

Abstract
Juvenile segmental progeroid syndromes are rare, heterogeneous disorders characterized by signs of premature aging affecting more than one tissue or organ starting in childhood. Hutchinson-Gilford progeria syndrome (HGPS), caused by a recurrent de novo synonymous LMNA mutation resulting in aberrant splicing and generation of a mutant product called progerin, is a prototypical example of such disorders. Here, we performed a joint collaborative study using massively parallel sequencing and targeted Sanger sequencing, aimed at delineating the underlying genetic cause of 14 previously undiagnosed, clinically heterogeneous, non-LMNA-associated juvenile progeroid patients. The molecular diagnosis was achieved in 11 of 14 cases (~ 79%). Furthermore, we firmly establish biallelic mutations in POLR3A as the genetic cause of a recognizable, neonatal, Wiedemann-Rautenstrauch-like progeroid syndrome. Thus, we suggest that POLR3A mutations are causal for a portion of under-diagnosed early-onset segmental progeroid syndromes. We additionally expand the clinical spectrum associated with PYCR1 mutations by showing that they can somewhat resemble HGPS in the first year of life. Moreover, our results lead to clinical reclassification in one single case. Our data emphasize the complex genetic and clinical heterogeneity underlying progeroid disorders.
Keywords: Juvenile segmental progeroid syndrome, Hutchinson–Gilford progeria syndrome, Wiedemann–Rautenstrauch progeroid syndrome, POLR3A, PYCR1
Introduction
Segmental progeroid syndromes are defined as a heterogeneous group of rare monogenic disorders which prematurely mimic physiological aging by simultaneously affecting multiple organs and tissues (Martin 1978). The best known form with onset in early childhood is Hutchinson-Gilford progeria syndrome (HGPS). HGPS is caused by a recurrent de novo synonymous alteration c.1824C > T, p.Gly608Gly, in exon 11 of LMNA (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003). Around 10% of similarly affected individuals, referred to as atypical HGPS, bear de novo LMNA mutations in the spliceosome recognition site of intron 11 (Gordon et al. 1993). These mutations result in increased usage of cryptic splice sites leading to an in-frame deletion of 150 nucleotides, and generation of a lamin A precursor lacking 50 amino acids at its C-terminus, termed prelamin AΔ50 or “progerin” (Eriksson et al. 2003). In addition to HGPS, other mutations in LMNA have been identified in a number of cases affected by a variety of progeroid symptoms (Chen et al. 2003; Garg et al. 2009; Saha et al. 2010; Soria-Valles et al. 2016), including cases of late-onset segmental progeroid syndromes bearing heterozygous mutations at the junction of exon 11 and intron 11. Notably, these latter mutations also result in aberrant splicing and production of progerin, however to a lower extent than observed in HGPS (Hisama et al. 2011).
Mutations in several other genes have also been connected with early-onset segmental progeroid syndromes sharing overlapping phenotypic features with HGPS such as growth retardation, sparse scalp hair, mandibular underdevelopment, thin-appearing and transparent skin with prominent veins, and lipodystrophy. Examples include Cutis Laxa Type IIB with progeroid features caused by biallelic mutations in PYCR1(Reversade et al. 2009), Nestor-Guillermo progeria syndrome caused by biallelic mutations in BANF1 (uente et al. 2011), ZMPSTE24-related progeroid cases (Navarro et al. 2014), Berardinelli-Seip congenital lipodystrophy (Van Maldergem 1993), mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome (Lessel et al. 2015; Nicolas et al. 2016; Weedon et al. 2013), marfanoid-progeroid-lipodystrophy syndrome caused by de novo mutations in FBN1 (Graul-Neumann et al. 2010; Passarge et al. 2016) as well as single cases bearing likely causative mutations in CAV1 (Schrauwen et al. 2015) and POLR3A (Jay et al. 2016).
Here, we describe results from systematic genetic analysis, including massively parallel sequencing and targeted Sanger sequencing, of 14 children referred either to The Progeria Research Foundation (PRF) or to the Institute of Human Genetics at the University Medical Center Hamburg-Eppendorf, with clinical diagnosis of an early-onset segmental progeroid syndrome in whom direct sequencing of LMNA and ZMPSTE24 gave normal results.
Materials and methods
Human subjects
We investigated 14 individuals who had been referred either to The Progeria Research Foundation (PRF, http://wwwprogeriaresearch.org ) or to the Institute of Human Genetics at the University Medical Center Hamburg-Eppendorf, with clinical diagnosis of a HGPS-like segmental progeroid syndrome, and in whom LMNA and ZMPSTE24 mutations were first excluded. All biological samples and images were obtained following written informed consent from affected individuals or their legal representatives. Lympho-blastoid cell lines (LCLs) of individual 4 and her parents were generated by standard procedures after an informed consent. The study has ongoing approval from the Hasbro Children’s Hospital and the University Medical Center Hamburg-Eppendorf Institutional Review Boards. The study was performed in accordance with the Declaration of Helsinki protocols.
Genetic analyses
DNA samples from whole blood were isolated by standard procedures. For candidate gene analysis, namely LMNA, ZMPSTE24, PYCR1 and POLR3A, we performed direct Sanger sequencing. Trio-whole exome sequencing experiments with DNA samples of both healthy parents and the index patients were performed in three different centers with slightly different procedures that have essentially been previously described. In more detail, in families 1–5 exome sequencing targeting ~ 44 Mb of the genome of proband DNAs was performed on exons captured by Roche Nimble-gen SeqCap EZ Human Exome Library v2.0 and sequenced using the Illumina HiSeq2500/4000 in the University of Michigan Sequencing Core. Individual DNAs were each run on a single lane using 100-bp paired-end sequencing. The average coverage ranged from 158× to 276×. Exome sequencing of parental DNA samples was performed at the University of Washington Center for Mammalian Genomics on exons captured by Roche Nimblegen SeqCap EZ Human Exome Library v2.0 and sequenced using the Illumina HiSeq2500/4000 as previously described (Chong et al. 2015). The average coverage ranged from 46× to 57×. In families 6–10, exome sequencing was performed at the Cologne Center for Genomics on two lanes of an Illumina GAIIx Sequencer using a single-read 150-bp protocol after enrichment of exonic and splice-site sequences with the Agilent SureSelect Human All Exon 50 Mb kit as previously described (Lessel et al. 2017). In families 11–14, only direct sequencing of candidate genes, i.e. PYCR1 or POLR3A, was performed.
Bioinformatic analysis
Variant annotation and bioinformatic filtering was performed using three independent in-house annotation pipelines.
For families 1–5, the exome sequencing data were included in a 734-exome joint call in GATK. After removing variants of low quality [GATK’s VQSR, Genotype Quality (GQ) < 20, Read Depth (RD) < 10], we annotated the remaining on-target variants by ANNOVAR. Further filtering was applied to keep (1) the splicing-altering sites, frameshift, nonsense and missense mutations predicted as damaging by Polyphen2 and SIFT, (2) those that are not common (MAF < 1%) in the European subset (n = 514) of the 1000 Genomes Project or in the Exome Sequencing Project (n = 4,300), and (3) not present in more than 5% of the in-house controls (N > 500). These variants were analyzed with a recessive model and a de novo model, with the top candidate genes further filtered to remove those with previous associations with human diseases and/or without OMIM annotations.
For families 6–10, in-house-developed scripts were applied to detect protein changes, affected splice sites and overlaps to known variations, with filtering against dbSNP build 138, the 1000 Genomes Project data build November 2014, ExAC Browser (status from August 2018) and the in-house database of exome variants (with data from > 200 exomes of individuals affected by different disorders). The criteria for a variation to be taken into account were: >6 reads, Phred scaled quality score > 15, population allele frequency < 1%, < 10 times seen in our in-house database and > 15% of the reads supporting the allele.
Candidate variants were validated by Sanger sequencing. For families 11–14, candidate genes, PYCR1 or POLR3A, were analyzed by Sanger sequencing. All primer pairs are available upon request.
RT-PCR analysis
Total RNA was extracted from LCLs of individual 4 and her parents using a Qiagen RNeasy Mini Kit. First-strand cDNA synthesis was carried out using the High-Capacity cDNA Reverse Transcription Kit (ThermoFisher Scientific), primed with an oligo in exon 31 of POLR3A (5’-TGTGAGCTGCAC CCTATCAG-3’) following the manufacturer’s protocol. The POLR3A cDNA was PCR amplified using primers in POLR3A exons 25 (5 ‘-GATCATCAACGCTTCCAAGG-3’) and 27 (5’-CCTTGGGGAGATCCTCTTTC-3’). Resulting products were electrophoresed, revealing bands of two sizes (287 bp, 380 bp), which were gel extracted, and subjected to Sanger sequencing. The larger band (380 bp) corresponded to the wild-type POLR3A sequence from exons 25 to 27, including exon 26. The smaller band (287 bp), found only in the proband and the father, corresponded to POLR3A mRNA lacking exon 26.
Results
Case reports
Individual 1
This male proband is the first child of healthy unrelated parents. He has three unaffected siblings. He was born after an uneventful pregnancy at 40 weeks of gestation. His birth weight was 1930 g (− 3.84 SD) with length of 40.5 cm (− 5.22 SD). This boy showed wrist contractures, kyphosis and poor feeding since birth, three natal teeth, and a generalized lipodystrophy. He further had thin-appearing and translucent skin with prominent veins, triangular face with midface retraction, frontal bossing, deep set and closely spaced eyes, low set ears and mandibular underdevelopment. However, he did not have sparse scalp hair (Fig. 1a; Table 1). Milestones of motor development and language development were unremarkable. Kyphosis improved by the age of 4 years. At clinical evaluation at the age 10 years, a small jaw and low set ears were observed. He had a weight of 25.4 kg (− 1.84 SD) and height of 128.3 cm (− 1.98 SD), still having severe feeding difficulties. His hair was sparse and thin. He had only five teeth. Further, he had global lipodystrophy, prominent veins and a nasal high-pitched voice.
Fig. 1.
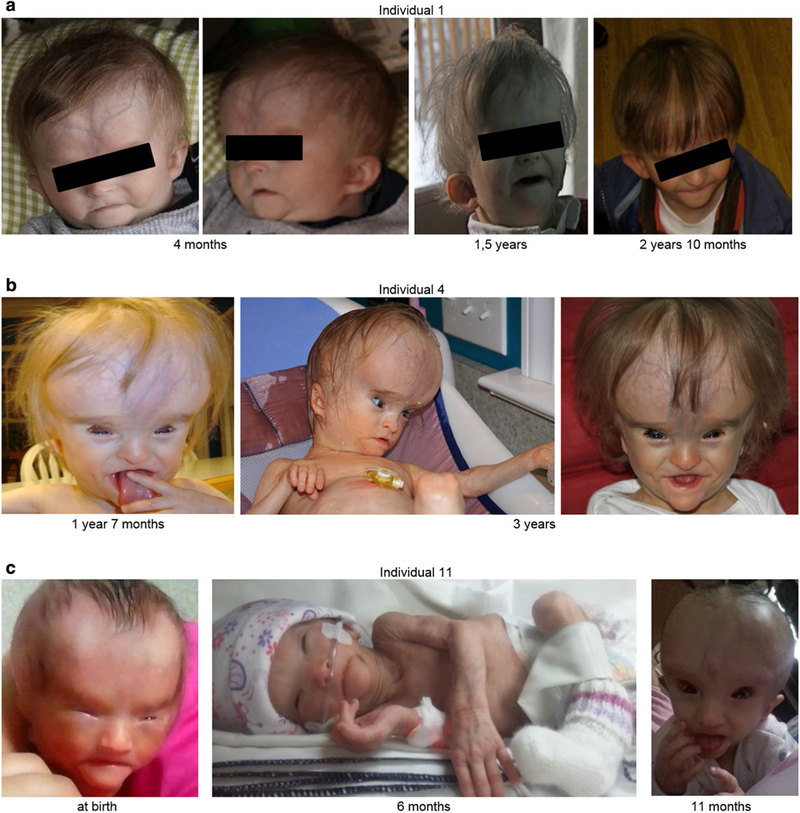
Clinical characteristics of POLR3A-mutated individuals. Facial images of individual 1 at age 4 months, 1.5 years and 2 years and 10 months (a), individual 4 at the age of 1 year and 7 months, and 3 years (b), and individual 11 from the birth till 11 months (c)
Table 1.
Comparison of clinical characteristics of Wiedemann-Rautenstrauch-like, POLR3A-mutated patients presented here, the POLR3A-patient from the previous study (Jay et al. 2016), and patients from the literature with suggestive diagnosis of Wiedemann-Rautenstrauch syndrome (Paolacci et al.)
| Individual 1 | Individual 4 | Individual 11 | Previous study, Jay et al. | WRS, Paolacci et al. | |
|---|---|---|---|---|---|
| Sex | Male | Female | Female | Female | 11 female |
| 7 male | |||||
| Age | 10 years | 12 years | 11 months | Deceased at 7 months of age | n.d |
| 9 months | |||||
| Ethnicity | European | European | Latino | European | Various |
| Intrauterine growth retardation | + | + | + | + | 9/16 |
| Triangular face | + | + | + | + | 18/18 |
| Sparse scalp hair | − | + | + | + | 17/18 |
| Thin/translucent skin | + | + | + | + | 15/15 |
| Prominent scalp veins | + | + | + | + | 17/18 |
| Frontal bossing | + | + | + | + | n.d |
| Closely spaced eyes | + | + | + | + | n.d |
| Midface retraction | + | + | + | + | n.d |
| Low set ears | + | + | + | + | n.d |
| Natal teeth | + | − | − | + | 17/18 |
| Mandibular underdevelopment | + | + | + | + | n.d |
| Joint contractures | + | + | − | + | n.d |
| Motor development delay | − | + | ? | ? | n.d |
| Impaired speech development | − | + | ? | ? | n.d |
| Delayed dentition/oligodontia | + | + | ? | ? | 8/16 |
| Lipodystrophy | + | + | + | + | 18/18 |
| Cardiac anomalies | − | + | + | − | n.d |
| Short stature | + | + | ? | ? | 6/7 |
| Kyphosis | + | − | − | − | n.d |
| Congenital respiratory complications | − | + | + | + | n.d |
| Brain anomalies US/MRI | n.d | n.d | Agenesis of corpus callosum | − | 2/7 |
| POLR3A mutation | |||||
| Paternal | c.3337-5T > A | c.3337-5T > A | p.M1I | c.1909 + 18G > A | n.d |
| Maternal | c.3337-1G > A | p.R254* | Not identified | p.R873* | n.d |
+ present, − absent, ? due to young age not applicable, n.d. not done, WRS Wiedemann-Rautenstrauch syndrome
Individual 2
This male proband is the only child of healthy unrelated parents. Pregnancy was complicated by oligohydramnios and frank breech position leading to birth by cesarean section at 32 weeks of gestation with APGAR scores of 7 at 1 min and 9 at 5 min. His birth weight was 935 g (− 2.38 SD), length 36.5 cm (− 2.16 SD), and a borderline occipitofrontal head circumference (OFC) of 26 cm (− 2.32 SD). A patent foramen ovale and large patent ductus arteriosis were observed after birth. In addition, due to pulmonary hypoplasia he received endotracheal intubation and ventilator support during the first few weeks of life. At the clinical evaluation at the age of 1 year and 5 months, he had a triangular face, sparse scalp hair, thin-appearing and translucent skin with prominent veins, frontal bossing, deep set eyes, somewhat low set ears and a mandibular underdevelopment. Moreover, he had bilateral corneal opacities, strabismus and nystagmus, global lipodystrophy, joint hypermobility, and had developed an inguinal hernia (Fig. 2a; Table 2). All milestones of motor development were delayed: rolling over at 12 months, sitting at 24 months, and walking at 3 years. Language development was significantly delayed as he spoke his first words at the age of 4 years. Clinical evaluation at the age of 3 years revealed the following facial dysmorphisms: frontal bossing, dolichocephaly, prominent occiput, mandibular prognathism (anterior open bite and mandibular overjet), mildly low set ears with right avulsion of pinna, short philtrum and deep set eyes. He had brittle teeth, requiring multiple dental restorations at age 4 years, and some secondary teeth were absent. He developed seizures at the age of 3 years (petit mal and grand mal). A brain MRI, performed at the age of 5 years, revealed mild enlargement of occipital horns with thinning of periventricular white matter. At the age of 6 years, an intracerebral abscess of the frontal lobe was observed. In addition, this boy had thin-appearing, transparent and wrinkled skin with easily visible vasculature. Several eye anomalies including mild optic nerve hypoplasia, small optic nerves, optic atrophy, nystagmus, strabismus, myopia, and dissociated vertical deviation (DVD) were present. He had muscular hypotonia with decreased muscle mass for age, and frequent myalgias. Further, severe pectus excavatum, osteoporosis, arthritis of the left leg, scoliosis, pes planus, advanced bone age, hyperflexibility in limbs and especially small joints (fingers), prominent interphalangeal (IP) joints in the hands, and hallux valgus deformity were documented. At the age of 8 years, a mildly prolapsed mitral valve was observed. In addition, he had conductive hearing loss and eustachian tube dysfunction (ETD), for which he received pressure equalization (PE) tubes. Moreover, he displayed global lipodystrophy, mild excessive posterior nuchal skin, widely spaced and underdeveloped nipples, and hypoplastic nails.
Fig. 2.
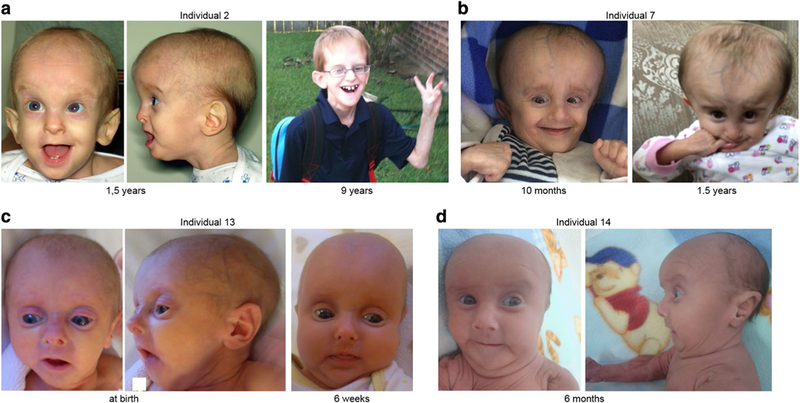
Clinical characteristics of PYCR1-mutated individuals. Facial images of individual 2 at 1.5 years and 9 years (a), individual 7 at the age of 10 months and 1.5 years (b), individual 13 at birth and 6 weeks (c), and individual 14 at 6 months (d)
Table 2.
Clinical characteristics of PYCR1-mutated patients presented here
| Individual | 2 | 5 | 6 | 7 | 10 | 12 | 13 | 14 |
|---|---|---|---|---|---|---|---|---|
| Sex | Male | Female | Male | Male | Male | Male | Female | male |
| Age | 9 years | 10 months | 4 years | 3.5 years | 2 years 9 months | 2 years 2 months | 13 months | 9 months |
| Ethnicity | European | European | African | Arabian | European | European | European | Latino |
| Intrauterine growth retardation | + | d.u | + | + | + | + | + | + |
| Postnatal growth retardation | + | d.u | + | + | − | − | ? | ? |
| Triangular face | + | + | + | + | + | + | + | + |
| Sparse scalp hair | + | + | + | + | + | + | + | + |
| Thin/translucent skin | + | + | + | + | + | + | + | + |
| Prominent scalp veins | + | + | + | + | + | + | + | + |
| Frontal bossing | + | + | − | + | + | + | − | − |
| Deep set eyes | − | + | + | + | − | − | + | + |
| Low set ears | + | + | − | − | − | − | − | |
| Mandibular underdevelopment | + | + | + | + | + | + | + | + |
| Joint hypermobility | + | d.u | − | − | + | + | + | + |
| Joint contractures | − | + | − | − | − | − | + | − |
| Motor development delay | + | d.u | + | + | + | + | + | ? |
| Muscular hypotonia | + | d.u | + | + | + | + | + | + |
| Impaired speech development | + | d.u | + | + | + | + | ? | ? |
| Dental anomalies | + | d.u | − | − | − | − | − | − |
| Cataract/corneal opacities | + | d.u | − | − | − | − | − | − |
| Strabismus | + | d.u | − | − | − | − | − | − |
| Myopia | + | d.u | + | + | ||||
| Osteopenia/osteoporosis | + | d.u | − | − | − | − | − | − |
| Lipodystrophy | + | + | + | + | + | + | + | + |
| Cardiac anomalies | + | d.u | − | − | − | − | − | + |
| Wrinkled skin | > 6 years | d.u | > 2.5 years | − | > 2.5 years | > 2 years | > 1 years | since birth |
| Microcephaly | + | d.u | + | + | + | + | + | + |
| Congenital respiratory problems | + | d.u | − | + | − | − | − | + |
| Hernias | + | d.u | − | − | − | + | − | + |
| Hearing loss | + | d.u | − | − | − | − | − | − |
| Brain anomalies US/MRI | White matter abnormalities | d.u | Agenesis of corpus callosum | Agenesis of corpus callosum | Cysts and micro-calci-fications | n.d | Agenesis of corpus callosum | Agenesis of corpus callosum |
| PYCR1 mutation | ||||||||
| Paternal | c.633 + 1G > C | p.K71Nfs*10 | p.R119C | p.I74Tfs* 10 | p.I74Tfs* 10 | p.I74Tfs* 10 | c.797 + 2_5 delTGGG | p.A257T |
| Maternal | p.A179T | p.R119H | p.R119C | p.I74Tfs* 10 | p.R313L | p.V258L | c.540 + 1G > A | p.A257T |
present
absent, d.u. data unavailable
due to young age not yet applicable
n.d. not done
Individual 3
This male proband is the first child of healthy unrelated parents. He has a younger unaffected sibling. He was born at 38 weeks of gestation by caesarean section due to in utero cracked ribs, broken femur and wide fontanelles. His birth weight was 2275 g ( −2.37 SD), length was 48 cm (− 1.35 SD) and OFC was 33.7 cm (− 0.86 SD). At birth, he displayed wrist and ankle contractures (bilateral talipes equi-novarus/club feet) that were surgically corrected soon after birth resulting in continued normal range of movement. In addition, the boy had no scalp hair, frontal bossing, prominent veins, poor ossification of the calvarium with open fontanelles and wide sutures. Further, he had a blepharo-phimosis-type appearance with telecanthus and epicanthus inversus with epiblepharon, unusual supraorbital rims that appeared hypoplastic and sloping (Fig. 3a). Muscular hypotonia characterized the postnatal period. X-ray at 4 months revealed poor ossification of the calvarium with widened sutures and fontanelles. Evidence of an old healed fracture at the junction of the proximal/mid left femoral diaphysis was seen. He underwent surgery for undescended testes at age 12 months. Milestones of motor development were delayed: he sat unaided at 12 months, exhibited bottom shuffling but no crawling at 18 months, and walked at around 2.5 years. Language development was significantly delayed; however, the speech dramatically improved after insertion of ventilation tubes at the age of 3 years. He had a high-pitched voice. First teeth erupted at the age of 18 months. He suffered from osteopenia and recurrent fractures: small fractures in right elbow at age 6 years and C2 vertebrae at age 8 years. At clinical evaluation at the age of 9 years, epiblepharon, astigmatism, light sensitivity, blue sclerae, and mandibular underdevelopment with dental crowding were observed. He had a weight of 13.7 kg (− 7.60 SD), a height of 104 cm (− 5.25 SD) and an OFC of 51 cm (− 1.68 SD). He had thin hair, global lipodystrophy, and muscular hypotonia.
Fig. 3.
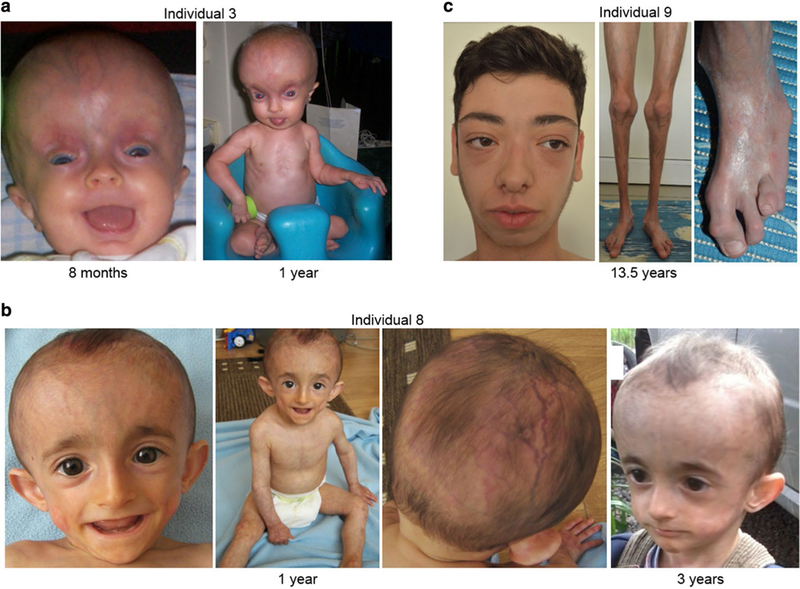
Clinical characteristics of COL1A1-mutated individual and the two unclassified juvenile progeroid individuals. Facial images of individual 3 bearing the de novo COL1A1 mutation at age 8 months and 1 year (a), individual 8 at the age of 1 year and 3 years (b), and individual 9 at 13.5 years (c)
Individual 4
This female proband is the second child of healthy unrelated parents. She was born at 34 weeks of gestation by cesarean section for intrauterine growth retardation and non-reassuring fetal heart tracing. Her birth weight was 1160 g (− 2.58 SD), length was 41.5 cm (− 1.41 SD) and OFC was 27.5 cm (− 2.47 SD). Apgars were 7 at 1 min and at 5 min. At delivery, she presented with poor respiratory effort initially that improved after continuous positive airway pressure (CPAP) therapy. In addition, a large patent ductus arteriosus (PDA), mild aortic insufficiency and mild tricuspid valve incompetency were observed. She had a feeding tube placed during the first year of life. At the age of 1 year and 7 months, she had a triangular face, sparse and thin hair, prominent scalp veins, frontal bossing, closely spaced eyes, midface retraction, mal-formed and low set ears, and mandibular underdevelopment, somewhat mimicking Wiedemann-Rautenstrauch syndrome (Fig. 1b; Table 1). She had severe knee flexion contracture and less severe bilateral hip flexion contractures that impeded her walking independently in the earlier years. She was walking independently at the age of years wearing a left knee brace. She had speech therapy in pre-school. Cardiac evaluation revealed a mildly eccentric aortic valve and mild left ventricular hypertrophy. She had a Chiari malformation, and received a ventriculoperitoneal shunt for intracranial hypertension. At the age of 5 years she had a weight of 8.3 kg (− 8.32 SD), a height of 87.6 cm (− 5.03 SD) and an OFC of 50.3 cm (− 1.06 SD). At the age of 12 years and 9 months, she exhibited a prematurely aged facial appearance. She still had partial alopecia, severe frontal bossing, a triangular face, small and deep set eyes, an entropion, a beaked nose, dental dysplasia with missing teeth, and mandibular underdevelopment. She had global lipodystrophy, thin arms and legs, and contractures of elbows and knees.
Individual 5
This female proband was referred at the age of 10 months due to a triangular face, sparse scalp hair, thin-appearing and translucent skin with prominent veins, frontal bossing, deep set eyes, somewhat low set ears and mandibular underdevelopment. In addition, she had a generalized lipodystrophy and joint hand contractures (Table 2). Unfortunately, the contact with the family and the referring physician was later lost.
Individual 6
This male proband is the second child of healthy consanguineous parents who are first cousins. His older brother is unaffected. He was born after an uneventful pregnancy at 40 weeks of gestation. Birth weight was 2550 g (− 2.44 SD), head circumference was 33 cm (− 2.00 SD), and length was not available. The neonatal period was notable for muscular hypotonia, hip dislocation, gastrointestinal reflux and mild jaundice. He had a triangular face, sparse scalp hair, thin-appearing and translucent skin with prominent veins and mandibular underdevelopment. All milestones of motor development were delayed: he sat unaided at 10 months, walked by age 1.5 years, verbal expression was restricted to a few words, and he understood only basic verbal commands. At the clinical evaluation at the age of 2 years and 7 months, he had microcephaly (OFC was 46.5 cm; − 2.50 SD), height of 90 cm (0.54 SD) and weight of 10 kg (− 1.73 SD). He had a triangular face with a progeroid appearance, sparse hair, prognathism, open mouth appearance and large ears. His skin was wrinkled on the dorsum of his hands and feet with visible subcutaneous veins. He had long fingers and flat feet. X-rays revealed atrophy of left femoral head. Osteopenia was not detected. Routine laboratory findings including lactates, creatine kinases (CK), and thyroid hormone were all within the normal range. ECG, echo Doppler cardiography, and abdominal ultrasound were normal. At the clinical visit at the age of 4 years, his growth parameters improved: OFC was 48.5 cm (− 2.12 SD), height was103 cm (− 0.26 SD), and weight was 15 kg (− 0.89). He had developed agitation, impaired concentration and gestural stereo-typies. MRI of the brain demonstrated agenesis of the corpus callosum (Table 2).
Individual 7
This male proband is the second child of healthy consanguineous parents who are first cousins. His older sibling was unaffected. He was born at 34 weeks of gestation with a birth weight of 900 g (− 3.39 SD), length of 37 cm (− 3.17 SD) and OFC of 26 cm (− 3.65 SD). He had respiratory problems in the first weeks of life and suffered from poor feeding since birth. Brain MRI, performed at the age of 8 months, revealed a prominent supratentorial ventricular system and agenesis of the corpus callosum. At the age of 1 year, he had a triangular face with a broad appearing cranium, midface retraction and relatively small facial bones, sparse scalp hair, thin-appearing and translucent skin with prominent veins, frontal bossing, deep set eyes, low set ears, a small mandible and lipoatrophy (Fig. 2b; Table 2). He started walking independently at age 3 years and spoke his first words by age 3.5 years. At 3.5 years of age he still suffered from a poor growth with a weight of 11 kg (− 2.52 SD) and height of 90 cm (− 2.49 SD), despite nutritional supplements (PediaSure®).
Individual 8
This male proband is the second child of healthy unrelated parents. His younger sibling is unaffected. He was born after an uneventful pregnancy at 40 weeks of gestation. His birth weight was 2800 g (− 1.87 SD) and length was 50 cm (− 1.09 SD). At birth, he presented with prominent scalp veins, prominent ears, small facial bones, generalized skin mottling and lipodystrophy. In addition, he had two ulcers on the scalp at birth. At the age of 9 months X-ray showed advanced bone age of 1.8 years. Chest ultrasound showed a patent foramen ovale. His psychomotor development was normal. By the age of 2 years he developed bilateral cataracts. At the age of 3 years, he had a triangular face with sparse scalp hair and prominent scalp veins, and mandibular underdevelopment. He still had generalized skin mottling, lipodystrophy, and poor weight gain (Fig. 3b). His weight was 10 kg (− 2.98 SD) and height was 87 cm (− 2.45 SD).
Individual 9
This proband is the first child of healthy unrelated parents. He was born after an uneventful pregnancy at 39 weeks of gestation. His birth weight was 3000 g (− 1.17 SD) and length was 48 cm (− 1.7 SD). His early development was unremarkable. He developed scleroderma-like skin changes by the age of 1 year. At the examination at 13 years and 7 months he had a height of 161 cm (+ 0.51 SD), weight of 38 kg (− 1.4 SD) and OFC of 54.3 cm (− 0.46 SD). He had hypertelorism, delimited nose wings, mandibular underdevelopment, scoliosis, delayed bone age, multiple muscle contractures (elbow, hands and feet), and generalized progressive muscular atrophy (Fig. 3c). CK was normal. X-ray of his calf revealed diminished muscles with multiple calcifications. Muscle biopsy showed phagocytosis with inflammation of some muscle fibers, unequal muscle fibers, and blood vessels with slightly thick walls, likely due to fibrosis in the perimysium. His skin was tight and he still had scleroderma-like skin changes. In addition, he had generalized lipodystrophy. Total blood cholesterol was 150 mg/dL, while he had low HDL of 24 mg/dL and high triglycerides of 370 mg/dL. Glucidic metabolism and glycated hemoglobin were normal. Moreover, acanthosis nigricans in the axillar and inguinal area, as well as on the flexion surface of the elbow and knee was observed. In addition, the skin was very thick in the axillar and inguinal area.
Individual 10
This male proband is the first child of healthy unrelated parents. Pregnancy was complicated by oligohydramnios and intrauterine growth retardation starting at 30 weeks of gestation. He was born after 38 weeks of gestation. His birth weight was 2300 g (− 2.32 SD), length was 43 cm (− 3.52 SD) and OFC was 32 cm (− 2.07 SD). At birth, he presented with plagiocephaly, frontal bossing, a sharp nasal tip, anteverted nostrils, micrognathia, generalized lipoat-rophy, and transparent skin with prominent veins. At the clinical evaluation at 9 months of age, his weight was 7 kg (− 1.89 SD), height was 70 cm (− 0.95 SD) and OFC was 42 cm (− 3.14 SD). He still had craniofacial abnormalities, generalized lipoatrophy, with tight and transparent skin and prominent veins. Cranial ultrasound revealed a choroid plexus cyst, a subependymal cyst and microcalcifications in the thalamo-caudate region. Extensive metabolic analyses gave normal results, apart from low HDL (32 mg/dL) and high total triglycerides (162.6 mg/dL). Several milestones were normal: he sat at age of 6 months, crawled at 9 months, and walked independently at age 1 year 3 months. However, at the clinical examination at 2 years, he had an intellectual disability with no speech, and a hyperkinetic movement disorder. His weight was 11 kg (− 0.69 SD), height was 86 cm (0.08 SD) and OFC was 47 cm (− 1.72 SD). He had a triangular facial gestalt, tight skin, sparse scalp hair, generalized lipoatrophy, muscular atrophy and joint hypermobility. At the last available clinical evaluation at 2 years and months of age, his weight was 11.7 kg (− 1.39 SD) and OFC was 48.5 cm (− 1.61 SD). He had transparent, lax and thin-appearing skin, with visible veins especially on his head and extremities. In addition, he had wrinkled skin on his hands. Moreover, he had joint hypermobility which affected his movements, and generalized lipodystrophy (Table 2). His gait was unsteady, with external rotated legs for better stability, and with flat feet. He had myopia. He spoke only single short syllables. Movements were hyperkinetic, and he displayed hand automatisms, taking objects to his mouth and touching them with his tongue.
Individual 11
This female proband is the only child of healthy unrelated parents. Prenatal ultrasound gave normal results in the first and second trimesters. However, ultrasound in the last trimester revealed a Dandy-Walker malformation, brachy-cephaly, a nasal bridge hypoplasia, and severe intrauterine growth retardation. Prenatal karyotype after amniocentesis at 28 weeks gave normal results, 46, XX. She was born at 37 weeks of gestation. Her weight was 1460 g (− 3.48 SD) and length was 43 cm (− 2.71). At birth, she had a triangular face, sparse scalp hair, frontal bossing, thin-appearing skin with prominent scalp veins, closely spaced eyes, midface retraction, low set ears, mandibular underdevelopment and generalized lipoatrophy (Fig. 1c; Table 1). In addition, she had a patent ductus arteriosus and mild renal pelvic ectasia. Due to low birth weight and severe tachypnea, she remained hospitalized for 6 weeks. She had a slow weight gain despite hyper-caloric nutritional supplements. All early milestones of motor development were unremarkable: rolling over at 6 months, sitting at 8 months, crawling at 9 months. She started babbling at 11 months. Brain MRI revealed agenesis of corpus callosum. At the clinical examination at 11 months of age, her weight was 3.6 kg (− 6.22 SD), height was 57 cm (− 5.99 SD) and OFC was 40 cm (− 5.11). She had trigo-nocephaly and brachycephaly, sparse scalp hair, low set ears with dysplastic auricular pavilions with bilateral grade 1 microtia, hypertelorism, marked enophthalmos, lagoph-thalmos and bilateral ectropion with blepharospasm resulting in incomplete closure of the eyes, midface retraction, a depressed and wide nasal bridge, hypoplastic nasal wings, mandibular underdevelopment, and a short neck. In addition, she had hypoplastic nipples, a small umbilical hernia, and generalized muscular atrophy.
Individual 12
This male proband is the only child of healthy unrelated parents. He was born at 38 weeks of gestation. His birth weight was 2300 g (− 2.32 SD) and length was 47 cm (− 1.78 SD). At birth, he presented with an inguinal hernia that was remedied by surgery. In addition, he presented with frontal bossing, a sharp nasal tip, anteverted nostrils, micrognathia, generalized lipodystrophy, and transparent skin with prominent veins. He sat independently at age 6 months, crawled at age 7 months, walked independently at age 1 year 9 months. At the clinical evaluation at 1 year and 8 months of age, his weight was 9.1 kg (− 2.04 SD), height was 79 cm (− 1.76 SD) and OFC was 46.5 cm (− 1.99 SD). He had a triangular facial gestalt, tight skin, sparse scalp hair, generalized lipodystrophy, muscular atrophy and joint hypermobility. At the last available clinical evaluation at 2 years and 2 months of age, his weight was 12 kg (− 0.52 SD), height 85 cm (− 1.35 SD) and OFC of 48.5 cm (− 1.12 SD). He still had thin-appearing and transparent skin, especially on the extremities, and developed wrinkled skin on his abdomen and hands (Table 2). He displayed a retraction of the sternum due to thoracic laxity that was accentuated by respiratory movements. He spoke only a few words but understood orders well. In addition, he is a hyperkinetic child.
Individual 13
This female proband is the only child of unrelated parents. Pregnancy was complicated by oligohydramnios and intrauterine growth retardation. She was born at 32 weeks of gestation. Her weight was 1400 g (− 0.98 SD), length was 42 cm (− 0.18 SD) and OFC was 28 cm (− 0.95 SD). She remained hospitalized until she reached 2000 g. She had an aged appearance with a triangular facial gestalt, sparse hair, pinched nose, hypoplastic alae nasi, sunken in eyes with lack of periorbital fat tissue, mandibular underdevelopment, thin-appearing and transparent skin with prominent veins, thin limbs distally, and the fat pads on her gluteal region and thighs were spared but saggy. Fingers exhibited mild contracture, with the thumbs overlapping the palms, and the 2nd and 5th digits overlapping the 3rd. Brain MRI revealed agenesis of the corpus callosum. Upon last available clinical evaluation at 13 months of age, her weight was 6 kg (− 3.79 SD), height was 75 cm (− 0.95 SD) and OFC was 42 cm (− 4.50 SD). She had a triangular face with broad forehead, protruding ears, and hypotelorism. Her skin was thin-appearing, atrophic, transparent, and wrinkled on distal extremities with sparse, thin scalp hair (Fig. 2c; Table 2). The muscles were atrophic and the joint hypermobile. Extensive metabolic analyses gave normal results. Her mother died following breast cancer, thus a maternal DNA sample was unavailable for genetic testing.
Individual 14
This male proband is the fourth child of healthy consanguineous parents who are second cousins. Pregnancy was complicated by breech presentation and premature rupture of membranes. He was born at 34 weeks of gestation. His weight was 1700 g (− 1.51 SD), length was 36.5 cm (− 3.34 SD) and OFC was 30 cm (− 1.29 SD). He remained hospitalized for 8 days due low birth weight, hyperbilirubinemia, mild renal pyelectasis, umbilical hernia and an inguinoscro-tal hernia. At last available clinical evaluation at 9 months of age, his weight was 4.3 kg (− 5.04 SD), height was 56 cm (− 6.03 SD) and OFC was 38 cm (− 6.94 SD). He had muscular hypotonia and an overall aged appearance with triangular face, sparse and fine hair, sunken in eyes, posteriorly rotated and low ears, thin lips, micrognathia, joint hypermobility and generalized lipodystrophy. His skin was thin appearing and transparent with prominent veins, wrinkled on the neck, trunk and especially pronounced on extremities (Fig. 2d; Table 2). Moreover, he had a linear sclerodermatous plaque skin over the proximal right thigh. His thumbs overlapped his palms, and his 3rd and 5th digits overlapped his 4th digit. His scrotum was hypoplasic. Moreover, brain MRI revealed agenesis of corpus callosum. Ultrasound further revealed mild aortic and pulmonary artery dilatation, and bilateral dysplasia of the hip. He suffered from poor feeding, and had moderate myopia (− 5.5 D).
Genetic analyses
Identification of biallelic PYCR1 mutations
Trio-whole exome sequencing (trio-WES), followed by bioinformatic filtering for biallelic variants in genes previously connected to well-established progeroid syndromes, identified in more than one individual, revealed likely pathogenic biallelic mutations in PYCR1 in five individuals. In individual 2, we identified compound heterozygosity for a paternally inherited splice donor alteration c.633 + 1G > C, and maternally inherited c.535G > A, p.(Ala179Thr). Individual 5 bears a paternally inherited c.213_214delGC, leading to a frameshift mutation p.(Lys71Asnfs*10), and maternally inherited c.356G > A, resulting in a missense alteration p.(Arg119His). Individual 6 bears homozygosity for c.355C > T, resulting in a missense alteration affecting the identical amino acid as in individual 5, p.(Arg119Cys). Individual 7 bears a homozygous 2-bp insertion c.219_220insAC, resulting in a frameshift mutation p.(Ile74Thrfs*10). Finally, individual 10 bears compound heterozygosity for the paternally inherited 2-bp insertion c.219_220insAC, resulting in a frameshift mutation p.(Ile74Thrfs*10), identical to the insertion identified in individual 7, and a maternally inherited c.938G > T, resulting in a missense alteration p.(Arg313Leu). Notably, the majority of these mutations have already been described in PYCR1-associated pathologies (p.(Arg119His), p.(Arg119Cys), c.633 + 1G > C and p.(Ala179Thr)) (Dimopoulou et al. 2013; Reversade et al. 2009), or result in a recurrent frameshift mutation (c.219_220insAC, p.(Ile74Thrfs*10)). Thus, there is a strong evidence for the pathogenicity of these mutations. The pathogenicity is not that clear for the missense alteration (p.(Arg313Leu)) that has not been found in dbSNP, 1000 Genomes, the ExAC or the gnomAD browser, is predicted to result in decreased PYCR1 stability (Table 4) and affects only a single PYCR1 isoform (ENST00000337943.9). We have, therefore, further evaluated all rare variants identified in individual 10 by trio-WES that was performed with sufficient coverage. Approximately, 94% of target sequences were covered at least 20-fold with a mean coverage of about 100×. Bioinformatic filtering for a rare candidate variant (minor allele frequency MAF < 0.02) did not identify any de novo variants nor variants following the X-linked recessive mode of inheritance, but only compound heterozygosity for two variants in DNER, a gene so far not connected to a human disease; namely a paternally inherited c.762T > A, resulting in missense alteration p.Asp254Glu, and a maternally inherited c.121C > T, resulting in missense alteration p.Pro41Ser (Supplementary Table 1). However, as both DNER variants have been deposited in the gnomAD browser in a homozygous state, we highly doubt their causality.
Table 4.
Population frequencies and bioinformatic predictions of the rare variants identified in this study
| ExAC | gnomAD | Polyphen-2 | SIFT | I-Mutant | CADD | BDGP | |
|---|---|---|---|---|---|---|---|
| POLR3A | |||||||
| p.M1I | 0/0/121360 | 0/1/246230 | Probably damaging | Damaging | Decreased stability | 33 | − |
| p.R254* | 0/3/121300 | 0/9/277188 | − | − | − | 37 | − |
| c.3337-5T > A | 0/1/108818 | 0/1/242920 | − | − | − | 8.67 | 0.79–>0.32 |
| c.3337-1G > A | 0/0/108818 | 0/0/242920 | − | − | − | 3.95 | 0.79–>0 |
| PYCR1 | |||||||
| p.K71Nfs*10 | 0/0/113986 | 0/1/30918 | − | − | − | 33 | − |
| p.I74Tfs*10 | 0/0/113986 | 0/0/245176 | − | − | − | 32 | − |
| p.R119C | 0/10/114118 | 0/34/273316 | Probably damaging | Damaging | Decreased stability | 28.8 | − |
| p.R119H | 0/3/114566 | 0/3/242550 | Probably damaging | Damaging | Decreased stability | 28.9 | − |
| p.A179T | 0/3/113196 | 0/5/272934 | Probably damaging | Damaging | Decreased stability | 33 | − |
| c.540 + 1G > A | 0/3/112300 | 0/5/242262 | − | − | − | 25.8 | 0.98–>0 |
| c.633 + 1G > C | 0/2/21260 | 0/6/199450 | − | − | − | 31 | 0.49–>0 |
| p.A257T | 0/5/109454 | 0/9/274864 | Probably damaging | Damaging | Decreased stability | 30 | − |
| p.V258L | 0/0/109454 | 0/0/274864 | Probably damaging | Damaging | Decreased stability | 29.5 | − |
| c.797 + 2_5delTGGG | 0/0/107108 | 0/0/243200 | − | − | − | 24.2 | 0.30–>0 |
| p.R313L | 0/0/120196 | 0/0/245668 | Benign | Tolerated | Decreased stability | 2.5 | − |
| COL1A1 | |||||||
| p.G22R | 0/0/118552 | 0/0/245568 | Probably damaging | Tolerated | Decreased stability | 23.9 | − |
| SMC2 | |||||||
| p.K921N | 0/0/120326 | 0/0/274588 | Benign | Tolerated | Decreased stability | 16.95 | − |
ExAC Exome Aggregation Consortium, gnomAD genome aggregation database, Polyphen2 polymorphism phenotyping v2, SIFT sorting intoler-ant from tolerant, I-Mutant neural network-based predictor of protein stability, CADD combined annotation-dependent depletion, BDGP splice site prediction by neural network
Identification of biallelic mutations in POLR3A in two individuals
In a further step, we searched for alterations in genes that have been previously connected to progeroid syndromes in single cases. This strategy revealed biallelic mutations in POLR3A (Jay et al. 2016) in individuals 1 and 4. Individual 1 bears compound heterozygosity for two variants within the splice acceptor site of intron 25, a paternally inherited c.3337-5T > A and a maternally inherited c.3337-1G > A. Individual 4 also bears compound heterozygosity, namely a maternally inherited c.760C > T, resulting in a nonsense mutation p.(Arg254*) and a paternally inherited c.3337-5T > A; the latter is notably the identical variant as also identified in individual 1. None of these variants were found in dbSNP, 1000 Genomes, the ExAC or the gnomAD browser in the homozygous state and the heterozygous carriers were extremely rare (Table 4). The p.(Arg254*) is predicted to lead to a premature termination codon likely activating non-sense-mediated mRNA decay. On the other hand, c.3337-1G > A and c.3337-5T > A most probably affect splicing as predicted by the Splice Site Prediction by Neural Network (Berkeley Drosophila Genome Project; http://www.fruitflyorg/seq_tools/splice.html ); namely this prediction tool suggests that the c.3337-1G > A completely abolishes the splice acceptor site, whereas c.3337-5T > A results in a weaker splice acceptor site with a score of 0.32 compared to a score of 0.79 that is assigned to the wild-type sequence. To test for the predicted splicing defect for the variant c.3337-5T > A, we performed RT-PCR analysis and Sanger sequencing with RNA obtained from LCLs of the individual 4 and his parents. This analysis revealed that the c.3337-5T > A indeed resulted in an alternate splice form in which POLR3A exon 26 was skipped. The mutation and exon skipping was found in both the proband and his father, but was absent in his mother (Fig. 4).
Fig. 4.
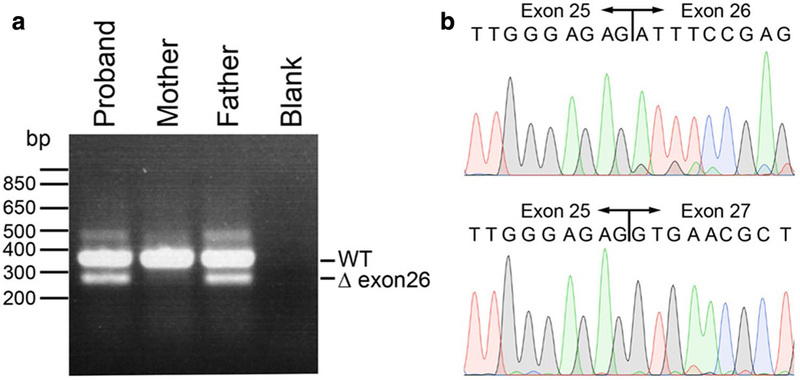
POLR3A splicing defect caused by c.3337-5T > A mutation results in skipping of exon 26. Agarose gel electrophoresis of POLR3A RT-PCR products. The proband and father both carry the c.3337-5T > A mutation, resulting in a smaller band of ~ 287 bp corresponding to a cDNA missing exon 26 compared to the size of the wild-type band of ~ 380 bp (a). Sanger sequence traces of gel-extracted POLR3A RT-PCR products, demonstrating wild-type sequence of the larger band (top) and exon 26 skipping, both from individual 4 (bottom) (b)
Other mutations and genetic heterogeneity of juvenile progeroid syndromes
Given that analysis of trio-WES data revealed no likely pathogenic variant in known progeroid genes in four individuals, we searched for rare candidate variants (minor allele frequency MAF < 0.005) following an autosomal recessive or X-linked recessive mode of inheritance, as well as those that might have arisen at de novo.
Individual 3 was found to have a de novo heterozygous mutation in COL1A1 c.64G > C, p.(Gly22Arg). Mutations in this gene are well known to cause osteogenesis imperfecta (OI) (Byers 1989) and some cases of Ehlers-Danlos syndrome (EDSARTH1) (Byers et al. 1997). The variant in individual 3 has been reported twice in the OI mutation database (http://www.le.ac.uk/genetics/collagen/). Both reports are from infants with OI type II, one of which was previously reported in the literature (Pollitt et al. 2006).
In individual 8, no de novo variant nor variants following autosomal recessive or X-linked mode of inheritance were identified.
In individual 9, bioinformatic filtering did not detect rare candidate variants following an autosomal recessive or X-linked recessive mode of inheritance. However, we identified a single non-annotated, de novo sequence change c.2763A > C, resulting in a missense alteration p.(Lys921Asn) in SMC2. To the best of our knowledge, mutations in SMC2 have so far not been connected to any human disease. Given the previous link between genomic instability and segmental progeroid syndromes (Lessel et al. 2014; Yamagata et al. 1998), SMC2 constitutes an interesting candidate gene for this progeroid disorder. However, as the identified amino acid substitution has a rather low Phred scaled CADD (v1.3) score of 16.95 (for de novo mutations), and is predicted to be “benign” by PolyPhen-2 and tolerated by SIFT (Table 4), we were reluctant at this point to perform functional analysis without identifying an additional patient with similar genotype and phenotype to elucidate the causality of this variant.
Identification of additional POLR3A and PYCR1 cases
After performing trio-WES analyses in the above-mentioned ten cases, four additional cases were referred with the putative clinical diagnosis of a juvenile progeroid syndrome. One of these cases, proband 11, highly resembled both probands 1 and 4 (Fig. 1), and the previously published Wiede-mann-Rautenstrauch-like case (Jay et al. 2016). For this reason, we performed Sanger sequencing of all coding exons and the exon-intron boundaries of POLR3A using genomic DNA of the affected individual and her parents. However, we were only able to identify a single heterozygous alteration in this proband, namely a start loss mutation, c.3G > T, p.(Met1Ile), that was transmitted from the father. Due to the remarkable clinical similarities to the other two patients bearing biallelic mutations in POLR3A, we speculate that the second, maternal mutation, is either a deep intronic one, a copy-number variant (CNV), a balanced translocation or even affects a regulatory region of POLR3A.
The other three individuals, 12, 13 and 14, all presented with intrauterine growth retardation, triangular facial gestalt, sparse scalp hair, mandibular underdevelopment, thin-appearing and transparent skin with prominent veins, generalized lipodystrophy and joint hypermobility in the first year of life. Thus, we hypothesized that they may bear pathogenic PYCR1 mutations. Indeed, this was confirmed by Sanger sequencing. Individual 12 bears a paternally inherited c.219_220insAC, p.(Ile74Thrfs*10). Notably, this is the identical frameshift mutation that we identified in individuals 7 and 10. In addition, he bears a maternally inherited c.772G > C, p.Val258Leu, that has been reported previously (Dimopoulou et al. 2013). Individual 13 bears a paternally inherited c.797 + 2_5delTGGG, and a likely maternally inherited c.540 + 1G > A, both of which have been reported previously (Dimopoulou et al. 2013; Reversade et al. 2009). We were unable to confirm that the second mutation was maternally inherited, as the mother deceased; however, this alteration was not identified in paternal DNA. Individual 14 bears a homozygous alteration c.769G > A, resulting in a missense mutation p.(Ala257Thr), and each of the parents is a heterozygous carrier. Again, the identical mutation has already been connected to PYCR1 pathologies before (Reversade et al. 2009).
Discussion
Juvenile progeroid syndromes constitute a group of diseases characterized by clinical signs of premature aging starting in childhood. The best known and most extensively studied example of such diseases is Hutchinson-Gilford progeria syndrome (HGPS) (De Sandre-Giovannoli et al. 2003; Eriksson et al. 2003; Gordon et al. 1993). Next-generation sequencing (NGS) is a powerful tool for delineating the genetic basis of monogenic diseases, including both the identification of novel disease genes and revealing the heterogeneous nature of Mendelian conditions. Given the rarity of these conditions, concerted efforts of open sharing NGS data from multiple groups are of utmost importance to dissect their genetic nature. Moreover, NGS-based studies have revealed several novel progeroid genes (Lessel et al. 2014, 2017; Puente et al. 2011). However, a full understanding of the genetic and clinical heterogeneity of juvenile progeroid syndromes is still lacking and the genetic cause of some of these syndromes, such as Wiedemann-Rautenstrauch syndrome (WRS), still remain largely unknown. Here, we present our joint collaborative efforts in the form of an extensive study of previously unclassified children affected by progeroid syndromes aimed at dissecting the genetic bases of their conditions. We, however, note that although all 14 studied cases were affected by an early-onset progeroid syndrome there was a considerable degree of clinical heterogeneity between them (Supplementary Table 2).
WRS, first described in 1977 by Rautenstrauch and Snigula (Rautenstrauch and Snigula 1977), has been suggested to constitute a syndrome characterized by neonatal progeroid features, including intrauterine growth retardation with subsequent failure to thrive, characteristic progeroid facial features, lipodystrophy and global developmental delay. Since the first description of two affected sisters which pointed to an autosomal recessive inheritance of the underlying genetic cause, more than 50 individuals have been clinically classified as WRS although with a huge phenotypic variability, as recently reviewed by Paolacci et al. (Paolacci et al. 2017). Despite tremendous progress in unravelling the genetics of several Mendelian disorders, which is mostly attributable to the rapid development of new technologies, the underlying genetic cause of WRS has still not been established yet. The latter is likely due to remarkable clinical heterogeneity of the documented cases. However, a recent study identified compound heterozygosity for two null mutations in POLR3A in a single WRS-like individual (Jay et al. 2016). Here, we identified two patients (individuals 1 and 4) with strikingly similar clinical characteristics and facial features to the published case which also bear compound heterozygosity for two POLR3A null mutations. Moreover, we report on another patient with comparable clinical characteristics and a remarkably similar facial gestalt. In this patient (individual 11), using direct Sanger sequencing of all coding exons and the exon-intron boundaries of POLR3A, only a single, paternally inherited, heterozygous mutation was identified. However, due to the remarkable similarities both to the previously published patient (Jay et al. 2016) and individuals 1 and 4 (Fig. 1; Table 1), we conjecture that the second, maternally inherited mutation, lies either within deep intronic or even regulatory regions of POLR3A, or might be a copy-number variant comprising more than a single exon or even a balanced translocation. Notably, the other WRS-like patients bear a pathogenic intronic variant at positions that do not commonly lead to aberrant splicing, namely c.1909 + 18G > A and c.3337-5T > A. Thus, deep intronic and regulatory variants that are usually not covered by WES analysis might explain why POLR3A has not previously been established as the underlying genetic cause of some WRS-like conditions. From the clinical perspective, the previously reported WRS-like POLR3A patient died at 7 months of age following respiratory complications (Jay et al. 2016). Noteworthy, two of the WRS-like patients presented here (4 and 11) also had severe respiratory problems, especially in the perinatal period. However, they both survived beyond the first year of life, and individual 4 actually reached 13 years of age at study closure, suggesting that this is not a uniformly early lethal condition. Notably, while this manuscript was under review a further study independently confirmed biallelic POLR3A mutations, most of which were intronic ones, as the cause of the WRS-like condition (Paolacci et al. 2018). Taken together, POLR3A mutations following an autosomal recessive mode of inheritance are now firmly established as the genetic cause of a recognizable WRS-like syndrome.
Notably, biallelic mutations in POLR3A are known to cause the 4H leukodystrophy syndrome (hypomyelination, hypogonadotropic hypogonadism and hypodontia). The condition is characterized by early-onset ataxia, delayed dentition, hypomyelination, and hypogonadotropic hypogonadism (Bernard et al. 2011). However, concerning the age of onset and progression of neurologic symptoms, 4H leukodystrophy is characterized by both high inter-and intra-familial variability (Wolf et al. 2014). Compared to the POLR3A- associated WRS-like condition, none of the 4H leukodystrophy patients bear biallelic truncating mutations, e.g. all bear at least one missense mutation (Jay et al. 2016; Wolf et al. 2014). The latter may explain both the earlier, neonatal onset of the WRS-like condition as well as the more pronounced growth retardation and lipodystrophy, distinct facial anomalies and congenital respiratory complications, as previously suggested (Jay et al. 2016; Paolacci et al. 2017). On the other hand, given that both entities are characterized by neurologic dysfunction and dental anomalies, POLR3A-associated WRS-like condition may be regarded as the extreme form of the 4H leukodystrophy.
Recessive mutations in PYCR1 were initially associated with Cutis Laxa Type IIB with progeroid features (Reversade et al. 2009). However, further descriptions of patients bearing biallelic PYCR1 mutations in a large geno-type-phenotype correlation study revealed a remarkable clinical heterogeneity and phenotypic continuum ranging from isolated wrinkly skin syndromes, gerodermia osteo-dysplastica, De Barsy syndrome to even patients classified as Hallermann-Streiff-like neonatal progeroid syndromes (Dimopoulou et al. 2013). Our initial trio-WES analysis of ten patients clinically classified as having a HGPS-like juvenile progeroid syndrome unexpectedly revealed biallelic PYCR1 mutations in 50% of probands (five cases). Notably, all of these cases did share some clinical characteristics with HGPS, albeit only in the first few years of life; namely all displayed triangular facial gestalt, sparse scalp hair, thin-appearing and translucent skin with prominent veins, mandibular underdevelopment and lipodystrophy. However, in contrast to HGPS, they displayed intrauterine growth retardation and later developed mild to moderate global developmental delay (GDD), and had various brain anomalies (Table 1). Importantly, in comparison with HGPS cases, which have a progressive and early fatal course of disease, progeroid and neurodevelopmental features of PYCR1, patients actually improve with time. This important clinical finding has already been documented in the literature (Dimo-poulou et al. 2013).
It was previously reported that PYCR1-related disorders are difficult to diagnose based on the huge clinical heterogeneity. However, the findings in these five patients suggested that in some cases PYCR1 mutations might result in a clinically recognizable progeroid syndrome. This hypothesis is further supported by findings in three further patients (individuals 12, 13 and 14) in whom, based on the above-mentioned clinical findings, we speculated the existence of biallelic PYCR1 mutations as their underlying genetic cause and then confirmed this by direct Sanger sequencing. Although the clinical manifestations in the eight PYCR1-mutated individuals presented here are highly similar to the previous studies (Table 3), it is somewhat striking that only 14% of patients presented here developed wrinkled skin before 1 year of age, and one individual (individual 7) actually never presented this clinical sign. This is in contrast to both the findings in the initial PYCR1 mutation description leading to the classification of the disease as autosomal recessive cutis laxa (ARCL) (Reversade et al. 2009) and the large genotype-phenotype study (Dimopoulou et al. 2013), in whom all of the 68 presented patients had wrinkled skin. Notably, these data suggest that the genetic analysis of PYCR1 should be extended to all patients with unclassified juvenile segmental progeroid syndromes even if wrinkled skin is not present.
Table 3.
Comparison of clinical findings in PYCR1-mutated patients presented here and patients from previous studies as summarized before (Dimopoulou et al. 2013)
| Our study | Previous studies Dimopoulou et al. (%) |
|
|---|---|---|
| Wrinkled skin | 86% (6/7) | ~ 100 |
| Joint hypermobility | 71% (5/7) | ~ 100 |
| Typical facial gestalt | 100% (8/8) | ~ 100 |
| Intrauterine growth retardation | 100% (7/7) | ~ 90 |
| Psychomotor retardation | 100% (6/6) | ~ 90 |
| Osteopenia | 14% (1/7) | ~ 75 |
| Thin/translucent skin | 100% (8/8) | ~ 75 |
| Microcephaly | 100% (7/7) | ~ 75 |
| Postnatal growth retardation | 60% (3/5) | ~ 75 |
| Muscular hypotonia | 100% (7/7) | ~ 70 |
| Contractures | 25% (2/8) | ~ 50 |
| Hernias | 43% (3/7) | ~ 35 |
| Blue sclera | 0% (0/7) | ~ 30 |
| Strabismus | 14% (1/7) | ~ 30 |
| Cataract/corneal opacities | 14% (1/7) | ~ 10 |
Regarding the spectrum of PYCR1 mutations, we identified two individuals bearing amino acid substitutions of the arginine at the position 119, p.(Arg119His) and p.(Arg119Cys). As substitutions of this arginine have previously been described in eight individuals (Dimopoulou et al. 2013; Reversade et al. 2009), including p.(Arg119Gly), our data further suggest that this arginine is a hot spot PYCR1 amino acid. Moreover, we identified a recurrent 2-bp insertion c.219_220insAC, resulting in a frameshift mutation p.(Ile74Thrfs*10) in three individuals. Interestingly, proband 7 bearing this frameshift mutation in the homozygous state was the most severely affected PYCR1 patient in our case series. However, the exact PYCR1 genotype-phenotype correlation still remains unknown (Dimopoulou et al. 2013). One explanation could be that some of the individuals documented in the literature, in addition to mutations in PYCR1, bear a further distinct genetic factor, likely in the genome region that is not covered by WES, which results in the somewhat different clinical outcome.
Interestingly, our trio-WES analysis identified a de novo mutation in COL1A1 in individual 3. The identical mutation was previously documented in the OI mutation database in two further infants with OI type II, a commonly perinatal lethal syndrome (Marini and Smith 2000; Van Dijk and Sillence 2014). Although, detailed clinical data on these two infants are not available, it is worth noting that individual 3 in our study survived beyond the ninth birthday. Moreover, in addition to common OI features such as intrauterine growth retardation, multiple prenatal and postnatal fractures, and blue sclerae, he additionally displayed several features not commonly observed in OI type II. The latter include multiple contractures of the large joints, global developmental delay, global lipodystrophy, blepharophimosis, sparse and thin hair, and mandibular underdevelopment (Van Dijk and Sillence 2014). Therefore, we cannot completely exclude the possibility that individual 3, in addition to the mutation in COL1A1, bears a further distinct genetic or epigenetic factor resulting in a somewhat different clinical outcome from the previously documented individuals. A further intriguing possibility is that individual 3 is actually a mosaic for the COL1A1 mutation. However, as further tissues from this individual were not available we were not able to further investigate this possibility.
Importantly, even using trio-WES analysis performed according to high standards, sufficient coverage and expert bioinformatic analysis, the study did not lead to a definitive molecular diagnosis in two individuals (individuals 8 and 9). In individual 8, we did not identify a single candidate rare variant, whereas in individual 9 a potential novel candidate gene, SMC2, was identified. However, bioinformatic predictions do not provide evidence for the pathogenicity of the latter (Table 4). Interestingly, both individuals, although not displaying identical clinical signs and symptoms, are affected by a progeroid lipodystrophy somewhat mimicking mandibular hypoplasia, deafness, progeroid features, and lipodystrophy syndrome, marfanoid-progeroid-lipodystrophy syndrome and Berardinelli-Seip congenital lipodystrophy (Table 5). Taken together, these data further emphasize genetic heterogeneity of progeroid syndromes suggesting the existence of further, yet uncovered, progeroid loci. Moreover, these data underscore the utility of whole-genome sequencing to delineate the underlying genetic cause of some progeroid cases. Thus, elucidation of the genetic cause in these two patients as well as delineating novel progeroid loci will require further work and will be the main focus of our future studies.
Table 5.
Comparison of clinical characteristics of individuals 8 and 9 to mandibular hypoplasia, deafness, progeroid features, and lipo-dystrophy syndrome (MDPL, Lessel et al. 2015), marfanoid-progeroid-lipodystrophy syndrome (MPLS, Passarge et al. 2016) and Berardinelli-Seip congenital lipodystrophy (BSCL, Van Maldergem L. 2016)
| Individual 8 | Individual 9 | MDPL Lessel et al. |
MPLS Passarge et al. |
BSCL Van Maldergem L. |
|
|---|---|---|---|---|---|
| Sex | Male | Male | 7 female and 6 male | 6 female and 1 male | Female and male |
| Age | 10 years | 12 years | 10–62 years | 3.5–27 years | Various |
| 9 months | |||||
| Ethnicity | European | European | Various | Various | Various |
| Intrauterine growth retardation | + | + | 0/13 | 7/7 | Rare |
| Triangular face | + | − | 0/13 | 0/7 | − |
| Sparse scalp hair | + | − | 0/13 | 0/7 | − |
| Greying or loss of hair | − | − | 3/13 | 0/7 | − |
| Prominent scalp veins | + | − | 0/13 | 0/7 | − |
| Dental crowding/irregular teeth | − | − | 10/13 | 0/7 | − |
| Myopia | − | − | 0/13 | 6/7 | − |
| Cataracts | + | − | 0/13 | 0/7 | − |
| Hearing impairment | − | − | 10/13 | 0/7 | − |
| Joint contractures | − | + | 5/13 | − | |
| Hyperextensible joints | − | − | 0/13 | 5/7 | − |
| Arachnodactyly | − | − | 0/13 | 7/7 | − |
| Bone age | Advanced | Delayed | n.r | n.r | Advanced |
| Osteopenia | − | − | 6/13 | n.r | − |
| Scoliosis | − | + | 0/13 | 2/6 | − |
| Osteopenia | − | − | 6/13 | − | |
| Congenital lipodystrophy | + | − | 0/13 | 7/7 | + |
| Lipodystrophy | + | + | 13/13 | 7/7 | + |
| Stature | Short | − | Short: 8/13 | tall | − |
| Acromegaly | − | − | 0/13 | n.r | + |
| Muscle wasting | − | + | 11/13 | n.r | − |
| Abnormal cognitive function | − | − | 1/13 | 0/7 | Mild to moderate |
| Cardiac problems | − | − | 0/13 | Aortic root dilatation 3/7 | Hypertrophic cardiomyopathy |
| Mitral valve prolapse 3/7 | |||||
present
absent
n.r. not reported
In conclusion, due to our joint collaborative efforts a molecular diagnosis was achieved in 11/14 (~ 79%) of previously undiagnosed progeroid patients. Our data confirm biallelic mutations in POLR3A as the genetic cause of a recognizable, neonatal, Wiedemann-Rautenstrauch-like progeroid syndrome, and suggest that the genetic analysis of POLR3A should be performed in WRS-like patients. Furthermore, our data further expand the clinical spectrum associated with PYCR1 mutations and suggest the clinical criteria for early diagnosis of this condition. Last but not least, speculation based on our data suggests the existence of further genetic loci for early-onset progeroid conditions that may be solved by application of whole-genome sequencing.
Supplementary Material
Acknowledgements
We thank the patients and their families for participation in this study. This work was funded in part by local funding (Forschungsförderungsfonds der Medizinischen Fakultät der Universitätsklinikum Hamburg-Eppendorf [FFM]) and the Deutsche Forschungsgemeinschaft (LE4223/1-1) to DL, The University of Michigan Center for Genomics in Health and Disease to TWG, and by The Progeria Research Foundation. UW-CMG Acknowledgement Statement: Sequencing of parental DNA from individuals 1–5 was provided by the University of Washington Center for Mendelian Genomics (UW-CMG) which was funded by the National Human Genome Research Institute and the National Heart, Lung and Blood Institute grant 2UM1HG006493 to Drs. Debbie Nickerson, Michael Bamshad, and Suzanne Leal.
Footnotes
Compliance with ethical standards
Conflict of interest The authors declare that they have no conflict of interest.
Research involving human participants The study has ongoing approval from the Hasbro Children’s Hospital and the University Medical Center Hamburg-Eppendorf Institutional Review Boards. The study was performed in accordance with the Declaration of Helsinki protocols.
Informed consent All biological samples were obtained following written informed consent from studied individuals or their legal representatives.
As this manuscript went to press, a further study independently confirmed biallelic POLR3A mutations as the cause of Wiedemann-Rautenstrauch syndrome. Moreover, that study report on the identical patient, their subject 3, as the herein presented individual 4 (Wambach et al. 2018).
Electronic supplementary material The online version of this article (https://doi.org/10.1007/s00439-018-1957-1) contains supplementary material, which is available to authorized users.
References
- Bernard G, Chouery E, Putorti ML, Tetreault M, Takanohashi A, Carosso G, Clement I, Boespflug-Tanguy O, Rodriguez D, Delague V, Abou Ghoch J, Jalkh N, Dorboz I, Fribourg S, Teich-mann M, Megarbane A, Schiffmann R, Vanderver A, Brais B (2011) Mutations of POLR3A encoding a catalytic subunit of RNA polymerase Pol III cause a recessive hypomyelinating leukodystrophy. Am J Hum Genet 89:415–423. https://doiorg/10.1016/j.ajhg.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers PH (1989) Inherited disorders of collagen gene structure and expression. Am J Med Genet 34:72–80. 10.1002/ajmg.1320340114 [DOI] [PubMed] [Google Scholar]
- Byers PH, Duvic M, Atkinson M, Robinow M, Smith LT, Krane SM, Greally MT, Ludman M, Matalon R, Pauker S, Quanbeck D, Schwarze U (1997) Ehlers-Danlos syndrome type VIIA and VIIB result from splice-junction mutations or genomic deletions that involve exon 6 in the COL1A1 and COL1A2 genes of type I collagen. Am J Med Genet 72:94–105 [DOI] [PubMed] [Google Scholar]
- Chen L, Lee L, Kudlow BA, Dos Santos HG, Sletvold O, Shafeghati Y, Botha EG, Garg A, Hanson NB, Martin GM, Mian IS, Kennedy BK, Oshima J (2003) LMNA mutations in atypical Werner’s syndrome. Lancet 362:440–445. 10.1016/S01406736(03)14069-X [DOI] [PubMed] [Google Scholar]
- Chong JX, McMillin MJ, Shively KM, Beck AE, Marvin CT, Armenteros JR, Buckingham KJ, Nkinsi NT, Boyle EA, Berry MN, Bocian M, Foulds N, Uzielli ML, Haldeman-Englert C, Hennekam RC, Kaplan P, Kline AD, Mercer CL, Nowaczyk MJ, Klein Wassink-Ruiter JS, McPherson EW, Moreno RA, Scheuerle AE, Shashi V, Stevens CA, Carey JC, Monteil A, Lory P, Tabor HK, Smith JD, Shendure J, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian G (2015) De novo mutations in NALCN cause a syndrome characterized by congenital contractures of the limbs and face, hypotonia, and developmental delay. Am J Hum Genet 96:462–473. 10.1016/ajhg.2015.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, Lyonnet S, Stewart CL, Munnich A, Le Merrer M, Levy N (2003) Lamin a truncation in Hutchinson-Gilford progeria. Science 300: 2055 10.1126/science.1084125 [DOI] [PubMed] [Google Scholar]
- Dimopoulou A, Fischer B, Gardeitchik T, Schroter P, Kayserili H, Schlack C, Li Y, Brum JM, Barisic I, Castori M, Spaich C, Fletcher E, Mahayri Z, Bhat M, Girisha KM, Lachlan K, Johnson D, Phadke S, Gupta N, Simandlova M, Kabra M, David A, Nijtmans L, Chitayat D, Tuysuz B, Brancati F, Mundlos S, Van Maldergem L, Morava E, Wollnik B, Kornak U (2013) Genotype-phenotype spectrum of PYCR1-related autosomal recessive cutis laxa. Mol Genet Metab 110:352–361. 10.1016/jymgme.2013.08.009 [DOI] [PubMed] [Google Scholar]
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, Erdos MR, Robbins CM, Moses TY, Berglund P, Dutra A, Pak E, Durkin S, Csoka AB, Boehnke M, Glover TW, Collins FS (2003) Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 423:293–298. 10.1038/nature01629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D’Apice MR, Novelli G, Crow Y (2009) Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab 94:4971–4983. 10.1210/jc.2009-0472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon LB, Brown WT, Collins FS (1993) Hutchinson–Gilford progeria syndrome. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews((R)), Seattle: [PubMed] [Google Scholar]
- Graul-Neumann LM, Kienitz T, Robinson PN, Baasanjav S, Karow B, Gillessen-Kaesbach G, Fahsold R, Schmidt H, Hoffmann K, Passarge E (2010) Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3’ terminus of the FBN1-gene. Am J Med Genet A 152A:2749–2755. 10.1002/ajmg.a.33690 [DOI] [PubMed] [Google Scholar]
- Hisama FM, Lessel D, Leistritz D, Friedrich K, McBride KL, Pastore MT, Gottesman GS, Saha B, Martin GM, Kubisch C, Oshima J (2011) Coronary artery disease in a Werner syndrome-like form of progeria characterized by low levels of progerin, a splice variant of lamin A. Am J Med Genet A 155A:3002–3006. https://doiorg/10.1002/ajmg.a.34336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay AM, Conway RL, Thiffault I, Saunders C, Farrow E, Adams J, Toriello HV (2016) Neonatal progeriod syndrome associated with biallelic truncating variants in POLR3A. Am J Med Genet A 170:3343–3346. 10.1002/ajmg.a.37960 [DOI] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, Cabrera E, Freire R, Pope K, Nahid A, Norris F, Leventer RJ, Delatycki MB, Barbi G, von Ameln S, Hogel J, Degoricija M, Fertig R, Burkhalter MD, Hofmann K, Thiele H, Altmuller J, Nurnberg G, Nurnberg P, Bahlo M, Martin GM, Aalfs CM, Oshima J, Terzic J, Amor DJ, Dikic I, Ramadan K, Kubisch C (2014) Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46:1239–1244. 10.1038/ng.3103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Hisama FM, Szakszon K, Saha B, Sanjuanelo AB, Salbert BA, Steele PD, Baldwin J, Brown WT, Piussan C, Plauchu H, Szilvassy J, Horkay E, Hogel J, Martin GM, Herr AJ, Oshima J, Kubisch C (2015) POLD1 germline mutations in patients initially diagnosed with werner syndrome. Hum Mutat 36:1070–1079. 10.1002/humu.22833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Wu D, Trujillo C, Ramezani T, Lessel I, Alwasiyah MK, Saha B, Hisama FM, Rading K, Goebel I, Schutz P, Speit G, Hogel J, Thiele H, Nurnberg G, Nurnberg P, Hammerschmidt M, Zhu Y, Tong DR, Katz C, Martin GM, Oshima J, Prives C, Kubisch C (2017) Dysfunction of the MDM2/p53 axis is linked to premature aging. J Clin Invest 127:3598–3608. https://doiorg/10.1172/JCI92171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marini J, Smith SM (2000) Osteogenesis imperfecta. In: De Groot LJ, Chrousos G, Dungan K, Feingold KR, Grossman A, Hersh-man JM, Koch C, Korbonits M, McLachlan R, New M, Purnell J, Rebar R, Singer F, Vinik A (eds) Endotext, South Dartmouth [Google Scholar]
- Martin GM, (1978)Genetic syndromes in man with potential relevance to the pathobiology of aging.Birth Defects Orig Artic Ser 14:5–39 [PubMed] [Google Scholar]
- Navarro CL, Esteves-Vieira V, Courrier S, Boyer A, Duong Nguyen T, Huong le TT, Meinke P, Schroder W, Cormier-Daire V, Sznajer Y, Amor DJ, Lagerstedt K, Biervliet M, van den Akker PC, Cau P, Roll P, Levy N, Badens C, Wehnert M, De Sandre-Giovannoli A (2014) New ZMPSTE24 (FACE1) mutations in patients affected with restrictive dermopathy or related progeroid syndromes and mutation update. Eur J Hum Genet 22:1002–1011. https://doiorg/10.1038/ejhg.2013.258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas E, Golemis EA, Arora S (2016) POLD1: Central mediator of DNA replication and repair, and implication in cancer and other pathologies. Gene 590:128–141. 10.1016/j.gene.2016.06.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolacci S, Bertola D, Franco J, Mohammed S, Tartaglia M, Wollnik B, Hennekam RC (2017) Wiedemann-rautenstrauch syndrome: a phenotype analysis. Am J Med Genet A. 10.1002/ajmg.a.38246 [DOI] [PubMed] [Google Scholar]
- Paolacci S, Li Y, Agolini E, Bellacchio E, Arboleda-Bustos CE, Carrero D, Bertola D, Al-Gazali L, Alders M, Altmuller J, Arboleda G, Beleggia F, Bruselles A, Ciolfi A, Gillessen-Kaesbach G, Krieg T, Mohammed S, Muller C, Novelli A, Ortega J, Sandoval A, Velasco G, Yigit G, Arboleda H, Lopez-Otin C, Wollnik B, Tartaglia M, Hennekam RC (2018) Specific combinations of biallelic POLR3A variants cause Wiedemann-Rautenstrauch syndrome. J Med Genet. 10.1136/jmedgenet-2018-105528 [DOI] [PubMed] [Google Scholar]
- Passarge E, Robinson PN, Graul-Neumann LM (2016) Marfanoid-prog-eroid-lipodystrophy syndrome: a newly recognized fibrillinopa-thy. Eur J Hum Genet 24:1244–1247. 10.1038/ejhg.2016.6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollitt R, McMahon R, Nunn J, Bamford R, Afifi A, Bishop N, Dalton A (2006) Mutation analysis of COL1A1 and COL1A2 in patients diagnosed with osteogenesis imperfecta type I-IV. Hum Mutat 27:716 10.1002/humu.9430 [DOI] [PubMed] [Google Scholar]
- Puente XS, Quesada V, Osorio FG, Cabanillas R, Cadinanos J, Fraile JM, Ordonez GR, Puente DA, Gutierrez-Fernandez A, Fanjul-Fernandez M, Levy N, Freije JM, Lopez-Otin C (2011) Exome sequencing and functional analysis identifies BANF1 mutation as the cause of a hereditary progeroid syndrome. Am J Hum Genet 88:650–656. 10.1016/j.ajhg.2011.04.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rautenstrauch T, Snigula F (1977) Progeria: a cell culture study and clinical report of familial incidence. Eur J Pediatr 124:101–111 [DOI] [PubMed] [Google Scholar]
- Reversade B, Escande-Beillard N, Dimopoulou A, Fischer B, Chng SC, Li Y, Shboul M, Tham PY, Kayserili H, Al-Gazali L, Shahwan M, Brancati F, Lee H, O’Connor BD, Schmidt-von Kegler M, Merriman B, Nelson SF, Masri A, Alkazaleh F, Guerra D, Ferrari P, Nanda A, Rajab A, Markie D, Gray M, Nelson J, Grix A, Sommer A, Savarirayan R, Janecke AR, Steichen E, Sillence D, Hausser I, Budde B, Nurnberg G, Nurnberg P, Seemann P, Kunkel D, Zambruno G, Dallapiccola B, Schuelke M, Robertson S, Hamamy H, Wollnik B, Van Maldergem L, Mundlos S, Kornak U (2009) Mutations in PYCR1 cause cutis laxa with progeroid features. Nat Genet 41:1016–1021. 10.1038/ng.413 [DOI] [PubMed] [Google Scholar]
- Saha B, Lessel D, Hisama FM, Leistritz DF, Friedrich K, Martin GM, Kubisch C, Oshima J (2010) A novel LMNA mutation causes altered nuclear morphology and symptoms of familial partial lipodystrophy (Dunnigan variety) with progeroid features. Mol Syndromol 1:127–132. 10.1159/000320166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen I, Szelinger S, Siniard AL, Kurdoglu A, Corneveaux JJ, Malenica I, Richholt R, Van Camp G, De Both M, Swaminathan S, Turk M, Ramsey K, Craig DW, Narayanan V, Huentelman MJ (2015) A frame-shift mutation in CAV1 is associated with a severe neonatal progeroid and lipodystrophy syndrome. PLoS One 10:e0131797. 10.1371/journal.pone.0131797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria-Valles C, Carrero D, Gabau E, Velasco G, Quesada V, Barcena C, Moens M, Fieggen K, Mohrcken S, Owens M, Puente DA, Asen-sio O, Loeys B, Perez A, Benoit V, Wuyts W, Levy N, Hennekam RC, De Sandre-Giovannoli A, Lopez-Otin C (2016) Novel LMNA mutations cause an aggressive atypical neonatal progeria without progerin accumulation. J Med Genet. 10.1136/jmedgenet-2015-103695 [DOI] [PubMed] [Google Scholar]
- Van Maldergem L (1993) Berardinelli-seip congenital lipodystrophy. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A (eds) GeneReviews((R)), Seattle Van Dijk FS, Sillence DO (2014) Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A 164A:1470–1481. 10.1002/ajmg.a.36545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wambach JA, Wegner DJ, Patni N, Kircher M, Willing MC, Baldridge D, Xing C, Agarwal AK, Vergano SAS, Patel C, Grange DK, Kenney A, Najaf T, Nickerson DA, Bamshad MJ, Cole FS, Garg A (2018) Bi-allelic POLR3A loss-of-function variants cause autosomal-recessive Wiedemann-Rautenstrauch syndrome. Am J Hum Genet. 10.1016/j.ajhg.2018.10.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weedon MN, Ellard S, Prindle MJ, Caswell R, Allen HL, Oram R, Godbole K, Yajnik CS, Sbraccia P, Novelli G, Turnpenny P, McCann E, Goh KJ, Wang Y, Fulford J, McCulloch LJ, Savage DB, O’Rahilly S, Kos K, Loeb LA, Semple RK, Hattersley AT (2013) An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet 45:947–950. 10.1038/ng.2670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf NI, Vanderver A, van Spaendonk RM, Schiffmann R, Brais B, Bugiani M, Sistermans E, Catsman-Berrevoets C, Kros JM, Pinto PS, Pohl D, Tirupathi S, Stromme P, de Grauw T, Fribourg S, Demos M, Pizzino A, Naidu S, Guerrero K, van der Knaap MS, Bernard G, Group HR (2014) Clinical spectrum of 4H leukodystrophy caused by POLR3A and POLR3B mutations. Neurology 83:1898–1905. 10.1212/WNL.0000000000001002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Kato J, Shimamoto A, Goto M, Furuichi Y, Ikeda H (1998) Bloom’s and Werner’s syndrome genes suppress hyperre-combination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci USA 95:8733–8738 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.