

Abstract
CAP256 is one of the highly potent, broadly neutralizing monoclonal antibodies (bNAb) designed for HIV-1 therapy. During the process development of one of the constructs, an unexpected product-related impurity was observed via microfluidics gel electrophoresis. A panel of complementary LC-MS analyses was applied for the comprehensive characterization of CAP256 which included the analysis of the intact and reduced protein, the middle-up approach, and a set of complementary peptide mapping techniques and verification of the disulfide bonds. The designed workflow allowed to identify a clip within a protruding acidic loop in the CDR-H3 region of the heavy chain, which can lead to the decrease of bNAb potency. This characterization explained the origin of the additional species reflected by the reducing gel profile. An intra-loop disulfide bond linking the two fragments was identified, which explained why the non-reducing capillary electrophoresis (CE) profile was not affected. The extensive characterization of CAP256 post-translational modifications was performed to investigate a possible cause of CE profile complexity and to illustrate other structural details related to this molecule’s biological function. Two sites of the engineered Tyr sulfation were verified in the antigen-binding loop, and pyroglutamate formation was used as a tool for monitoring the extent of antibody clipping. Overall, the comprehensive LC-MS study was crucial to (1) identify the impurity as sequence clipping, (2) pinpoint the clipping location and justify its susceptibility relative to the molecular structure, (3) lead to an upstream process optimization to mitigate product quality risk, and (4) ultimately re-engineer the sequence to be clip-resistant.
Keywords: HIV-1 bNAb, Comprehensive LC-MS/MSE, Peptide mapping, Sequence clipping, Subunit, IdeS, Pyroglutamination, Vaccine product quality risk
Graphical Abstract:
Introduction
Studies of HIV-1 broadly neutralizing antibodies (bNAbs) provide valuable information for vaccine design and HIV-1 treatment [1, 2]. Among multiple antigenic epitopes of HIV-1, the variable region loops are a target for one of the new highly potent bNAbs, CAP256-VRC26.25 [3, 4]. This CAP256 bNAb contains unique structural features, such as a protruding tyrosine-sulfated, anionic antigenbinding loop in the complementarity-determining region CDR-H3, and an extra intra-CDR-H3 disulfide bond. Furthermore, these features have been known to contribute to tenfold greater neutralization potency compared to previously described lineage members [5-7], which makes this bNAb an attractive candidate for clinical development: either as a preventative agent for infection or as part of an antiretroviral regimen to prevent HIV-1 infection.
During the CAP256 bNAb cell culture development and switching from the human embryonic kidney (HEK) to the Chinese hamster ovary (CHO) cell line [8], a panel of analytical assays was applied to determine critical quality attributes (CQAs) of the bNAb, such as titer, level of aggregation, and fragmentations. Most CQAs showed the expected characteristics, except for a high impurity level observed by reducing microfluidic capillary electrophoresis with sodium dodecylsulfate (CE-SDS, hereafter as “CE”). The reducing gel profile revealed two extra peaks, in addition to the two commonly observed heavy and light chains. To investigate the integrity of this bNAb and to identify the two extra peaks, a liquid chromatography separation coupled with mass spectrometry detection (LC-MS) was employed [9-11]. In this work, a comprehensive LC-MS analysis was applied to fully characterize this molecule, including subunit mass analysis as well as peptide and disulfide mapping. 3D-crystallography analysis of CAP256 was also aligned with the LC-MS results to better understand the structural features of this molecule.
Herein, LC-MS analysis of the intact CAP256 was used to survey its integrity. Subunit LC-MS analysis was also applied for characterizing the light and heavy chains, screening for degradants or impurities, and glycosylation and modification profiling. The molecule was also digested with the IdeS proteolytic enzyme as an orthogonal subunit analysis technique, to narrow down the search for the clipping location. A complementary set of peptide mapping analyses was used to confirm the targeted protein structure, characterize post-translational modifications (PTMs), obtain the glycosylation profile per amino acid site, and ultimately locate the exact clipping site. Lastly, disulfide bond mapping was essential for providing an insight on the differences of the reducing and non-reducing CGE profiles. Altogether, the comprehensive LC-MS studies provided a strong characterization and a complete understanding of an unexpected CAP256 sequence clipping, which supported the findings of the routine analytical assessment.
Experimental
Materials and Reagents
Sample Process and Purification
CAP256 bNAb materials (CAP256-VRC 26.25LS) were produced in-house at the Vaccine Production Program Lab in NIH (Bethesda, MD). Purified, non-clipped transient CAP256 material was produced in the HEK cell line; the stable clone material was produced in the CHO cell line and exhibited various degrees of clipping. The purity of CAP256 was assessed by a gel electrophoresis chip technology (CE) (Perkin-Elmer, Waltham, MA) using DTT as the reducing agent [12]. The CE assay was further developed and optimized to determine the % clipping ranging between 1 and 31%. “Medium-clipped” (17%, per CE) CHO CAP256 sample was chosen for the purpose of this investigation and was compared to the HEK-produced (control) bNAb.
LC-MS Reagents
High purity LC-MS grade water and acetonitrile containing formic acid used for mobile phase preparation, and ammonium bicarbonate reagent were purchased from J.T. Baker (Phillipsburg, NJ). RapiGest™ surfactant and [Glu1]-Fibrinopeptide B lock mass standard (GluFib) were purchased from Waters (Milford, MA). Formic acid, urea, and Zeba™ spin desalting column (7K MWCO) were purchased from Pierce (Rockford, IL). Dithiothreitol (DTT) and iodoacetamide were purchased from ThermoFisher Life Technologies (Grand Island, NY) and Sigma-Aldrich (St. Louis, MO), respectively. Trypsin (modified sequencing grade), chymotrypsin, IdeS (immunoglobulin G-degrading enzyme of Streptococcus pyogenes), and PNGase F glycosidase were purchased from Promega (Madison, WI).
Sample Preparation for the LC-MS Analyses
All samples were buffer-exchanged in ammonium bicarbonate using Zeba™ spin desalting columns, 7K MWCO (Pierce, Rockford, IL), and diluted to 1 mg/mL with 50 mM ammonium bicarbonate buffer, pH 7.2.
For the intact protein analysis, the samples were loaded on the column immediately following the procedure described above. For the subunit analysis, 1.0 mg/mL protein solution was denatured using 8 M urea and reduced with 40 mM DTT for 30 min at 60 °C, cooled to room temperature, alkylated with 80 mM iodoacetamide for 30 min (protected from light), and acidified with formic acid to final concentration of 0.1%. For glycan removal, the reduced samples were incubated overnight at 37 °C with 0.1 units/mg protein PNGase F.
To perform IdeS digestion, the samples were buffer-exchanged into ammonium bicarbonate and incubated with 0.6 units/mg protein IdeS and PNGase F for 2 h at 37 °C. The samples were then denatured and reduced using 8 M urea and 40 mM DTT and then alkylated using IAM as previously described. The samples were buffer-exchanged into ammonium bicarbonate, underwent a second deglycosylation by the addition of 0.3 units/mg protein PNGase F, and incubated overnight at 37 °C. The samples were then acidified with formic acid to a final concentration of 0.1%.
For peptide mapping analysis using tryptic or chymotryptic digestion, the samples were denatured using a 0.5% RapiGest solution in 50 mM ammonium bicarbonate, reduced with 40 mM DTT, alkylated with 80 mM iodoacetamide, and incubated overnight with trypsin at 37 °C or chymotrypsin at 25 °C using an enzyme/protein ratio of 1:50 (w/w). For disulfide bond mapping, the same procedure was used, bypassing DTT reduction. The digestion was quenched by acidifying the solution with 0.1% formic acid, which also served to hydrolyze RapiGest detergent. The samples were spun down and the supernatant was analyzed by LC-MS.
LC-MS Setup
LC-MS analyses were performed using an Acquity H-Class chromatography system without an optical detector, coupled with mass spectrometry detection on a SYNAPT G2 QTof, both from Waters (Milford, MA). A reversed phase C18 column was used for the separation of the tryptic and chymotryptic peptide digests and for the disulfide bond mapping, whereas a C4 column was used for loading the intact CAP256 sample and for separation of all subunits (reduced mAb subunits, IdeS digested mAb subunits, and reduced and deglycosylated mAb subunits).
For all types of the LC separation, the mobile phase consisted of the aqueous solution of 0.1% formic acid as solvent A and 0.1% formic acid in acetonitrile as solvent B. The MS data were acquired in positive ion mode. Lock mass correction was done by means of 20 μL/min infusion of 100 fmol/μL GluFib in water/acetonitrile 1:1 with 0.1% formic acid (785.837 m/z peak, 30 s intervals, 3 scans to average). Alternatively, for higher mass range, 1971.615 m/z peak of NaI was used as a lock mass (2 μg/mL in water/iso-propanol, 1:1).
For the peptide mapping analyses, reduced or non-reduced protein digests were separated on a UPLC Peptide BEH C18 column (300 Å, 1.7 μm, 2.1 mm × 50 mm) (Waters), with the column temperature set to 65 °C, at a 0.2-mL/min flow rate. Gradient: 0 min—3%, 1 min—3%, 91 min—57%, 91.5 min— 85%, 102 min—85%, 103 min—3%, 105 min—3%. Acquisition range: 100–2000 m/z, acquired in resolution analyzer mode, with high energy channel acquired in MSE mode: 30–45 V collisional energy ramp, 0.5 s scan time. Capillary 3.0 kV, cone 35 V, source 120 °C, desolvation 350 °C, desolvation gas 800 L/h.
For the reduced protein analysis—with and without deglycosylation—the samples were separated on a UPLC Protein BEH C4 column (300 Å, 1.7 μm, 2.1 × 50 mm) (Waters), with the column temperature set to 80 °C, at 0.3 mL/min flow rate. Gradient: 0 min—5%, 2 min—5%, 2.1 min—25%, 17 min—35%, 17.1 min— 90%, 19 min—90%, 20 min—10%, 25 min—10%. Acquisition range: 600–4500 m/z, in sensitivity analyzer mode. Capillary 3.0 kV, cone 35 V, source 100 °C, desolvation 350 °C, desolvation gas 800 L/h.
Same LC-MS conditions were applied for the analysis of the IdeS-generated subunit (with deglycosylation), with an exception of the optimized gradient: 0 min—10%, 2 min—10%, 2.1 min—26%, 35 min—33%, 35.1 min—95%, 40 min—95%, 43 min—10%, 48 min—10%.
Data Acquisition and Processing
The acquisition of the intact bNAb and its subunit data was performed using a single MS channel. For bottom-up analyses, a low energy MS channel was acquired simultaneously with the corresponding high energy MSE channel, allowing to obtain the information on the precursor ion and its ion fragments for each chromatogram peak. MassLynx, v. 4.1, with maximum entropy algorithms (MaxEnt1 and MaxEnt3) and BiopharmaLynx software, v.1.3, were used for data processing, with MassLynx applied for peptide de novo sequencing and BiopharmaLynx to streamline the large bulk of data processing. The BiopharmaLynx search included semidigested and miscleaved peptides, with 5 ppm mass accuracy of the precursor ions and 15 ppm for the fragment ions. 3D-crystallographic analysis of the CAP256 molecule (Protein Databank sequence entry 5DT1) was performed using PyMOL Molecular Graphic System, v. 1.8.2.3.
Results and Discussion
Following the practice for routine product quality assessment, reducing CE analysis was performed to assess purity of CAP256. As shown in Figure 1, the HEK-expressed control sample demonstrated a typical gel-separated mAb profile, with light and heavy chain peaks only. However, two extra peaks were observed in the electropherogram of the CHO-expressed material. While analyzing the same samples by non-reducing CE, HEK- and CHO-produced CAP256 showed no difference in their profiles. Therefore, in order to assign the additional peaks discovered by reducing CE and to thoroughly characterize all possible structural changes of the CHO-expressed CAP256, a panel of LC-MS analyses was strategized, shown in Scheme 1. The set of orthogonal LC-MS techniques included the following: (1) intact protein mass analysis to probe for sample heterogeneity; (2) bNAb reduction with and without deglycosylation for PTM and glycoforms profiling, and the accurate mass assignment of the individual subunits; (3) a middle-up approach using IdeS enzymatic digestion to complement the subunit mass analysis; and (4) full characterization of the primary structure and PTMs by complementary peptide mapping techniques and disulfide bond mapping. As a result of such a comprehensive approach, a clipping in the CAP256 was successfully identified, and a full structural characterization of this molecule was achieved.
Figure 1.

CE profiles of the reduced CAP256 bNAb: HEK cell line (top) and CHO (bottom). The CHO cell line platform resulted in two additional peaks, in addition to the expected light and heavy chains
Scheme 1.

Various LC-MS workflows used to characterize the CAP256 bNAb
Analysis of the Non-reduced and Reduced bNAb Samples
The CHO-expressed CAP256 sample containing 17% impurity (per reducing CE data) was used for the subsequent LC-MS characterization. Prior to subunit analysis, intact analysis of CAP256 was performed as an initial screening. The mass chromatogram of the intact CAP256 bNAb revealed a highly heterogeneous glycosylation profile of overlapping peaks, which was expected since this molecule contains multiple glycosylation sites (data not shown). However, no unusual fragments or impurities were detected, which was consistent with the results of the non-reducing CE data.
LC-MS analysis of the reduced CAP256 was subsequently performed in order to characterize peaks observed in the reducing gel electropherogram. A reversed phase gradient that was previously optimized for the separation of the heavily glycosylated subunits of various HIV-mAb materials was adopted for the separation of CAP256 produced in the CHO cell line. Unlike a traditional chromatographic profile of a mAb, which has two peaks reflecting the glycosylated light chain and glycosylated heavy chain, the complex separation pattern of CHO-expressed CAP256 suggested a higher sample heterogeneity. As shown in Figure 2, a cluster of peaks around 4.5 min and a very broad peak at 6.5 min (indicating heterogeneity) was observed, followed by the apparent glycoforms. Deglycosylation was used to simplify the LC profile (chromatographic data not shown) by removing the glycosylated pattern, yet it did not eliminate the early eluting peak cluster or the peak broadness. In comparison, for the CAP256 produced in the HEK cell line, the reduced and deglycosylated LC-MS analysis showed a typical chromatographic profile (Figure 2, inset) of a bNAb—with acetylation of the light chain also being observed and later confirmed by peptide mapping. No other major impurities or unidentified peaks were discovered in the HEK material, which was consistent with the CE results, whereas four major species were found for the deglycosylated reduced CHO CAP256 material.
Figure 2.

TIC of the reduced CAP256 with medium clipping, showing separation of the subunits, the subunit fragments with modifications, and the light chain glycoforms. Inset: TIC of the reduced and deglycosylated CAP256 produced in HEK cell line
CAP256 contains two N-glycosylated sites—one in the Fc region and one in the light chain. As demonstrated in Figure 3, the mass spectra were extracted from multiple chromatographic peaks eluting in the 7–9-min range and were identified as various light chain glycoforms. The glycosylation profile included fucosylated, heavily sialylated species, and evidently the extra branch of lactosamine added to the complex N-glycan structure (i.e., G0F, G1F, G2F, G2F + SA, G2F + 2SA, G2F + 2SA, G2F + GlcNAc + 3SA). Acetylation (+ 42 Da) was also observed in the light chain and was later confirmed by peptide mapping results, similarly to the HEK cell line material (Figure 3d). Deglycosylation was applied to confirm the accurate mass of the light chain (Figure 3e), and the observed mass matched the theoretical sequence of the light chain.
Figure 3.

Light chain characterization: TIC of the reduced CAP256 (inset) with the individual components of the light chain numbered, corresponding to the deconvoluted spectra of the light chain glycoforms (a–d) and deconvoluted spectrum of the deglycosylated light chain (e). An additional peak on the mass spectrum (e) corresponds to incomplete cysteine alkylation, a sample preparation artifact
Two sets of closely eluting components appeared in the mass spectrum after extraction from 6.4 ± 0.3 min chromatogram range (Figure 4a): a 54-kDa cluster in a form of a fucosylated glycosylation pattern of G0F, G1F, and G2F, typical for the Fc region of a IgG1-type bNAb, was identified as the glycosylated heavy chain variants. The second (40 kDa) peak also contained the same glycosylation profile as the 54-kDa cluster peak [13]. These 40-kDa species were subsequently identified to be related to a C-terminal heavy chain fragment. As demonstrated in Figure 4b, the mass spectra, extracted from the 4.5–5.2-min time range, revealed a set of 14-kDa peaks, which was identified as the complementary non-glycosylated N-terminal fragment of the heavy chain. In summary, these two fragments (14 kDa and 40 kDa) originated from the heavy chain (54 kDa), and it was concluded that a clipping occurred within the heavy chain of the CAP256 bNAb. The clipping phenomenon was further confirmed via complementary peptide mapping and IdeS digestion techniques; therefore, the terms “large fragment” and “small fragment” will be applied for description of clipped species throughout the text.
Figure 4.

Deconvoluted mass spectra. (a) The heavy chain and its ~ 40-kDa fragment, extracted from the 6.4 ± 0.3-min time range and (b) the small (~ 14 kDa) fragment of the heavy chain (4.5–5.2-min time range)
Discovering the clipping phenomenon was essential for deciphering the rest of the chromatographic heterogeneity. The heavy chain characterization was further completed by reporting PTMs, in addition to glycosylation and its clipped fragments. Both intact heavy chain and its large fragment had the same extent of C-terminal Lys cleavage. The smaller heavy chain fragment and the intact subunit also demonstrated three peaks that were 80 Da apart (Figure 4). Considering that the CAP256 structure was designed specifically to contain O-sulfated tyrosines (Tyr112, Tyr113) in the CDR-H3 region [7], these 80-Da mass shifts were tentatively assigned as sulfation and were later confirmed by peptide mapping analysis. This assignment correlates with the biological growth path of CAP256 [7]. The non-sulfated variant was present in small quantity while the mono-sulfated variant was the most abundant form. Among other PTMs, N-terminal pyroglutamination and carbamylation of the NH2-terminus of Lys or Cys (+ 43 Da) were also observed, the latter resulting from the reaction of CAP256 with urea-derived cyanate [14]. The trace amounts of incomplete carbamidomethylation, a sample preparation artifact, are denoted on the spectrum (Figure 4b).
Peptide Mapping Analysis
Following a bottom-up approach for routine mAb characterization, the structure of CAP256 was investigated by LC-MS peptide mapping, which is commonly used to verify protein sequence and identify PTMs. The discovery of sequence clipping by subunit mass analysis presented another challenge for peptide mapping analysis. To provide sufficient evidence for a clipping phenomenon, the peptide mapping portion of the MS characterization started with primary sequence confirmation and common PTM profiling.
Separate digests using trypsin and chymotrypsin were performed to improve the sequence coverage of CAP256 and to help confirm the clipping location. Identification of the peptides was verified by a strict set of filtering criteria including the characteristic ion fragments generated by high-energy MSE fragmentation. For the MS peaks which failed to get assigned by the automatic data processing, a de novo manual assignment was performed using the characteristic ion fragment of the MSE spectra. The tryptic digest alone resulted in 96% sequence coverage; in combination with chymotryptic digestion, the results of the cumulative coverage increased to 98% (Figure 5). This was sufficient for CAP256 identification, complementing the results of the subunit LC-MS analysis. The PTMs were identified and quantified, as shown in Figure 6. Common PTMs, such as oxidation, deamidation, and N-pyroglutamination, could occur at a wide range of sample manufacturing, storage, and sample preparation conditions. Further details of the PTM analysis can be found in Figure 5 and the Supplementary Information. The only critical PTM which required detailed confirmation was the + 80-Da mass shift observed during the subunit analysis. This delta mass corresponds to either sulfation or phosphorylation. Since these isobaric modifications differ by only 9.3 mDa, subunit analysis cannot differentiate these two PTMs. Therefore, peptide mapping analysis (Supplementary Figure S1) was employed, and the results favored sulfation, judging by the mass accuracy (1 ppm for sulfation for a 2600-Da peptide, vs. 5 ppm for phosphorylation) and the characteristic high-energy fragmentation. Collectively, sulfation was verified and expected to be present in the protruding antigen-binding loop of the CAP256 molecule [7].
Figure 5.

Summary of the identified portion of the CAP256 primary sequence: tryptic coverage (solid underline); chymotryptic coverage (dotted underline) complementing the missing portions of the tryptic coverage; both digest types contributing to a cumulative 98% sequence coverage. PTMs with % abundance (when applicable) denoted above the modified site marked in bold
Figure 6.

TIC chromatogram of the CAP256 bNAb after IdeS digestion, reduction, and deglycosylation with PNGase F, demonstrating the clipped heavy chain fragments among other IdeS digested bNAb major products
The glycosylation profiles on the light and heavy chains were also probed by peptide mapping analysis. Expected sialylation of the light chain was confirmed, whereas the glycosylation profile of the heavy chain was determined to exhibit the typical IgG1-type mAb glycosylation profile, as described earlier. Overall, typical PTMs including the engineered sulfation were identified; however, several high-intensity peaks from the peptide mapping results were still observed, which automatic data processing failed to identify and needed further investigation.
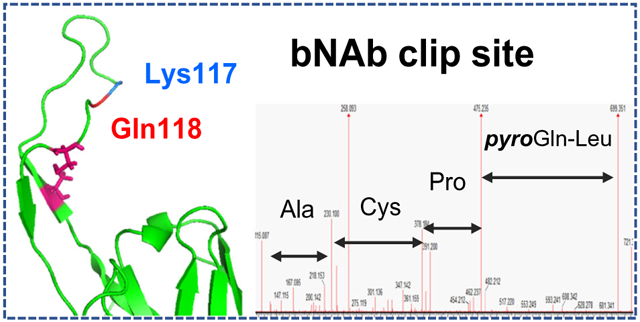
The clipping of the CAP256 heavy chain phenomenon was proposed based on the results of the subunit LC-MS analysis; however, the observed masses included PTMs and terminal modifications which complicated the data interpretation. Therefore, the exact location of the clipping needed to be verified using a more precise technique, and de novo sequencing of the unassigned peptide components was performed using their high-energy MSE spectra. Subsequently, this de novo analysis helped identify a very high degree of Q-pyroglutamination present in one of the internal tryptic peptides (pyroQLPCAK) of the heavy chain, as demonstrated in Figure 7. This modification can only be produced on a terminal Gln or Glu residue, indicating that glutamine formed an N-terminus in some portion of the CAP256 protein. The cleavage occurred at a Lys117-Gln118 site in the Fab region of the heavy chain and therefore was assigned as the clipping location. The non-modified QLPCAK peptide and its pyroglutaminated analog pyroQLPCAK were well resolved chromatographically, as shown by the extracted ion chromatogram of 699.3 Da (pyroQLPCAK) and 716.3 Da (QLPCAK) (Figure 7, insets). The reason the data processing software could not provide the correct assignment for this particularly peptide was because pyroglutamination is not expected to be present in internal peptides. Once the QLPCAK component was entered in the search as a separate peptide entity, the automatic processing was able to assign its components as “Pyroglutamic Acid Q N-term” (Supplementary Figure S2). A related dimer peptide and semidigested species were identified and reported as well. Altogether, the clipping site at the Lys117-Gln118 residues was substantiated since the molecular weight of the unmodified large and small fragments identified in the subunit analysis matched these theoretical, clipped sequences.
Figure 7.

Identification of the clipping location based on MSE de novo sequencing of the unassigned tryptic component. Top: unassigned peak on the chromatogram. Middle: assignment of pyroQLPCAK (a) and its non-modified analog QLPCAK (b); XIC of the corresponding peptides: 699.3 Da (pyroQLPCAK) (a, inset), 716.3 Da (QLPCAK) (b, inset), (0.1 Da extracted mass window). Bottom: Fab portion of CAP256 heavy chain showing the clipping site and the identified tryptic peptide marked in bold
Formation of pyroglutamic acid from N-terminal glutamic acid is a commonly observed mAb modification. Pyroglutamination is a non-enzymatic reaction, which typically occurs at a wide range of sample manufacturing, storage, and sample preparation conditions [15-17]. However, there is a possibility that pyroglutamate formation can occur shortly after tryptic digestion: trypsin cleaves at the Lys117-Gln118 site, with subsequent conversion of Gln to pyroGlu. Therefore, a chymotrypsin digest of CAP256 was performed as a secondary peptide mapping analysis. It also confirmed the presence of the internal pyroQLPCAK peptide (Supplementary Figure S3), supporting the results of the tryptic digest analysis. Therefore, the pyroglutamination was not a sample preparation artifact, but rather a PTM originally present in the provided CHO cell line CAP256 samples. Moreover, this was proven by the subunit LC-MS analysis, with the masses corresponding to the pyroGlu-formation in the small and large fragments of the heavy chain.
Middle-Up Approach Using IdeS Digestion
A middle-up LC-MS approach was also employed as a complimentary analysis to further probe the heavy chain and its fragments. The use of IdeS has become increasingly popular for middle-up/down approaches because the enzyme specifically cleaves antibodies at a single site below the hinge region of the heavy chain (between GG residues), yielding F(ab’)2 and Fc/2 subunits [18, 19]. For most LC-MS applications though, a reduction/alkylation step is used, which allows for characterization of three ~ 25-kDa-sized subunits: Fd, Fc/2, and the light chain. In comparison to reduced-only antibodies, which yields free heavy chains (50 kDa) and the light chain (25 kDa), these 25-kDa IdeS-generated subunits better meet the challenges for the LC-MS analysis of complex bNAbs at the subunit level. Therefore, an IdeS middle-up LC-MS approach was employed to further confirm the identity of CAP256 and its clipped products.
As shown in Figure 6, CAP256 was successfully digested using IdeS and deglycosylated using PNGase F. Experimental masses of the intact Fd, Fc/2, and LC subunits were found to be in good agreement with the theoretical masses. In addition, small and large fragments of the Fd subunit were also observed, confirming the results of the clipping phenomenon and subsequently its location. Specifically, the large fragment of the Fd subunit had an observed molecular weight of 15,377 Da and matched the molecular weight of amino acid residues Gln118-Gly264 with a N-terminal pyroglutamination modification. These results further proved that the N-terminal pyroglutamination modification was not caused by trypsin digestion during the peptide mapping analysis. The smaller fragment of the Fd subunit was identified as amino acid residues Gln1-Lys117, and some mass heterogeneity could be explained by the presence of N-terminal pyroglutamination and sulfation, as described earlier. Two peaks were observed for the small Fd fragment, which corresponded to one and two 80-Da mass shifts.
Low-clipped (< 1% by reductive CGE) CAP256 development-grade material was also analyzed for comparison purpose. Only three major peaks were detected, which matched the intact Fd, Fc/2, and the light chain theoretical subunit masses (data now shown). In summary, reductive IdeS digestion generated Fc/2, intact Fd, and LC subunits of CAP256. Small and large fragments of the Fd subunit were easily identified in the medium clipped CAP256 sample, as well as some variants of the subunits and their fragments. These results helped confirm the clipping location and were complimentary to the subunit and peptide mapping analyses.
Disulfide Bond Mapping and 3D Structural Alignment
The non-reducing CE did not reveal unexpected peaks, and the same results were obtained by MS analysis of the intact CAP256, for both HEK and CHO cell line materials. Disulfide bond mapping helped elucidate this observation. To capture the arrangement of the disulfide linkage within the clipped CHO material, a non-reduced CAP256 sample was prepared and characterized by peptide mapping analysis [20]. All non-reduced peptides of the heavy chain were identified (Figure 8a, marked in red), with their structures verified by high-energy MSe spectra. Among the non-reduced peptides, DLREDEC105 EEWWSDYYDFGKQLPC121AK dimers were identified, which represented miscleaved peptides containing the Lys117-Gln118 clipping site: one was a 3122.3-Da peptide—which is part of the intact heavy chain—and the other was a 3123.3-Da peptide—which was a part of the clipped fragments with pyroglutamic acid at the terminal Gln (Table 1). The mass difference of 1 Da was sufficient to differentiate between these two peptides, and high-energy MS spectra confirmed their identity (Supplementary Figure S4). Therefore, the disulfide bond linkage was verified, demonstrating that the two clipped portions of the heavy chain were connected under nonreducing conditions. This explained why the non-reducing CE and the LC-MS analysis ofthe intact bNAb (not shown) did not show fragments and resulted in one homogeneous monomer peak (~ 150 kDa). Once the protein was reduced, the clipped fragments became disconnected, resulting in three heavy chain-related peaks: an intact heavy chain, 14-kDa N-terminal fragment, and 40-kDa C-terminal glycosylated fragment. No clipped peptide dimer (a tryptic component held together by disulfide bond) was observed without a pyroglutaminated terminal modification (3140.3 Da), assuming that all clipped species were 100% modified. Compared to the low level of pyroglutamination commonly observed in Q-terminal peptides of tryptic digests of various mAbs, 100% conversion suggested that the protein degradation occurred prior to the sample preparation, with sufficient time for spontaneous cyclization.
Figure 8.

Clipping site identification. (a) A protruding loop in the CDR-H3 region (left); clipping location with a disulfide bond preventing the non-reduced bNAb to fall apart into two fragments (right). (b Disulfide bond arrangement, planar view: Cys residues are numbered, verified disulfide bonds marked in red. The sequence of the heavy chain Fab region is shown, along with the corresponding schematic view zoomed into the clipping site: the tryptic semidigested peptide (highlighted in orange) identified as a peptide dimer (tryptic component held together by a disulfide bond), covering the clipping site area
Table 1.
Possible Non-reduced Dimer Peptides Based on DLREDECEEWWSDYYDFGKQLPCAK Sequence at the Clipping Site
| Dimer peptide | Mass, Da |
|---|---|
| 3122.295 | |
| 3140.306 | |
 |
3123.303 |
To align the LC-MS data with the proposed unique structural features of this bNAb, a 3D structure of the CAP256 Fab region was constructed using the PyMOL software, based on the Protein Databank sequence entry 5DT1 [21]. According to the designed structure of CAP256, this bNAb has an additional intra-CDR-H3 disulfide bond, contributing to the total of three intra-disulfide bridges in the heavy chain. This disulfide bond arrangement of the Fab region was consistent with the experimental MS data, showing that the Lys117-Gln118 clip is located in the unusually long CDR-H3 portion (particularly, within the anionic antigen-binding sequence loop protruding away from the mAb coil surface) which serves to facilitate the bNAb penetration of the HIV-1 glycan shield (Figure 8b) [7]. This loop is exposed to the environment and may be susceptible to residual enzymes in the cell culture supernatant. A clarification of the disulfide bond arrangement within this unique bNAb explained the different purity observations of the reducing and non-reducing CE methods, which was further supported the LC-MS analysis findings.
Conclusions
A combination of complementary LC-MS-based approaches was applied for HIV-1 bNAb structure elucidation and to assist the process/product investigations. During a development investigation of CHO-expressed CAP256, a clipping in the heavy chain was discovered and the clipping site was identified by parallel subunit and peptide mapping approaches. Disulfide bond mapping analysis confirmed that an intra-disulfide bond linked the two clipped fragments, explaining why the clipping-caused fragmentation was only observed under reducing condition.
As a result of this comprehensive LC-MS study for characterization of the highly potent bNAb, the primary amino acid sequence was confirmed, both the light chain and heavy chain glycosylation profiles were characterized, and lastly, the critical disulfide bonds were verified. Various post-translational modifications were reported, including sulfation, acetylation, N-terminal pyroglutamination (also used as indication of a clipping site), and C-terminal Lys variants. The clipping in the heavy chain was found to be located in the CDR-H3 region, at the Lys117-Gln118 site.
This information has led to process improvement to further develop this bNAb candidate and prompted the re-engineering designs to mutate out the clipping susceptible amino acid motif. LC-MS characterization of this CAP256 represents an analytical advancement for product quality control, as it was demonstrated to be a powerful investigation tool for providing direction to monitor CQA and to mitigate product quality risk.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dr. Kuang-Chuan Cheng and Kevin Carlton from the Vaccine Production Program for scientific discussions and Dr. Daniel Gowetski for contributing to the 3D structural analysis of CAP256.
Funding Information
This work was supported by the intramural research program of the Vaccine Research Center, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s13361-018-1968-0) contains supplementary material, which is available to authorized users.
References
- 1.Mascola JR, Haynes BF: HIV-1 neutralizing antibodies: understanding nature’s pathways. Immunol. Rev 254(1), 225–244 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kwong PD, Mascola JR: Human antibodies that neutralize HIV-1: identification, structures, and B cell ontogenies. Immunity. 37(3), 412–425 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O’Rourke SM, Schweighardt B, Phung P, Mesa KA, Vollrath AL, Tatsuno GP, To B, Sinangil F, Limoli K, Wrin T, Berman PW: Sequences in glycoprotein gp41, the CD4 binding site, and the V2 domain regulate sensitivity and resistance of HIV-1 to broadly neutralizing antibodies. J. Virol 86(22), 12105–12114 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Montero M, van Houten NE, Wang X, Scott JK: The membrane-proximal external region of the human immunodeficiency virus type 1 envelope: dominant site of antibody neutralization and target for vaccine design. Microbiol Mol Biol Rev 72(1), 54–84 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Doria-Rose NA, Bhiman JN, Roark RS, Schramm CA, Gorman J, Chuang GY, Pancera M, Cale EM, Ernandes MJ, Louder MK, Asokan M, Bailer RT, Druz A, Fraschilla IR, Garrett NJ, Jarosinski M, Lynch RM, McKee K, O’Dell S, Pegu A, Schmidt SD, Staupe RP, Sutton MS, Wang K, Wibmer CK, Haynes BF, Abdool-Karim S, Shapiro L, Kwong PD, Moore PL, Morris L, Mascola JR: New member of the V1V2-directed CAP256-VRC26 lineage that shows increased breadth and exceptional potency. J. Virol 90(1), 76–91 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wagh K, Bhattacharya T, Williamson C, Robles A, Bayne M, Garrity J, Rist M, Rademeyer C, Yoon H, Lapedes A, Gao H, Greene K, Louder MK, Kong R, Karim SA, Burton DR, Barouch DH, Nussenzweig MC, Mascola JR, Morris L, Montefiori DC, Korber B, Seaman MS: Optimal combinations of broadly neutralizing antibodies for prevention and treatment of HIV-1 clade C infection. PLoS Pathog. 12(3), 1–27 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Doria-Rose NA, Schramm CA, Gorman J, Moore PL, Bhiman JN, DeKosky BJ, Ernandes MJ, Georgiev IS, Kim HJ, Pancera M, Staupe RP, Altae-Tran HR, Bailer RT, Crooks ET, Cupo A, Druz A, Garrett NJ, Hoi KH, Kong R, Louder MK, Longo NS, McKee K, Nonyane M, O’Dell S, Roark RS, Rudicell RS, Schmidt SD, Sheward DJ, Soto C, Wibmer CK, Yang Y, Zhang Z, Comparative Sequencing Program, N.I.S.C., Mullikin JC, Binley JM, Sanders RW, Wilson IA, Moore JP, Ward AB, Georgiou G, Williamson C, Abdool Karim SS, Morris L, Kwong PD, Shapiro L, Mascola JR: Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature. 509(7498), 55–62 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hossler P, Khattak SF, Li ZJ: Optimal and consistent protein glycosylation in mammalian cell culture. Glycobiology. 19(9), 936–949 (2009) [DOI] [PubMed] [Google Scholar]
- 9.Beck A, Wagner-Rousset E, Ayoub D, Van Dorsselaer A, Sanglier-Cianferani S: Characterization of therapeutic antibodies and related products. Anal. Chem 85(2), 715–736 (2013) [DOI] [PubMed] [Google Scholar]
- 10.Rehder DS, Dillon TM, Pipes GD, Bondarenko PV: Reversed-phase liquid chromatography/mass spectrometry analysis of reduced monoclonal antibodies in pharmaceutics. J. Chromatogr. A 1102, 164–175 (2006) [DOI] [PubMed] [Google Scholar]
- 11.Zhang Z, Pan H, Chen X: Mass spectrometry for structural characterization of therapeutic antibodies. Mass Spectrom. Rev 28(1), 147–176 (2009) [DOI] [PubMed] [Google Scholar]
- 12.Gollapudi D, Wycuff DL, Schwartz RM, Cooper JW, Cheng KC: Development of high-throughput and high sensitivity capillary gel electrophoresis platform method for Western, Eastern, and Venezuelan equine encephalitis (WEVEE) virus like particles (VLPs) purity determination and characterization. Electrophoresis. 38, 2610–2621 (2017) [DOI] [PubMed] [Google Scholar]
- 13.Sinha S, Pipes G, Topp EM, Bondarenko PV, Treuheit MJ, Gadgil HS: Comparison of LC and LC/MS methods for quantifying N-glycosylation in recombinant IgGs. J. Am. Soc. Mass Spectrom 19(11), 1643–1654 (2008) [DOI] [PubMed] [Google Scholar]
- 14.Lippincott J, Apostol I: Carbamylation of cysteine: a potential artifact in peptide mapping of hemoglobins in the presence of urea. Anal. Biochem 267(1), 57–64 (1999) [DOI] [PubMed] [Google Scholar]
- 15.Kumar M, Chatterjee A, Khedkar AP, Kusumanchi M, Adhikary L: Mass spectrometric distinction of in-source and in-solution pyroglutamate and succinimide in proteins: a case study on rhG-CSF. J. Am. Soc. Mass Spectrom 24(2), 202–212 (2013) [DOI] [PubMed] [Google Scholar]
- 16.Dick LW Jr., Kim C, Qiu D, Cheng KC: Determination of the origin of the N-terminal pyro-glutamate variation in monoclonal antibodyies using model peptides. Biotechnol. Bioeng 97(3), 544–553 (2007) [DOI] [PubMed] [Google Scholar]
- 17.Purwaha P, Silva LP, Hawke DH, Weinstein JN, Lorenzi PL: An artifact in LC-MS/MS measurement of glutamine and glutamic acid: in-source cyclization to pyroglutamic acid. Anal. Chem 86(12), 5633–5637 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fornelli L, Ayoub D, Aizikov K, Beck A, Tsybin YO: Middle-down analysis of monoclonal antibodies with electron transfer dissociation orbitrap fourier transform mass spectrometry. Anal. Chem 86(6), 3005–3012(2014) [DOI] [PubMed] [Google Scholar]
- 19.Sjogren J, Olsson F, Beck A: Rapid and improved characterization of therapeutic antibodies and antibody related products using IdeS digestion and subunit analysis. Analyst. 141(11), 3114–3125 (2016) [DOI] [PubMed] [Google Scholar]
- 20.Liu H, May K: Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs. 4(1), 17–23 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huang J, Kang BH, Ishida E, Zhou T, Griesman T, Sheng Z, Wu F, Doria-Rose NA, Zhang B, McKee K, O’Dell S, Chuang GY, Druz A, Georgiev IS, Schramm CA, Zheng A, Joyce MG, Asokan M, Ransier A, Darko S, Migueles SA, Bailer RT, Louder MK, Alam SM, Parks R, Kelsoe G, Von Holle T, Haynes BF, Douek DC, Hirsch V, Seaman MS, Shapiro L, Mascola JR, Kwong PD, Connors M: Identification of a CD4-binding-site antibody to HIV that evolved near-Pan neutralization breadth. Immunity. 45(5), 1108–1121 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.