ectoderm and telencephalon induces craniofacial phenotypes distinct from those induced by p53 activation in NCCs, suggesting that the specific set of craniofacial defects that manifests in a given p53-associated syndrome depends on the particular tissues in which p53 is activated.
Figure 3. Activation of p53 in the facial ectoderm and telencephalon induces craniofacial defects.

(A) P21 Foxg1-Cre;Trp53LSL−25,26,53,54/+ mice have slightly smaller eyes than controls (100%, n=5).
(B) Foxg1-Cre;Trp53LSL−25,26/+ embryos are viable at E18.5 (i) and display the following phenotypes at E9.5–18.5: small eyes (ii-v, 100%, n=15), retinal coloboma (arrow, ii-v, 93%, n=15), an absence of a lens (L, v, 100%, n=6), hypoplastic nasal passages (vi, 100%, n=6), a blocked nasal airway and nasopharynx (vii-viii, 100%, n=4), hypoplastic inner ears (dotted outline, ix, 100%, n=3), small otic vesicles (Ov, x, 100%, n=6), and small telencephalons (Te, xi-xii, 100%, n=6). NPh: nasopharynx, PS: palatal shelves, T: tongue.
(C) Foxg1-Cre;Mdm2flox/flox embryos have no eyes and lack the entire frontonasal region, including the nasal septum (NS), at E12.5–14.5 (i-iii, 100%, n=4), display exencephaly at E12.5–14.5 (Ex, ii, 25%, n=4), have no discernable otic vesicle (Ov) at E9.5 (iv, 100%, n=6), and have a telencephalon (Te) that is smaller than controls at E9.5 (*, iv, 100%, n=6) and largely absent at E10.5–12.5 (v-vi, 100%, n=4). T: tongue.
Scale bar: 5mm (A,Bi), 1mm (Bxi;Ci,ii,iv,v), 400µm (Biii-x,xii;Ciii,vi). Sectioning plane: coronal (Bv,vii,xii), transverse (Bvi,ix;Ciii,vi), sagittal (Bviii). See also Figure S1, Figure S2, Table S1.
Inappropriate activation of p53 in mesodermal populations triggers cardiovascular defects
While our data indicate that p53 activation in NCCs can trigger heart defects (Figure 2Bix–x), NCCs only contribute to part of the heart, with the remainder deriving from the mesodermal heart fields (Figure 1G) (Brade et al., 2013). To better understand the causes of congenital heart defects, we sought to determine whether p53 activation in mesodermal populations also induces heart defects. To this end, we first used mice carrying Mesp1-Cre, which is expressed broadly in the mesoderm by embryonic day 6.5 (E6.5) (Saga et al., 1999) (Figure 1F,G). Mice with mild p53 activation in the mesoderm were viable with no overt developmental defects, whereas embryos with moderate or severe p53 activation died at E10.5 or were resorbed by E8.5, respectively (Figure 4A; Table S1). Thus, activating p53 above a certain threshold in the mesoderm dramatically perturbs embryogenesis, rather than leading to specific heart defects. To activate p53 in a more restricted fashion, we next used mice carrying Mef2c-Cre, which is expressed in the anterior heart field (AHF) that contributes to the outflow tract, ventricular septum, and right ventricle (Verzi et al., 2005), and Tie2-Cre, which is expressed in endothelial cells, including those that give rise to the endocardial cushions, which contribute to the outflow tract and ventricular septum (Kisanuki et al., 2001) (Figure 1F,G). Embryos with severe p53 activation in either of these populations were hypoplastic and died at ~E10.5 (Figure 4B) (Zhang et al., 2012). In contrast, embryos with moderate p53 activation in the AHF were viable at E18.5 and exhibited both VSDs and PTA (Figure 4C, Table S1), and embryos with moderate p53 activation in endothelial cells survived until E12.5 and displayed hypoplastic ventricular septa but normally septated outflow tracts (Figure 4D, Table S1). Together with previous observations that mild p53 activation in endothelial cells triggers incompletely penetrant VSDs (Zhang et al., 2012), these observations define the mesodermal populations in which p53 acts to drive heart defects, with p53 activation in the AHF inducing both PTA and VSDs, and p53 activation in endothelial cells only triggering VSDs. Collectively, our findings suggest that the etiology of p53-driven developmental defects is multifaceted, with contributions from multiple cell compartments. Moreover, by defining how the spatiotemporal pattern and degree of p53 activation influence the phenotypic outcome, our data provide a fundamental framework for understanding phenotypic diversity in developmental syndromes (Figure 4E).
Figure 4. Activation of p53 in mesodermal populations triggers cardiovascular defects.

(A) Mesp1-Cre;Trp53LSL−25,26/+ embryos are hypoplastic at E9.5 (i, 100%, n=7) and are severely hypoplastic with no heart beat (50%) or a weak heart beat (50%) at E10.5 (ii, n=8).
(B) Mef2c-Cre;Mdm2flox/flox embryos are viable at E9.5 (i, 100%, n=4) and are hypoplastic with no heart beat (25%) or a weak heart beat (75%) at E10.5 (ii, n=4).
(C) Mef2c-Cre;Trp53LSL−25,26/+ embryos are viable at E18.5 (i, 100%, n=7), have hypoplastic right ventricles (RV) at E11.5 (ii, 100%, n=8), and display persistent truncus arteriosus (PTA, iii-iv, 100%, n=3), ventricular septal defects (VSD, v, 100%, n=2) and hypoplastic right ventricles (RV, v, 100%, n=3) at E18.5. Ao: aorta, PA: pulmonary artery, DA: ductus arteriosus, LV: left ventricle.
(D) Tie2-Cre;Trp53LSL−25,26/+ embryos are viable at E9.5–12.5 (i-ii), are dead with extensive hemorrhages at E13.5 (iii, 100%, n=7), and have a hypoplastic heart at E12.5 (iv-v, 100%, n=3). Note that the outflow tract has properly septated into the aorta (Ao) and pulmonary artery (PA) but the interventricular septum (IVS) and endocardial cushion (EC) appear hypoplastic.
(E) Developmental defects that arise following different spatiotemporal patterns and degrees of p53 activation.
Scale bar: 1mm (A;B;Ci,iii;Di-iii), 400µm (Cii,iv,v;Div,v). Sectioning plane: transverse (Civ,v;Div,v). See also Figure S2, Table S1.
Inappropriate p53 activation induces apoptosis or dampens proliferation in a cell-type-dependent and timing-dependent manner
Having decoded the spatiotemporal and threshold requirements for p53 activation to drive different developmental defects, we next sought to gain insight into the molecular pathways underlying these defects. Based on our observation that NCCs play a key role in various p53-driven developmental phenotypes (Figure 4E), we chose to define the transcriptional response to p53 hyperactivation in NCCs. By isolating NCCs from E10.5 embryos and performing RNA-sequencing (RNA-seq), we identified 448 and 209 genes that were up- and downregulated, respectively, in NCCs with moderate p53 activation (Trp5325,26/+ compared to Trp53+/+; Figure 5A). 63% of the upregulated genes had been identified previously as p53-induced genes in other cell types, validating our approach (Table S2) (Kenzelmann Broz et al., 2013; Lee et al., 2010; Li et al., 2012; Tonelli et al., 2015). Notably, the top signatures enriched in the upregulated genes were those related to apoptosis and cell cycle arrest (Figure 5A; Table S3), suggesting that these processes may be important for promoting p53-driven developmental defects.
Figure 5. Moderate p53 activation triggers apoptosis in certain cell populations and restrains proliferation in others.

(A). RNA-seq experiment. n=5 embryos per genotype. FDR: false discovery rate, FC: fold change.
(B-D) Immunofluorescence staining for p53, BrdU and Cleaved Caspase 3 (CC3) on transverse sections of E9.5–10.5 embryos counterstained with DAPI (blue). Arrows indicate CC3-positive cells. Bar graphs represent mean ± SD. AHF: anterior heart field, RV: right ventricle. n=3–4 per genotype. *p<0.05. Scale bar: 100µm.
(E) Choice of cell fate following moderate p53 activation in different cell compartments.
See also Figure S3, Figure S4, Figure S5, Figure S6, Table S2, Table S3.
p53 activation can trigger apoptosis, restrain proliferation, or have no obvious effect on cell survival or proliferation, depending on the context (Gudkov and Komarova, 2003). To determine whether p53-driven NCC-based developmental defects are associated with increased apoptosis and/or decreased proliferation, we performed immunostaining for cleaved-Caspase 3 (CC3) and 5-bromodeoxyuridine (BrdU) after BrdU-pulse labeling, respectively. Moderate p53 activation triggered apoptosis in NCCs without affecting their proliferation (Figure 5B). Intriguingly, this induction of apoptosis was observed in the dorsal neural tube containing pre-migratory NCCs and in migratory NCCs, but not in NCCs that had reached the pharyngeal arches (PA) (Figure 5B). Thus, NCCs are susceptible to p53-mediated apoptosis during a narrow developmental time window, providing mechanistic insight into our prior observation that subtle differences in the timing of p53 activation in NCCs substantially alters the phenotypic outcome (Figure 4E).
We next asked whether increased apoptosis and/or decreased proliferation underlie p53-driven developmental defects of ectodermal and mesodermal origin. Moderate p53 activation triggered apoptosis in the otic and optic vesicles and the AHF, but restrained proliferation without inducing apoptosis in the right ventricle and endocardial cushions (Figure 5C–D; Figure S3). Thus, the susceptibility of mesodermal cells to p53-driven apoptosis also changes during a narrow developmental time window, with the choice of cell fate following p53 activation switching from increased apoptosis to decreased proliferation as cells from the AHF migrate into and populate the right ventricle and endocardial cushions. Overall, our data indicate that p53 activation triggers apoptosis and restrains proliferation in a cell type- and timing-dependent manner during embryogenesis (Figure 5E).
We next asked whether the degree of p53 activation influences the choice of cellular response. We found that the susceptibility of a given cell compartment to p53-driven apoptosis was not dependent on the degree of p53 activation, with severe p53 activation triggering apoptosis in the same tissues as moderate p53 activation (dorsal neural tube, the otic and optic vesicles, and the AHF, but not the right ventricle; Figure S4). However, increasing the degree of p53 activation did alter the amplitude of the apoptotic response, with severe p53 activation triggering more extensive apoptosis in apoptosis-susceptible tissues than moderate p53 activation (Figure S4). Conversely, decreasing the degree of p53 activation dampened the apoptotic response (Figure S5A). Interestingly, no level of p53 activation (mild, moderate or severe) affected cellular proliferation in the dorsal neural tube (Figure S5; Figure 5B), Thus, our data suggest that the degree of p53 activation dictates the amplitude rather than the choice of the cellular response.
Neuronal derivatives of the neural crest are hypersensitive to p53 activation
To further explore the mechanisms underlying p53-driven developmental defects, we next analyzed the RNA-seq data for genes that were downregulated upon p53 activation in NCCs (Figure 5A). Gene Ontology analysis of these genes revealed a strong enrichment for genes expressed in neurons (Figure 6A; Table S3). Because we had performed RNA-seq at E10.5, a time point at which some NCCs are initiating differentiation programs for downstream lineages (e.g. neurons and chondrocytes), we reasoned that reduced neuronal gene expression could reflect a decrease in the number of NCCs contributing to neuronal lineages relative to other NCC lineages. Indeed, NCC lineage tracing experiments showed that moderate p53 activation in either pre-migratory or migratory NCCs dramatically decreased the number of NCCs contributing to the dorsal root ganglia (DRG), the enteric nervous system, and the sympathetic ganglia (Figure 6Bi-iii; Figure S2G, Figure S6I). Whole mount staining for the pan-neuronal marker Neurofilament-M (NFM) also revealed that moderate p53 activation led to a striking decrease in the size of the neuronal DRG population (Figure 6Ci–ii). In contrast, moderate p53 activation did not dramatically disrupt the formation of the PAs (Figure 6Biv) or the initiation of chondrogenesis in the craniofacial region (Figure 6Ciii). Flow cytometry-based quantification revealed that activating p53 in migrating NCCs led to a ~60% reduction in the size of the trunk NCC population at E11.5, yet only a ~20% reduction in the size of the cranial NCC population (Figure 6E–F). Together, these data suggest that different NCC lineages have different sensitivities to p53 hyperactivation, with trunk neuronal lineages being more sensitive than cranial chondrocytic lineages.
Figure 6. Neuronal populations are particularly sensitive to p53 activation.

(A) Left: Top 3 most significantly enriched Gene Ontology (GO) terms amongst the genes down-regulated in Trp5325,26/+ NCCs. Right: Heat map showing relative expression values for genes with GO classifications related to neuronal biology.
(B) (i-iii) NCC lineage tracing performed by Xgal staining of embryos carrying the R26LSL-LacZ reporter. In Wnt1-Cre;Trp53LSL−25,26/+;R26LSL-LacZ embryos, fewer Xgal-positive cells contribute to the DRG at E10.5–12.5 (100%, n=6) and to the stomach and intestine at E12.5 (100%, n=4) than in controls. (iv) NCC lineage tracing performed by visualizing GFP fluorescence in embryos carrying the R26mTmG reporter. In Wnt1-Cre;Trp53LSL−25,26/+;R26mTmG embryos, the contribution of GFP-positive cells to the PA and FNP is modestly decreased (100%, n=3).
(C) (i-ii) In Wnt1-Cre;Trp53LSL−25,26/+ embryos, whole mount neurofilament (NFM) staining is markedly decreased in the DRG at E10.5 (100%, n=4) and E11.5 (100%, n=3) but is not dramatically affected in the ventral roots (VR), which are not derived from NCCs. (iii) In Wnt1-Cre;Trp53LSL−25,26/+ embryos, Alcian blue staining (cartilage) in the NCC-derived cranial regions (arrow) appears relatively normal (n=3).
(D) Immunohistochemical staining (brown) for p53 and CC3 on transverse sections from whole mount Xgal-stained (blue) embryos. In Wnt1-Cre;Trp53LSL−25,26/+;R26LSL-LacZ embryos, CC3-positive cells (arrows) are observed within the DRG but not the first PA. n=3.
(E) NCC lineage tracing performed by visualizing tdTomato (Tom) fluorescence in embryos carrying the R26LSL-tdTomato reporter. In Sox10-Cre;Trp53LSL−25,26/+;R26LSL-tdTomato embryos, the contribution of tdTomato-positive cells to the DRG is markedly decreased (100%, n=3).
(F) Flow cytometry-based quantification (mean±SD) of the percentage of tdTomato-positive cells in the head and trunk of Sox10-Cre;Trp53LSL−25,26/+;R26LSL-tdTomato embryos relative to Sox10-Cre;Trp53+/+;R26LSL-tdTomato littermate controls. n=3 per genotype. *p<0.05.
(G) Flow cytometry-based quantification (mean±SD) of the percentage of head or trunk tdTomato-positive cells that are AnnexinV-positive. n=3 per genotype. *p<0.05.
DRG: dorsal root ganglia. PA: pharyngeal arch. FNP: frontonasal prominence. Scale bar: 1mm (Bi-iii;C;E), 400µm (Aiv). 100µm (D). See also Figure S1, Figure S6, Table S2, Table S3.
We next asked whether defects in the formation of neuronal derivatives were also caused by p53-driven apoptosis in these structures. Indeed, moderate p53 activation triggered apoptosis in the developing DRG (Figure 6D). In contrast, the majority of NCCs that reached the PAs and frontonasal process did not undergo apoptosis in response to moderate p53 activation, with increased apoptosis only being observed in the lateral mandibular process of the first PA but not in other craniofacial compartments (Figure 6D, Figure S6). Furthermore, quantification of apoptosis by flow cytometry indicated that moderate p53 activation triggered higher levels of apoptosis in trunk NCCs than in cranial NCCs (Figure 6G). Thus, while the majority of NCCs that migrate to the PAs become resistant to p53-driven apoptosis, NCCs that migrate to the DRG retain their sensitivity to p53-driven apoptosis. Collectively, our data indicate that although inappropriate p53 activation in NCCs leads to developmental defects affecting multiple tissues, including the craniofacial skeleton and heart (Figure 4E), particularly severe developmental defects occur in neuronal NCC derivatives, which is due, at least in part, to their continued sensitivity to p53-mediated apoptosis. Importantly, our finding that neuronal development is hypersensitive to p53 activation provides a basis for the neurodevelopmental phenotypes, such as microcephaly, cerebellar defects, intellectual disability and cranial nerve defects, observed in p53-associated syndromes (Blake et al., 2008; Chrzanowska et al., 2012; Sir et al., 2011; Sohn et al., 2016).
Both apoptosis-resistant and apoptosis-susceptible embryonic cells have highly primed mitochondria
To better understand the molecular underpinnings of the observed developmental phenotypes, we next sought to determine why p53 activation induces apoptosis only in certain embryonic cell types. We leveraged a human NCC (hNCC) system in which we could study homogeneous populations undergoing synchronous responses to p53 activation (Bajpai et al., 2010). Specifically, hNCCs undergo efficient apoptosis when exposed to p53-activating drugs such as Nutlin3a or Doxorubicin, but become strikingly resistant to p53-driven apoptosis when differentiated toward the smooth muscle cell lineage (“early-hSMCs”; Figure 7A–B; Calo et al., 2018). We supported our analysis of the factors underlying the nature of the p53 response by microdissecting the embryonic mouse heart and neuroepithelium, which comprise cells that are resistant and sensitive to p53-driven apoptosis, respectively (Figure 5B,D, Figure S6).
Figure 7. Early-hSMCs are resistant to p53-driven apoptosis, are highly primed, and exhibit a dampening of the transcriptional response to p53 activation.

(A) Outline of experiments.
(B) Flow cytometry-based quantification (mean±SD) of viable cells (AnnexinV- and propidium iodide (PI)-negative) in hNCCs and early-hSMCs treated with DMSO, Nutlin3a or Doxorubicin (Dox) for 16 hours. n=3 per treatment. *p<0.05.
(C) Change in mitochondrial potential (JC-1 fluorescence) over time in hNCCs and early-hSMCs treated with DMSO, FCCP, or Bim and Bid peptides.
(D) Percent mitochondrial depolarization (mean±SD), calculated from the JC-1 kinetic traces as the area under the curve relative to DMSO (100%) and FCCP (0%). n=3.
(E) Heat map (upper) and histograms (lower) of all genes that were Nutlin3a-responsive in either hNCCs or early-hSMCs. The heatmaps and histograms depict the fold change (log2) when comparing Nutlin3a-treated samples (n=3) to DMSO-treated samples (n=3).
(F) Counts per million (CPM) for p53 target genes encoding components of the extrinsic or intrinsic apoptotic pathways (mean±SD).
(G) Upper: representative immunoblot of lysates from hNCCs and early-hSMCs 8hrs after treatment with DMSO or Nutlin3a. Lower: quantification of p53 protein levels relative to GAPDH levels in 3 independent immunoblots (mean±SD). In each experiment, data were normalized such that the p53 levels in the Nutlin3a-treated hNCC sample was equal to 1. *p<0.05.
(H) Counts per million (CPM) for genes encoding p53 repressors and apoptosis inhibitors (mean±SD). *p<0.05.
(I) Model depicting similarities and differences between early-hSMCs and hNCCs.
Recent studies have suggested that the propensity of a given cell type to undergo apoptosis in response to stimuli is determined by its degree of mitochondrial priming, a property dictated by the balance of proapoptotic and antiapoptotic proteins at the mitochondria (Potter and Letai, 2016). Mitochondrial priming is measured using BH3 profiling assays, in which cells are exposed to proapoptotic peptides and the permeabilization of the mitochondrial outer membrane is measured using the JC-1 dye (Ryan and Letai, 2013). Surprisingly, we found that the degree of mitochondrial priming in early-hSMCs was as high as that in hNCCs, and the degree of priming in the E9.5 heart was only slightly lower than that of the E9.5 neuroepithelium (Figure 7C–D, Figure S7A). Indeed, each of these cell populations exhibited a degree of priming similar to that of the adult mouse spleen and far greater than that of the adult mouse brain (Figure S7B), tissues known to be highly primed and unprimed, respectively (Sarosiek et al., 2017). These findings suggest that factors other than mitochondrial priming likely play the predominant role in determining the resistance of the E9.5 mouse heart and early-hSMCs to p53-driven apoptosis.
Apoptosis-resistant populations display lower induction of p53 pro-apoptotic target genes
The similar degree of mitochondrial priming in cells prone to undergoing apoptosis and those resistant to apoptosis suggested that the differences in cell responses related more specifically to the p53 gene expression programs activated in the two contexts. To test this hypothesis, we performed RNA-seq to define the transcriptional response to p53 activation in hNCCs and early-hSMCs (Figure 7A). We first identified p53-responsive genes by selecting genes that were induced or repressed in response to Nutlin3a-treatment in either cell type (Figure 7E; Table S4). We found that 309 (97%) of the 320 Nutlin3a-induced genes were significantly upregulated or trended toward upregulation (log2FC>0) upon Nutlin3a treatment in both cell types. Likewise, 124 (93%) of the 133 Nutlin3a-repressed genes were significantly downregulated or trended toward downregulation upon Nutlin3a treatment in both cell types. These results indicate that p53 regulates the same set of genes in hNCCs and early-hSMCs and that the differential apoptotic sensitivities of these two cell populations is not due to differences in the repertoire of p53 target genes. Interestingly, the magnitude of the induction or repression of these p53-responsive genes did differ substantially between cell types. Specifically, the Nutlin3a-induced genes had a median fold induction of 2.5 fold in hNCCs but only 1.6 fold in early-hSMCs (Figure 7E). Similarly, the Nutlin3a-repressed genes had a median fold repression of 1.7 fold in hNCCs but only 1.3 fold in early-hSMCs (Figure 7E). These results suggest an overall dampening of the transcriptional response to p53 activation in early-hSMCs relative to hNCCs. We also observed less p53 protein accumulation following Nutlin3a treatment in early-hSMCs than in hNCCs (Figure 7G), suggesting that the diminished p53 transcriptional response is likely at least in part due to reduced p53 levels.
To understand the responses of these cell types in more detail, we assessed the expression of specific genes. We found first that p53 pro-apoptotic target genes, including PIDD1, PMAIP1/NOXA and TNFRSF10A, were notably less efficiently induced in early-hSMCs than in hNCCs (Figure 7F). Similarly, Pidd1 was less efficiently induced and Noxa and Bax trended toward less efficient induction upon p53 activation in the mouse embryonic heart than in the neuroepithelium (Figure S7C). Together, these data suggest that in both mouse and human embryonic cells, certain pro-apoptotic target genes are less efficiently induced in tissues that are resistant to p53-driven apoptosis. We next explored whether genes that change in expression as hNCCs differentiate towards hSMCs (Table S5) could influence the p53 response. We identified two p53 repressors, HDAC5 and iASPP, whose gene expression increased as hNCCs differentiate toward hSMCs (Figure 7H). Both HDAC5, which deacetylates p53, and iASPP have been shown to inhibit p53’s transactivation of pro-apoptotic target genes (Bergamaschi et al., 2006; Sen et al., 2013) and therefore could contribute to the dampened p53 transcriptional response in early-hSMCs. We also examined the expression of genes encoding members of the inhibitor of apoptosis (IAP) family, which can suppress apoptosis by inhibiting caspases (Kocab and Duckett, 2016). Two of these genes, BIRC2 and BIRC7, displayed enhanced expression as hNCCs differentiate towards hSMCs, suggesting that their increased expression could contribute to the resistance of early-hSMCs to p53-driven apoptosis (Figure 7H). Thus, reduced p53 protein accumulation, diminished p53 pro-apoptotic target gene induction, and enhanced expression of p53 repressors and anti-apoptotic proteins could coordinately contribute to the resistance of certain embryonic populations to p53-mediated apoptosis (Figure 7I).
DISCUSSION
Congenital defects affecting craniofacial, cardiovascular and neuronal tissues occur in a wide range of developmental syndromes. As a common underlying mechanism – namely hyperactivation of p53 – may contribute to the developmental phenotypes in a number of these syndromes, the basis for their phenotypic differences has remained elusive. Here, we provide a critical framework for understanding how p53 hyperactivation drives diverse sets of developmental phenotypes. Our findings indicate that p53-driven developmental defects arise due to the sensitivity of a few key embryonic cell compartments to p53 activation, and that variations in the strength of p53 activation, as well as subtle differences in the timing of p53 activation, can substantially alter the spectrum and severity of symptoms. Thus, our results suggest that differences in the spatiotemporal pattern and degree of p53 activation alone could account for many of the phenotypic differences between p53-associated syndromes.
The factors that dictate the spatiotemporal pattern of p53 activation in p53-associated syndromes are not well understood. One likely contributing factor is that the genes mutated in these syndromes are themselves expressed in a tissue-specific fashion, which in turn would trigger p53 activation in those tissues. For example, Chd7, Tbx1, and Pax3, which are associated with CHARGE, 22q11.2 deletion, and Waardenburg syndromes respectively, encode transcription factors or chromatin remodelers with highly tissue-specific expression patterns during embryogenesis (Bosman et al., 2005; Epstein et al., 2000; Vitelli et al., 2002). Other p53-associated syndromes are caused by mutations in genes involved in ribosome biogenesis, DNA repair, RNA processing and centriole duplication (Figure 1A), and although these genes are involved in global cellular processes, they are not uniformly expressed. For example, expression of the Treacher Collins syndrome-associated gene Tcof1 is enriched in NCCs (Dixon et al., 2006), expression of ribosomal genes mutated in Diamond Blackfan anemia is enhanced in craniofacial and hematopoietic tissues (Robson et al., 2016), and expression of the Richieri-Costa-Pereira syndrome-associated gene Eif4a3 and the microcephaly-associated gene Cenpj is strongest in the developing brain (Bond et al., 2005; Mao et al., 2016). Another factor that could dictate the spatiotemporal pattern of p53 activation is that different cell types differ in their sensitivities to specific cellular perturbations. For example, NCCs exhibit high levels of endogenous oxidative stress, potentially explaining an increased propensity to activate p53 in response to mutations that disrupt pathways needed to repair oxidative stress-induced DNA damage (Sakai et al., 2016). Thus, both differences in the expression patterns of genes mutated in p53-associated syndromes and differences in the sensitivities of diverse cell types to specific cellular defects could contribute to differences in the spatiotemporal patterns of p53 hyperactivation in developmental syndromes.
Our findings suggest that phenotypic differences between syndromes could also arise due to differences in the degree of p53 activation. Different types of p53-activating signals associated with diverse syndromes likely activate p53 to different extents, according to the different intracellular pathways and p53 post-translational modifications triggered in a given context (Meek, 2015). Moreover, even patients with the same syndrome could exhibit different degrees of p53 activation, due to genetic polymorphisms that modify p53 activity or in utero exposure to p53-activating stressors such as ionizing radiation (Quintens et al., 2015; Whibley et al., 2009). It will be of interest to determine whether the high degree of variability in the penetrance and severity of developmental phenotypes in p53-associated syndromes arises due to genetic or environmental factors that modify p53 activity levels.
To understand the basis of the phenotypes observed in our models, we delineate the role for p53 in triggering apoptosis and restraining proliferation in a cell type- and timing-dependent manner during embryogenesis. Our results reveal striking differences in the p53 response even in closely-related lineages. Our finding that neuronal lineages are particularly susceptible to undergoing p53-driven apoptosis provides a rational for the range of neurodevelopmental defects observed in p53-associated syndromes, as well as the neurodegenerative defects observed in diseases such as Alzheimer’s and Parkinson’s disease and the microcephaly phenotype induced by Zika virus infection, which have also been associated with increased p53 activity (Slomnicki et al., 2017; Szybińska and Leśniak, 2017).
Why p53 activation only triggers apoptosis in certain cell types is an intriguing question. Given that p53 can induce pro-apoptotic target genes, such as Bax and Puma, in both apoptosis-sensitive and apoptosis resistant cells (Attardi et al., 2000; Fischer, 2017), it has been proposed that the extent of mitochondrial priming determines the apoptotic cell fate (Liu et al., 2013). Surprisingly, we show here that the degree of mitochondrial priming does not differ between hNCCs and early-hSMCs, and only differs slightly between the embryonic mouse heart and neuroepithelium. This important observation indicates that differences in mitochondrial priming are not sufficient to explain different sensitivities to p53-driven apoptosis in all contexts. Instead, we find that differences in gene expression programs, including in the extent of induction of p53 pro-apoptotic target genes, may underlie the different apoptotic susceptibilities of these two cell populations.
Therapeutic strategies to dampen p53-activating stress signals or inhibit components of the p53 pathway may have clinical potential for mitigating the manifestation of developmental phenotypes. For example, maternal antioxidant supplementation can limit the accumulation of DNA damage in syndromes associated with impaired DNA repair, thereby attenuating p53 activation and mitigating developmental phenotypes (Sakai et al., 2016). Direct pharmacological inhibition of p53 can also prevent developmental defects (Jones et al., 2008; Morgan et al., 2008), but would have to be performed with care given the increased risk for tumorigenesis. Instead, delineating the specific pathways through which p53 drives developmental defects may allow the possibility of targeting downstream components, such as specific apoptotic factors, without completely perturbing p53. To provide further insight into potential therapeutic targets, it will be important to determine the relative contributions of other p53-regulated cellular processes – such as cellular migration, epithelial-to-mesenchymal transition, and differentiation (Kaiser and Attardi, 2018) – to p53-driven developmental defects. Furthermore, for some developmental syndromes, it may be necessary to also target p53-independent pathways. Indeed, in mouse models of some p53-associated syndromes, the developmental phenotypes are only partially rescued by loss of p53, suggesting that both p53-dependent and p53-independent pathways contribute to their pathogenesis (Mao et al., 2016; Van Nostrand et al., 2014). Parsing the relative contributions of p53-dependent and -independent pathways to each of the phenotypes in p53-associated syndromes will be an important step toward developing novel strategies for their prevention.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents and resources should be directed to and will be fulfilled by the Lead Contact, Dr. Laura D. Attardi (attardi@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse husbandry
All mouse work was approved and performed in compliance with the Stanford University Administrative Panel on Laboratory Animal Care (APLAC). Mice were maintained on a mixed 129/Sv-C57BL/6 background and the following previously described alleles were used: Trp53LSL−25,26 (Johnson et al., 2005), Trp53LSL−25,26,53,54 (Brady et al., 2011), Trp53-null (Jacks et al., 1994), Wnt1-Cre (Danielian et al., 1998), Foxg1-Cre (Hébert and McConnell, 2000), Tie2-Cre (Kisanuki et al., 2001), Mef2c-AHF-Cre (referred to here as Mef2c-Cre) (Verzi et al., 2005), Mesp1-Cre (Saga et al., 1999), Sox2-Cre (Hayashi et al., 2002), Sox10-Cre (Matsuoka et al., 2005), Rosa26LSL-LacZ (Soriano, 1999), Rosa26mTmG (Muzumdar et al., 2007), Rosa26LSL-tdTomato (Madisen et al., 2010), Mdm2flox (Grier et al., 2002), Mdm2-null (Montes de Oca Luna et al., 1995), and Mdm4-null (Parant et al., 2001).
To generate embryos, a male mouse was housed with 1–2 females overnight, and the day a vaginal plug was observed was considered E0.5. Embryos were harvested from pregnant females that had been euthanized via CO2 exposure, according to APLAC-approved methods. PCR genotyping was performed using tail or yolk sac DNA extracted using the HotSHOT method (Truett et al., 2000). Genotyping primers described in each aforementioned publication were used, with the exception that all Cre transgenes were genotyped using generic Cre primers (Marques et al., 2005). For 5’-Bromo-2’-deoxyuridine (BrdU) incorporation experiments, pregnant females were injected with BrdU (Millipore) prepared in phosphate buffered saline (PBS) at a dose of 0.1 mg per gram body weight 10 minutes before the embryos were harvested.
Data from male and female mice were pooled for the analysis of postnatal phenotypes. For characterization of embryonic phenotypes, gender was not determined, with two exceptions: 1) gender was determined for embryos used for RNA-seq to ensure that both genders were equally represented; 2) gender was determined for Wnt1-Cre;Mdm2flox/flox embryos, to determine whether the exencephaly phenotype was female-specific, as had previously been reported for the exencephaly phenotype in Trp53-null embryos (Sah et al., 1995). We found that 2 out of 6 male Wnt1-Cre;Mdm2flox/flox embryos had exencephaly (33%), and 6 out of 11 female Wnt1-Cre;Mdm2flox/flox embryos had exencephaly (55%), which was not significantly different based on a Chi-squared test. Gender was determined by PCR amplification of the male-specific Zfy gene (Sah et al., 1995).
Mouse cell culture
MEFs were generated from E13.5 embryos and were cultured in Dulbecco’s Modified Eagle Medium (DMEM; ThermoFisher) with 10% fetal calf serum at 5% oxygen and 5% carbon dioxide. Infection with Adenoviral Cre (AdCre) or an empty Adenoviral vector (AdEmpty) (Viral Vector Core, University of Iowa) was performed at a multiplicity of infection of 100 and cells were harvested after 24 hours. Some cells were treated with 0.2 μg/ml Doxorubicin hydrochloride (Millipore Sigma) 8 hours prior to harvesting. The gender of the MEF lines was not determined as it was not expected to impact the results.
Human cell culture
H9 human embryonic stem cells (WiCell) were differentiated into cranial neural crest cells (CNCCs) as previously described (Prescott et al., 2015). Briefly, H9 colonies were detached using collagenase (2mg/ml, diluted in KnockOut DMEM) and clusters of 100–200 cells were plated in CNCC differentiation medium (1:1 Neurobasal medium and D-MEM F-12 medium [Invitrogen]) supplemented with 0.5x B-27 supplement with Vitamin A (50x stock, Gemini Bio-Products), 0.5x N-2 supplement (100x stock, Gemini Bio-Products), 0.5x Glutamax-I supplement (100x stock, ThermoFisher), 20 ng/ml bFGF (Peprotech), 20 ng/ml EGF (Sigma-Aldrich) and 5 µg/ml bovine insulin (Sigma-Aldrich)) to form neuroepithelial spheres. After seven days of differentiation, neuroepithelial spheres attached to the dish and migratory CNCCs began to form at the periphery of the spheres. After a further 3–4 days of differentiation, cells were passaged using Accutase onto fibronectin-coated plates in CNCC early maintenance media (1:1 Neurobasal medium and D-MEM F-12 medium (Invitrogen) supplemented with 0.5x B-27 supplement with Vitamin A (50x stock, Gemini Bio-Products ##400-160), 0.5x N-2 supplement (100x stock, Gemini Bio-Products #400-163), 0.5x Glutamax-I supplement (100x stock, Invitrogen), 20 ng/ml bFGF (Peprotech), 20 ng/ml EGF (Sigma-Aldrich) and 1 mg/ml bovine serum albumin (serum replacement grade, Gemini Bio-Products #700-104P)). After 1–2 passages, CNCCs were transitioned into long term maintenance media (CNCC early maintenance media supplemented with 3µM ChIRON 99021 (Selleck, CHIR-99021) and 50pg/ml BMP2 (Peprotech)). After 4 passages, CNCCs were either maintained in long term maintenance media for an additional 2 days, or were differentiated towards the smooth muscle cell lineage for 2 days in DMEM (Thermo Fisher Scientific) supplemented with 10% fetal bovine serum (Omega), as previously described (Calo et al., 2018). Cells were then harvested for BH3 profiling assays or were treated with 0.2 µg/ml Doxorubicin hydrochloride (Millipore Sigma), 10 µM Nutlin-3a (Millipore Sigma), or 0.001% DMSO (vehicle control), and harvested after 8 hours for RNA-seq or Western blots, or after 14–16 hours for AnnexinV/PI staining. The gender of the H9 line is female.
METHOD DETAILS
Tissue processing and histology
Embryos were fixed in 4% paraformaldehyde (PFA) at 4°C for 1–3 hours (E10.5 and below) or overnight (E11.5 and above). For histological analysis, embryos were dehydrated in graded ethanol, cleared in xylene, paraffin-embedded, and sectioned at 5 microns. Hematoxylin and Eosin (H&E) staining was performed by the Stanford Comparative Medicine Histology Facility.
Immunofluorescence and immunohistochemistry
For immunofluorescence staining on 5 micron paraffin sections, slides were deparaffinized in xylene, re-hydrated in graded ethanol and subjected to heat-mediated antigen retrieval in a pressure cooker for 5 min in Tris-EDTA buffer (10 mM Tris, 1 mM EDTA, 0.05% Tween 20, pH 9.0). Slides were permeabilized with tris-buffered saline containing 0.025% Triton X (TBS-TX), blocked in 10% goat serum and 1% bovine serum albumin (BSA) in TBS-TX for 1 hour, and incubated overnight at 4°C in the primary antibody diluted in TBS-TX with 1% BSA. Slides were rinsed in TBS-TX and incubated for 1 hour at room temperature in the secondary antibody diluted in TBS-TX with 1% BSA. Slides were incubated in 1μg/ml DAPI for 5 minutes and were mounted in mounting medium prepared from glycerol, Mowiol 4–88 (Sigma-Aldrich) and DABCO (Sigma-Aldrich). For immunohistochemical staining on paraffin sections, slides were stained using the VECTASTAIN Elite ABC HPR kit (Vector laboratories) according to the manufacturer’s instructions, with hematoxylin counterstaining. The following primary antibodies and dilutions were used: rabbit anti-p53 CM5 (Leica) 1:1000, mouse anti-BrdU (BD pharmigen) 1:800, rabbit anti-CC3 (Cell Signaling) 1:800, and rabbit anti-RFP (Rockland) 1:1000. The following secondary antibodies and dilutions were used: Fluorescein goat anti-rabbit (Vector Laboratories, 1:200) and Alexa Fluor 546 goat anti-mouse (Thermo Fischer Scientific, 1:200).
Whole mount staining
Whole mount immunohistochemistry on E10.5-E11.5 embryos was performed as described (Joyner and Wall, 2008) with mouse anti-neurofilament 2H3-s (Developmental Studies Hybridoma Bank) used at 1:20, and the DAB Peroxidase (HRP) Substrate Kit with Nickel (Vector Laboratories). Whole mount Xgal staining and Alcian blue staining were performed as described (Gierut et al., 2014; Nagy et al., 2009).
FACS isolation and RNA-seq library preparation
E10.5 embryos were harvested in cold PBS and were digested in 0.1% Trypsin for 15 min at 37 °C with intermittent agitation. Cells wer e resuspended in sorting buffer (PBS, 1% fetal calf serum, 1 mM EDTA), filtered through a 35 µm nylon mesh (Corning), and stained with the viability dye 7AAD (Biolegend). FACS-based isolation of GFP-positive cells was performed on instruments at the Stanford Shared FACS Facility. After sorting, cells were pelleted and lysed in TRIzol (ThermoFisher) and RNA was extracted using the RNeasy mini kit (Qiagen). RNA quality was assessed using a Bioanalyzer, and only samples with an RNA integrity number (RIN) greater than 8.0 were used. RNA-seq libraries were prepared using the TruSeq RNA Sample Preparation Kit (Illumina). Each library was prepared from 300ng to 1µg of RNA that had been obtained from FACS-isolated cells from a single E10.5 embryo, and 5 libraries were generated per genotype. The quality and concentration of libraries was assessed using a High Sensitivity Bioanalyzer. Libraries were pooled in equimolar quantities and were sequenced on one lane of an Illumina HiSeq 4000 at the Stanford Functional Genomics Facility.
qRT-PCR
RNA was extracted from whole embryos or microdissected tissues using TRIzol (ThermoFisher), cDNA synthesis was performed using M-MLV reverse transcriptase (ThermoFisher), and qRT-PCR was performed using Power SYBR Green Master Mix (ThermoFisher). Changes in transcript abundance were calculated according to the 2-∆∆Ct method using B2m or Ubl5 as reference transcripts. The following forward and reverse primers were used: B2m (ATT CAC CCC CAC TGA GAC TG, TGC TAT TTC TTT CTG CGT GC), Ubl5 (TCG AGC TAC GAG TTG TGT CG, GCA CTT AAC GCG GAC TTT CT), Cdkna1/p21 (CAC AGC TCA GTG GAC TGG AA, ACC CTA GAC CCA CAA TGC AG), Pmaip1/Noxa (GCA GAG CTA CCA CCT GAG TTC, CTT TTG CGA CTT CCC AGG CA), Phlda3 (TTC GCC CGC ATC AAA GCC GT, AGG GGG CAG CGG AAG TCG AT), Perp (GAC CCC AGA TGC TTG TTT TC, CAG CAG GGT TAT CGT GAA GC), Mdm2 (CTG TGT CTA CCG AGG GTG CT, CGC TCC AAC GGA CTT TAA CA), Crip2 (CCC AAA GGC GTG AAC ATC GGG G, GGG ACC TC GAT GGG GCC AGT), Pidd1 (TCG CTG TCG TGA GGT AGT TG, GAG AAG TGC TCC CTC TGG TG), Apaf1 (GTT CAA AGC CGA GAC AGG AG, ATT GAC TTG CTC CGA GTG CT), Bbc3/Puma (GCG GCG GAG ACA AGA AGA, AGT CCC ATG AAG AGA TTG TAC ATG AC), and Bax (TGA AGA CAG GGG CCT TTT TG, AAT TCG CCG GAG ACA CTC G).
Western blots
Cells were lysed with NP-40 Lysis Buffer (1%NP-40; 200mM NaCl; 50mM Tris Base pH8.0; 10% Glycerol; in dH2O) or RIPA buffer (50 mM Tris pH 8.0, 150 mM NaCl, 1% NP-40, 0.5% Sodium Deoxycholate, 0.1% SDS) with freshly added Complete Protease Inhibitor Cocktail (Roche). Protein concentration was determined using the Pierce bicinchoninic acid (BCA) assay kit (ThermoFisher). Protein extracts were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and were transferred to Immobilon-P Polyvinylidene Difluoride (PVDF) membranes (Millipore). Membranes were blocked in 5% nonfat dry milk prepared in Tris buffered saline with 0.1% Tween-20 (TBST). Washes were performed in TBST and the following primary and secondary antibodies diluted in 5% milk in TBST were used: mouse anti-Mdm2 (Abcam ab16895, 1:500), rabbit anti-p53 (CM5, Leica, 1:1000), mouse anti-wild-type p53 (pAB242, provided by D. Lane and B. Vojtesek (Yewdell et al., 1986), 1:150), mouse anti-p53 (D01, Santa Cruz sc-126, 1:1000), mouse anti-GAPDH (Fitzgerald 10R-G109a, 1:10000), Peroxidase AffiniPure goat anti-mouse (Jackson ImmunoResearch 115-035-146, 1:5000), and Peroxidase AffiniPure goat anti-rabbit (Jackson ImmunoResearch 111-035-144, 1:5000). Immunodetection was performed using ECL Prime (Pierce).
Fluorescence-based BH3 profiling
BH3 profiling assays were performed as described (Ryan and Letai, 2013; Sarosiek et al., 2017). Custom peptides were synthesized by Elim Biopharm (Hayward, CA) as TFA salts at >95% purity with the following sequences: hBIM (Acetyl-MRPEIWIAQELRRIGDEFNA-Amide) and hBID-Y (Acetyl-EDIIRNIARHLAQVGDSMDRY-Amide). Peptides were dissolved in DMSO, aliquoted and stored at −80°C. 2x staining solution was prepared by supplementing MEB buffer (150 mM Mannitol, 10 mM HEPES-KOH pH 7.5, 50 mM KCl, 5 mM Succinate, 20 µM EGTA, 20 µM EDTA, 0.1% BSA) with 20 µg/mL Oligomycin, 50 µg/mL Digitonin, 2 µM JC-1 and 10 mM 2-mercaptoethanol. Peptide treatments were prepared at 2x concentration in 2x staining solution and arrayed at 15 µl per well in a 384 well black flat bottom plates (Mansion Labs). Controls for no depolarization (1% DMSO) and complete depolarization with the mitochondrial oxidative phosphorylation uncoupler FCCP (10 µM) were also included.
Single cell suspensions were prepared from mouse tissues and human cells. The heart and neuroepithelium were isolated from wild-type mouse embryos under a dissecting microscope in cold PBS. For microdissection of the neuroepithelium, the neuroepithelium of the midbrain was isolated and surrounding ectodermal and mesenchymal tissue was discarded. For microdissection of the heart, the outflow tract and the left and right ventricles were isolated, and the atria and neighboring anterior heart field were discarded. The heart ventricles were dissected open and washed in PBS to remove blood cells inside the heart. Microdissected tissues from 2 to 5 embryos were pooled to obtain sufficient cells. Tissue samples from the spleen and brain from the pregnant female mouse were also harvested to be run in parallel. Tissues were digested in 0.1% Tryspin for 10 min at 37 °C. The Trypsin was neutralized by adding media (DMEM with 10% FBS) and the tissues were repeatedly pipetted until completely dissociated. To prepare hNCCs and hSMCs, Accutase was used for cell dissociation. Cells were counted and resuspended at a concentration of 6.7×105 cells/ml in MEB buffer. Cells (15 µl for 10,000 cells per well) were added to the 384 well plate containing peptide treatments.
Fluorescence at Ex545/Em590 was recorded at 0, 15, 30, 60, 90 and 120 minutes. In between recordings, cells were incubated at 30 °C. The area under the curve was calculated and the percentage depolarization was calculated relative to the DMSO (no depolarization) and FCCP (complete depolarization) controls.
AnnexinV/PI staining
Cells were treated for 14–16 hours with 0.2 µg/ml Doxorubicin hydrochloride (Millipore Sigma) or 10 µM Nutlin-3a (Millipore Sigma). Cells were dissociated using Accutase. The cell media was also collected, as this may contain non-adherent apoptotic cells, and was combined with the Accutase-dissociated adherent cells. Cells were washed in PBS and resuspended at 1-5×106 cells/ml in 1x binding buffer (10 mM Hepes, pH 7.4, 140 mM NaCl, 2.5 mM CaCl2). Cells were filtered through a 35 µm nylon mesh (Corning). 100µl of cells in binding buffer were stained with AnnexinV-FITC (Biolegend) and propidium iodide (PromoKine) for 15 minutes. 400µl of binding buffer was then added to each sample and the samples were placed on ice. Samples were analyzed by flow cytometry.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
Excel (Microsoft) and R software (R Core Team, 2016) were used for analysis and graphical representations. In all bar charts, means are graphed with error bars representing standard deviation. The n number is included in the figure legends and represents the number of embryos analyzed per genotype. Unpaired T-tests were performed when assessing a mutant genotype compared to littermate controls. Chi-Square tests were used to determine whether genotype frequencies were significantly different from the expected Mendelian ratios.
RNA-seq data analysis
Reads were aligned to the GRCm38 mouse genome or the GRCh38 human genome using HISAT2 (Kim et al., 2015). Sorted BAM files were generated using SAMtools (Li et al., 2009). The number of reads mapping to each gene in the mouse Ensembl database (version 87) or human Ensembl database (version 93) (Aken et al., 2016) was counted using HTSeq-count (Anders et al., 2015). Differential gene expression was performed using DESeq2 (Love et al., 2014). Genes were classified as differentially expressed in they exhibited a fold change of at least 1.5 and an adjusted p value < 0.01. When classifying genes as Nutlin3a-responsive in hNCCs or early-hSMCs, only genes with a counts per million (CPM) value of at least 1 in at least 9 out of 12 samples were included. For comparison to published datasets, the lists of genes bound by p53 (ChIP-seq / ChIP-chip) or responsive to p53 activation (RNA-seq / microarray) were obtained from the supplementary data files associated with the original publications (Fischer, 2017; Kenzelmann Broz et al., 2013; Lee et al., 2010; Li et al., 2012; Tonelli et al., 2015). Gene Ontology and pathway analysis was performed using Enrichr (Kuleshov et al., 2016) using the categories “KEGG 2016”, “GO Biological Process 2017”, “GO Cellular Compartment 2017”. Given that Enrichr performs the Gene Ontology analysis using human gene names, we first used Biomart (Smedley et al., 2015) to obtain the human orthologs for our differentially expressed mouse genes.
DATA AVAILABILITY
RNA-seq data have been deposited in the Gene Expression Omnibus under series number GSE111753 (mouse NCCs) and GSE128191 (hNCCs and hSMCs).
Supplementary Material
Table S1. Related to Figure 2–4. Observed and expected numbers of mouse embryos and pups with tissue-specific p53 activation
Table S4. Related to Figure 7. Genes differentially expressed following Nutlin3a treatment in either hNCCs or early-hSMCs
Table S5. Related to Figure 7. Genes differentially expressed between hNCCs and early-hSMCs
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Peroxidase AffiniPure goat anti-mouse IgG | Jackson ImmunoResearch | Cat# 115-035-146, RRID:AB_2307392 |
| Peroxidase AffiniPure goat anti-rabbit IgG | Jackson ImmunoResearch | Cat# 111-035-144, RRID:AB_2307391 |
| Fluorescein goat anti-rabbit IgG | Vector Laboratories | Cat# FI-1000, RRID:AB_2336197 |
| Alexa Fluor 546 goat anti-mouse IgG | Thermo Fischer Scientific | Cat# A-11003, RRID:AB_2534071 |
| Rabbit anti-Cleaved Caspase 3 | Cell Signaling | Cat# 9661, RRID:AB_2341188 |
| Mouse anti-BrdU | BD Pharmigen | Cat# 555627, RRID:AB_395993 |
| Rabbit anti-RFP | Rockland | Cat#600-401-379, RRID:AB_2209751 |
| Rabbit anti-p53 CM5 | Leica Biosystems | Cat# NCL-p53-CM5p, RRID:AB_563933 |
| Mouse anti-p53 PAb242 | Yewdell et al 1986 | N/A |
| Mouse anti-p53 DO1 | Santa Cruz | Cat#sc-126, RRID:AB_628082 |
| Mouse anti-GAPDH | Fitzgerald Industries International | Cat# 10R-G109a, RRID:AB_1285808 |
| Mouse anti-Mdm2 | Abcam | Cat# ab16895, RRID:AB_2143534 |
| Mouse anti-Neurofilament (2H3) | Developmental Studies Hybridoma Bank | Cat# 2H3, RRID:AB_531793 |
| Bacterial and Virus Strains | ||
| Adenoviral Cre (Ad5CMVCre) | University of Iowa Viral Vector Core | Cat#VVC-U of Iowa-5 |
| Adenoviral empty (Ad5CMVempty) | University of Iowa Viral Vector Core | Cat#VVC-U of Iowa-272 |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| 5-bromo-4-chloro-3-indolyl-β-D-galactosidase (X-Gal) | ThermoFisher | Cat#15520034 |
| 7-amino-actinomycin D (7AAD) | BioLegend | Cat#420403 |
| 5-Bromo-2’-deoxyuridine (BrdU) | Millipore Sigma | Cat#203806 |
| Alcian Blue 8GX | Millipore Sigma | Cat#A5268 |
| Doxorubicin hydrochloride | Millipore Sigma | Cat#D1515 |
| Nutlin-3a | Millipore Sigma | Cat#SML0580 |
| Dulbecco’s Modified Eagle Medium (DMEM), high glucose, pyruvate | ThermoFisher | Cat#11995065 |
| KnockOut DMEM | ThermoFisher | Cat#10829018 |
| Neurobasal Medium | ThermoFisher | Cat#21103049 |
| B-27 supplement with Vitamin A | Gemini Bio-Products | Cat#400-160 |
| N-2 supplement | Gemini Bio-Products | Cat#400-163 |
| GlutaMAX supplement | ThermoFisher | Cat#35050061 |
| bFGF | Peprotech | Cat#100-18B 1MG |
| EGF | Sigma-Aldrich | Cat#AF-100-15 1MG |
| Bovine insulin | Sigma-Aldrich | Cat#700-112P |
| Bovine serum albumin, serum replacement grade | Gemini Bio-Products | Cat#700-104P |
| ChIRON 99021 | Selleckchem | Cat#S2924 |
| BMP2 | Peprotech | Cat#120-02 |
| JC-1 | Enzo Life Sciences | Cat#ENZ-52304 |
| Digitonin | Sigma | Cat#D141-100MG |
| Oligomycin | Sigma | Cat#O4876-5MG |
| FCCP | Sigma | Cat#C2920-10MG |
| AnnexinV-FITC | Biolegend | Cat#640906 |
| Propidium iodide | PromoKine | Cat#PK-CA707-40017 |
| Critical Commercial Assays | ||
| VECTASTAIN Elite ABC-HRP kit | Vector Laboratories | Cat#PK-6100 |
| DAB Peroxidase (HRP) Substrate Kit (with Nickel), 3,3’-diaminobenzidine | Vector Laboratories | SK-4100 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat#RS-122-2001 |
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat#4367660 |
| TRIzol reagent | ThermoFisher | Cat#15596026 |
| GoTaq Green Master Mix | Promega | Cat#M7123 |
| M-MLV Reverse Transcriptase | TermoFisher | Cat#28025013 |
| ECL prime Western Blotting System | Millipore Sigma | Cat#GERPN2232 |
| RNeasy Mini Kit | Qiagen | Cat#74104 |
| Deposited Data | ||
| RNA-seq data from E10.5 Wnt1-Cre;Trp53LSL−25,26/+ FACS-sorted neural crest cells | This paper | GEO: GSE111753 |
| RNA-seq data from hNCCs and early hSMCs | This paper | GEO: GSE128191 |
| Experimental Models: Cell Lines | ||
| Human embryonic stem cells: WA09 H9 | WiCell | Cat#WA09 |
| Mouse embryonic fibroblasts: Trp53+/− | This paper | N/A |
| Mouse: embryonic fibroblasts: Trp53−/− | This paper | N/A |
| Mouse: embryonic fibroblasts: Trp53LSL−25,26/+ | This paper | N/A |
| Mouse: embryonic fibroblasts: Trp53LSL−25,26,53,54/+ | This paper | N/A |
| Mouse: embryonic fibroblasts: Mdm2flox/flox | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Trp53−: B6.129S2-Trp53tm1Tyj/J | Jackson Laboratories, Jacks et al., 1994 | JAX:002101 |
| Mouse: Trp53LSL−25,26/+: 129-Trp53tm1Att/J | Jackson Laboratories, Johnson et al., 2005 | JAX:022070 |
| Mouse: Trp53LSL−25,26,53,54/+: 129-Trp53tm4Att/J | Jackson Laboratories, Brady et al., 2011 | JAX:021984 |
| Mouse: Mdm2flox | Grier et al., 2002 | N/A |
| Mouse: Mdm4− | Parant et al., 2001 | N/A |
| Mouse: Mdm2− | Montes et al., 1995 | N/A |
| Mouse: R26mTmG: Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J | Jackson Laboratories, Muzumdar et al., 2007 | JAX:007576 |
| Mouse: R26LSL-LacZ: B6.129S4-Gt(ROSA)26Sortm1Sor/J | Jackson Laboratories, Soriano, 1999 | JAX:003474 |
| Mouse: tdTomato: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | Jackson Laboratories, Madisen et al., 2010 | JAX:007914 |
| Mouse: Wnt1-Cre: Tg(Wnt1-Cre)11Rth Tg(Wnt1-GAL4)11Rth/J | Jackson Laboratories, Danielian et al., 1998 | JAX:003829 |
| Mouse: Foxg1-Cre: B6.129P2(Cg)-Foxg1tm1(cre)Skm/J | Jackson Laboratories, Hébert and McConnell, 2000 | JAX:006084 |
| Mouse: Sox10-Cre: B6;CBA-Tg(Sox10-cre)1Wdr/J | Jackson Laboratories, Matsuoka et al., 2005 | JAX:035807 |
| Mouse: Mef2c-Cre | Verzi et al., 2005 | N/A |
| Mouse: Mesp1-Cre | Saga et al., 1999 | N/A |
| Mouse: Tie2-Cre: B6.Cg-Tg(Tek-cre)1Ywa/J | Jackson Laboratories, Kisanuki et al., 2001 | JAX:008863 |
| Mouse: Sox2-Cre: B6N.Cg-Tg(Sox2-cre)1Amc/J | Jackson Laboratories, Hayashi et al., 2002 | JAX:014094 |
| Oligonucleotides | ||
| qRT-PCR primers, see method details | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Hisat2 version 2.0.4 | Kim et al., 2015 | https://ccb.jhu.edu/software/hisat2/index.shtml |
| SAMtools version 1.3.1 | Li et al., 2009 | http://www.htslib.org/ |
| HTSeq-count version 0.6.1 | Anders et al. 2015 | https://htseq.readthedocs.io/en/release_0.9.1/count.html#count |
| DESeq2 version 1.14.1 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| Biomart | Smedley et al., 2015 | https://www.ensembl.org/biomart/martview/ |
| Enrichr | Kuleshov et al., 2016 | amp.pharm.mssm.edu/Enrichr/ |
| R version 3.3.2 | R Core Team, 2016 | www.r-project.org |
| Other | ||
HIGHLIGHTS.
Inappropriate p53 activation during embryogenesis triggers developmental defects
Spectrum of defects depends on degree and spatiotemporal pattern of p53 activation
p53 activation drives apoptosis and cell cycle arrest in a context-dependent manner
Gene expression rather than mitochondrial priming dictates apoptotic responses
ACKNOWLEDGMENTS
We thank Guillermina Lozano, Michael Longaker, Kristy Red-Horse, and Sean Wu for mouse strains and Shady Saad, Donna Martin, Kristy Red-Horse, and Chantal Thibert for technical advice. We also thank Brittany Flowers, Alyssa Kaiser, and Stephano Mello for critical reading of the manuscript. This work was supported by a March of Dimes Foundation grant #6-FY15-189 (L.D.A.), R35 grant CA197591 (L.D.A.), Jane Coffin Childs Fund Postdoctoral Fellowship (M.E.B.), Stanford Child Health Research Institute Postdoctoral Fellowship (M.E.B.), Stanford Dean’s Postdoctoral Fellowship (M.E.B.), and Sir Henry Wellcome Postdoctoral Fellowship 106051/Z/14/Z (H.K.L.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Aken BL, Ayling S, Barrell D, Clarke L, Curwen V, Fairley S, Fernandez Banet J, Billis K, García Girón C, Hourlier T, et al. (2016). The Ensembl gene annotation system. Database 2016, baw093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attardi LD, Reczek EE, Cosmas C, Demicco EG, McCurrach ME, Lowe SW, and Jacks T (2000). PERP, an apoptosis-associated target of p53, is a novel member of the PMP-22/gas3 family. Genes Dev 14, 704–718. [PMC free article] [PubMed] [Google Scholar]
- Bajpai R, Chen DA, Rada-Iglesias A, Zhang J, Xiong Y, Helms J, Chang C-P, Zhao Y, Swigut T, and Wysocka J (2010). CHD7 cooperates with PBAF to control multipotent neural crest formation. Nature 463, 958–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, Del Sal G, Syed N, Smith P, Gasco M, et al. (2006). iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat. Genet 38, 1133–1141. [DOI] [PubMed] [Google Scholar]
- Bergman JEH, Janssen N, Hoefsloot LH, Jongmans MCJ, Hofstra RMW, and van Ravenswaaij-Arts CMA (2011). CHD7 mutations and CHARGE syndrome: the clinical implications of an expanding phenotype. J. Med. Genet 48, 334–342. [DOI] [PubMed] [Google Scholar]
- Blake KD, Hartshorne TS, Lawand C, Dailor AN, and Thelin JW (2008). Cranial nerve manifestations in CHARGE syndrome. American Journal of Medical Genetics Part A 146A, 585–592. [DOI] [PubMed] [Google Scholar]
- Bond J, Roberts E, Springell K, Lizarraga SB, Lizarraga S, Scott S, Higgins J, Hampshire DJ, Morrison EE, Leal GF, et al. (2005). A centrosomal mechanism involving CDK5RAP2 and CENPJ controls brain size. Nat. Genet 37, 353–355. [DOI] [PubMed] [Google Scholar]
- Bosman EA, Penn AC, Ambrose JC, Kettleborough R, Stemple DL, and Steel KP (2005). Multiple mutations in mouse Chd7 provide models for CHARGE syndrome. Hum. Mol. Genet 14, 3463–3476. [DOI] [PubMed] [Google Scholar]
- Bowen ME, and Attardi LD (2019). The role of p53 in developmental syndromes. Journal of Molecular Cell Biology [DOI] [PMC free article] [PubMed]
- Brade T, Pane LS, Moretti A, Chien KR, and Laugwitz K-L (2013). Embryonic Heart Progenitors and Cardiogenesis. Cold Spring Harbor Perspectives in Medicine 3, a013847– a013847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady CA, Jiang D, Mello SS, Johnson TM, Jarvis LA, Kozak MM, Kenzelmann Broz D, Basak S, Park EJ, McLaughlin ME, et al. (2011). Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell 145, 571–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronner ME, and Simões-Costa M (2016). The Neu ral Crest Migrating into the Twenty-First Century. In Current Topics in Developmental Biology, (Elsevier; ), pp. 115–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo E, Gu B, Bowen ME, Aryan F, Zalc A, Liang J, Flynn RA, Swigut T, Chang HY, Attardi LD, et al. (2018). Tissue-selective effects of nucleolar stress and rDNA damage in developmental disorders. Nature 554, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrzanowska KH, Gregorek H, Dembowska-Bagińska B, Kalina MA, and Digweed M (2012). Nijmegen breakage syndrome (NBS). Orphanet Journal of Rare Diseases 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corsten-Janssen N, Kerstjens-Frederikse WS, du Marchie Sarvaas GJ, Baardman ME, Bakker MK, Bergman JEH, Hove HD, Heimdal KR, Rustad CF, Hennekam RCM, et al. (2013). The cardiac phenotype in patients with a CHD7 mutation. Circ Cardiovasc Genet 6, 248–254. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, and McMahon AP (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol 8, 1323–1326. [DOI] [PubMed] [Google Scholar]
- Dixon J, Jones NC, Sandell LL, Jayasinghe SM, Crane J, Rey J-P, Dixon MJ, and Trainor PA (2006). Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. U.S.A 103, 13403–13408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egbe AC (2015). Birth Defects in the Newborn Population: Race and Ethnicity. Pediatrics & Neonatology 56, 183–188. [DOI] [PubMed] [Google Scholar]
- Epstein JA, Li J, Lang D, Chen F, Brown CB, Jin F, Lu MM, Thomas M, Liu E, Wessels A, et al. (2000). Migration of cardiac neural crest cells in Splotch embryos. Development 127, 1869–1878. [DOI] [PubMed] [Google Scholar]
- Fischer M (2017). Census and evaluation of p53 target genes. Oncogene [DOI] [PMC free article] [PubMed]
- Gierut JJ, Jacks TE, and Haigis KM (2014). Whole-Mount X-Gal Staining of Mouse Tissues. Cold Spring Harbor Protocols 2014, pdb.prot073452-pdb.prot073452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore EC, and Walsh CA (2013). Genetic causes of microcephaly and lessons for neuronal development. Wiley Interdisciplinary Reviews: Developmental Biology 2, 461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grier JD, Yan W, and Lozano G (2002). Conditional allele of mdm2 which encodes a p53 inhibitor. Genesis 32, 145–147. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, and Komarova EA (2003). The role of p53 in determining sensitivity to radiotherapy. Nat. Rev. Cancer 3, 117–129. [DOI] [PubMed] [Google Scholar]
- Hari L, Miescher I, Shakhova O, Suter U, Chin L, Taketo M, Richardson WD, Kessaris N, and Sommer L (2012). Temporal control of neural crest lineage generation by Wnt/ -catenin signaling. Development 139, 2107–2117. [DOI] [PubMed] [Google Scholar]
- Hayashi S, Lewis P, Pevny L, and McMahon AP (2002). Efficient gene modulation in mouse epiblast using a Sox2Cre transgenic mouse strain. Mech. Dev 119 Suppl 1, S97–S101. [DOI] [PubMed] [Google Scholar]
- Hébert JM, and McConnell SK (2000). Targeting of cre to the Foxg1 (BF-1) Locus Mediates loxP Recombination in the Telencephalon and Other Developing Head Structures. Developmental Biology 222, 296–306. [DOI] [PubMed] [Google Scholar]
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, and Weinberg RA (1994). Tumor spectrum analysis in p53-mutant mice. Curr. Biol 4, 1–7. [DOI] [PubMed] [Google Scholar]
- Jiang D, Brady CA, Johnson TM, Lee EY, Park EJ, Scott MP, and Attardi LD (2011). Full p53 transcriptional activation potential is dispensable for tumor suppression in diverse lineages. Proceedings of the National Academy of Sciences 108, 17123–17128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson TM, Hammond EM, Giaccia A, and Attardi LD (2005). The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nature Genetics 37, 145–152. [DOI] [PubMed] [Google Scholar]
- Jones NC, Lynn ML, Gaudenz K, Sakai D, Aoto K, Rey J-P, Glynn EF, Ellington L, Du C, Dixon J, et al. (2008). Prevention of the neurocristopathy Treacher Collins syndrome through inhibition of p53 function. Nat. Med 14, 125–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner A, and Wall N (2008). Immunohistochemistry of Whole-Mount Mouse Embryos. Cold Spring Harbor Protocols 2008, pdb.prot4820-pdb.prot4820. [DOI] [PubMed] [Google Scholar]
- Kaiser AM, and Attardi LD (2018). Deconstructing networks of p53-mediated tumor suppression in vivo. Cell Death and Differentiation 25, 93–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenhuber ER, and Lowe SW (2017). Putting p53 in Context. Cell 170, 1062–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenzelmann Broz D, Spano Mello S, Bieging KT, Jiang D, Dusek RL, Brady CA, Sidow A, and Attardi LD (2013). Global genomic profiling reveals an extensive p53-regulated autophagy program contributing to key p53 responses. Genes Dev 27, 1016–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokha MK, Mitchell LE, and Wallingford JB (2017). White paper on the study of birth defects: Birth Defects White Paper. Birth Defects Research 109, 180–185. [DOI] [PubMed] [Google Scholar]
- Kim D, Langmead B, and Salzberg SL (2015). HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, and Yanagisawa M (2001). Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev. Biol 230, 230–242. [DOI] [PubMed] [Google Scholar]
- Kocab AJ, and Duckett CS (2016). Inhibitor of apoptosis proteins as intracellular signaling intermediates. FEBS Journal 283, 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Research 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K-H, Li M, Michalowski AM, Zhang X, Liao H, Chen L, Xu Y, Wu X, and Huang J (2010). A genomewide study identifies the Wnt signaling pathway as a major target of p53 in murine embryonic stem cells. Proceedings of the National Academy of Sciences 107, 69–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, Saha B, Hisama FM, Rading K, Goebel I, et al. (2017). Dysfunction of the MDM2/p53 axis is linked to premature aging. Journal of Clinical Investigation 127, 3598–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, He Y, Dubois W, Wu X, Shi J, and Huang J (2012). Distinct Regulatory Mechanisms and Functions for p53-Activated and p53-Repressed DNA Damage Response Genes in Embryonic Stem Cells. Molecular Cell 46, 30–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JC, Guan X, Ryan JA, Rivera AG, Mock C, Agrawal V, Agarwal V, Letai A, Lerou PH, and Lahav G (2013). High mitochondrial priming sensitizes hESCs to DNA-damage-induced apoptosis. Cell Stem Cell 13, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nature Neuroscience 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, McMahon JJ, Tsai Y-H, Wang Z, and Silver DL (2016). Haploinsufficiency for Core Exon Junction Complex Components Disrupts Embryonic Neurogenesis and Causes p53-Mediated Microcephaly. PLOS Genetics 12, e1006282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques MR, Horner JS, Ihrie RA, Bronson RT, and Attardi LD (2005). Mice lacking the p53/p63 target gene Perp are resistant to papilloma development. Cancer Res 65, 6551–6556. [DOI] [PubMed] [Google Scholar]
- Matsuoka T, Ahlberg PE, Kessaris N, Iannarelli P, Dennehy U, Richardson WD, McMahon AP, and Koentges G (2005). Neural crest origins of the neck and shoulder. Nature 436, 347–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JAS, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, et al. (2015). 22q11.2 deletion syndrome. Nature Reviews Disease Primers 15071 [DOI] [PMC free article] [PubMed]
- Meek DW (2015). Regulation of the p53 response and its relationship to cancer. Biochemical Journal 469, 325–346. [DOI] [PubMed] [Google Scholar]
- Mello SS, Valente LJ, Raj N, Seoane JA, Flowers BM, McClendon J, Bieging-Rolett KT, Lee J, Ivanochko D, Kozak MM, et al. (2017). A p53 Super-tumor Suppressor Reveals a Tumor Suppressive p53-Ptpn14-Yap Axis in Pancreatic Cancer. Cancer Cell 32, 460–473.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montes de Oca Luna R, Wagner DS, and Lozano G (1995). Rescue of early embryonic lethality in mdm2-deficient mice by deletion of p53. Nature 378, 203–206. [DOI] [PubMed] [Google Scholar]
- Morgan SC, Lee H-Y, Relaix F, Sandell LL, Levorse JM, and Loeken MR (2008). Cardiac outflow tract septation failure in Pax3-deficient embryos is due to p53-dependent regulation of migrating cardiac neural crest. Mech. Dev 125, 757–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer SM, Larsson CA, and Lozano G (2017). Mdm proteins: critical regulators of embry ogenesis and homeostasis. Journal of Molecular Cell Biology mjx004. [DOI] [PMC free article] [PubMed]
- Muzumdar MD, Tasic B, Miyamichi K, Li L, and Luo L (2007). A global double-fluorescent Cre reporter mouse. Genesis 45, 593–605. [DOI] [PubMed] [Google Scholar]
- Nagy A, Gertsenstein M, Vintersten K, and Behringer R (2009). Alcian Blue Staining of the Mouse Fetal Cartilaginous Skeleton. Cold Spring Harbor Protocols 2009, pdb.prot5169-pdb.prot5169. [DOI] [PubMed] [Google Scholar]
- Parant J, Chavez-Reyes A, Little NA, Yan W, Reinke V, Jochemsen AG, and Lozano G (2001). Rescue of embryonic lethality in Mdm4-null mice by loss of Trp53 suggests a nonoverlapping pathway with MDM2 to regulate p53. Nat. Genet 29, 92–95. [DOI] [PubMed] [Google Scholar]
- Petrini J, Damus K, Russell R, Poschman K, Davidoff MJ, and Mattison D (2002). Contribution of birth defects to infant mortality in the United States. Teratology 66 Suppl 1, S3–6. [DOI] [PubMed] [Google Scholar]
- Potter DS, and Letai A (2016). To Prime, or Not to Prime: That Is the Question. Cold Spring Harbor Symposia on Quantitative Biology 81, 131–140. [DOI] [PubMed] [Google Scholar]
- Prescott SL, Srinivasan R, Marchetto MC, Grishina I, Narvaiza I, Selleri L, Gage FH, Swigut T, and Wysocka J (2015). Enhancer Divergence and cis-Regulatory Evolution in the Human and Chimp Neural Crest. Cell 163, 68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintens R, Verreet T, Janssen A, Neefs M, Leysen L, Michaux A, Verslegers M, Samari N, Pani G, Verheyde J, et al. (2015). Identification of novel radiation-induced p53-dependent transcripts extensively regulated during mouse brain development. Biology Open 4, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2016). R: A language and environment for statistical computing
- Robson A, Owens NDL, Baserga SJ, Khokha MK, and Griffin JN (2016). Expression of ribosomopathy genes during Xenopus tropicalis embryogenesis. BMC Developmental Biology 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan J, and Letai A (2013). BH3 profiling in whole cells by fluorimeter or FACS. Methods 61, 156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J. i, and Inoue T (1999). MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development 126, 3437–3447. [DOI] [PubMed] [Google Scholar]
- Sah VP, Attardi LD, Mulligan GJ, Williams BO, Bronson RT, and Jacks T (1995). A subset of p53-deficient embryos exhibit exencephaly. Nat. Genet 10, 175–180. [DOI] [PubMed] [Google Scholar]
- Sakai D, Dixon J, Achilleos A, Dixon M, and Trainor PA (2016). Prevention of Treacher Collins syndrome craniofacial anomalies in mouse models via maternal antioxidant supplementation. Nature Communications 7, 10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarosiek KA, Fraser C, Muthalagu N, Bhola PD, Chang W, McBrayer SK, Cantlon A, Fisch S, Golomb-Mello G, Ryan JA, et al. (2017). Developmental Regulation of Mitochondrial Apoptosis by c-Myc Governs Age- and Tissue-Specific Sensitivity to Cancer Therapeutics. Cancer Cell 31, 142–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen N, Kumari R, Singh MI, and Das S (2013). HDAC5, a Key Component in Temporal Regulation of p53-Mediated Transactivation in Response to Genotoxic Stress. Molecular Cell 52, 406–420. [DOI] [PubMed] [Google Scholar]
- Sir J-H, Barr AR, Nicholas AK, Carvalho OP, Khurshid M, Sossick A, Reichelt S, D’Santos C, Woods CG, and Gergely F (2011). A primary microcephaly protein complex forms a ring around parental centrioles. Nature Genetics 43, 1147–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slomnicki LP, Chung D-H, Parker A, Hermann T, Boyd NL, and Hetman M (2017). Ribosomal stress and Tp53-mediated neuronal apoptosis in response to capsid protein of the Zika virus. Scientific Reports 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smedley D, Haider S, Durinck S, Pandini L, Provero P, Allen J, Arnaiz O, Awedh MH, Baldock R, Barbiera G, et al. (2015). The BioMart community portal: an innovative alternative to large, centralized data repositories. Nucleic Acids Research 43, W589–W598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohn YB, Ko JM, Shin CH, Yang SW, Chae J-H, and Lee K-A (2016). Cerebellar vermis hypoplasia in CHARGE syndrome: clinical and molecular characterization of 18 unrelated Korean patients. Journal of Human Genetics 61, 235–239. [DOI] [PubMed] [Google Scholar]
- Soriano P (1999). Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Steventon B, Mayor R, and Streit A (2014). Neural crest and placode interaction during the development of the cranial sensory system. Developmental Biology 389, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szybińska A, and Leśniak W (2017). P53 Dysfunction in Neurodegenerative Diseases - The Cause or Effect of Pathological Changes? Aging Dis 8, 506–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T, Wang Y, Van Pelt CS, Box NF, Travis EL, and Lozano G (2007). Haploinsufficiency of Mdm2 and Mdm4 in tumorigenesis and development. Mol. Cell. Biol 27, 5479–5485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toki T, Yoshida K, Wang R, Nakamura S, Maekawa T, Goi K, Katoh MC, Mizuno S, Sugiyama F, Kanezaki R, et al. (2018). De Novo Mutations Activating Germline TP53 in an Inherited Bone-Marrow-Failure Syndrome. The American Journal of Human Genetics 103, 440– 447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonelli C, Morelli MJ, Bianchi S, Rotta L, Capra T, Sabò A, Campaner S, and Amati B (2015). Genome-wide analysis of p53 transcriptional programs in B cells upon exposure to genotoxic stress in vivo. Oncotarget 6, 24611–24626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truett GE, Heeger P, Mynatt RL, Truett AA, Walker JA, and Warman ML (2000). Preparation of PCR-quality mouse genomic DNA with hot sodium hydroxide and tris (HotSHOT). BioTechniques 29, 52, 54. [DOI] [PubMed] [Google Scholar]
- Tsilou ET, Giri N, Weinstein S, Mueller C, Savage SA, and Alter BP (2010). Ocular and Orbital Manifestations of the Inherited Bone Marrow Failure Syndromes: Fanconi Anemia and Dyskeratosis Congenita. Ophthalmology 117, 615–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, and Attardi LD (2014). Guilty as CHARGED: p53’s expanding role in disease. Cell Cycle 13, 3798–3807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand JL, Brady CA, Jung H, Fuentes DR, Kozak MM, Johnson TM, Lin C-Y, Lin C-J, Swiderski DL, Vogel H, et al. (2014). Inappropriate p53 activation during development induces features of CHARGE syndrome. Nature [DOI] [PMC free article] [PubMed]
- Verzi MP, McCulley DJ, De Val S, Dodou E, and Black BL (2005). The right ventricle, outflow tract, and ventricular septum comprise a restricted expression domain within the secondary/anterior heart field. Dev. Biol 287, 134–145. [DOI] [PubMed] [Google Scholar]
- Vincent M, Geneviève D, Ostertag A, Marlin S, Lacombe D, Martin-Coignard D, Coubes C, David A, Lyonnet S, Vilain C, et al. (2016). Treacher Collins syndrome: a clinical and molecular study based on a large series of patients. Genetics in Medicine 18, 49–56. [DOI] [PubMed] [Google Scholar]
- Vitelli F, Morishima M, Taddei I, Lindsay EA, and Baldini A (2002). Tbx1 mutation causes multiple cardiovascular defects and disrupts neural crest and cranial nerve migratory pathways. Hum. Mol. Genet 11, 915–922. [DOI] [PubMed] [Google Scholar]
- Whibley C, Pharoah PDP, and Hollstein M (2009). p53 polymorphisms: cancer implications. Nature Reviews Cancer 9, 95–107. [DOI] [PubMed] [Google Scholar]
- Yewdell JW, Gannon JV, and Lane DP (1986). Monoclonal antibody analysis of p53 expression in normal and transformed cells. J. Virol 59, 444–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaman A, Capper R, and Baddoo W (2015). Waardenburg syndrome: more common than you think! Clinical Otolaryngology 40, 44–48. [DOI] [PubMed] [Google Scholar]
- Zhang G, Xie Y, Zhou Y, Xiang C, Chen L, Zhang C, Hou X, Chen J, Zong H, and Liu G (2017). p53 pathway is involved in cell competition during mouse embryogenesis. Proceedings of the National Academy of Sciences 114, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, He X, Chen L, Zhang C, Gao X, Yang Z, and Liu G (2012). Synergistic regulation of p53 by Mdm2 and Mdm4 is critical in cardiac endocardial cushion morphogenesis during heart development. J. Pathol 228, 416–428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Related to Figure 2–4. Observed and expected numbers of mouse embryos and pups with tissue-specific p53 activation
Table S4. Related to Figure 7. Genes differentially expressed following Nutlin3a treatment in either hNCCs or early-hSMCs
Table S5. Related to Figure 7. Genes differentially expressed between hNCCs and early-hSMCs
Data Availability Statement
RNA-seq data have been deposited in the Gene Expression Omnibus under series number GSE111753 (mouse NCCs) and GSE128191 (hNCCs and hSMCs).