

Abstract
Microbial communities have not been studied using molecular approaches at high elevations on the African continent. Here we describe the diversity of microbial communities from ice and periglacial soils from near the summit of Mt. Kilimanjaro by using both Illumina and Sanger sequencing of 16S and 18S rRNA genes. Ice and periglacial soils contain unexpectedly diverse and rich assemblages of Bacteria and Eukarya indicating that there may be high rates of dispersal to the top of this tropical mountain and/or that the habitat is more conducive to microbial life than was previously thought. Most bacterial OTUs are cosmopolitan and an analysis of isolation by geographic distance patterns of the genus Polaromonas emphasized the importance of global Aeolian transport in the assembly of bacterial communities on Kilimanjaro. The eukaryotic communities were less diverse than the bacterial communities and showed more evidence of dispersal limitations and apparent endemism. Cercozoa dominated the 18S communities, including a high abundance of testate amoebae and a high diversity of endemic OTUs within the Vampyrellida. These results argue for more intense study of this unique high-elevation “island of the cryosphere” before the glaciers of Kilimanjaro disappear forever.
Subject terms: Soil microbiology, Biodiversity
Introduction
High-elevation microbial diversity has been studied in several of the highest mountain ranges on Earth1–3, however microbial communities have not been studied at high elevation in the African continent and in particular, on the climate sensitive glaciers and periglacial soils on the top of Mt. Kilimanjaro. Kilimanjaro, Africa’s highest mountain and the tallest free standing mountain on Earth, is located on the Kenya-Tanzania border, about 370 km south of the equator and approximately the same distance from the Indian ocean (3 04′S, 37 21′E). This massive stratovolcano (about 80 by 50 km) consists of 3 main peaks, of which Kibo (5893 m) is the highest and the only one still retaining glaciers. Its summit has collapsed to form a caldera, enclosing the Reusch crater, which is about 800 m across and still presents a continuous geothermal heat flux. In recent years Kilimanjaro and its dramatic ice loss have become an “icon” of climate change, attracting broad interest in its fate4. The three remaining ice fields on the plateau and slopes are both shrinking laterally and rapidly thinning5 leaving behind just the ragged fringe of an ice cap, which is believed to have once covered the entire summit of the mountain6. Of the ice cover recorded in 1912, 85% has disappeared, with a peak reduction rate registered from 1989 to 2007 of about 2.5% per year5. A drastic drop in atmospheric moisture starting at the end of the 19th century and an increase of shortwave incoming radiation due to decreased cloudiness are currently believed to be the main drivers of glacier retreat (through sublimation) on Kilimanjaro, as well as other equatorial east African glaciers7. It is believed that under present conditions, rapid glacier shrinking at the top of Kilimanjaro will continue unabated and the entire summit of the mountain is expected to be devoid of ice for the first time in 11000 years by mid-century4.
As the glaciers on top of Kilimanjaro continue to recede, unique microbial communities and paleodiversity archives will be gradually compromised and lost. Glaciers are known to function as reservoirs of airborne microorganisms that can preserve information about their relationship with climatic and environmental changes through time8. To date there have been no investigations published on the microbiology of the habitats at the top of Kilimanjaro and the expected dramatic change of these environments in the next decades makes it important to study the biological communities dwelling within the ice and soil. Its high elevation and its considerable isolation from any other mountain range also makes it an ideal site to further advance our knowledge of microbial endemicity and biogeographic patterns. Aerial deposition and post-depositional selection are the main drivers of microbial community composition at high elevations8 but relatively little is known about their specific roles in the establishment of microbial communities.
The top of Mt. Kilimanjaro is also of interest to the field of astrobiology. Kilimanjaro’s periglacial soils may be some of the most extreme environments on Earth and are therefore considered as potential analogues for habitable zones on Mars9. Microbial life at the top of this mountain has to cope with a complex interplay of parameters that are similar to those that could be found on Mars or other planetary bodies, such as a high UV flux, extreme diurnal freeze-thaw cycles, low atmospheric pressure and an extremely low nutrient content and water activity9. For this reason, these oligotrophic soils and glaciers have received some attention as Martian analogues prior to the launch of the Mars Science Laboratory (MSL) Curiosity Rover and as a field test for a planned Mars 2018 mission9. Preliminary data from Ponce et al.9 showed that Kilimanjaro soils in proximity to glacier walls may be nearly sterile with a total organic carbon (TOC) content of less than 1000 µg/g. TOC has been traditionally correlated with viable microbial biomass and the value observed for Kilimanjaro soils are slightly higher than TOC levels of oligotrophic mineral soils found in extreme environments in Antarctica, high elevation sites in the Andes and the Atacama Desert1,10,11. In the present study we follow up on the preliminary work of Ponce et al.9 to provide the first culture-independent analysis of the microbial communities in ice and soil on top of Mt. Kilimanjaro.
Materials and Methods
Site location and sample collection
The study area at the top of Mt. Kilimanjaro was accessed from the Machame trail on June 4th, 2012, at the beginning of the dry season. Prevailing weather conditions at the top were clear, windy and cold, with most of the surface covered in fresh snow. Samples were collected at the border of the tabular-shaped plateau glacier in the Southern Ice Field (SIF) at 5772 m elevation (S 03 04. 839′ E 037 21. 628′) (Fig. 1). The collection site was far off the main trail with no apparent foot traffic. A total of 8 samples were collected along a transect from the base of the glacier wall: 2 from the ice wall (N3 and N8) and 6 from soils at increasing distance from the base of the glacier wall at distances of 0.4 m (N1), 2.5 m (N2), 5 m (N4), 7 m (N6), 7.5 m (N5) and 8 m (N8) (Table 1). Latitude, longitude and elevation data were collected using a handheld GPS device. The original plan to collect soil samples along a longer perpendicular transect was made impossible by deep snow on the ground, thus samples were collected from areas close to the glacier that were devoid of snow. Ice samples were collected by scraping the ice wall with a sterile spatula, while soil samples (50 g each) were collected from the top 0–4 cm of soil. Both types of sample were placed in sterile 50 ml centrifuge tubes (Fisher Brand, Hampton, NH, USA) and kept on ice for 1 day during the descent from the mountain. They were subsequently stored at −20 °C for a week and then shipped to the University of Colorado at Boulder. Upon arrival they were stored at −80 °C for later use in molecular and chemical analysis. A subset of collected samples has been archived at −80 °C for long-term storage to be available for potential future studies.
Figure 1.

Location of Kilimanjaro sampling sites. (A) A map of Eastern Africa showing the location of Mt. Kilimanjaro on the Tanzania-Kenya border (red triangle). Mt. Kilimanjaro is located 370 km south of the Equator. (B) Google Earth imagery captured after field collections were made (Map data: Google, DigitalGlobe). Red arrow indicates sampling location at the border of the tabular-shaped plateau glacier in the Southern Ice Field at 5772 m elevation (S 03 04. 839′ E 037 21. 628′). (C) This panel features the tabular-shaped plateau glaciers of the Southern Icefield facing the sampling location and shows the large amount of snow cover that was present on the sampling day.
Table 1.
Kilimanjaro sample sites and soil characteristics, sampled June 2012.
| Sample | Location | Elevation | Distance from Glacier | Type | pH | DOCa (µg/g soil) | TDNb (µg/g soil) | % of H2O |
|---|---|---|---|---|---|---|---|---|
| N3 | S 03° 04.839′ E 037° 21. 628 | 5772 m | on glacier wall | Ice | ||||
| N8 | S 03° 04.837′ E 037° 21. 638′ | 5772 m | on glacier wall | Ice | ||||
| N1 | S 03° 04.839′ E 037° 21. 628′ | 5771 m | 0.4 m | Soil | 7.5 | 44.8 | 25.3 | 28.1 |
| N2 | S 03° 04.839′ E 037° 21. 637′ | 5770 m | 2.5 m | Soil | 7.61 | 174.8 | 14.1 | 19.8 |
| N4 | S 03° 04.839′ E 037° 21. 636′ | 5772 m | 5 m | Soil | 7.49 | 22.3 | 9.5 | 13.6 |
| N5 | S 03° 04.839′ E 037° 21. 635′ | 5772 m | 7.5 m | Soil | 7.69 | 11.7 | 1.5 | 7.2 |
| N6 | S 03° 04.836′ E 037° 21. 634′ | 5772 m | 7 m | Soil | 7.93 | 7 | 3.1 | 7.7 |
| N7 | S 03° 04.835′ E 037° 21. 632′ | 5771 m | 8 m | Soil | 7.6 | 8.7 | 2.4 | 6.9 |
aDOC, Dissolved organic carbon.
bTDN, Total dissolved nitrogen.
DNA extraction, PCR and 16S/18S rRNA gene clone library construction
Total environmental genomic DNA from soil and ice samples was extracted using PowerSoil® and PowerWater® DNA Isolation Kits respectively (MO BIO, Carlsbad, CA USA), according to the manufacturer’s instructions. PCR of 3 soil (N1, N5 and N7) and ice samples was performed using 1–3 µl of template DNA, 12.5 µl of AmpliTaq Gold® 360 MasterMix 5 Units/µL (Applied Biosystems, Foster City, CA, USA) and 1 µl of Forward and Reverse primers (10 µM) in a G-Storm GS2 thermal cycler (GRI Ltd., Essex, UK). Bacterial 16S rRNA genes were amplified using the bacterial domain-specific primer 8F (5′-AGA GTT TGA TCC TGG CTC AG-3′) and universal primer 1391 R (5′-GAC GGG CGG TGW GTR CA-3′). Eukaryotic 18S rRNA and archaeal 16S rRNA genes were amplified using the universal primer pair 4 Fa-short (5′-ATT CCG GTT GAT CCT GC-3′) and 1492r (5′-GGT TAC CTT GTT ACG ACT T-3′). PCR amplification for 16S rRNA genes was carried out using a program of 95 °C for 10 min followed by 35 cycles of 95 °C, 1 min; 53 °C, 30 sec; 72 °C, 2 min and 30 sec; with a final elongation step of 72 °C for 10 min. 18S eukaryotic and 16S archaeal rRNA genes were amplified by a program of 95 °C for 10 min followed by 35 cycles of 95 °C, 1 min; 49 °C, 30 sec; 72 °C, 2 min and 30 sec; and a final elongation step of 72 °C for 10 min. PCR products of the appropriate length were excised from agarose gels and purified following the protocol of the QIAquick Gel Extraction Kit (Qiagen, Valencia, CA USA), with Hyperladder IITM as a reference. PCR products were ligated into TOPO TA® cloning vectors and transformed into OneShotTM E. coli cells (Invitrogen, Carlsbad, CA, USA). Transformed cells were grown on Ampicillin 50 µg/mL agar plates overnight at 37 °C. Single colonies containing the inserts were pelleted and randomly arrayed on 96-well plates. Functional Biosciences (Madison, WI, USA) performed Sanger sequencing bi-directionally using vector-targeted T7 (5′-AAT ACG ACT CAC TAT AG-3′) and M13R-9 (5′-GCT ATG ACC ATG ATT ACG-3′) primers.
Sanger sequencing and phylogenetic analysis
Geneious (Biomatters, Auckland, New Zealand) was used to edit sequences, trim primers and assemble contigs. Edited sequences were aligned using the SINA aligner tool12 and imported into ARB13 where they were manually fine-tuned. Putative chimeras were identified using a combination of Bellerophon14 and the Mallard Program15. Once putative chimeras were removed, remaining sequences were used to generate a phylip-formatted distance matrix and cluster analysis was performed with MOTHUR16 using the average neighbor algorithm implementation to define OTUs at the minimum threshold of 97% sequence identity. Phylogenetic affiliation of OTUs and related sequences were found using both the basic local alignment search tool (BLAST) and ARB through the parsimony insertion function. Phylogenetic trees for both bacterial and eukaryotic OTUs were inferred using maximum likelihood (ML). The ML analysis was conducted using a general time reversible (GTR) model of evolution with a gamma distribution (g) and a proportion of invariant sites (I) with MEGA6.0617. Node support was estimated using 300 bootstrap replicates.
PCR and MiSeq Illumina sequencing
Amplification of the bacterial V4-V5 16S rRNA gene region and eukaryotic 18S rRNA gene for all soil and ice samples was performed using the oligonucleotide primers sets 515F/806R and Euk_1391f/EukBr respectively (Earth Microbiome Project, accessible at http://www.earthmicrobiome.org/emp-standard-protocols/16s-18s/). All forward and reverse primers were modified to include a unique 12 nucleotide barcode. PCR reaction mixtures contained 0.5 μL of forward primer (10 µM), 0.5 μL of reverse primer (10 µM), 1 μL of template and 12.5 μL of MM Gotaq Hot start Colorless Master Mix (Promega Corporation, Madison, WI, USA). The reaction volume was adjusted to a total of 25 μL with ultrapure DNase/RNase free water. Thermal cycles for 16S rRNA gene amplification consisted of an initial denaturation of 94 °C for 3 min, followed by 35 cycles of 94 °C for 1 min; 50 °C for 1 min; and 72 °C for 105 sec; with a final elongation step of 72 °C for 10 min. The program for 18S rRNA gene amplification consisted of an initial denaturation of 94 °C for 3 min, followed by 35 cycles of 94 °C for 45 sec; 57 °C for 1 min; and 72 °C for 1.5 min; with a final elongation step of 72 °C for 10 min. To prepare amplicons for sequencing, amplicon purification and normalization was done with Invitrogen SequalPrep Normalization Kit (Invitrogen Inc., CA, USA). Amplicons were combined into a single pool and sequenced using the Illumina MiSeq platform (BioFrontiers Institute, Boulder, CO) using pair-end 2 × 150 bp chemistry.
Forward-oriented sequences were demultiplexed, quality filtered and processed using the QIIME pipeline18. Paired-end sequences did not work for eukaryotic reads and only the read corresponding to the 1391 F primer was used because it overlaps more with sequences in the NCBI and SILVA databases19. Singletons were excluded from further analysis and sequences with >97% SSU rRNA gene sequence identity were clustered into an OTU via UCLUST. Representative sequences for each OTU were chosen for classification and the Greengenes and Silva 104 reference database for 16S and 18S rRNA gene sequences respectively were employed to assign taxonomy identification to each OTU. Sequences were aligned with PyNAST20 and a phylogeny was built with the FastTree algorithm21. OTU tables were rarified to the level of the lowest number of sequences in the lowest populous sample and were used to assess alpha diversity and relative abundance of all taxa.
Diversity measures and statistical analysis
Diversity within individual communities (alpha diversity) was evaluated by measures of phylogenetic dominance, evenness and richness with MOTHUR16. Dominance was estimated through the Berger and Parker index22, which calculates the ratio between the abundance of the most represented OTU and the total OTU abundance. The Shannon Index was used to estimate community diversity taking into account OTUs richness and evenness. Phylotype richness was determined through the Schao1 estimator and an estimate of how well sampling had covered true community diversity was obtained with the non-parametric estimator Good’s coverage.
Weighted UniFrac Analysis, which quantifies shared evolutionary history through shared branch length23 was used to construct beta diversity matrices for both the ice and soil datasets (both 16S rRNA and 18S rRNA genes) and a one-way Analysis of Similarity test (ANOSIM) from the vegan R package24 was used to test significance of phylogenetic difference in beta diversity between ice and soil samples. Rarefaction curves were plotted for both bacterial and eukaryotic sequences with a sequence similarity cut-off value of 3% to evaluate if sampling effort had revealed true phylogenetic diversity within the site. Additionally, Principal Coordinate Analysis (PCoA) ordination was constructed based on weighted Unifrac distance matrices in order to visualize differences among community compositions of soil and ice samples.
Environmental classification of OTUs
Environmental classification of long-read bacterial OTUs containing 2 or more sequences was done following a modified version of the method of Herbold et al.25. Each OTU representative sequence was queried against the NCBI nucleotide database using BLAST. OTUs were classified as “non-endemic” if closest matches to OTUs were >97% identical, “endemic” if closest matching sequences were <97% identical (Supp. Table 1). Within the “non-endemic” group, reads were classified as “cryophilic” if all database entries that matched the representative unique sequence with >97% identity had been previously observed in perennially cold environments, as “non-cryophilic” if all matches were observed in temperate environments and as “polythermal” if the matches were observed in both perennially cold and temperate environments. Reads for the endemic OTUs were classified as “subnovel” or “novel” if the nearest match was between 97% and 95.5% or 95.5–88.5% respectively25.
Biogeographic analysis
In order to examine the biogeographic distribution within the cryosphere of the most abundant phylotypes retrieved on top of Mt. Kilimanjaro, a genetic isolation by geographic distance analysis was performed on the 2 most abundant long-read OTUs for which geographic coordinates of closely matching sequences were available in Genbank: a Polaromonas clade within the Bacteria (Supp. Fig. 1) and a Chlamydomonas clade within the Eukarya. Other long-read sequences from glacial and periglacial environments (Supp. Figs 2 and 3) closely matching the sequences of the clades were downloaded from GenBank (Supp. Tables 2 and 3). Geographic distances between sample sites were computed in R (R Development Core Team, 2012) using the Fields package (http://CRAN.R‐project.org/package=fields) and an uncorrected genetic distance matrix was created using the Ape package (http://CRAN.R‐project.org/package=ape). To test for a correlation between the matrices, Mantel tests were performed in R using 1000 randomized permutations per test. A Mantel correlogram was constructed and the application of Sturge’s rule resulted in data being partitioned in distance classes. Further Mantel tests were carried out on each distance class with a Bonferroni correction.
Soil analyses
Soil pH was determined according to the method of King et al.26. Specifically, 2 g of soil and 2 mL of DI water were placed into 15 mL centrifuge tubes and shaken horizontally for 1 hr at 175 rpm. Soil pH was then measured with an Oakton benchtop pH meter (OAKTON Instruments, Vernon Hill, IL, USA). Total water content of soil samples was measured by placing 1 g of soil of each sample in 15 mL conical sterile tubes left open to dry at 60 °C in an oven for 24 hrs. Water content was determined as the percentage of the ratio between the water loss and dry weight of the sample.
Dissolved organic carbon (DOC) and total dissolved nitrogen (TDN) were measured using a modification of the method described in Weintraub et al.27. 2 g of soil of each sample was shaken with 25 mL of 0.5 M K2SO4 for 1 hour. Solutions were then filtered using a 0.2 µm IsoporeTM membrane filter (Millipore, Darmstadt, Germany). DOC and TDN were measured using a Shimadzu TOC-V CSN Total Organic Carbon Analyzer with TNM-1 module.
Nucleotide sequence accession numbers
The SSU rRNA gene sequences from Sanger libraries from this study were deposited in the GenBank database under accession numbers KX771236 to KX772158 while the SSU rRNA gene sequences from Illumina MiSeq libraries were deposited in the SRA (Short Read Archive) database under Bioproject ID PRJNA340181 and PRJNA340027.
Results
Alpha and beta diversity
Bacterial richness (Chao 1) and Alpha Diversity (PD) were comparable for soil and ice samples (Table 2), whereas the eukaryotic community showed more diversity in ice compared to soils. The Shannon Index implied than evenness was similar in soils and ice for both bacterial and eukaryotic communities. The Berger Parker Index revealed relatively low dominance in both ice and soil bacterial communities, meaning that the most abundant OTUs in the samples are only represented by a small number of sequences. Eukaryotic communities in soil and ice displayed a higher Berger Parker Index than that seen for bacterial communities showing that the most abundant OTUs constitute 40–50% of total abundance.
Table 2.
Diversity and dominance indices, coverage, number of observed OTUs and phylogenetic diversity index (PD) for bacterial and eukaryotic communities in soil and ice samples.
| Sample | Chao1 | Shannon | Berger-Parker | Good’s Coverage | Observed OTUs | PD |
|---|---|---|---|---|---|---|
| Bacteria | ||||||
| Soil | 603.37 | 6.37 | 0.12 | 0.97 | 418 | 20.74 |
| Ice | 646.83 | 6.32 | 0.09 | 0.97 | 400 | 19.89 |
| Eukarya | ||||||
| Soil | 184.36 | 2.65 | 0.47 | 0.99 | 113.3 | 38.48 |
| Ice | 284.85 | 2.73 | 0.39 | 0.98 | 143.5 | 45.59 |
Rarefaction curves (Supp. Fig. 4) yielded similar patterns as those estimated by Chao1 index and were close to saturation at a 97% clustering level. The number of OTUs observed was ~3.5 times higher for Bacteria compared to Eukarya. Within the Eukarya, sampling coverage for ice communities was about twice as high as soil communities. Good’s coverage index demonstrated that the estimated proportion of phylotypes represented in our libraries was very high (>0.97), however, “species-level” sampling coverage was not complete as the observed number of OTUs was less than that estimated with the Chao1 richness.
ANOSIM analysis revealed that there was a significant difference in composition (beta diversity) between ice and the soil samples for Bacteria (R = 0.8, P = 0.03), but there was not a significant difference between ice and soil eukaryotic assemblages, perhaps indicating that the soil Eukarya came from the ice. Interestingly both the bacterial and eukaryotic composition of the soil sample closest to the ice wall (N1) was more similar to that of ice samples compared to soil (Supp. Fig. 5). Ice assemblages showed a lower weighted UniFrac dissimilarity value to the soil sample closest to the glacier wall than the other soil samples for both Bacteria (0.13 vs 0.202) and Eukarya (0.212 vs 1.49).
Community composition and phylogenetic analysis
Bacterial OTUs from soil and ice were distributed among 18 phyla, including 4 candidate divisions and were dominated in both ice and soil by Proteobacteria, Bacteroidetes, Actinobacteria and Acidobacteria (Fig. 2). Betaproteobacteria represented the dominant fraction of the community in both ice (25%) and soil (22%) and most of these were in the Comamonadaceae (Polaromonas, Variovorax, Rhodoferax, Curvibacter, Methylibium) and Oxalobacteraceae in the soil and Rhodocyclales in the ice. Bacteroidetes accounted for 9 and 22% in ice and soil, respectively, with the majority in the Chitinophagaceae family (Ferruginibacter, Segetibacter and Sediminibacterium). Actinobacteria accounted for 5 and 8% of the total sequences in ice and soil, respectively, with most being in the Actinomycetales (Frigobacterium, Nocardioides and Pseudonocardia). Cyanobacteria only represented 4% of sequences in the soil, whereas they were more abundant in the ice (10%), with most sequences in the Oscillatoriales and Chroococcales. Overall soil communities harbored higher abundances of Actinobacteria, Bacteroidetes and Acidobacteria than ice, while sequences related to Betaproteobacteria, Alphaproteobacteria and Cyanobacteria displayed higher abundance within ice communities (Fig. 2).
Figure 2.

Phylogenetic analysis of 16S rRNA gene sequences obtained from ice and soil samples close to the summit of Mt. Kilimanjaro. (A) Cluster analysis of the phylogenetic structure and the relative abundance of bacterial phyla in the ice and soil samples as assigned by Greengenes to Illumina MiSeq sequences. Proteobacteria were split into classes for greater detail. The asterisk indicates statistical significant difference in composition (ANOSIM, P < 0.05). (B) Classification at lower taxonomic level is reported for Alphaproteobacteria, Bacteroidetes and Betaproteobacteria.
Illumina sequencing data revealed that both soil and ice 18S communities were limited to 9 phyla. Ice communities were heavily dominated by Cercozoa, representing 72% of total sequences (Fig. 3), while in the soil they were the most abundant taxonomic group together with green algae (Chlorophyta), at about 34% abundance each. The Chlorophyceae comprised about 9% and 34% of total sequences in the ice and soil respectively and were most closely related to the genera Chlamydomonas, Chloromonas and Stigeoclonium.
Figure 3.

Phylogenetic analysis of 18S rRNA gene sequences obtained from ice and soil samples close to the summit of Mt. Kilimanjaro. (A) Cluster analysis of the phylogenetic structure and the relative abundance of eukaryotic phyla in the ice and soil samples as assigned by Silva 104 database to Illumina MiSeq sequences. Cercozoa were split into classes (Thecofilosea), orders (Vampyrellida) and families (Trinematidae) for greater detail (B) Classification at lower taxonomic level is reported for Chlorophyta and Fungi.
Within the Cercozoa, most sequences fell within the Trinematidae and the Vampirellida. Longer reads from Sanger sequencing showed that the Trinematidae family clustered in very few OTUs closely related to the known organisms Trinema lineare and Trinema enchelis (Fig. 4). In contrast, long-read sequences within the Vampirellida order were very diverse and all fell into a previously undescribed clade (wedge in Fig. 4).
Figure 4.
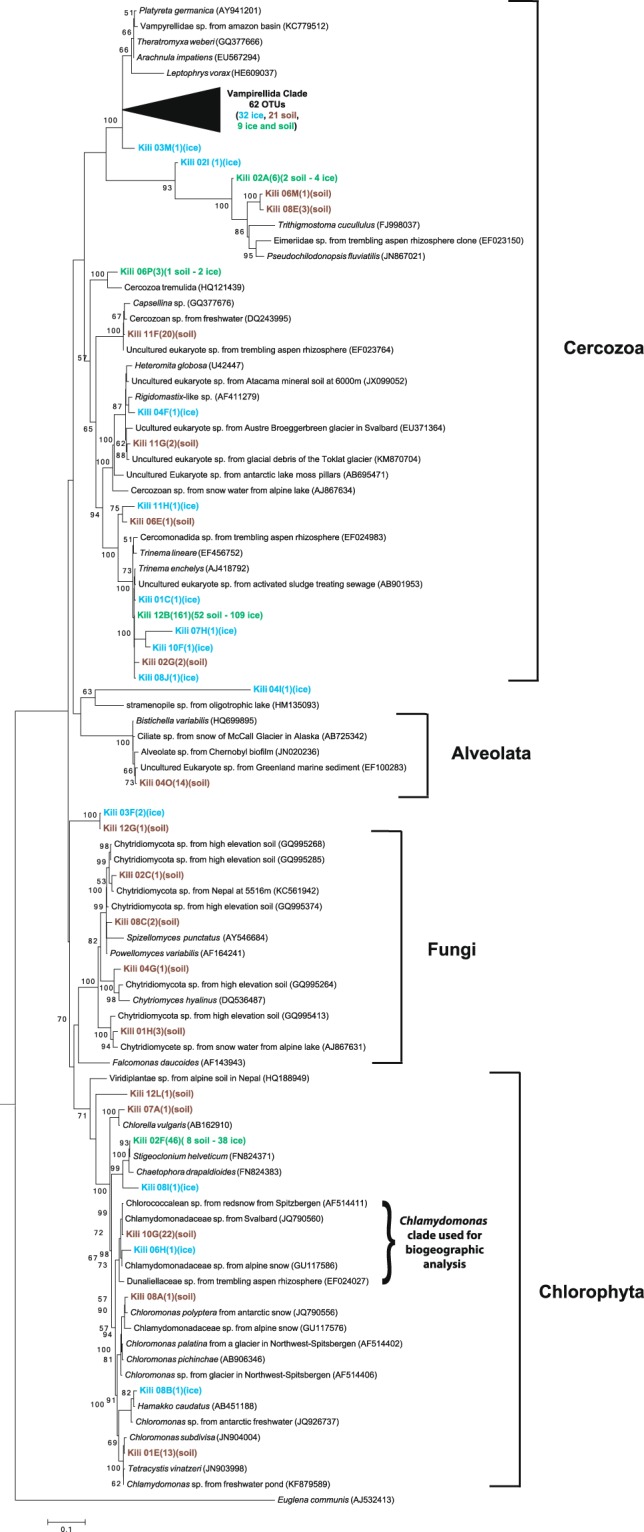
Phylogenetic analysis of 18S rRNA gene long-read sequences retrieved in ice and soil samples close to Mt. Kilimanjaro summit. Maximum Likelihood consensus phylogenetic tree includes 18S rRNA gene sequences from ice and soil close to the summit of Mt. Kilimanjaro and their closest GenBank BLAST and ARB matches. The accession numbers of most closely related taxa are listed parenthetically. Tree is rooted with the sequence of Euglena communis (AJ532413). Kilimanjaro phylotypes are bolded and followed by the number of sequences in each phylotype. Kilimanjaro phylotypes color code is as follows: blue, sequences retrieved in ice: brown, sequences retrieved in soil: green, sequences retrieved in both ice and soil. Node support is given as maximum likelihood values (n. of bootstrap replicates) when equal or greater than 50%. The scale bar corresponds to 0.1 substitutions per site. Major groups are shown to the right. The parenthesis indicates the Chlamydomonas clade that was used for the biogeographic analysis.
A total of 15 archaeal Sanger sequences that clustered into 3 OTUs were also retrieved in one of the soil samples (N7) with closest relatives being uncultured Haloarchaea from alkaline-saline desert and Antarctic soils (Supp. Fig. 6), however we did not use Archaea-specific primers in this study.
Environmental classification of OTUs
We used the approach of Herbold et al.25 to classify bacterial phylotypes into “endemic” and “non-endemic” and within the non-endemic whether they are “cryophilic”, “non-cryophilic” or “polythermal” (Supp. Table 1). Of the 60 non-endemic bacterial OTUs found on Mt. Kilimanjaro summit, 46 (77%) were found to be cosmopolitan, meaning that all database entries that matched the representative sequence for that OTU had been previously observed in both perennially cold and temperate environments. Only 6 bacterial OTUs appeared to be endemic, meaning that they were less than 97% identical to any phylotype in the NCBI database.
Biogeographic analysis
Within the Bacteria and the Eukarya, two genera common to glacial and periglacial environments were selected to test their biogeographical distribution by comparison of genetic distance versus geographic distance. The analysis of the Polaromonas clade revealed that there was a slight (rM = 0.08) but significant (Mantel test, P = 0.002) increase in genetic distance with geographic distance, however spatial structuring was not evident for 11 out of 14 distance classes (Fig. 5). An expanded dataset of Darcy et al. 2011 was used (Supp. Table 2) including 96 long-read sequences from 25 geographically well separated glacial and periglacial environments (Supp. Fig. 2), which makes it the most inclusive long-read Polaromonas biogeographical analysis performed to date. The biogeographic analysis on the Chlamydomonas clade showed a slightly stronger (rM = 0.125, P = 0.001) increase in genetic distance with geographic distance, but spatial structuring was not evident for 8 out of 11 distance classes (Fig. 6).
Figure 5.
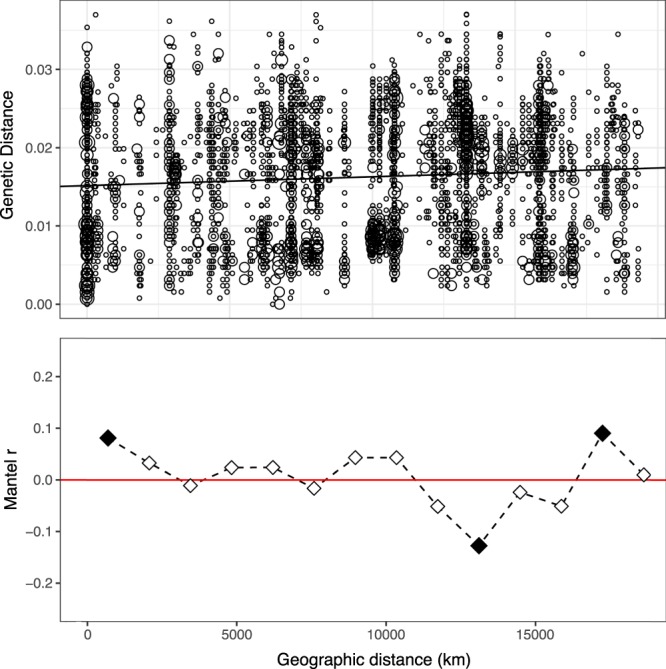
Genetic distance by geographical distance relationships for the dominant Polaromonas clade from Mt. Kilimanjaro summit. Upper panel shows pairwise comparisons of genetic distance among Polaromonas sequences (n = 4560) and geographic distance among different glacial environments (n = 25). The largest geographical distance in this study was between John Evans Glacier, Nunavut and Kamb Ice Stream, Antarctica (19307 km). There was a slight (rM = 0.08) but significant (Mantel test, P = 0.002) increase in genetic distance with geographic distance. Circle size is proportional to the number of pairwise comparisons at each point on the plot. Lower panel shows Mantel correlogram of genetic distance among the same sequences separated into 14 distance classes. The midpoint of each distance class is plotted. Shaded squares indicate significant tests in the Mantel correlogram after applying the Bonferroni correction. Spatial structuring was not evident for 11 out of 14 distance classes, supporting the contention that Polaromonas phylotypes are globally distributed.
Figure 6.
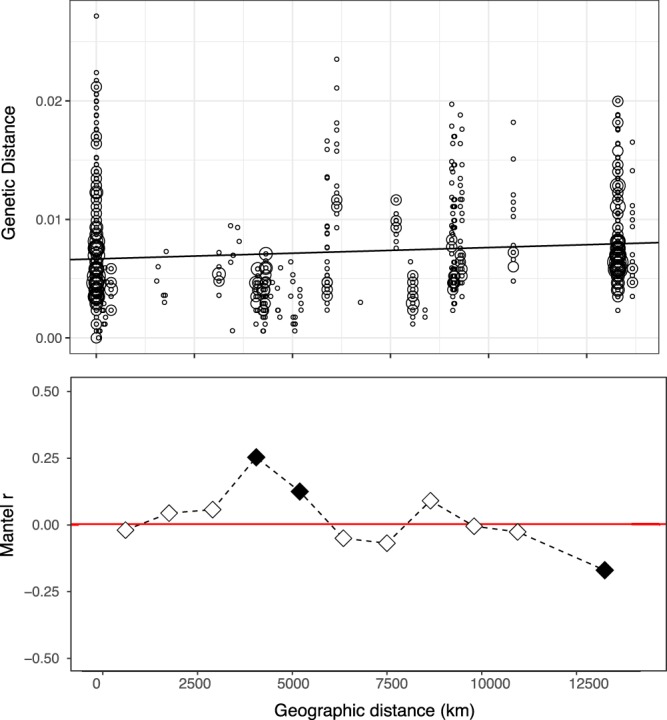
Genetic distance by geographical distance relationships for the dominant Chlamydomonas clade from Mt. Kilimanjaro summit. Upper panel shows pairwise comparisons of genetic distance among Chlamydomonas sequences (n = 1326) and geographic distance among different glacial environments (n = 15). The largest geographical distance in this study was between Kilimanjaro and Harding icefield, Alaska (13667 km). There was a slight (rM = 0.125) but significant (Mantel test, P = 0.001) increase in genetic distance with geographic distance. Circle size is proportional to the number of pairwise comparisons at each point on the plot. Lower panel shows Mantel correlogram of genetic distance among the same sequences separated into 11 distance classes. The midpoint of each distance class is plotted. Shaded squares indicate significant tests in the Mantel correlogram after applying the Bonferroni correction. Spatial structuring was not evident for 8 out of 11 distance classes.
Soil characteristics
Soil moisture levels decreased with distance from the glacier wall (Table 1) and are comparable with other high elevation and periglacial sites1,28. Soil pH did not show a trend and ranged from 7.5 to 7.9 across the transect (Table 1). DOC and TDN away from the glacier (>5 m) are similar to levels found for Antarctic bare, fell-field soils29 and high elevation sites in Peru and Colorado30. In general, nutrient levels were higher in samples taken close to the glacier, especially TDN (Table 1).
Discussion
Despite their ecological importance to downstream ecosystems, the study of microbial diversity at high elevations has lagged behind the study of other ecosystems. In particular, no work, using modern molecular approaches, has been done at high elevations in Africa. The present study was initiated to probe the diversity of soil- and ice-dwelling microbes near the top of Mt. Kilimanjaro. To our knowledge, this is the first investigation of the microbial diversity on Mt. Kilimanjaro or the African continent at elevations above 5000 m. This investigation provides a reference for future studies of microbial community structure and function in this extreme environment and other high-elevation sites in Africa.
Data from Illumina and Sanger libraries demonstrated that communities in both soil and ice are represented by a phylogenetically broad spectrum of microorganisms. Richness was high for both soil and ice communities for Bacteria (Table 2) and was close to that previously reported for other cold environments such as Antarctica and the Himalayas31,32. Glaciers can support very active microbial communities that sequester nutrients from the atmosphere33–35, which may explain the high diversity encountered in ice samples. The majority of bacterial OTUs were not closely related to cultured microorganisms and among the closest matching sequences were environmental sequences from cold (high-elevation and high-latitude), arid and heavy metal contaminated environments. These comprised putative psychrophilic, xerophilic, endolithic, radioresistant and halophilic microorganisms (Supp. Fig. 1). Diversity of Eukarya was a third of that for Bacteria according to the Shannon Index and Observed OTUs (Table 2), but was comparable to Eukaryotic diversity in polar waters36 and sea ice37.
Bacterial communities in ice and soil samples were significantly different (ANOSIM, P < 0.05). However, the soil sample closest to the ice wall harbored bacterial and eukaryotic communities closely resembling those of the ice samples. This may suggest that the main microbial input for the soils adjacent to the glaciers is the glacier itself and that these ice microbes are replaced by a soil community as the soils age. Ice microorganisms may not survive as well in soils because of exposure to environmental stressors such as freeze-thaw cycles and desiccation, stressors that are partially alleviated in the more constant ice environment. A likely consequence may therefore be the establishment in the soil of new phylotypes from the atmosphere.
Taxonomic diversity of bacteria
Overall, both Illumina and Sanger technologies yielded similar patterns of taxonomic diversity (Supp. Fig. 7) as has been noted in other studies38 with the taxonomic resolution achieved dependent on the abundance of reference sequences in the particular region of the rRNA tree of life. Sanger sequences were much longer (1345–1715 vs 150–250 bp) and were therefore used to identify sequences at a finer taxonomic level.
Betaproteobacteria (especially members of the Comamonadaceae) are the dominant members of the community similar to other recently deglaciated soils39–41. Comamonadaceae are phylogenetically similar in soil and ice (Supp. Fig. 1), indicating that ice may be seeding the youngest soils as previously seen in the High Andes of Peru40. Many Comamonadaceae are heterotrophs that are able to metabolize both recalcitrant42 and more labile carbon sources43 and could therefore use multiple aeolian deposited carbon sources. It is still unclear if many Comamonadaceae (e.g. Polaromonas) are indigenous to ice or if they are transients from the upper atmosphere. They are found in almost all glacial habitats studied to date39, perhaps indicating that they are being constantly deposited in glacial systems, but not necessarily growing there.
The other most abundant bacterial OTUs in soil were in the Chitinophagaceae (Bacteroidetes) but these were in lower relative abundance in the ice. Closely related sequences have been previously retrieved from similar environments such as oligotrophic volcanic cave sediments from Mt. Erebus, Antarctica44, fumarole soils at high elevation in the Andes1, pyroclastic deposits in Alaska45, and debris-covered glaciers in the Alps46. Chitinophagaceae have the ability to degrade complex polymers such as chitin, which provides microbes with C and N47. Chitin may come from Fungi and Arthropods, which often occur in recently deglaciated soils48. Members of the Chitinophagaceae may be opportunists, relying on Aeolian inputs and some species have a dormant stage49, which may allow them to persist in the atmosphere and survive in periglacial habitats once they are deposited.
All cyanobacteria sequences retrieved in ice and soil are closely related to non-heterocystous nitrogen fixers (Oscillatoria, Microcoleus, Leptolyngbya) commonly found in cold ecosystems50. Nitrogen is often a limiting nutrient in poorly developed early successional soils51. Other sites at similar elevations in Peru and Nepal have also recently been shown to have similar cyanobacteria3,40,52 and cyanobacteria have long been known to colonize barren habitats in the Arctic and Antarctica53. However, very dry, high-elevation sites like the stratovolcanoes in the Atacama Desert completely lack cyanobacteria, except in wet areas near active fumaroles1,2,54. Cyanobacteria were more abundant in the ice wall and the soil adjacent to the glacier than soil, possibly indicating that they depend on access to a constant source of water (provided by meltwater of the glacier), which is not available in soils further from the glacier.
Among the Alphaproteobacteria, the most abundant Hyphomicrobiaceae family was only found within ice samples and the soil closest to the ice wall and are known for being oligocarbophilic, thriving only in the presence of low carbon concentrations55, which can explain their being restricted to the ice and soils near the ice. Sphingomonadaceae were the second most abundant family within the Alphaproteobacteria and were more abundant in soils. Sequences from this family are commonly detected in glacial habitats and have been reported in Arctic cryoconite56, Antarctic ice sheet57 and Arctic glaciers58. Members of the Deinococcus-Thermus group were present in our site, as well as Antarctica Dry Valleys59 and sites over 5400 meters in the Andes60.
Taxonomic diversity of eukarya
Among the Eukarya, Cercozoa dominated the community and the most unexpected diversity found was in the order Vampyrellida (family Leptophryidae). These sequences are distinctly different from any taxa in Genbank (Fig. 4), suggesting that this unique environment harbors a previously unknown Vampyrellida community. Vampyrellida are only rarely recovered in environmental DNA surveys61 and have only been previously found at high elevations in soils from fumaroles on Volcán Socompa62. Our sequences were however less than 95% identical to those found on Volcán Socompa suggesting high endemicity of this group on Kilimanjaro. Given the presence of fumarolic activity on Mt. Kilimanjaro, it is possible that this group is associated with thermal environments. The Vampyrellida are very diverse and are generally known from aquatic habitats and soil where they are predators of algae, fungi, protozoa and small metazoans63. Vampyrellida have multiple nuclei63, which could lead to a bias and overestimation in their relative abundance. However, our Sanger clone libraries showed many distinct OTUs suggesting that multiple representation of the same organism was minimal. Many Cercozoa (including some Vampyrellida) are very resistant to drought64 and can respond quickly to periods of transient nutrient and water availability63, which may account for their abundance in the soils studied here. Food preferences may also be an important basis for lineage differentiation in Vampyrellida65. Their relative abundance was significantly higher in the ice samples (Fig. 3 and Supp. Fig. 8), suggesting that those in the soil may have come from the ice.
The most abundant OTU in the Eukarya was very closely affiliated with the known Cercozoans Trinema lineare and Trinema enchelis (order Euglyphida)66, common testatae amebae in moist environments. The same taxa of testatae amebae have recently been found for the first time in glaciers in the Andes67 and in glacier forefield soils in the Alps68. Heterotrophic Cercozoa seem to be more abundant than their potential food sources in our samples. However, inverted trophic pyramids have been seen before at high elevation where producers are scarce1 or absent2. The high abundance of these predatory microeukaryotes could be sustained by feeding on fungi and algae in addition to bactivory69.
These soils, like other even higher-elevation volcanoes1,2, harbored some of the simplest fungal communities yet described compared to, for example the 15 lineages recovered in the Dry Valleys of Antarctica70. Environmental extremes characteristic of high elevation, such as freeze-thaw cycles and high UV levels may limit fungal communities on Kilimanjaro compared to polar regions. Chytrids dominated our Sanger fungal libraries similar to previous studies of high-elevation and high-latitude soils71. Most chytrids were in the Spizellomycetes, which are commonly found in harsh soil environments such as arid grasslands72, and glacial till73. They can resist extremes of temperature, pH, and desiccation74 and can decompose pollen or be parasitic. These features may explain their presence on Mt. Kilimanjaro and their wide geographical distribution.
Green algae sequences found in ice and soils of Kilimanjaro are closely related to those found in similar environments such as alpine snow75, Arctic glaciers76 and Antarctic snow77. Their high relative abundance and the scarcity of cyanobacterial sequences in these soils suggests that they may play a significant role as primary producers in this habitat and in early stages of succession following glacial retreat as has been previously suggested for green algae in high-altitude ecosystems78.
Taxonomic diversity of archaea
Archaea sequences distantly related (92%) to the Haloarchaea class were only retrieved with Sanger sequencing in one of the soil samples (N7) (Supp. Fig. 6). Scarcity of liquid water at this elevation can drive the formation of salty pockets within the soils that can explain the presence of Haloarchaea but more work is needed to understand the true phylogenetic diversity of Archaea on the top of Mt. Kilimanjaro. It is known that Archaea are more difficult to detect in cold79,80 and hot deserts81, which may explain our findings.
Biogeography and spatial scale
Kilimanjaro is the most isolated high-altitude peak on Earth82 and approximately 5700 km from the closest mountain range with similar and higher elevation peaks (the Himalayas) (Google earth data, last accessed on 10 March 2018) making this site ideal for testing ideas related to microbial dispersal and biogeography. Despite being separated by large distances and climatic barriers, it has been proposed that cold high-elevation and high-latitude environments are connected through the upper atmosphere39 but data is lacking for Africa. Small microbes are easily dispersed over long distances, but their long-term survival requires a number of distinct adaptations, similar to those required to survive in icy environments83. The top of Mt. Kilimanjaro may be an oasis where microbes in transit in the upper atmosphere persist, but more work is needed to determine if they can grow on Kilimanjaro. The present study is a first step towards gaining an understanding of the global dispersal of microbial phylotypes to high elevation sites in Africa.
It is currently unknown how Aeolian deposition affects microbial composition and if Kilimanjaro’s great distance from similar environments is reflected in more speciation. Our survey showed that most bacterial diversity at our site is cosmopolitan, as defined by Herbold et al.25, with the vast majority of OTUs having close matches with sequences from very distant both cold and temperate environments showing little endemism in this cold isolated environment. More evidence of minimal endemism of this isolated site comes from the biogeographical analysis of one of the most abundant clades within the Bacteria, Polaromonas (Fig. 5). Our analysis showed remarkably little increase in genetic diversity with geographic distance, pointing towards a uniform distribution of Polaromonas phylotypes across the cryosphere. These results confirm the conclusion of previous work that Polaromonas phylotypes are globally distributed in the cryosphere39. Interaction with local microbiota may drive microevolution and post-selection niche separation within Polaromonas on the top of Mt. Kilimanjaro.
The finding in Kilimanjaro samples of both cosmopolitan and endemic microorganisms helps corroborate the hypothesis that the distribution of microbial species in the cryosphere is the outcome of the continuum between two opposing theories in microbial ecology: endemism and cosmopolitanism84. Moreover, the closest matches of 14% of sequences retrieved from our samples were from heavy metal contaminated sites. Since our site is not known to contain relevant quantities of heavy metal, it may be possible that tolerance and resistance mechanisms to heavy metals overlap with those employed by microorganisms to cope with stresses found in extreme environments such as high radiation, freeze-thaw cycles, low nutrient and water levels81,85.
It is likely that larger eukaryotic microbes might be more dispersal limited than smaller Bacteria. Previous work in the cryosphere has shown that larger microbes like algae and zoosporic fungi show distinct biogeographic patterns at global and regional scales3,86,87 and this could be the case for the most abundant phylum retrieved from our samples, the Cercozoa, which showed a highly diverse new clade within the Vampyrellida. Successful dispersal of small microbes is due to their high abundance and long residence time in the atmosphere88, which may not be the case for eukaryotic microorganisms due to their average larger size. Evidence of a pattern of genetic isolation by genetic distance was revealed by the phylo-geographic analysis of a highly abundant Chlamydomonas clade. However, despite the large distances, the pattern revealed was weak (Mantel rM = 0.125, P = 0.001) and it is in accordance with recent molecular studies that suggest that red snow algae may be readily dispersed across the Earth89,90. Our analyses suggest that Chlamydomonas may be successful at global dispersal, however in a more limited fashion compared to most bacteria.
Conclusions and implications
Microbial diversity in the extreme environments on top of Mt. Kilimanjaro proved to be higher than expected compared to high volcanoes in drier regions1,2,54, especially for the Bacteria. In addition, the ice bacterial community was significantly different from the soil community, suggesting that soils are predominately seeded by wind-transported terrestrial sources. The combination of cosmopolitan bacterial diversity and weak biogeographical patterns for the clades analyzed suggest that the effect of distance and local environmental conditions is overwhelmed by continuous dispersal. Taken together, considering the isolation of Mt. Kilimanjaro from other perennially cold environments, our results strongly support the concept of a global distribution of microbial ecotypes throughout the cold terrestrial biosphere as has been suggested by other studies of Bacteria3,39,50. This finding suggests that there may be gene flow between the polar regions or that the rate of evolutionary divergence has been slow relative to the timescales of isolation. In depth molecular comparisons of microbial diversity with other distant high elevation sites could provide further insights into dispersal mechanisms and may help elucidate how microbes adapt and diversify in response to environmental pressures. Unravelling the full extent of diversity in these environments would provide a unique opportunity to investigate microbial endemism and evolution as well as roles of inoculum from the atmosphere, fumaroles and glaciers for microbial communities in recently deglaciated soils.
As glacier recession continues unabated, understanding the biodiversity and function of organisms in these glaciers and periglacial soils is important, before these ecosystems are changed forever. One of the main concerns is the loss of biodiversity and potentially the loss from the biosphere of a pool of genes adapted to surviving and thriving in the cold91. Our work adds to a growing body of knowledge about microbial diversity in high elevation ecosystems that are currently responding to a rapidly changing climate. The presence of high bacterial and eukaryotic diversity indicates that specific life forms have evolved to thrive in this extreme environment and may provide a model for viable habitats for life on early Mars since similar conditions may have existed or still exist in sediments adjacent to and beneath the Martian north polar ice cap9,92.
Supplementary information
Acknowledgements
We thank Eli Gendron, Jessica Henley and Courtney Naff for laboratory and bioinformatics assistance and Sky Beard and Ezio Melotti for help in the field. We also thank our climbing guide Philemon Chacha and the chief park warden of Kilimanjaro National Park for permission to take the samples used in this study. This work, including the efforts of Lara Vimercati and Steve Schmidt was funded by NSF grants for studying microbial community assembly and migration to higher elevations in response to global warming (DEB-1258160 and DEB-1457827) and a grant from the USAF Office of Scientific Research for studying microbial adaptation to extreme elevations (FA9550-14-1-0006).
Author Contributions
L.V. determined the study design and collected and processed the samples. L.V., J.D. and S.K.S. contributed to analysis and interpretation of community data. L.V. and S.K.S. primarily wrote the manuscript and prepared figures, and all authors contributed text and revisions to the manuscript.
Competing Interests
The authors declare no competing interests.
Footnotes
Publisher’s note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Supplementary information accompanies this paper at 10.1038/s41598-019-46521-0.
References
- 1.Costello EK, Halloy SRP, Reed SC, Sowell P, Schmidt SK. Fumarole-supported islands of biodiversity within a hyperarid, high-elevation landscape on Socompa Volcano, Puna de Atacama, Andes. Appl. Environ. Microbiol. 2009;75:735–747. doi: 10.1128/AEM.01469-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lynch RC, et al. The potential for microbial life in the highest‐elevation (>6000 masl) mineral soils of the Atacama region. J. Geophys. Res. Biogeochem. 2012;117:G02028. [Google Scholar]
- 3.Schmidt SK, et al. Phylogeography of microbial phototrophs in the dry valleys of the high Himalayas and Antarctica. Proc. Roy. Soc. B. 2011;278:702–708. doi: 10.1098/rspb.2010.1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaser G, Hardy DR, Mölg T, Bradley RS, Hyera TM. Modern glacier retreat on Kilimanjaro as evidence of climate change: observations and facts. Int. J. Climatol. 2004;24:329–339. doi: 10.1002/joc.1008. [DOI] [Google Scholar]
- 5.Thompson LG, Brecher HH, Mosley-Thompson E, Hardy DR, Mark BG. Glacier loss on Kilimanjaro continues unabated. Proc. Natl. Acad. Sci. 2009;106:19770–19775. doi: 10.1073/pnas.0906029106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Humphries DW. Preliminary notes on the glaciology of Kilimanjaro. J. Glaciol. 1959;3:475–479. doi: 10.1017/S0022143000017226. [DOI] [Google Scholar]
- 7. Mölg, T., Hardy, D. R. & Kaser, G. Solar‐radiation‐maintained glacier recession on Kilimanjaro drawn from combined ice‐radiation geometry modeling. J. Geophys. Res. Atmos. 108 (D23) (2003).
- 8.Xiang SR, Shang TC, Chen Y, Yao TD. Deposition and post deposition mechanisms as possible drivers of microbial population variability in glacier ice. FEMS Microbiol. Ecol. 2009;70:165–176. doi: 10.1111/j.1574-6941.2009.00759.x. [DOI] [PubMed] [Google Scholar]
- 9.Ponce, A., Anderson, R. C., McKay, C. P. Microbial habitability in periglacial soils of Kilimanjaro. In Analogue sites for Mars missions: MSL and beyond. 1612 (2011).
- 10.Parsons AN, Barrett JE, Wall DH, Virginia RA. Soil carbon dioxide flux in Antarctic dry valley ecosystems. Ecosystems. 2004;7:286–295. doi: 10.1007/s10021-003-0132-1. [DOI] [Google Scholar]
- 11.Drees KP, et al. Bacterial community structure in the hyperarid core of the Atacama Desert, Chile. Appl. Environ. Microbiol. 2006;72:7902–7908. doi: 10.1128/AEM.01305-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pruesse E, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007;35:7188–7196. doi: 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ludwig W, et al. ARB: a software environment for sequence data. Nucleic Acids Res. 2004;32:1363–1371. doi: 10.1093/nar/gkh293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Huber T, Faulkner G, Hugenholtz P. Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics. 2004;20:2317–2319. doi: 10.1093/bioinformatics/bth226. [DOI] [PubMed] [Google Scholar]
- 15.Ashelford KE, Chuzhanova NA, Fry JC, Jones AJ, Weightman AJ. New screening software shows that most recent large 16S rRNA gene clone libraries contain chimeras. Appl. Environ. Microbiol. 2006;72:5734–5741. doi: 10.1128/AEM.00556-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schloss PD, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013;30:2725–2729. doi: 10.1093/molbev/mst197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Caporaso JG, et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods. 2010;7:335–336. doi: 10.1038/nmeth.f.303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Darcy JL, Schmidt SK. Nutrient limitation of microbial phototrophs on a debris-covered glacier. Soil Biol. Biochem. 2016;95:156–163. doi: 10.1016/j.soilbio.2015.12.019. [DOI] [Google Scholar]
- 20.Caporaso JG, et al. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics. 2010;26:266–267. doi: 10.1093/bioinformatics/btp636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price MN, Dehal PS, Arkin AP. FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol. Biol. Evol. 2009;26:1641–1650. doi: 10.1093/molbev/msp077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Berger WH, Parker FL. Diversity of planktonic foraminifera in deep-sea sediments. Science. 1970;168:1345–1347. doi: 10.1126/science.168.3937.1345. [DOI] [PubMed] [Google Scholar]
- 23.Lozupone C, Hamady M, Knight R. UniFrac–an online tool for comparing microbial community diversity in a phylogenetic context. BMC Bioinformatics. 2006;7:1. doi: 10.1186/1471-2105-7-371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oksanen, J. et al. Vegan: Community Ecology Package. R Package version 2.0–10, http://CRAN.R-project.org/package=vegan (2013).
- 25.Herbold CW, Lee CK, McDonald IR, Cary SC. Evidence of global-scale aeolian dispersal and endemism in isolated geothermal microbial communities of Antarctica. Nature Commun. 2014;5:3875. doi: 10.1038/ncomms4875. [DOI] [PubMed] [Google Scholar]
- 26.King AJ, et al. Biogeography and habitat modelling of high-alpine bacteria. Nature Commun. 2010;1:53. doi: 10.1038/ncomms1055. [DOI] [PubMed] [Google Scholar]
- 27.Weintraub MN, Scott-Denton LE, Schmidt SK, Monson RK. The effects of tree rhizodeposition on soil exoenzyme activity, dissolved organic carbon, and nutrient availability in a subalpine forest ecosystem. Oecologia. 2007;154:327–338. doi: 10.1007/s00442-007-0804-1. [DOI] [PubMed] [Google Scholar]
- 28.Niederberger TD, et al. Microbial community composition in soils of Northern Victoria Land, Antarctica. Environ. Microbiol. 2008;10:1713–1724. doi: 10.1111/j.1462-2920.2008.01593.x. [DOI] [PubMed] [Google Scholar]
- 29.Roberts P, Newsham KK, Bardgett RD, Farrar JF, Jones DL. Vegetation cover regulates the quantity, quality and temporal dynamics of dissolved organic carbon and nitrogen in Antarctic soils. Polar Biol. 2009;32:999–1008. doi: 10.1007/s00300-009-0599-0. [DOI] [Google Scholar]
- 30.King AJ, Meyer AF, Schmidt SK. High levels of microbial biomass and activity in unvegetated tropical and temperate alpine soils. Soil Biol. Biochem. 2008;40:2605–2610. doi: 10.1016/j.soilbio.2008.06.026. [DOI] [Google Scholar]
- 31.Rhodes, M. et al. Alpine and arctic soil microbial communities. The Prokaryotes, 43–55 (Springer Berlin Heidelberg, 2013).
- 32.Yergeau E, Newsham KK, Pearce DA, Kowalchuk GA. Patterns of bacterial diversity across a range of Antarctic terrestrial habitats. Environ. Microbiol. 2007;9:2670–2682. doi: 10.1111/j.1462-2920.2007.01379.x. [DOI] [PubMed] [Google Scholar]
- 33.Anesio AM, Hodson AJ, Fritz A, Psenner R, Sattler B. High microbial activity on glaciers: importance to the global carbon cycle. Global Change Biol. 2009;15:955–960. doi: 10.1111/j.1365-2486.2008.01758.x. [DOI] [Google Scholar]
- 34.Steven B, Pollard WH, Greer CW, Whyte LG. Microbial diversity and activity through a permafrost/ground ice core profile from the Canadian high Arctic. Environ. Microbiol. 2008;10:3388–3403. doi: 10.1111/j.1462-2920.2008.01746.x. [DOI] [PubMed] [Google Scholar]
- 35.Vincent WF, Mueller DR, Bonilla S. Ecosystems on ice: the microbial ecology of Markham Ice Shelf in the high. Arctic. Cryobiol. 2004;48:103–112. doi: 10.1016/j.cryobiol.2004.01.006. [DOI] [PubMed] [Google Scholar]
- 36.Luo W, et al. Molecular diversity of microbial eukaryotes in sea water from Fildes Peninsula, King George Island, Antarctica. Polar Biol. 2016;39:605–616. doi: 10.1007/s00300-015-1815-8. [DOI] [Google Scholar]
- 37.Majaneva M, Rintala JM, Piisilä M, Fewer DP, Blomster J. Comparison of wintertime eukaryotic community from sea ice and open water in the Baltic Sea, based on sequencing of the 18S rRNA gene. Polar Biol. 2012;35:875–889. doi: 10.1007/s00300-011-1132-9. [DOI] [Google Scholar]
- 38.Harris J, et al. Phylogenetic stratigraphy in the Guerrero Negro hypersaline microbial mat. ISME J. 2013;7:50–60. doi: 10.1038/ismej.2012.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darcy JL, Lynch RC, King AJ, Robeson MS, Schmidt SK. Global distribution of Polaromonas phylotypes – Evidence for a highly successful dispersal capacity. PLOS ONE. 2011;6(8):e23742. doi: 10.1371/journal.pone.0023742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nemergut DR, et al. Microbial community succession in an unvegetated, recently deglaciated soil. Microb. Ecol. 2007;53:110–122. doi: 10.1007/s00248-006-9144-7. [DOI] [PubMed] [Google Scholar]
- 41.Sattin SR, et al. Functional shifts in unvegetated, perhumid, recently-deglaciated soils do not correlate with shifts in soil bacterial community composition. J. Microbiol. 2009;47:673–681. doi: 10.1007/s12275-009-0194-7. [DOI] [PubMed] [Google Scholar]
- 42.Park IH, Ka JO. Isolation and characterization of 4-(2, 4-dichlorophenoxy) butyric acid-degrading bacteria from agricultural soils. J. Microbiol. Biotechnol. 2003;13:243–250. [Google Scholar]
- 43.Willems Anne. The Prokaryotes. Berlin, Heidelberg: Springer Berlin Heidelberg; 2014. The Family Comamonadaceae; pp. 777–851. [Google Scholar]
- 44.Tebo BM, et al. Microbial communities in dark oligotrophic volcanic ice cave ecosystems of Mt. Erebus, Antarctica. Frontiers Microbiol. 2015;6:179. doi: 10.3389/fmicb.2015.00179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeglin LH, Wang B, Waythomas C, Rainey F, Talbot SL. Organic matter quantity and source affects microbial community structure and function following volcanic eruption on Kasatochi Island, Alaska. Environ. Microbiol. 2016;18:146–158. doi: 10.1111/1462-2920.12924. [DOI] [PubMed] [Google Scholar]
- 46.Franzetti A, et al. Bacterial community structure on two alpine debris-covered glaciers and biogeography of Polaromonas phylotypes. ISME J. 2013;7:1483–1492. doi: 10.1038/ismej.2013.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Moorhead DL, Lashermes G, Sinsabaugh RL. A theoretical model of C- and N-acquiring exoenzyme activities, which balances microbial demands during decomposition. Soil Biol. Biochem. 2012;53:133–141. doi: 10.1016/j.soilbio.2012.05.011. [DOI] [Google Scholar]
- 48.Zumsteg A, et al. Bacterial, archaeal and fungal succession in the forefield of a receding glacier. Microb. Ecol. 2012;63:552–564. doi: 10.1007/s00248-011-9991-8. [DOI] [PubMed] [Google Scholar]
- 49.Kämpfer P, et al. Transfer of [Flexibacter] sancti, [Flexibacter] filiformis, [Flexibacter] japonensis and [Cytophaga] arvensicola to the genus Chitinophaga and description of Chitinophaga skermanii sp. nov. Int. J. Syst. Evol. Microbiol. 2006;56:2223–2228. doi: 10.1099/ijs.0.64359-0. [DOI] [PubMed] [Google Scholar]
- 50.Jungblut AD, Lovejoy C, Vincent WF. Global distribution of cyanobacterial ecotypes in the cold biosphere. ISME J. 2010;4:191–202. doi: 10.1038/ismej.2009.113. [DOI] [PubMed] [Google Scholar]
- 51.Knelman JE, et al. Nutrient addition dramatically accelerates microbial community succession. PLOS ONE. 2014;9(7):e102609. doi: 10.1371/journal.pone.0102609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Schmidt SK, et al. The earliest stages of ecosystem succession in high-elevation (5000 metres above sea level), recently deglaciated soils. Proc. Roy. Soc. B. 2008;275:2793–2802. doi: 10.1098/rspb.2008.0808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Quesada, A. & Vincent, W. F. Cyanobacteria in the cryosphere: snow, ice and extreme cold. Ecology of cyanobacteria II, 387–399. (Springer Netherlands, 2012).
- 54.Solon AJ, et al. Microbial Communities of High-Elevation Fumaroles, Penitentes, and Dry Tephra “Soils” of the Puna de Atacama Volcanic Zone. Microb. Ecol. 2018;76(2):340–351. doi: 10.1007/s00248-017-1129-1. [DOI] [PubMed] [Google Scholar]
- 55. Oren, A. & Xu, X. W. The family Hyphomicrobiaceae. The Prokaryotes, 247–281. (Berlin Heidelberg, 2014).
- 56.Edwards A, et al. Possible interactions between bacterial diversity, microbial activity and supraglacial hydrology of cryoconite holes in Svalbard. ISME J. 2011;5:150–160. doi: 10.1038/ismej.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alekhina IA, et al. Molecular analysis of bacterial diversity in kerosene-based drilling fluid from the deep ice borehole at Vostok, East Antarctica. FEMS Microbiol. Ecol. 2007;59:289–299. doi: 10.1111/j.1574-6941.2006.00271.x. [DOI] [PubMed] [Google Scholar]
- 58.Zeng YX, et al. Diversity of bacteria in surface ice of Austre Lovénbreen glacier, Svalbard. Arch. Microbiol. 2013;195:313–322. doi: 10.1007/s00203-013-0880-z. [DOI] [PubMed] [Google Scholar]
- 59.De la Torre JR, Goebel BM, Friedmann EI, Pace NR. Microbial diversity of cryptoendolithic communities from the McMurdo Dry Valleys, Antarctica. Appl. Environ. Microbiol. 2003;69:3858–3867. doi: 10.1128/AEM.69.7.3858-3867.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schmidt SK, et al. Microbial activity and diversity during extreme freeze-thaw cycles in periglacial soils, 5400 m elevation, Cordillera Vilcanota, Perú. Extremophiles. 2009;13:807–816. doi: 10.1007/s00792-009-0268-9. [DOI] [PubMed] [Google Scholar]
- 61.Berney C, Fahrni J, Pawlowski J. How many novel eukaryotic ‘kingdoms’? Pitfalls and limitations of environmental DNA surveys. BMC biology. 2004;2(1):13. doi: 10.1186/1741-7007-2-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Schmidt SK, et al. Life at extreme elevations on Atacama volcanoes: the closest thing to Mars on Earth? Antonie van Leeuwenhoek. 2018;20:1–3. doi: 10.1007/s10482-018-1066-0. [DOI] [PubMed] [Google Scholar]
- 63.Berney C, et al. Vampires in the oceans: predatory cercozoan amoebae in marine habitats. ISME J. 2013;7:2387–2399. doi: 10.1038/ismej.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Harder CB, et al. Local diversity of heathland Cercozoa explored by in-depth sequencing. ISME J. 2016;10:2488–2497. doi: 10.1038/ismej.2016.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hess S, Sausen N, Melkonian M. Shedding light on vampires: the phylogeny of vampyrellid amoebae revisited. PLOS ONE. 2012;7(2):e31165. doi: 10.1371/journal.pone.0031165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heger TJ, et al. Molecular phylogeny of euglyphid testate amoebae (Cercozoa: Euglyphida) suggests transitions between marine supralittoral and freshwater/terrestrial environments are infrequent. Mol. Phylogenet. Evol. 2010;55:113–122. doi: 10.1016/j.ympev.2009.11.023. [DOI] [PubMed] [Google Scholar]
- 67.Santibáñez PA, Kohshima S, Scheihing RA, Silva R, Casassa G. First record of testate amoebae on glaciers and description of a new species Puytoracia jenswendti nov. sp. (Rhizaria, Euglyphida) Acta Protozool. 2015;50:1–14. [Google Scholar]
- 68.Lazzaro A, Risse-Buhl U, Brankatschk R. Molecular and morphological snapshot characterisation of the protist communities in contrasting Alpine glacier forefields. Acta Protozool. 2015;54(2):143–154. [Google Scholar]
- 69.Krashevska V, et al. Microorganisms as driving factors for the community structure of testate amoebae along an altitudinal transect in tropical mountain rain forests. Soil Biol. Biochem. 2008;40:2427–2433. doi: 10.1016/j.soilbio.2008.06.004. [DOI] [Google Scholar]
- 70.Chan Y, Van Nostrand JD, Zhou J, Pointing SB, Farrell RL. Functional ecology of an Antarctic dry valley. Proc. Natl. Acad. Sci. 2013;110:8990–8995. doi: 10.1073/pnas.1300643110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Freeman KR, et al. Evidence that chytrids dominate fungal communities in high-elevation soils. Proc. Natl. Acad. Sci. 2009;106:18315–18320. doi: 10.1073/pnas.0907303106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lozupone CA, Klein DA. Molecular and cultural assessment of chytrid and Spizellomyces populations in grassland soils. Mycologia. 2002;94:411–420. doi: 10.1080/15572536.2003.11833206. [DOI] [PubMed] [Google Scholar]
- 73.Wakefield WS, et al. A molecular phylogenetic evaluation of the Spizellomycetales. Mycologia. 2010;102:596–604. doi: 10.3852/09-120. [DOI] [PubMed] [Google Scholar]
- 74.Gleason FH, Schmidt SK, Marano A. Can zoosporic true fungi grow or survive in extreme or stressful environments? Extremophiles. 2010;14:417–425. doi: 10.1007/s00792-010-0323-6. [DOI] [PubMed] [Google Scholar]
- 75.Remias D, Karsten U, Lütz C, Leya T. Physiological and morphological processes in the Alpine snow alga Chloromonas nivalis (Chlorophyceae) during cyst formation. Protoplasma. 2010;243:73–86. doi: 10.1007/s00709-010-0123-y. [DOI] [PubMed] [Google Scholar]
- 76.Leya T, Müller T, Ling HU, Fuhr G. Snow algae from north-western Spitsbergen (Svalbard). The coastal ecosystem of Kongsfjorden, Svalbard. Synopsis of biological research performed at the Koldewey Station in the years 1991–2003. Berichte zur Polar-und Meeresforschung (Reports on Polar and Marine Research) 2004;492:46–54. [Google Scholar]
- 77.Remias D, Wastian H, Lütz C, Leya T. Insights into the biology and phylogeny of Chloromonas polyptera (Chlorophyta), an alga causing orange snow in Maritime Antarctica. Antarct. Sci. 2013;25:648–656. doi: 10.1017/S0954102013000060. [DOI] [Google Scholar]
- 78.Schmidt SK, Darcy JL. Phylogeny of ulotrichalean algae from extreme high-altitude and high-latitude ecosystems. Polar Biol. 2015;38:689–697. doi: 10.1007/s00300-014-1631-6. [DOI] [Google Scholar]
- 79.Ayton J, Aislabie J, Barker GM, Saul D, Turner S. Crenarchaeota affiliated with group 1.1 b are prevalent in coastal mineral soils of the Ross Sea region of Antarctica. Environ. Microbiol. 2010;12:689–703. doi: 10.1111/j.1462-2920.2009.02111.x. [DOI] [PubMed] [Google Scholar]
- 80.Cary SC, McDonald IR, Barrett JE, Cowan DA. On the rocks: the microbiology of Antarctic Dry Valley soils. Nat. Rev. Microbiol. 2010;8:129–138. doi: 10.1038/nrmicro2281. [DOI] [PubMed] [Google Scholar]
- 81.Chanal A, et al. The desert of Tataouine: an extreme environment that hosts a wide diversity of microorganisms and radiotolerant bacteria. Environ. Microbiol. 2006;8:514–525. doi: 10.1111/j.1462-2920.2005.00921.x. [DOI] [PubMed] [Google Scholar]
- 82.Duane WJ, Pepin NC, Losleben ML, Hardy DR. General characteristics of temperature and humidity variability on Kilimanjaro, Tanzania. Arct. Antarct. Alp. Res. 2008;40:323–334. doi: 10.1657/1523-0430(06-127)[DUANE]2.0.CO;2. [DOI] [Google Scholar]
- 83.Pearce DA, et al. Microorganisms in the atmosphere over Antarctica. FEMS Microbiol. Ecol. 2009;69:143–157. doi: 10.1111/j.1574-6941.2009.00706.x. [DOI] [PubMed] [Google Scholar]
- 84.Martiny JB, et al. Microbial biogeography: putting microorganisms on the map. Nat. Rev. Microbiol. 2006;4:102–112. doi: 10.1038/nrmicro1341. [DOI] [PubMed] [Google Scholar]
- 85.Dib J, Motok J, Zenoff VF, Ordoñez O, Farías ME. Occurrence of resistance to antibiotics, UV-B, and arsenic in bacteria isolated from extreme environments in high-altitude (above 4400 m) Andean wetlands. Curr. Microbiol. 2008;56:510–517. doi: 10.1007/s00284-008-9103-2. [DOI] [PubMed] [Google Scholar]
- 86.De Wever A, et al. Hidden levels of phylodiversity in Antarctic green algae: further evidence for the existence of glacial refugia. Proc. Roy. Soc. B. 2009;276:3591–3599. doi: 10.1098/rspb.2009.0994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naff CS, Darcy JL, Schmidt SK. Phylogeny and biogeography of an uncultured clade of snow chytrids. Environ. Microbiol. 2013;15:2672–2680. doi: 10.1111/1462-2920.12116. [DOI] [PubMed] [Google Scholar]
- 88.Schmidt SK, Nemergut DR, Darcy JL, Lynch R. Do bacterial and fungal communities assemble differently during primary succession? Mol. Ecol. 2014;23:254–258. doi: 10.1111/mec.12589. [DOI] [PubMed] [Google Scholar]
- 89.Lutz S, et al. The biogeography of red snow microbiomes and their role in melting arctic glaciers. Nature communications. 2016;7:11968. doi: 10.1038/ncomms11968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Brown SP, Ungerer MC, Jumpponen A. A community of clones: Snow algae are diverse communities of spatially structured clones. International Journal of Plant Sciences. 2016;177(5):432–439. doi: 10.1086/686019. [DOI] [Google Scholar]
- 91.Anesio AM, Laybourn-Parry J. Glaciers and ice sheets as a biome. Trends Ecol. Evol. 2012;27:219–225. doi: 10.1016/j.tree.2011.09.012. [DOI] [PubMed] [Google Scholar]
- 92.Preston LJ, Dartnell LR. Planetary habitability: lessons learned from terrestrial analogues. Int. J. Astrobiol. 2014;13:81–98. doi: 10.1017/S1473550413000396. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.