

Abstract
Cortical spreading depression (CSD) is a wave of neuronal depolarization thought to underlie migraine aura. Calcitonin gene-related peptide (CGRP) is a potent vasodilator involved in migraine pathophysiology. Evidence for functional connectivity between CSD and CGRP has triggered scientific interest in the possibility that CGRP antagonism may disrupt vascular responses to CSD and the ensuing plasma protein extravasation (PPE). Using imaging tools that allow us to generate continuous, live, high-resolution views of spatial and temporal changes that affect arteries and veins in the dura and pia, we determined the extent to which CGRP contributes to the induction of arterial dilatation or PPE by CSD in female rats, and how these events are affected by the anti-CGRP monoclonal antibody (anti-CGRP-mAb) fremanezumab. We found that the CSD-induced brief dilatation and prolonged constriction of pial arteries, prolonged dilatation of dural arteries and PPE are all unaffected by fremanezumab, whereas the brief constriction and prolonged dilatation of pial veins are affected. In comparison, although CGRP infusion gave rise to the expected dilatation of dural arteries, which was effectively blocked by fremanezumab, it did not induce dilatation in pial arteries, pial veins, or dural veins. It also failed to induce PPE. Regardless of whether the nociceptors become active before or after the induction of arterial dilatation or PPE by CSD, the inability of fremanezumab to prevent them suggests that these events are not mediated by CGRP, a conclusion with important implications for our understanding of the mechanism of action of anti-CGRP-mAbs in migraine prevention.
SIGNIFICANCE STATEMENT The current study identifies fundamental differences between two commonly used models of migraine, CSD induction and systemic CGRP infusion. It raises the possibility that conclusions drawn from one model may not be true or relevant to the other. It sharpens the need to accept the view that there is more than one truth to migraine pathophysiology and that it is unlikely that one theory will explain all types of migraine headache or the mechanisms of action of drugs that prevent it. Regarding the latter, it is concluded that not all vascular responses in the meninges are born alike and, consequently, that drugs that prevent vascular dilatation through different molecular pathways may have different therapeutic outcomes in different types of migraine.
Keywords: aura, CGRP, CSD, migraine, PPE, vasodilation
Introduction
Cortical spreading depression (CSD) is a wave of neuronal depolarization thought to be the phenomenon that underlies migraine aura (Lauritzen, 1994; Nozari et al., 2010). Associated with this wave of depolarization is a brief (1–2 min) vasodilatation that is followed by a prolonged vasoconstriction of cerebral (pial/cortical) arteries (Colonna et al., 1994; Wahl et al., 1994; Reuter et al., 1998; Brennan et al., 2007). As evidence for the role of calcitonin gene-related peptide (CGRP) in migraine continues to mount (Russo, 2015; Edvinsson et al., 2018), so do attempts to determine whether CGRP is involved in the process by which CSD activates the trigeminovascular pathway (Close et al., 2019) and how drugs that intercept CGRP or interfere with its ability to bind to its receptors prevent the initiation of the headache (Dodick et al., 2014, 2018; Tepper et al., 2017; Skljarevski et al., 2018; Stauffer et al., 2018).
To date, there is no direct evidence implicating a role for CGRP in CSD. In the absence of such evidence, presumed connections between CSD and CGRP are based on studies that show that both CSD induction and CGRP infusion dilate cortical surface arterioles (Hong et al., 1994; Jansen-Olesen et al., 2003; Brennan et al., 2007; Chang et al., 2010; Ayata and Lauritzen, 2015; Hoffmann et al., 2019), and that CSD-induced dilatation of pial arteries is mediated by CGRP (Wahl et al., 1994; Reuter et al., 1998) and intact meningeal nociceptors (Reuter et al., 1998; Bolay et al., 2002).
The mounting evidence for functional connectivity between CSD, vascular response to CSD, and CGRP has triggered scientific interest in the possibility that CGRP antagonism may disrupt the neurovascular coupling in CSD to the extent that it may minimize or prevent either the neuronal or the vascular or both events from occurring. To date, there are two peer-reviewed studies that tested the role of CGRP receptors in vitro using CGRP receptor antagonists (Tozzi et al., 2012; Wang et al., 2016), and three in vivo (Colonna et al., 1994; Wahl et al., 1994; Reuter et al., 1998)—all focusing on events that occur in the cortex rather than the meninges. The in vitro studies showed that CGRP receptor antagonists reduce the overall magnitude of cortical (and retinal) spreading depression (Tozzi et al., 2012; Wang et al., 2016), whereas the in vivo studies showed that direct administration of CGRP receptor antagonists to the pia reduces CSD-induced dilatation of cerebral arterioles (Colonna et al., 1994; Wahl et al., 1994; Reuter et al., 1998) or cerebral blood flow (Colonna et al., 1994; Wahl et al., 1994; Reuter et al., 1998).
Based on clinical evidence for the role of the dura in the initiation of the headache phase of migraine with aura (Penfield and McNaughton, 1940; Ray and Wolff, 1940), preclinical evidence for the activation of meningeal nociceptors by CSD (Zhang et al., 2010, 2011), and the consequential release of vasoactive neuropeptides (Huang et al., 1993; Moskowitz and Macfarlane, 1993; Moskowitz et al., 1993), we sought to determine whether anti-CGRP monoclonal antibodies (anti-CGRP-mAbs) can alter the vascular response to CSD or the ensuing plasma protein extravasation (PPE) in the dura, and additionally whether it could affect the vascular response to CSD in the pia. Our working hypothesis was that the selective blockade of activation of meningeal nociceptors by CSD may be mediated in part by a direct effect of reduced CGRP (sequestered by the mAbs) on the nociceptors, which we showed recently (Melo-Carrillo et al., 2017b) and in part through prevention of the vascular (arterial dilatation) and neurogenic (plasma protein extravasation) responses to CSD. Using powerful imaging tools and techniques that allow us to generate continuous, live, high-resolution views of spatial and temporal changes in both arteries and veins in the dura and pia, the current study reveals novel aspects of these responses to CSD in female rats, the extent to which vascular responses to CSD differ from vascular responses to systemic infusion of CGRP, and the extent to which vascular responses to CSD and CGRP infusion are altered by fremanezumab, a fully humanized IgG2Δa/κ-monoclonal antibody that selectively neutralizes CGRP.
Materials and Methods
Animals.
All procedures involving animals were in compliance with the experimental protocol approved by the Institutional Animal Care and Use Committee of Beth Israel Deaconess Medical Center and Harvard Medical School and adhered to the guidelines of the Committee for Research and Ethical Issues of the International Association for the Study of Pain. Only female Sprague Dawley Rats (Taconic) weighing 230–310 g were used throughout the experiments. Female mice expressing COP4 and enhanced yellow fluorescent protein (EYFP) under the thy1 promoter (strain 007612, The Jackson Laboratory) were used for fluorescent fremanezumab injections. Rats and mice were kept in 12 h light/dark cycles and were housed two rats or four mice per cage.
Experimental design.
Four hours before the beginning of the imaging session, rats were anesthetized and intubated, their femoral vein was cannulated, and saline or fremanezumab (total volume, 1 ml) was injected. We have previously determined that 4 h was enough time for intravenous fremanezumab to have an effect on the responses of sensory neurons in the dura to CSD (Melo-Carrillo et al., 2017b). Two hours later, the skull was thinned over the parietal cortex and a craniotomy was performed over the frontal cortex. An hour later, rats were placed into the microscope and an image stack was taken at 800 nm excitation for the identification of and differentiation between arteries and veins. Four hours after the injection of saline or fremanezumab, we injected 2 MDa FITC dextran into the femoral cannula to label the blood and began imaging at 890 nm excitation. After at least 2 min at baseline, we induced a single wave of CSD by pinpricking the frontal cortex and then continued to image for 45 min. In some experiments, we waited until the dural artery diameter had returned to baseline (for at least 10 min post-CSD), then imaged for at least 2 min to define a new baseline before injecting 2 μg/kg CGRP into the femoral vein. We continued imaging for 10–15 min post-CGRP infusion. In two rats, CGRP was injected first, and then CSD was induced with no changes to the results. Figure 1 illustrates the experimental timetable. Additionally, in a separate experiment, we injected fluorescently labeled fremanezumab into rats and mice to determine the extent that it penetrates into the dura.
Figure 1.

Experiment timeline. Diagram of the timing of injections and imaging for experimental rats.
Anesthesia.
Rats were briefly anesthetized using isoflurane, and rats or mice were injected with urethane (1.5 g/kg, i.p.) and atropine (0.3 ml, s.c.). Rats were intubated with an endotracheal tube and provided oxygen through a nose cone. Core temperature was maintained at 37°C, and blood oxygen levels were monitored with a PhysioSuite (Kent Scientific) throughout the length of the experiment.
Injections.
The femoral vein was cannulated in rats for dye and drug infusions. FITC- or Texas Red-conjugated 2 MDa dextran (Thermo Fisher Scientific) was used to label the blood (150 μl of 5 mg/ml in saline). Fremanezumab and Alexa Fluor 594-conjugated fremanezumab were obtained from Teva Pharmaceuticals and were injected at 30 mg/kg. Rat CGRP peptide was obtained from Sigma-Aldrich and was injected at 2 μg/kg diluted in saline. All injections into the femoral vein were followed by at least 1 ml of saline to flush the dead volume within the cannula and attached tube. Mice were given 30 mg/kg Alexa Fluor 594-conjugated fremanezumab via tail vein injection. Fremanezumab was fluorescently conjugated via reaction with a succinimidyl ester of Alexa Fluor 594 (catalog #A20004, Thermo Fisher Scientific) in PBS, pH 9.0, and mixed in a 1:2 antibody-to-dye molar ratio for 4 h at room temperature. Unreacted dye was removed by size exclusion chromatography through a Zeba desalting column (catalog #89891, Thermo Fisher Scientific).
Skull thinning.
To create an imaging window, rats were placed on a modified stereotaxic frame with a movable, rotatable ball joint to tilt the head. The scalp was shaved and sterilized, and the skin above the superficial skull was removed. The periosteum was removed over the right parietal and frontal skull plates, a layer of cyanoacrylate was placed around the perimeter of the plates for its hydrophobicity, and an area of 3 mm in diameter was thinned on the right parietal skull plate above a branch of the middle meningeal artery to a thickness of 70–100 μm. A high-speed drill was used to thin the skull. For placement of the CSD-inducing electrode, a similar area of ∼1 mm was thinned in the frontal plate, and a small craniotomy was performed using an 18 gauge needle. To allow the dura to recover from any stress of thinning, imaging started at least 1 h after all surgical procedures were completed.
In vivo imaging.
Fully anesthetized rats were imaged in an Olympus FV1000MPE-E multiphoton Imaging System using a Spectra-Physics Mai Tai DeepSee laser (pulse width, ∼70 fs) and a PLAN 25× 1.05 numerical aperture objective. Three color channels were collected simultaneously (420–460 nm blue in a PMT; 495–540 nm green and 575–630 nm red in GaAsP detectors). A wavelength of 890 nm was used to excite all fluorophores and produce a second harmonic generation at 445 nm (blue). An excitation of 800 nm was used for the detection of intrinsic fluorescence (green) emitted by the outermost tissue layer of arteries, presumably representing the smooth muscle layer. Time-lapse three-dimensional image stacks were taken covering a space from the skull through the meninges to the brain. Image resolution was 512 × 512 pixels covering an area of 509 × 509 μm. Custom software was written to allow special imaging parameters that ensured fast imaging while still correcting for any up-down motion of the rat brain: two sets of three image slices were taken 10 μm apart, one set in the dura, and one in the pia, for each time point. This allowed us to skip slices in the 50–100 μm of subarachnoid space that we were not interested in for the sake of time, yielding time points ∼10 s apart.
In vivo imaging of Alexa Fluor 594-conjugated fremanezumab.
Imaging of rats was performed with a laser wavelength of 850 nm and otherwise was as described above. Mice were placed on a stereotaxic frame and the scalp was cut, and they were injected with fluorescently conjugated fremanezumab via tail vein, and images were taken through intact (not thinned) periosteum, skull, and dura.
CSD initiation.
For CSD initiation, a metal electrode was inserted into the cortex through the dura to ∼3 mm, for 20 s. The CSD was initiated from a site in the frontal cortex that was sufficiently distant (∼1.5 cm) from the imaging site to minimize the possibility that the craniotomy or electrode insertion would affect the imaged area.
Image analysis.
Images were analyzed using Fiji (http://www.fiji.sc), a version of ImageJ (http://imagej.net). Blood vessel diameter was determined programmatically with a custom-written plugin that took a line ROI across the blood vessel as an input, averaged the brightness over the adjacent ∼50 lines, then, using a threshold at the mean brightness to count pixels, determined the width of the blood vessel for each time point. Quantification of plasma protein extravasation events was inspected manually and visually, where one event was counted at the first detection of an increase of brightness next to a blood vessel (see Results). The frequency of these events was counted per 250 s bins.
Determination of arrival of CSD wave.
The time of the arrival of the CSD wave under the imaging window was determined by changes in the diameter of pial arteries, typically occurring 2–3 min after CSD initiation. Time point 0 for “time post-CSD” was chosen as three time points (1 min) previous to the time point with the largest pial artery dilation.
Fixed tissue imaging.
Rats were injected with fluorescently conjugated fremanezumab or unconjugated fremanezumab via femoral vein cannulation, then given oxygen for 4 h. They were then fixed via transcardial perfusion with 4% paraformaldehyde and placed in PBS. The dura was removed and mounted on a slide with PermaFluor Mounting Media (Thermo Fisher Scientific). Slides were imaged using a Leica DM5500 Fluorescent Microscope with two channels (green, GFP; red, Texas Red fluorescence blocks) and persistent acquisition settings. Images were further normalized to the green channel, which represents background fluorescence.
Statistics.
Multiple two-tailed paired Student's t tests with Holm–Bonferroni correction were used to compare individual time points after CSD to baseline for blood vessel diameter. A two-tailed Student's t test was performed to compare maximum dilations or constrictions between treatment groups, as well as average normalized vein diameter throughout 500–1500 s post-CSD between treatment groups. Two-way ANOVA was also performed to compare blood vessel diameters, with time post-CSD/CGRP infusion as one variable, and treatment group as the other. Unless otherwise noted, all data is reported with ± standard error, and all error bars in figures represent ± standard error.
Results
Anatomical visualization and functional characterization of dural and pial blood vessels in rats
In vivo two-photon imaging through a thinned skull enabled us to visualize the effect of CSD on arteries and veins in both the pia and dura nearly simultaneously. We took image stacks ranging from the bottom of the skull, through the dura, and covering the pia (Fig. 2A). In an orthogonal reconstruction (Fig. 2B), the distinct blood vessel network on the outer surface of the rat dura could be easily differentiated from the vasculature within the pia. We noticed that rat vasculature had a strong intrinsic fluorescence, and furthermore, vessels that behaved as arteries (from previous reports) had an outer layer of tissue with strong intrinsic green emission, likely originating from smooth muscle. This emission was particularly noticeable using an 800 nm excitation laser wavelength (Fig. 2C, top, arrows). Therefore, before every experiment, we took one image stack at 800 nm to aid in the selection of an area that contained both dural and pial arteries and veins. Because the parenchyma also emitted autofluorescence at 800 nm excitation, the paravascular space surrounding pial arteries, which we have previously shown to be larger than the space around pial veins (Schain et al., 2017), also aided in the distinction between arteries and veins (Fig. 2C, right top, arrowheads). Once we identified arteries and veins in the dura and pia, we labeled the vessel lumen with 2 MDa FITC-dextran. We then quantified the diameter of each blood vessel type as it changed in response to CSD.
Figure 2.

In vivo imaging of meningeal vasculature in the rat. A, Example images from an image stack including the skull, dura, and pia of the rat. Intravenous FITC-dextran is green, the second harmonic generation from skull and dura is blue, and intrinsic fluorescence (from lipofuscin or other molecules) is red. B, Orthogonal reconstruction of image stack in A, showing the locations of blood vessels in relationship to skull, dura, pia, and brain. C, Top images were taken before FITC injection and show only intrinsic fluorescence using an excitation wavelength of 800 nm, in the dura (left images) and pia (right images). Bottom images are taken at 890 nm after FITC injection. Note the green border (arrows) at 800 nm around arteries but not veins, as well as a darkened paravascular space surrounding pial arteries (arrowheads in top right image). D, Representative images of a dural artery (top), pial artery (middle), and pial vein (bottom) during CSD. E, Quantification of the fold change in diameter of pial and dural arteries and veins during CSD in a representative rat. Scale bars: A–D, 50 μm.
The effects of CSD on dural and pial arteries and veins
To test the effects of CSD on the vasculature, we induced a single wave of CSD with a pinprick through a small craniotomy in the frontal cortex while imaging through a thinned skull window above the parieto-occipital cortex (∼4 mm caudally). Figure 2, D and E, shows an example of the vascular changes induced by CSD. Cortical spreading depression caused a brief but robust dilatation and prolonged constriction in the pial artery; a delayed, prolonged, and fairly robust dilatation in the dural artery; and, surprisingly, in addition to diameter changes in arteries, we also observed an immediate but brief dilatation and constriction that was followed by prolonged dilatation and constriction in the pial vein. Dural veins, however, showed no changes. We found these responses to be very characteristic of CSD in the rat.
Arterial responses to CSD are unaffected by fremanezumab
Pretreatment with fremanezumab had no effect on responses of pial or dural arteries to CSD (Fig. 3A,B). Pial arteries dilated within 40 s to a maximum of 38 ± 3.8% in the saline group and 43 ± 5.6% in the fremanezumab group (not significantly different, p = 0.49), and were compared with their original diameter, constricted ∼7.9 ± 0.95% in the saline group and 6.1 ± 1.2% in the fremanezumab group (p = 0.25), and remained constricted for nearly an hour of imaging. There was no significant interaction between treatment group and time post-CSD (two-way ANOVA: F(92,1) = 0.9, p = 0.72). Dural arteries dilated later compared with pial arteries (saline group, 188 ± 47 s; fremanezumab group, 150 ± 49 s; not significantly different between treatment groups, p = 0.58), and more slowly than pial arteries, reaching a maximum of 28 ± 2.6% in the saline group, and 29 ± 2.4% in the fremanezumab group (p = 0.79 between treatment groups). Both treatment groups stayed dilated for similar amounts of time, with the dilatation lasting approximately 2160 ± 230 s in saline, and 1690 ± 370 s in fremanezumab (p = 0.28). According to a two-way ANOVA, there was no significant interaction between treatment group and response to CSD for dural arteries (F(92,1) = 0.7, p = 0.97). Since the groups were not different from each other, we further analyzed the combined data for greater statistical power and compared each time point post-CSD to baseline. The pia arteries were significantly dilated from 20 to 50 s, and significantly constricted from 110 through 2000 s, and the dura arteries were significantly dilated from 200 to 2240 s (p < 4.63 × 10−4; multiple t tests with Bonferroni correction, with each time point compared with baseline).
Figure 3.

Effects of fremanezumab on CSD-induced vasculature changes. A–D, Plots of the average fold change in pial arteries (A), dural arteries (B), pial veins (C), and dural veins (D) during CSD in rats injected with drug (n = 7) and vehicle (n = 9). The p value in C indicates the significant difference between fremanezumab and saline for the normalized vein diameters averaged per rat over the time indicated in blue and red (500–1500 s).
Fremanezumab prevents dilatation of pial veins
Surprisingly, fremanezumab inhibited a previously undescribed CSD-induced dilatation of pial veins (Fig. 3C). In saline-treated rats, CSD induced an immediate and brief dilatation (maximum of 9.1 ± 1.3%, lasting 215 ± 45 s) and constriction (maximum of 11.2 ± 1.7%), and a secondary prolonged dilation (maximum of 11.1 ± 2.6%) that lasted an average 1080 ± 230 s, ending in a moderately constricted state (6.0 ± 2.0%). In contrast, in the fremanezumab-treated rats, CSD induced the immediate and brief dilatation (maximum dilatation = 12 ± 4.6%, p = 0.30; lasting 201 ± 34 s, p = 0.11), but not the secondary prolonged dilatation seen in the saline group. Instead, the brief dilatation was followed by a prolonged constriction (maximum constriction, 9.1 ± 0.9%) lasting the duration of the imaging session (3000 s). The average normalized diameter of pial veins between 500 and 1500 s post-CSD between the two treatment groups was significantly different (p = 0.01), with a 4.6 ± 1.9% dilatation in the saline group and a 5.5 ± 1.6% constriction in the fremanezumab group. A two-way ANOVA showed a significant interaction between treatment groups and post-CSD pial diameter (F(68,1) = 2.3, p = 5.5 × 10−8).
Dural veins did not respond to CSD (Fig. 3D) in both saline- and fremanezumab-treated rats (two-way ANOVA: F(92,1) = 1.1, p = 0.17).
CSD-induced plasma protein extravasation events are unaffected by fremanezumab
PPE is a leakage of proteins from blood vessels into the surrounding tissue. Previously, PPE has been demonstrated to occur in the dura in tissue fixed immediately after the occurrence of CSD (Bolay et al., 2002). Using in vivo two-photon imaging, we visualized leakage of 2 MDa FITC-dextran from dural arteries and veins. Such leakages did not occur equally along all of the endothelium, but rather occurred as discreet events at particular locations along a blood vessel. Each event was marked by a cloud of fluorescent expulsion that spreads from a small point along the endothelium, covering a small area nearby (half circle with ∼25 μm radius), and partially diffusing away (Fig. 4A) over 30–60 s. The distribution of PPE events (PPEEs) appeared to occur primarily along smaller blood vessels, rather than the larger artery (Fig. 4B). The frequency of PPEEs followed to some extent the dilatation of the dural arteries (Fig. 4C). PPEEs were significantly increased for the first 1250 s (five 250 s bins) post-CSD compared with baseline (p < 0.05 multiple t tests with Holm–Bonferroni correction; F(15,160) = 3.62, p = 2.1 × 10−5 one-way ANOVA), and reached their maximum frequency in the 500–750 s bin (averaging the time of maximum bin for each rat, 806 ± 139 s), close to the time of the dural artery maximum diameter, which occurred at 765 ± 95 s post-CSD across animals (Fig. 4C). Also, the average first PPEE occurred ∼210 ± 52 s post-CSD, which is close to the average start time of dura arterial dilation (when the artery first starts to dilate), which occurred at 162 ± 33 s.
Figure 4.

Effects of fremanezumab on CSD-induced PPEEs. A, Example images of a PPEE occurring on the border of a dural blood vessel (red arrow). B, Representative image showing location of PPEEs (red circles) during CSD in one rat. C, Quantification of average frequency of PPEEs per 250 s bins (red) overlaid with a plot of average dural artery diameter (green) post-CSD. D, Quantification of the average number of PPEEs during CSD in rats injected with drug (red) and vehicle (blue).
As with dural artery dilation, treatment with fremanezumab did not significantly change the onset, frequency, or duration of PPEEs induced by CSD (Fig. 4D). Before CSD (baseline), the number of PPEEs per 250 s bin was 1.3 ± 1.0 in the saline group and 1.5 ± 1.5 in the fremanezumab group (p = 0.90). In the first 1250 s after CSD, the number of PPEEs was 6.0 ± 2.3 in the saline group and 6.4 ± 2.6 in the fremanezumab group (p = 0.92). A two-way ANOVA did not show any interaction between treatment groups and time post-CSD (F(15,1) = 0.7, p = 0.74, n = 5/group).
Effects of CGRP infusion on dural and pial arteries and veins
CGRP infusion caused dilation in dural arteries, but not pial arteries, pial veins, or dural veins (Fig. 5). Intravenous infusion of 2 mg/kg caused an immediate dilatation in dural arteries, reaching a peak dilatation of ∼25.9 ± 3.3%, with time points of 20 s through 260 s significantly dilated (Fig. 5B; p < 0.002, multiple t tests with Bonferroni correction; n = 8).
Figure 5.

Effects of fremanezumab on CGRP infusion-induced vasculature changes. CGRP was infused into the blood after recovery from CSD in most rats. In two vehicle-treated rats, CGRP was infused before CSD was induced. A–D, Plots of the average fold change in pial arteries (A) pial veins (B), dural arteries (C), and dural veins (D) during CGRP infusion in rats treated with (n = 5) and vehicle (n = 8). The p value in B indicates significant differences between fremanezumab and saline for the normalized dural artery diameter averaged per rat across the time frame indicated in blue and red (20–300 s).
Fremanezumab prevents the dilation of dural arteries post-CGRP infusion
Although in the saline-treated group, the infusion of CGRP gave rise to a distinct dilatation of the dural artery, in the fremanezumab-treated group, such dilation was not seen (maximum of 6.1 ± 1.2% dilation in the fremanezumab-treated group). In the fremanezumab-treated group, no time points between 0 and 800 s were statistically significantly different from baseline (p > 0.002 multiple t test with Bonferroni correction, F(9,40) = 1.72, p = 0.12, one-way ANOVA; n = 5). The average diameter over the first 300 s was significantly different between groups (p = 4.9 × 10−4; n = 7, saline; n = 5, fremanezumab). Two-way ANOVA showed a significant interaction with treatment and postinfusion dilation (F(19,1) = 6.3, p = 8.2 × 10−12; n = 5/group).
CGRP infusion does not induce PPE events
Using the same method described above for the quantification of PPE events, we found no evidence for increased occurrence of PPE events after the infusion of CGRP (Fig. 6) at levels sufficient to induce arterial dilatation (F(4,60) = 0.60, p = 0.66, one-way ANOVA; n = 13). In contrast to CSD, no rats had more than five PPEEs take place per bin post-CGRP infusion at any time. This was true in both saline- and fremanezumab-treated animals (F(4,1) = 1.3, p = 0.30, two-way ANOVA; n = 5/group).
Figure 6.

Effects of fremanezumab on CGRP infusion-induced PPEEs. A, Quantification of the average frequency of PPEEs in 250 s bins during CGRP infusion in fremanezumab- and saline-injected rats. B, Representative image showing locations of PPEEs (red circle) during CGRP infusion in one rat (the same rat as in Fig. 2G).
Fremanezumab is present in the dura postinfusion
Intravenous infusion of fluorescently conjugated fremanezumab produced intense labeling in the dura but not pia or brain (Fig. 7). These observations were seen in thinned skulls (Fig. 7A) and intact skulls (Fig. 7B,C) of rats (Fig. 7A,B) and mice (Fig. 7C) in vivo (Fig. 7A,C) as well as in fixed tissue (Fig. 7B). To test for presence of fremanezumab at the time experiments were performed above, in vivo scans of the dura, pia, and brain were taken 3–4 h after the infusion of labeled fremanezumab in rats in which frontal craniotomy was performed (without induction of CSD). As shown in the Figure 7, 4 h after infusion fremanezumab is clearly seen in the dura but not in pia or brain of rats (Fig. 7A). Four hours after infusion, it is also seen in fixed dura of a rat that received no craniotomy or skull thinning (Fig. 7B). Because repeated in vivo imaging of dura and pia of intact (not thinned) skull could not be performed in rats (as their skull is too thick), we performed these experiments in 2-month-old mice. As shown in Figure 7C, infusion of fremanezumab produced immediate (within seconds) labeling in pial and dura blood vessels (Fig. 7C, left and middle panels), and in extravascular labeling in the dura but not pia 3 h later (Fig. 7C, right column).
Figure 7.
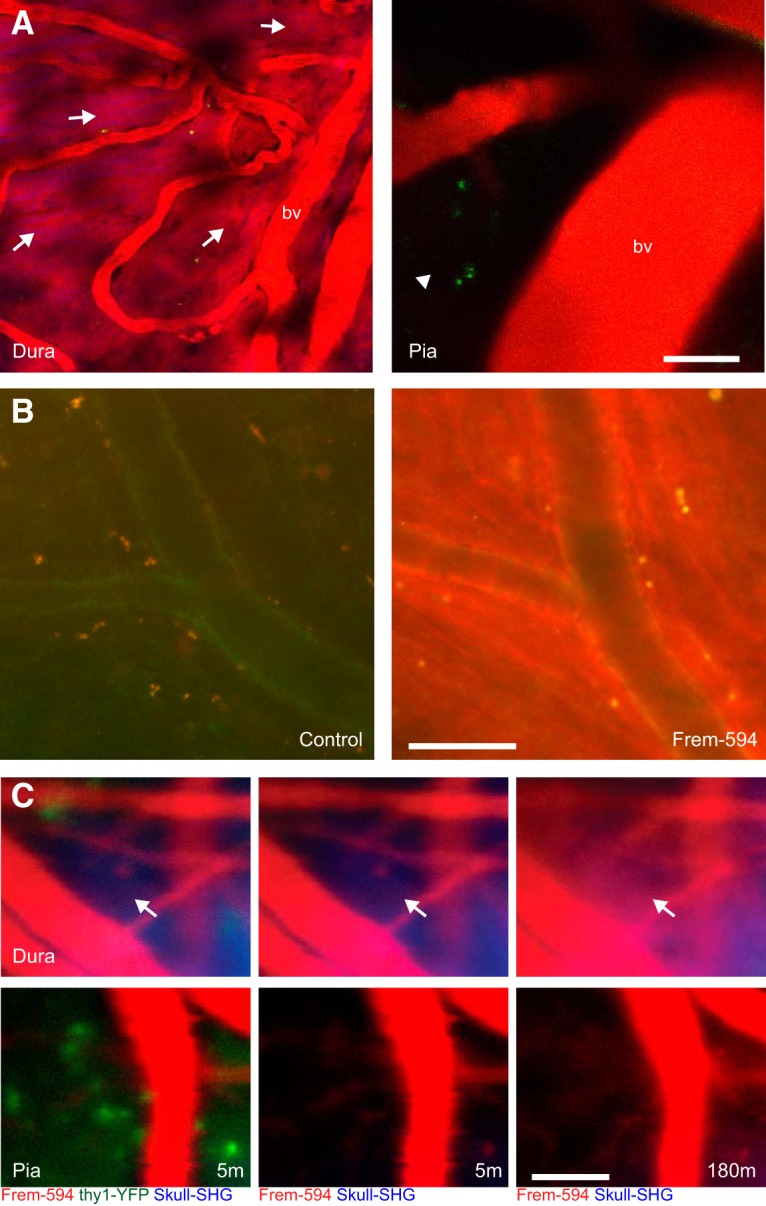
Presence of fremanezumab in the dura. A, In vivo images of the dura (left) and pia (right) of a rat 4 h after infusion with Alexa Fluor 594-conjugated fremanezumab (Fr594; representative of n = 2 rats). Labeled fremanezumab is indicated as red fluorescent areas (arrows) in the dura outside of blood vessels (bv). Note the lack of labeling outside of blood vessels in the pia (arrowhead). B, Fluorescent images of fixed dura removed from rats that did not undergo skull thinning and were injected with control nonfluorescent fremanezumab (left) or Fr594 (right; representative of n = 2 rats). C, In vivo images of a mouse expressing EYFP in the brain (thy1-EYFP), after injection with Fr594. The mouse scalp was cut, but the skull was not thinned. Images are shown at 5 m post injection (left and middle), with the EYFP channel displayed only in the left images, and at 180 m post injection (right) (representative of n = 2 mice). Note the presence of fremanezumab in the dura after 180 m (arrow) but not the pia or cortex. Scale bars: A–C, 50 μm.
Discussion
This is the first study where two-photon imaging has been used to document with high temporal and spatial resolution the simultaneous responses to CSD of arteries and veins in the pia and dura. Our main findings are that the CSD-induced brief dilatation and prolonged constriction of pial arteries and prolonged dilatation of dural arteries are unaffected by the anti-CGRP monoclonal antibody fremanezumab, whereas the brief constriction and prolonged dilatation of pial veins in response to CSD—a novel finding on its own—is prevented by the anti-CGRP monoclonal antibody. To the best of our knowledge, this is also the first documentation of CSD-induced dural PPE in real time. Temporally, we found that PPE events become more frequent within 2 min of the occurrence of CSD, that they reach maximal frequency at ∼12–13 min, that they subside 30 min later, and that this timeline correlates broadly with the onset and duration of the dura arterial dilation. As with the vascular response, fremanezumab did not alter the CSD-induced PPE. The reason for the lack of effects of fremanezumab in the dura is not any failure of the antibody to reach there, as we show here that it permeates the dura 3–4 h after injection. In fact, our study is the first to provide direct anatomical evidence that these antibodies enter the dura but not the pia or the cerebral cortex.
We also investigated the effect of CGRP infusion on pial and dural arteries and veins. Whereas CGRP infusion gave rise to the expected dilatation of dural arteries, which was effectively blocked by fremanezumab, it did not induce any dilatation in pial arteries, pial veins, or dural veins. Unlike the CSD scenario, whereby dilatation of dural arteries was tightly correlated with occurrence of PPE, the CGRP-induced robust dilatation of dural arteries was not followed by PPE.
Relevant to our understanding of migraine pathophysiology, the findings suggest that CGRP is not involved in the induction of dilatation of pial or dural arteries or the occurrence of PPE after CSD; that dilatation of dural arteries is not sufficient for the induction of PPE (as it is not followed by PPE if induced by CGRP rather than CSD); and that not all arterial dilatation in the dura are born alike. Regarding fremanezumab, it is reasonable to suggest that the mechanism of action of this class of drugs in migraine prevention is more likely to be mediated by an ability to reduce or block activation of peripheral nociceptors rather than the prevention of arterial dilatation.
While the occurrence of arterial dilatation in response to aura in patients or CSD in animals is well documented (Bolay et al., 2002; Asghar et al., 2011), the mechanism that drives this response is unclear. One of the longest-held hypotheses asserts that CSD activates meningeal nociceptors, which in turn release CGRP from their peripheral nerve endings, which then acts on dural arteries to induce arterial dilatation and PPE (Moskowitz et al., 1989; Olesen et al., 1990; Moskowitz, 1993). Because this scenario predicts that meningeal nociceptors become active before the dilatation and PPE (i.e., almost immediately after occurrence of CSD), it may be true for those whose headache and aura begins simultaneously (Hansen et al., 2012), and cases in which meningeal nociceptor activation and CSD occur nearly simultaneously (Zhang et al., 2010, 2011; Melo-Carrillo et al., 2017a). Our findings argue against the role played by CGRP in this scenario because the neutralization of CGRP by fremanezumab did not prevent the arterial dilatation or PPE (Fig. 8). Another scenario, which until now received far less attention, is that CSD directly induces arterial dilatation and PPE that leads to mast cell degranulation, and the release of inflammatory or algogenic molecules capable of activating the nociceptors (Dimitriadou et al., 1992). Because this scenario predicts that meningeal nociceptors become active last rather than first (i.e., after the occurrence of dilatation and PPE, events that last 30–40 min), it may be true for those whose headaches begin 30–60 min after the aura (Hansen et al., 2012), and cases in which activation of meningeal nociceptors begin >30 min after the occurrence of CSD (Zhang et al., 2010, 2011; Melo-Carrillo et al., 2017a). The sequence of events predicted by this scenario is consistent with our finding that CGRP is not involved in the induction of dilatation and PPE by CSD in animals as these events were not blocked by fremanezumab. If CGRP is not involved, it is reasonable to suggest that the release of other molecules from autonomic fibers mediate the arterial dilatation and the PPE by CSD. These may include adenosine (Honey et al., 2002), histamine (Lassen et al., 2003; Yuan and Silberstein, 2018), pituitary adenylate cyclase-activating polypeptide (Seki et al., 1995; Schytz et al., 2009; Amin et al., 2012; Syed et al., 2012), nitric oxide (Moncada et al., 1988; Iversen et al., 1989; Buchanan and Phillis, 1993) and a respective localized activation/inhibition of parasympathetic/sympathetic fibers innervating the dura (Brennan et al., 2007; Busija et al., 2008).
Figure 8.

Summary diagram. Diagram of the CGRP-dependent and CGRP-independent mechanisms in two scenarios involving CSD-induced headache.
The cerebral venous system consists of large dural sinuses and cerebral veins (Schaller, 2004; Kiliç and Akakin, 2008). Cerebral veins, which lack valves (Kiliç and Akakin, 2008), consist of superficial cortical and deep penetrating veins. The superficial cortical veins are located in the pia matter and drain the cerebral cortex. So far, no attention has been given to the functional role played by pial veins in the pathophysiology of migraine aura or CSD. Therefore, the finding that CSD induces distinct delayed and prolonged dilatation of pial veins that was prevented by fremanezumab, is novel and puzzling. It is novel because pial vein dilatation by CSD, although small, was not reported before. It is puzzling because pial veins are located inside the blood–brain barrier and as such, are not expected to be affected by an anti-CGRP-mAb. This last statement is based not only on our observations of the lack of fluorescence seen within the brain in fluorescently conjugated fremanezumab-injected animals, but also on previously reported measurements that show that only 0.1–0.5% of anti-CGRP-mAbs cross the intact blood–brain barrier, and that even if sporadic disruption of the blood–brain barrier exists, the amount of antibody penetrating the barrier during migraine attacks would be substantially lower than what would be required for meaningful CGRP inhibition within the brain (Bigal et al., 2015).
There are several potential explanations as to why pial veins exhibit the second prolonged dilation phase after CSD. Dilatation may be secondary to the activation of parasympathetic pathways. By definition, this possibility depends on how much smooth muscle is surrounding the veins that we studied. If sufficient muscle is present, then a scenario by which CSD induces this dilatation by the activation of parasympathetic pathways is possible. Abolishment of certain aspects of vascular dilatation by CSD in animals in which the sphenopalatine ganglion (SPG) was lesioned (Bolay et al., 2002) may support this scenario. If, however, the veins or venules do not have enough smooth muscle surrounding them, the parasympathetic hypothesis becomes irrelevant. Another explanation may be that the dilatation is passively driven by changes in pressure in the brain that are secondary to the transient cortical swelling that occurs in the wake of CSD (i.e., a CGRP-independent event).
Further complicating the discussion over this finding is the fact that fremanezumab prevented the dilatation of pial veins. In the absence of data that can explain this finding, we propose three different scenarios for future consideration. (1) Fremanezumab reduces pial vein dilatation by modulating the activation of postganglionic parasympathetic neurons in the sphenopalatine ganglion by CSD (Bolay et al., 2002). The presence of CGRP-positive fibers in the SPG (Ivanusic et al., 2011) may support this possibility. (2) Fremanezumab may affect the wall of the pial vein intraluminally, a scenario that may explain the effect of fremanezumab on pial blood vessels but not on cortical neurons. (3) If CGRP was released within the cortex during CSD, and then cleared via the glymphatic system, which removes solutes from the interstitial space by an outflow through the paravascular space surrounding pial veins, it is reasonable to speculate that such an explanation of flow will result in a preferential concentration of CGRP around pial veins but not around pial arteries. Perhaps in this case, the small amount of fremanezumab that can cross the BBB is then able to reduce this dilatation. However, we must make several unproven assumptions in this case, including that CSD releases CGRP in the brain, that this brain CGRP is able to dilate pial veins abluminally, that the small amount of antibody that crosses into the brain occurs near veins, and that it is of sufficient amount to counter the CGRP released in the brain.
Evidence that trigeminal nerve stimulation results in PPE (Markowitz et al., 1987) and that the anti-migraine drug sumatriptan blocks it (Moskowitz and Buzzi, 1991), lend support to the notion that CSD-induced PPE in the meninges of rodents (Bolay et al., 2002; Karatas et al., 2013) may be critical for the activation of the meningeal nociceptors and the initiation of headache after aura. Our finding that the induction of PPE by CSD is not blocked by fremanezumab, an anti-CGRP-mAb that successfully inhibits the activation of Aδ meningeal nociceptors (Melo-Carrillo et al., 2017b) and high-threshold trigeminovascular neurons (Melo-Carrillo et al., 2017a) by CSD in rats and effectively prevents migraine (Silberstein et al., 2017) in humans, questions this notion. Further supporting the conclusion that CGRP has little or no role in dural PPE is the finding that CGRP infusion, although giving rise to an immediate and very distinct dilatation of dural arteries, triggers no PPE in the dura. A similar conclusion was drawn by Markowitz et al. (1987).
While CGRP appears to be uninvolved in the dilatation of dural and pial arteries or the neurogenic (PPE) responses to CSD, it can, on its own, trigger a massive arterial dilatation in the dura but not the pia. Because CGRP infusion triggers delayed migraine in susceptible migraineurs (Lassen et al., 2002), it is reasonable to propose that the dilatation of dural arteries by CGRP may be necessary for the activation of meningeal nociceptors and initiation of headache. However, if we take into consideration the enigmatic observation that while the arterial dilatation is immediate (i.e., it begins seconds after CGRP infusion) and brief (i.e., it lasts ∼10 min), the headache begins nearly 4 h later (Lassen et al., 2002), we must also consider the possibility that the induction of migraine-like headache by systemic CGRP infusion is mediated mainly by the modulatory effects of CGRP on meningeal nociceptors, potentially involving yet unidentified environmental changes triggered at least partially by the dilatation of the dural arteries.
In summary, this study identifies fundamental differences between two commonly used animal models of migraine: CSD induction and CGRP infusion. It thus raises the possibility that conclusions drawn from one model may not be true or relevant to the other. It sharpens the need to accept the view that there is more than one truth to migraine pathophysiology, that it is unlikely that one theory will explain all types of migraine, or the mechanisms of action of anti-migraine drugs. In regard to the latter, it is concluded that similar vascular responses (in the meninges) may be mediated by different mechanisms and, consequently, that it is possible that a combination of drugs that prevent dilatation will have to be used or that different such therapeutic agents will be used in different types of migraine headache.
Footnotes
This study was supported by a grant from Teva Pharmaceutical Industries, and by National Institutes of Health Grants R37-NS-079678, R01-NS-069847, and R01-NS-094198 (to R.B.). We thank Jason Tien for the preparation of Alexa Fluor-labeled fremanezumab.
Teva Pharmaceutical Industries holds the patent for treating episodic and chronic migraine with fremanezumab and funded parts of the study. J.S. is an employee of and R.B. is a consultant to Teva Pharmaceutical Industries. The authors declare no other competing financial interests.
References
- Amin FM, Asghar MS, Guo S, Hougaard A, Hansen AE, Schytz HW, van der Geest RJ, de Koning PJ, Larsson HB, Olesen J, Ashina M (2012) Headache and prolonged dilatation of the middle meningeal artery by PACAP38 in healthy volunteers. Cephalalgia 32:140–149. 10.1177/0333102411431333 [DOI] [PubMed] [Google Scholar]
- Asghar MS, Hansen AE, Amin FM, van der Geest RJ, Koning Pv, Larsson HB, Olesen J, Ashina M (2011) Evidence for a vascular factor in migraine. Ann Neurol 69:635–645. 10.1002/ana.22292 [DOI] [PubMed] [Google Scholar]
- Ayata C, Lauritzen M (2015) Spreading depression, spreading depolarizations, and the cerebral vasculature. Physiol Rev 95:953–993. 10.1152/physrev.00027.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigal ME, Edvinsson L, Rapoport AM, Lipton RB, Spierings EL, Diener HC, Burstein R, Loupe PS, Ma Y, Yang R, Silberstein SD (2015) Safety, tolerability, and efficacy of TEV-48125 for preventive treatment of chronic migraine: a multicentre, randomised, double-blind, placebo-controlled, phase 2b study. Lancet Neurol 14:1091–1100. 10.1016/S1474-4422(15)00245-8 [DOI] [PubMed] [Google Scholar]
- Bolay H, Reuter U, Dunn AK, Huang Z, Boas DA, Moskowitz MA (2002) Intrinsic brain activity triggers trigeminal meningeal afferents in a migraine model. Nat Med 8:136–142. 10.1038/nm0202-136 [DOI] [PubMed] [Google Scholar]
- Brennan KC, Beltrán-Parrazal L, López-Valdés HE, Theriot J, Toga AW, Charles AC (2007) Distinct vascular conduction with cortical spreading depression. J Neurophysiol 97:4143–4151. 10.1152/jn.00028.2007 [DOI] [PubMed] [Google Scholar]
- Buchanan JE, Phillis JW (1993) The role of nitric oxide in the regulation of cerebral blood flow. Brain Res 610:248–255. 10.1016/0006-8993(93)91408-K [DOI] [PubMed] [Google Scholar]
- Busija DW, Bari F, Domoki F, Horiguchi T, Shimizu K (2008) Mechanisms involved in the cerebrovascular dilator effects of cortical spreading depression. Prog Neurobiol 86:379–395. 10.1016/j.pneurobio.2008.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Shook LL, Biag J, Nguyen EN, Toga AW, Charles AC, Brennan KC (2010) Biphasic direct current shift, haemoglobin desaturation and neurovascular uncoupling in cortical spreading depression. Brain 133:996–1012. 10.1093/brain/awp338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Close LN, Eftekhari S, Wang M, Charles AC, Russo AF (2019) Cortical spreading depression as a site of origin for migraine: role of CGRP. Cephalalgia 39:428–434. 10.1177/0333102418774299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colonna DM, Meng W, Deal DD, Busija DW (1994) Calcitonin gene-related peptide promotes cerebrovascular dilation during cortical spreading depression in rabbits. Am J Physiol 266:H1095–H1102. 10.1152/ajpheart.1994.266.3.H1095 [DOI] [PubMed] [Google Scholar]
- Dimitriadou V, Buzzi MG, Theoharides TC, Moskowitz MA (1992) Ultrastructural evidence for neurogenically mediated changes in blood vessels of the rat dura mater and tongue following antidromic trigeminal stimulation. Neuroscience 48:187–203. 10.1016/0306-4522(92)90348-6 [DOI] [PubMed] [Google Scholar]
- Dodick DW, Goadsby PJ, Silberstein SD, Lipton RB, Olesen J, Ashina M, Wilks K, Kudrow D, Kroll R, Kohrman B, Bargar R, Hirman J, Smith J (2014) Safety and efficacy of ALD403, an antibody to calcitonin gene-related peptide, for the prevention of frequent episodic migraine: a randomised, double-blind, placebo-controlled, exploratory phase 2 trial. Lancet Neurol 13:1100–1107. 10.1016/S1474-4422(14)70209-1 [DOI] [PubMed] [Google Scholar]
- Dodick DW, Ashina M, Brandes JL, Kudrow D, Lanteri-Minet M, Osipova V, Palmer K, Picard H, Mikol DD, Lenz RA (2018) ARISE: a phase 3 randomized trial of erenumab for episodic migraine. Cephalalgia 38:1026–1037. 10.1177/0333102418759786 [DOI] [PubMed] [Google Scholar]
- Edvinsson L, Haanes KA, Warfvinge K, Krause DN (2018) CGRP as the target of new migraine therapies—successful translation from bench to clinic. Nat Rev Neurol 14:338–350. 10.1038/s41582-018-0003-1 [DOI] [PubMed] [Google Scholar]
- Hansen JM, Lipton RB, Dodick DW, Silberstein SD, Saper JR, Aurora SK, Goadsby PJ, Charles A (2012) Migraine headache is present in the aura phase: a prospective study. Neurology 79:2044–2049. 10.1212/WNL.0b013e3182749eed [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann J, Baca SM, Akerman S (2019) Neurovascular mechanisms of migraine and cluster headache. J Cereb Blood Flow Metab 39:573–594. 10.1177/0271678X17733655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honey AC, Bland-Ward PA, Connor HE, Feniuk W, Humphrey PP (2002) Study of an adenosine A1 receptor agonist on trigeminally evoked dural blood vessel dilation in the anaesthetized rat. Cephalalgia 22:260–264. 10.1046/j.1468-2982.2002.00345.x [DOI] [PubMed] [Google Scholar]
- Hong KW, Pyo KM, Lee WS, Yu SS, Rhim BY (1994) Pharmacological evidence that calcitonin gene-related peptide is implicated in cerebral autoregulation. Am J Physiol 266:H11–H16. 10.1152/ajpheart.1994.266.1.H11 [DOI] [PubMed] [Google Scholar]
- Huang Z, Byun B, Matsubara T, Moskowitz MA (1993) Time-dependent blockade of neurogenic plasma extravasation in dura mater by 5-HT1B/D agonists and endopeptidase 24.11. Br J Pharmacol 108:331–335. 10.1111/j.1476-5381.1993.tb12805.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanusic JJ, Kwok MMK, Ahn AH, Jennings EA (2011) 5-HT(1D) receptor immunoreactivity in the sphenopalatine ganglion: implications for the efficacy of triptans in the treatment of autonomic signs associated with cluster headache. Headache 51:392–402. 10.1111/j.1526-4610.2011.01843.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iversen HK, Olesen J, Tfelt-Hansen P (1989) Intravenous nitroglycerin as an experimental model of vascular headache. basic characteristics. Pain 38:17–24. 10.1016/0304-3959(89)90067-5 [DOI] [PubMed] [Google Scholar]
- Jansen-Olesen I, Jørgensen L, Engel U, Edvinsson L (2003) In-depth characterization of CGRP receptors in human intracranial arteries. Eur J Pharmacol 481:207–216. 10.1016/j.ejphar.2003.09.021 [DOI] [PubMed] [Google Scholar]
- Karatas H, Erdener SE, Gursoy-Ozdemir Y, Lule S, Eren-Koçak E, Sen ZD, Dalkara T (2013) Spreading depression triggers headache by activating neuronal Panx1 channels. Science 339:1092–1095. 10.1126/science.1231897 [DOI] [PubMed] [Google Scholar]
- Kiliç T, Akakin A (2008) Anatomy of cerebral veins and sinuses. Front Neurol Neurosci 23:4–15. 10.1159/000111256 [DOI] [PubMed] [Google Scholar]
- Lassen LH, Haderslev PA, Jacobsen VB, Iversen HK, Sperling B, Olesen J (2002) CGRP may play a causative role in migraine. Cephalalgia 22:54–61. 10.1046/j.1468-2982.2002.00310.x [DOI] [PubMed] [Google Scholar]
- Lassen LH, Christiansen I, Iversen HK, Jansen-Olesen I, Olesen J (2003) The effect of nitric oxide synthase inhibition on histamine induced headache and arterial dilatation in migraineurs. Cephalalgia 23:877–886. 10.1046/j.1468-2982.2003.00586.x [DOI] [PubMed] [Google Scholar]
- Lauritzen M. (1994) Pathophysiology of the migraine aura. the spreading depression theory. Brain 117:199–210. 10.1093/brain/117.1.199 [DOI] [PubMed] [Google Scholar]
- Markowitz S, Saito K, Moskowitz MA (1987) Neurogenically mediated leakage of plasma protein occurs from blood vessels in dura mater but not brain. J Neurosci 7:4129–4136. 10.1523/JNEUROSCI.07-12-04129.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Carrillo A, Noseda R, Nir RR, Schain AJ, Stratton J, Strassman AM, Burstein R (2017a) Selective inhibition of trigeminovascular neurons by fremanezumab: a humanized monoclonal anti-CGRP antibody. J Neurosci 37:7149–7163. 10.1523/JNEUROSCI.0576-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo-Carrillo A, Strassman AM, Nir RR, Schain AJ, Noseda R, Stratton J, Burstein R (2017b) Fremanezumab-A humanized monoclonal anti-CGRP antibody-inhibits thinly myelinated (Aδ) but not unmyelinated (C) meningeal nociceptors. J Neurosci 37:10587–10596. 10.1523/JNEUROSCI.2211-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA (1988) The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension 12:365–372. 10.1161/01.HYP.12.4.365 [DOI] [PubMed] [Google Scholar]
- Moskowitz MA. (1993) Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology 43:S16–S20. [PubMed] [Google Scholar]
- Moskowitz MA, Buzzi MG (1991) Neuroeffector functions of sensory fibres: implications for headache mechanisms and drug actions. J Neurol 238 [Suppl 1]:S18–S22. [DOI] [PubMed] [Google Scholar]
- Moskowitz MA, Macfarlane R (1993) Neurovascular and molecular mechanisms in migraine headaches. Cerebrovasc Brain Metab Rev 5:159–177. [PubMed] [Google Scholar]
- Moskowitz MA, Buzzi MG, Sakas DE, Linnik MD (1989) Pain mechanisms underlying vascular headaches. Progress report 1989. Rev Neurol 145:181–193. [PubMed] [Google Scholar]
- Moskowitz MA, Nozaki K, Kraig RP (1993) Neocortical spreading depression provokes the expression of c-fos protein-like immunoreactivity within trigeminal nucleus caudalis via trigeminovascular mechanisms. J Neurosci 13:1167–1177. 10.1523/JNEUROSCI.13-03-01167.1993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozari A, Dilekoz E, Sukhotinsky I, Stein T, Eikermann-Haerter K, Liu C, Wang Y, Frosch MP, Waeber C, Ayata C, Moskowitz MA (2010) Microemboli may link spreading depression, migraine aura, and patent foramen ovale. Ann Neurol 67:221–229. 10.1002/ana.21871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen J, Friberg L, Olsen TS, Iversen HK, Lassen NA, Andersen AR, Karle A (1990) Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol 28:791–798. 10.1002/ana.410280610 [DOI] [PubMed] [Google Scholar]
- Penfield W, McNaughton F (1940) Dural headache and innervation of the dura mater. Arch Neurol Psychiatr 44:43–75. 10.1001/archneurpsyc.1940.02280070051003 [DOI] [Google Scholar]
- Ray BS, Wolff HG (1940) Experimental studies on headache. pain-sensitive structures of the head and their significance in headache. Arch Surg 41:813–856. 10.1001/archsurg.1940.01210040002001 [DOI] [Google Scholar]
- Reuter U, Weber JR, Gold L, Arnold G, Wolf T, Dreier J, Lindauer U, Dirnagl U (1998) Perivascular nerves contribute to cortical spreading depression-associated hyperemia in rats. Am J Physiol 274:H1979–H1987. 10.1152/ajpheart.1998.274.6.H1979 [DOI] [PubMed] [Google Scholar]
- Russo AF. (2015) Calcitonin gene-related peptide (CGRP): a new target for migraine. Annu Rev Pharmacol Toxicol 55:533–552. 10.1146/annurev-pharmtox-010814-124701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schain AJ, Melo-Carrillo A, Strassman AM, Burstein R (2017) Cortical spreading depression closes paravascular space and impairs glymphatic flow: implications for migraine headache. J Neurosci 37:2904–2915. 10.1523/JNEUROSCI.3390-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaller B. (2004) Physiology of cerebral venous blood flow: from experimental data in animals to normal function in humans. Brain Res 46:243–260. 10.1016/j.brainresrev.2004.04.005 [DOI] [PubMed] [Google Scholar]
- Schytz HW, Birk S, Wienecke T, Kruuse C, Olesen J, Ashina M (2009) PACAP38 induces migraine-like attacks in patients with migraine without aura. Brain 132:16–25. 10.1093/brain/awn307 [DOI] [PubMed] [Google Scholar]
- Seki Y, Suzuki Y, Baskaya MK, Kano T, Saito K, Takayasu M, Shibuya M, Sugita K (1995) The effects of pituitary adenylate cyclase-activating polypeptide on cerebral arteries and vertebral artery blood flow in anesthetized dogs. Eur J Pharmacol 275:259–266. 10.1016/0014-2999(95)00011-9 [DOI] [PubMed] [Google Scholar]
- Silberstein SD, Dodick DW, Bigal ME, Yeung PP, Goadsby PJ, Blankenbiller T, Grozinski-Wolff M, Yang R, Ma Y, Aycardi E (2017) Fremanezumab for the preventive treatment of chronic migraine. N Engl J Med 377:2113–2122. 10.1056/NEJMoa1709038 [DOI] [PubMed] [Google Scholar]
- Skljarevski V, Matharu M, Millen BA, Ossipov MH, Kim BK, Yang JY (2018) Efficacy and safety of galcanezumab for the prevention of episodic migraine: results of the EVOLVE-2 phase 3 randomized controlled clinical trial. Cephalalgia 38:1442–1454. 10.1177/0333102418779543 [DOI] [PubMed] [Google Scholar]
- Stauffer VL, Dodick DW, Zhang Q, Carter JN, Ailani J, Conley RR (2018) Evaluation of galcanezumab for the prevention of episodic migraine: the EVOLVE-1 randomized clinical trial. JAMA Neurol 75:1080–1088. 10.1001/jamaneurol.2018.1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed AU, Koide M, Braas KM, May V, Wellman GC (2012) Pituitary adenylate cyclase-activating polypeptide (PACAP) potently dilates middle meningeal arteries: implications for migraine. J Mol Neurosci 48:574–583. 10.1007/s12031-012-9851-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tepper S, Ashina M, Reuter U, Brandes JL, Doležil D, Silberstein S, Winner P, Leonardi D, Mikol D, Lenz R (2017) Safety and efficacy of erenumab for preventive treatment of chronic migraine: a randomised, double-blind, placebo-controlled phase 2 trial. Lancet Neurol 16:425–434. 10.1016/S1474-4422(17)30083-2 [DOI] [PubMed] [Google Scholar]
- Tozzi A, de Iure A, Di Filippo M, Costa C, Caproni S, Pisani A, Bonsi P, Picconi B, Cupini LM, Materazzi S, Geppetti P, Sarchielli P, Calabresi P (2012) Critical role of calcitonin gene-related peptide receptors in cortical spreading depression. Proc Natl Acad Sci U S A 109:18985–18990. 10.1073/pnas.1215435109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl M, Schilling L, Parsons AA, Kaumann A (1994) Involvement of calcitonin gene-related peptide (CGRP) and nitric oxide (NO) in the pial artery dilatation elicited by cortical spreading depression. Brain Res 637:204–210. 10.1016/0006-8993(94)91234-3 [DOI] [PubMed] [Google Scholar]
- Wang Y, Li Y, Wang M (2016) Involvement of CGRP receptors in retinal spreading depression. Pharmacol Rep 68:935–938. 10.1016/j.pharep.2016.05.001 [DOI] [PubMed] [Google Scholar]
- Yuan H, Silberstein SD (2018) Histamine and migraine. Headache 58:184–193. 10.1111/head.13164 [DOI] [PubMed] [Google Scholar]
- Zhang X, Levy D, Noseda R, Kainz V, Jakubowski M, Burstein R (2010) Activation of meningeal nociceptors by cortical spreading depression: implications for migraine with aura. J Neurosci 30:8807–8814. 10.1523/JNEUROSCI.0511-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Levy D, Kainz V, Noseda R, Jakubowski M, Burstein R (2011) Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol 69:855–865. 10.1002/ana.22329 [DOI] [PMC free article] [PubMed] [Google Scholar]