

Abstract
Formaldehyde is a ubiquitous DNA damaging agent, with human exposures occurring from both exogenous and endogenous sources. Formaldehyde exposure can result in multiple types of DNA damage, including DNA-protein crosslinks and thus, is representative of other exposures that induce DNA-protein crosslinks such as cigarette smoke, automobile exhaust, wood smoke, metals, ionizing radiation, and certain chemotherapeutics. Our objective in this study was to identify the genes necessary to mitigate formaldehyde toxicity following chronic exposure in human cells. We used siRNAs that targeted 320 genes representing all major human DNA repair and damage response pathways, in order to assess cell proliferation following siRNA depletion and subsequent formaldehyde treatment. Three unrelated human cell lines frequently used in genotoxicity studies (SW480, U-2 OS and GM00639) were used to identify common pathways involved in mitigating formaldehyde sensitivity. Although there were gene-specific differences among the cell lines, four inter-related cellular pathways were determined to mitigate formaldehyde toxicity: homologous recombination, DNA double-strand break repair, ionizing radiation response and DNA replication. Additional insight into cell line-specific response patterns was obtained by using a combination of exome sequencing and Cancer Cell Line Encyclopedia genomic data. The results of this DNA damage repair pathway-focused siRNA screen for formaldehyde toxicity in human cells provide a foundation for detailed mechanistic analyses of pathway-specific involvement in the response to environmentally-induced DNA-protein crosslinks and, more broadly, genotoxicity studies using human and other mammalian cell lines.
Keywords: DNA Damage Response, DNA Repair, formaldehyde, double-strand break repair, siRNA screen
1. Introduction
Endogenous and environmental exposure to formaldehyde are correlated with an increased risk of cancer, asthma, and other diseases (1–4). Formaldehyde is produced endogenously as an essential component of human cellular metabolism, including one carbon pool, amino acid and alcohol metabolism, lipid peroxidation and P450-dependent demethylation (1). In humans, the steady state level of formaldehyde in whole blood or plasma is remarkably high, ranging between 22–87 μM (5–7). However, these reported blood levels may be an overestimate considering a recent study that failed to detect any endogenous formaldehyde-serum albumin adducts (summarized in (8)). Some cell lineages, e.g., hematopoietic stem cells, may receive higher exposure via lineage-specific generation of formaldehyde from histone demethylation as part of chromatin remodeling during differentiation (9,10). Human tumor cells can be stimulated to generate formaldehyde in response to treatment with widely used chemotherapeutic agents, e.g., anthracyclines (11,12). Environmental exposure to formaldehyde includes occupational exposures and sources such as automobile exhaust, cigarettes and e-cigarettes, cosmetic products, forest fires and manufactured wood products (13–19).
The genotoxicity and ubiquitous nature of formaldehyde exposure have driven efforts to better understand the cellular pathways that mitigate formaldehyde toxicity. Specific cellular processes reported to promote cell survival include Nucleotide Excision Repair (NER) (20–22), proteasomal degradation (23), metalloproteases (24,25), the Fanconi Anemia pathway (26–29), and Homologous Recombination (HR) (20,22,30,31). We and others have also shown that formaldehyde can perturb the cell cycle and alter gene expression (21,30,32–35). However, based on prior literature, it was unclear to what extent the different DNA damage response and repair pathways protect cells from chronic formaldehyde exposures. To systematically analyze the role of DNA damage repair (DDR) pathways in modulating formaldehyde toxicity, we selectively depleted each of 320 proteins representing major DNA damage response and repair, cell cycle telomere maintenance and mitotic cell division pathways. We chose to focus this screen on these pathways in order to dissect the role of these inter-related pathways in mitigating formaldehyde-mediated DNA damage. Gene depletion was followed by quantification of cell proliferation suppression as a function of formaldehyde dose. The resulting library is referred to herein as the 320 DDR (320 gene DNA damage repair) library. This library was used to screen three well-characterized, though otherwise unrelated, human cell lines to identify genes that modified cell proliferation in response to chronic formaldehyde exposure. The cell lines, GM00639, SW480, and U-2 OS, were derived respectively from primary human fibroblasts, an epithelial adenocarcinoma and an osteosarcoma, and were chosen due to their wide-spread use in genotoxic studies (30,36–38). This work is the first screen for DDR-related determinants of formaldehyde toxicity in human cells.
2. Materials and methods
2.1. Cell lines
Three well-characterized human cell lines were used for screens: GM00639, SW480, and U-2 OS. GM00639 is a widely used human fibroblast cell line that was derived by SV40 transformation of primary fibroblasts from an 8-year-old galactosemic male (39). SW480 is an epithelial colorectal adenocarcinoma cell line (40). U-2 OS is an osteosarcoma cell line derived from a 15-year-old female (41). GM00639 and U-2 OS cells were kind gifts from Dr. Robb Moses, and SW480 from Dr. Owen McCarty (both at Oregon Health & Science University). Cells were grown in DMEM supplemented with 10% fetal bovine serum and antibiotic/antimycotic (penicillin, streptomycin, and Amphotericin B, Gibco) at 37°C in a humidified, 5% CO2 ambient oxygen incubator.
2.2. Genomic analyses
Coding region variant calls for the SW480 and U-2 OS cell lines were downloaded from the Cancer Cell Line Encyclopedia (CCLE) Project web-based data portal (42) (merged variant maf file: CCLE_DepMap_18Q1_maf_20180207.txt). Briefly, this merged file integrates variant calls from CCLE whole genome and exome sequencing (WGS, WES), CCLE RNA sequencing (43) and WES generated by the Sanger Institute as part of the COSMIC project and is available at the European Genome-phenome Archive (EGAD00001001039). We filtered out variant calls sourced from the RNA-Seq data alone, and those with low alternate allele counts from any of the WES/WGS platforms.
To generate comparable data for GM00639, we carried out exome capture and sequencing of a clonal derivative, GM639-CC1 (39,44). Exome capture was performed using Nimblegen SeqCap EZ HGSC VCRome kit (V2), with sequencing performed on an Illumina HiSeq to generate 200 bp read lengths. Sequence reads were aligned to the hg19 human reference genome using BWA (BWA-MEM) (45). We applied GATK (46) indel realignment and base quality score recalibration according to GATK best practices recommendations (47,48). Variant calling by GATK UnifiedGenotyper was restricted to ±1000 bp around the capture regions (VariantFiltration module). Median read depth was 109 and prior to filtering variants were annotated using in-house scripts including the ANNOVAR pipeline (49). We filtered variants using six criteria: coverage ≥ 30, GATK hard filter pass, read quality ≥ 20 (Phred-based), read depth ≥15, variant allele frequency ≥15% and exclusion of common variants using a population minor allele frequency threshold of 1%. Population allele frequencies were mined from the Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP) (http://evs.gs.washington.edu/EVS/) [ESP6500], 1000 Genomes Project (phase three)(50), The Exome Aggregation Consortium (ExAC)(51) and two internal exome collections consisting of 650 and 945 exomes. To enable joint analysis across three cell lines, we merged and re-annotated variant calls using the Oncotator (52) pipeline, generated a unified variant annotation, and then performed additional filtering to exclude synonymous mutations.
Copy number data for the SW480 and U-2 OS cell lines were downloaded from the CCLE web portal (42). Gene-level copy number estimates (normalized log ratios) were inferred from genome-wide Affymetrix SNP6.0 array data as previously described (42). Using a strategy similar to that used by Kim et al. (53), we determined genes that were amplified or deleted using a threshold of ± 0.7 to identify approximate two-fold changes for mean segment values that estimate copy number.
2.3. Dose-dependent formaldehyde-induced suppression of cell proliferation
We determined dose-dependent formaldehyde toxicity by quantifying cell proliferation suppression after 5 days of continuous formaldehyde exposure. We had previously determined that 5-day exposure elicited a robust formaldehyde-dependent DNA damage response. Assays were performed in triplicate in 96-well plates: cells were plated at sub-confluent density, allowed to attach overnight, and then treated with formaldehyde (Fisher Scientific) at indicated doses. Viable cell number was assessed on Day 5 using Cell Titer-Glo® (CTG) following the manufacturer’s instructions. CTG assesses viable cell numbers by quantifying ATP generated by metabolically active cells. Briefly, 100 µl of CTG reagent was added to each well prior to mixing the plate on a shaker for 10–15 min. Luminescence output for each well was quantified on a Tecan plate reader (Infinite M200). The suppression of cell proliferation as a function of formaldehyde dose (expressed as GI25–75) was calculated for each cell line using Graph Pad Prism 7 software (La Jolla, CA, USA) with a sigmoidal, 4PL log curve fit.
2.4. RNAi screens
A custom RNAi library was used for screens that consisted of sets of four pooled siRNAs specific for each target gene where individual siRNAs that had been previously verified for depletion effectiveness in human cells by the supplier (Qiagen). The 320 targeted genes represented all major DNA repair pathways, together with additional genes involved in the DNA damage response, cell cycle regulation, telomere maintenance and mitotic cell division (54). The 1280 siRNAs in this library, termed the 320DDR library considering target gene annotations, were synthesized on a 0.25 μmol scale prior to pooling each gene-specific siRNA set in a separate master plate well (Qiagen). Each gene-specific pool was then tested in triplicate, on separate plates, to establish experimental variability, cell-to-cell variability, statistical validity, and to identify potential batch effects. The full 320DDR RNAi library and sequences of gene-specific and control siRNAs is given in Supplementary Table 1 of Kehrli et al 2016 (54). This siRNA library is available for both academic and commercial use as the ‘320DDR (DNA Damage Repair) Library’ through the University of Washington Quellos High Throughput Screening Core (http://depts.washington.edu/iscrm/quellos/rnai-screens).
RNAi-targeted genes that when depleted led to formaldehyde dose-dependent growth inhibition were identified in 384-well format screens on the Quellos High Throughput Screening Core platform. Transfection conditions for siRNA were first optimized for each cell line using an siRNA that targets the KIF11 kinesin family member 11 protein that arrests mitotic division. A minimum target of at least 50% growth suppression with <25% absolute deviation upon KIF11 siRNA transfection versus a control siRNA and mock-transfected cells (cells plus media and Optimem only) were used as controls to establish assay thresholds. Transfection optimization and screens were performed using Lipofectamine RNAi max according to manufacturer (Invitrogen) instructions. Mock transfections and a non-targeting universal siRNA control were used in addition to KIF11 positive control as transfection and RNAi pathway-dependent controls.
AKIF11 positive control siRNA (SASI_Hs01_00161689) and the siRNA negative control siRNA (MISSION siRNA SIC001 Universal Control #1) were both purchased from Sigma-Aldrich. Optimal transfection and treatment conditions for each cell line were determined using a simple Z-factor score (all scores ≥ 0.5) to determine differences and variability among replicates. We identified siRNA-specific toxic effects in the absence of formaldehyde using ≥ 20% cell viability (≤ 80% cell death) as a cut off to remain within assay detection limits and biological plausibility (data not shown). Growth inhibitory concentrations of formaldehyde leading to no (i.e., GI0) or 10–90% (i.e., GI10–90) growth inhibition were determined for each cell line by formaldehyde titration over a 0 – 100 μM range with continuous exposure over 5 days. We verified reproducibility across replicates for each formaldehyde dose, then calculated the standard deviation to determine the variation across the sample replicates. Z Scores for the standard deviation were calculated using the equation,
| Equation 1 |
where μ is the mean for standard deviations, and σ standard deviation for the mean standard deviations. This was used to reproducible formaldehyde dose ranges for our assays. Genes with z-scores ≥ +2.0 or those for which an untreated data point did not meet the quality minimums outlined above were excluded.
These results were used to design RNAi screens in which cells were plated in opaque 384-well plates and transfected 24 hrs prior to the addition of formaldehyde or PBS (control/untreated), followed by growth for an additional 5 days prior to determining viability on an Envision Multilable detector/plate reader (Perkin Elmer). CTG reagent alone (blank) was subtracted from all wells to establish final luminescence values. A non-targeting universal siRNA negative control (MISSION siRNA SIC001 Universal Control #1) was used to monitor transfection efficiency and the effects of siRNA delivery, with results standardized as percent proliferation of siRNA-transfected compared to mock-transfected wells on the same plate. Compound additions were performed using peri-pumps, which more reproducibly and rapidly deliver 5 μL sample volumes than capillary pins in order to minimize cell exposure times and generate more reproducible signal intensities.
2.5. RNAi screen data analyses
Treated cell growth inhibition means (μt) were calculated from three replicate cultures for each gene and dose across each cell line. The mean for each dose treatment (μt) was then normalized to the untreated mean (μu) to calculate normalized growth inhibition or mean (μn).
| Equation 2 |
Normalized data were then used to calculate area under the curve (AUC) using GraphPad Prism 7 software (La Jolla, CA, USA). Next, we determined the p-value for AUCs and used this to identify the subset of siRNAs that lead to statistically significant alterations in growth (p-value < 0.05), which was further assessed using Z-scores. Z scores for AUC were calculated using the equation below, where μa is the mean for the AUCs, and σa standard deviation for the AUCs.
| Equation 3 |
In each cell line, all genes with Z scores ≤ 0 were considered sensitive. This cut-off score was chosen because we used an siRNA library already highly enriched for genes known to modulate cellular response to genotoxic agents, and for which sensitivities were expected to be less variable across genes within a cell line. Highly sensitizing genes were classified as genes with Z score ≤ −1.0 for each cell line, whereas genes with a Z score ≥ +2.0 were classified as protective.
Hierarchical clustering with complete linkage and Euclidean distance was used to identify concordant growth inhibition results among genes across cell lines. This analysis was performed on a 98-gene matrix where we had high quality relative viability measurements across all cell lines with no missing data. Based on visual examination of the resulting heatmap, we selected six gene clusters and used the ‘cutree’ R function to generate groups of genes informed by dendrogram height. For each cluster, we applied the multiple protein search query in String DB (Version 10.5) to assess the extent of protein-protein interactions (PPI) within these gene clusters (55). The STRING Analysis module was then used to assess genome-wide KEGG pathway enrichment within clusters.
2.6. Data Availability
Exome sequencing data are deposited in the Sequence Read Archive (SRA), accession number SRP131620.
3. Results
3.1. Genes that suppress cell proliferation following formaldehyde exposure
We first determined formaldehyde dose-dependent suppression of cell proliferation for the human cell lines GM00639, SW480, and U-2 OS after five days of continuous formaldehyde exposure. All three cell lines displayed formaldehyde dose-dependent inhibition of proliferation (Figure 1A), with similar formaldehyde doses leading to a 50% decrease in proliferation (expressed as GI50)(Figure 1B). Among the three lines, SW480 was the least sensitive and GM00639 the most sensitive to proliferation suppression at a specific formaldehyde concentration.
Figure 1. GM00639, U-2 OS, and SW480 have similar dose response curves following chronic formaldehyde exposure.

(A) Dose response curves and (B) GI25–75 doses for each cell line following continuous formaldehyde exposure over 5 days.
We transfected each cell line 24 h prior to formaldehyde exposure with siRNAs that targeted each gene in the 320DDR library (54), then assessed cell proliferation by CTG assay after 5 days of continuous formaldehyde exposure. Using calculations for area under the curve (3 doses in triplicate) a total of 23 genes were identified that when depleted conferred formaldehyde sensitivity across all three cell lines (Figure 2, Supplementary Table S1). These 23 genes represent pathways that may plausibly limit formaldehyde toxicity by promoting the repair of formaldehyde-induced DNA damage. Of note, no individual gene when depleted was either highly sensitizing (Z-score ≤ −1) or protective (Z-score ≥ +2) (Supplementary Figure S1A and B, respectively and Supplementary Table S1).
Figure 2. Twenty-three genes sensitize three human cell lines to formaldehyde.
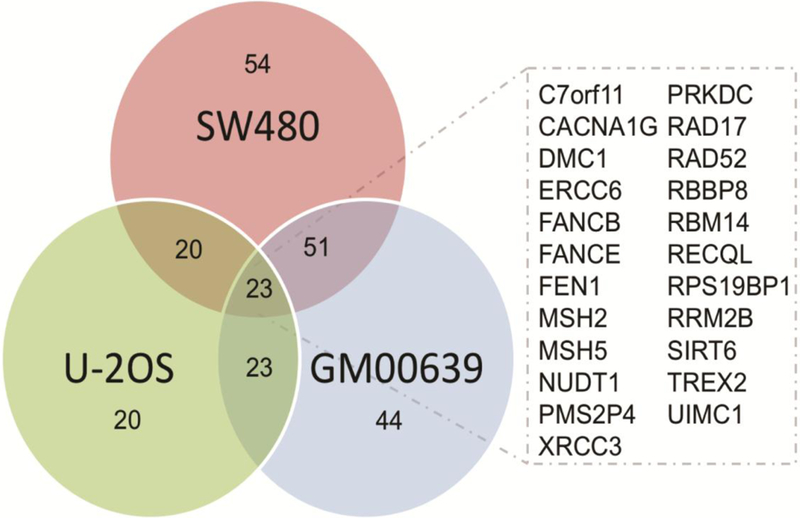
Venn diagram of the number of sensitizing genes (Z-score ≤ 0) in all three cell lines. Inset: List of 23 genes that were sensitizing in all three cell lines.
3.2. Genomic alterations in the cell lines do not contribute to formaldehyde sensitivity
We analyzed non-synonymous mutations across cell lines to determine whether cell line-specific variability in the response to formaldehyde might be explained by cell line-specific genetic variation (Figure 3A and Supplementary Table S2). All three cell lines shared mutations in 2 genes (PKHD1L1, TTN) that were not included in our siRNA library. According to the ExAC database, both PKHD1L1 and TTN fall in the top 5% (z-score < −1.7) of genes over-represented for synonymous variation and both also have loss-of-function probabilities of 0. These results indicate that PKHD1L1 and TTN are tolerant to variation, and thus unlikely determinants of cellular fitness (51).
Figure 3. Prevalence of mutations in GM00639, SW480 and U-2 OS cell lines.
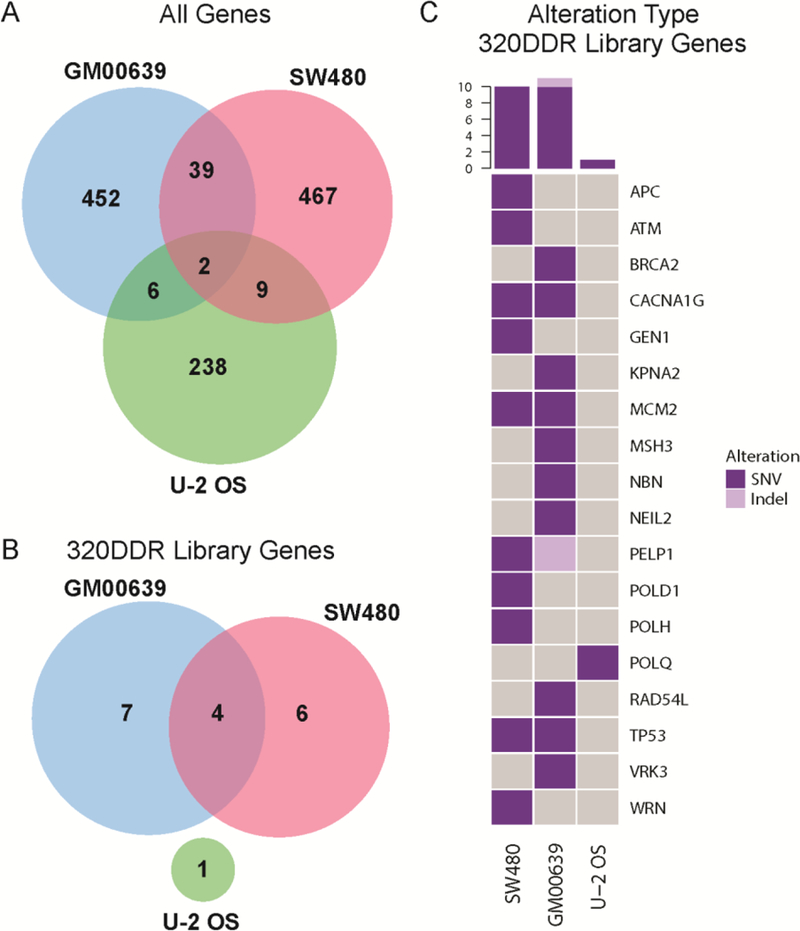
Venn diagrams depicting the number of genes with nonsynonymous mutations in (A) all genes, versus (B) genes included in the 320 DDR library across cell lines. Mutations include single nucleotide variants (SNVs) and small insertions and deletions (indels). (C) Oncoprint visualizing mutations in 320 DDR library genes where each row represents a gene and each column a cell line. Colors indicate a true event, i.e., a gene that is mutated in a given cell line. Gray indicates that no alteration was observed. The histogram (top) summarizes the number of genes affected for a given cell line.
No mutations were identified in siRNA target genes in all three lines (Figure 3B). However, 18 (or 6%) siRNA library-targeted genes were mutated in one cell line (Figure 3C) with no significant enrichment in specific DDR pathways (data not shown). These gene alterations may reflect a combination of donor-specific germline, or in the case of SW480 and U-2 OS cells somatic, variants in the tumors from which these two lines were isolated. Thus, no common genomic alterations in siRNA-targeted genes could fully explain the formaldehyde sensitivity or resistance.
Analysis of copy number variation for the SW480 and U-2 OS cell lines were mined from the CCLE data portal and are based on SNP genotyping. These data were not available for GM00639 cell line, and thus whole exome sequencing was performed. In the two cell lines represented in the CCLE, we identified 269 genes in regions of copy number gain and 111 genes in regions of copy number loss that were shared between the SW480 and U-2 OS cell lines (Figure S2A-B, Table S3). Thirty-three (10%) genes targeted in the 320 DDR library were in regions of significant copy number variation (Figure S2C). We found a modest enrichment for amplified genes in cytokinesis pathway genes (CETN2, KIF4A) and for deleted genes in chromatin modifiers (ATRX, CHAF1B) in U-2 OS cells (Fisher’s exact test, p-value < 0.05). Apart from these associations, we identified no additional pathway enrichment for alterations in siRNA-targeted genes compared to all other genes assessed across all three cell lines. Of note, there was no enrichment in genomic alterations, either non-synonymous mutations or copy number changes, in genes known to modulate formaldehyde response when depleted versus control siRNAs. These results argue that genomic alterations in genes included in the 320 DDR library are not strong drivers of cell line-specific differences in formaldehyde response.
3.3. Formaldehyde response pathways identified across cell lines
We extended our analyses by mapping genes targeted by the 320 DDR siRNA library to functional pathways. These were enriched for DNA damage repair and response or DNA metabolism, reflecting the initial design of the 320 DDR library (54). Twenty-two functional gene groups were identified for siRNA library-targeted genes by combining Gene Ontology (GO) consortium terms, Reactome pathway data, and manual literature searches (Figure 4A and Table S4). Genes were assigned to multiple functional groups or pathways when appropriate (Figure 4B). Genes that when depleted sensitized cells to formaldehyde (i.e., Z-score ≤ 0) were significantly associated with eight functional pathways: HR, DSB Repair, Chromatin Modification, Cell Cycle, DNA Damage Checkpoints, Response to Oxidative Stress, Response to IR, and DNA replication (Figure 4C). An additional quantitative analysis of these associations, performed by bootstrapping with replacement, identified four functional pathways that were significantly over-represented among the sensitizing gene set: HR, DSB Repair, Response to IR, and DNA replication (Figure 4D).
Figure 4. Enrichment analysis identifies pathways that mitigate proliferation suppression following formaldehyde exposure.
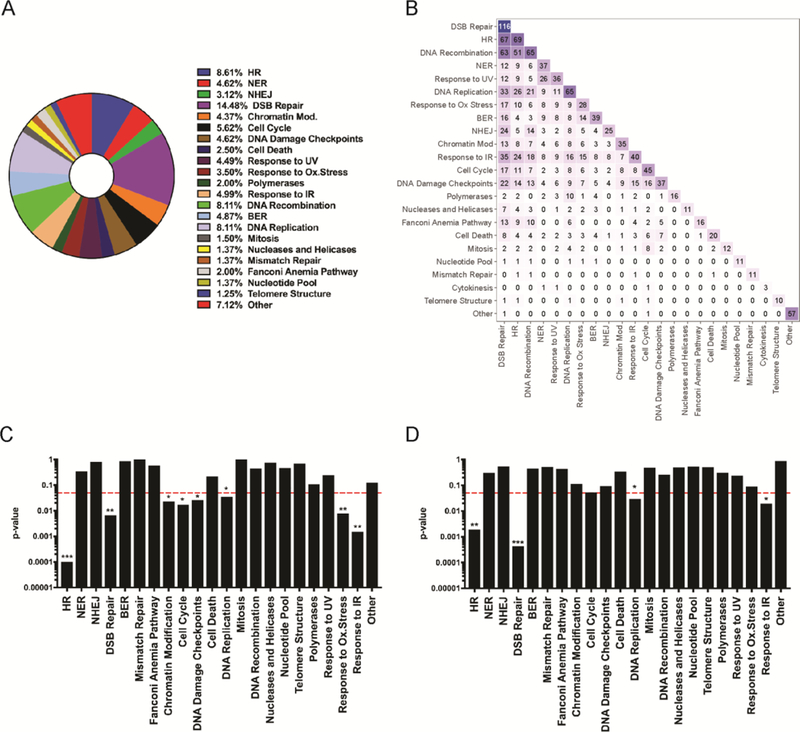
(A) Fractional representation of genes included in 22 functional pathways that are represented by one or more genes targeted by our 320 DDR siRNA library. Genes present in more than one pathway were counted multiple times for these percentages. Percentages are not disambiguated for genes annotated to multiple pathways. (B) Heatmap summarizing the number of shared genes between annotated pathways. The diagonal represents the size of a given pathway annotation. Barplots depicting statistical significance (y-axis) of pathway enrichment testing by (C) Fisher’s exact test, and (D) bootstrapping with replacement with 1,000 iterations for the set of annotated pathways (x-axis). The dotted red line represents a p-value ≤ 0.05, threshold for significance.
The 23 sensitizing gene depletions did not belong to a single cellular pathway, and were not genomically altered in a way that directly contributed to formaldehyde sensitivity. Thus, we interrogated gene product interaction networks to identify system-level cellular processes that might mitigate formaldehyde sensitivity. Hierarchical clustering of relative cell viabilities across cell lines for varying doses of formaldehyde identified two main clusters. The high dose formaldehyde samples (doses 3 and 4) primarily formed one cluster and low dose samples (doses 1 and 2) formed another cluster (Figure 5A). Genes in cluster 1 and 2 appear to mediate formaldehyde sensitivity across all cell lines at high doses, except for the U-2 OS cell line. Clusters 3–6 contained genes that when depleted led to cell line-specific variation in formaldehyde sensitivity. Pathway enrichment analysis showed an enhancement of genes in clusters 1 and 2 in 3 of the same KEGG pathways: NER, HR, and BER. Each cluster also displayed unique pathway enrichments, e.g., mismatch repair in cluster 2, or cell cycle and basal transcription factors in cluster 6 (Figure 5B). This approach identifies a concordance across cell lines in the pathways, but not individual gene products, necessary to mitigate formaldehyde-induced proliferation suppression.
Figure 5. Relative cell proliferation reveals global and cell-line specific patterns of formaldehyde sensitivity.
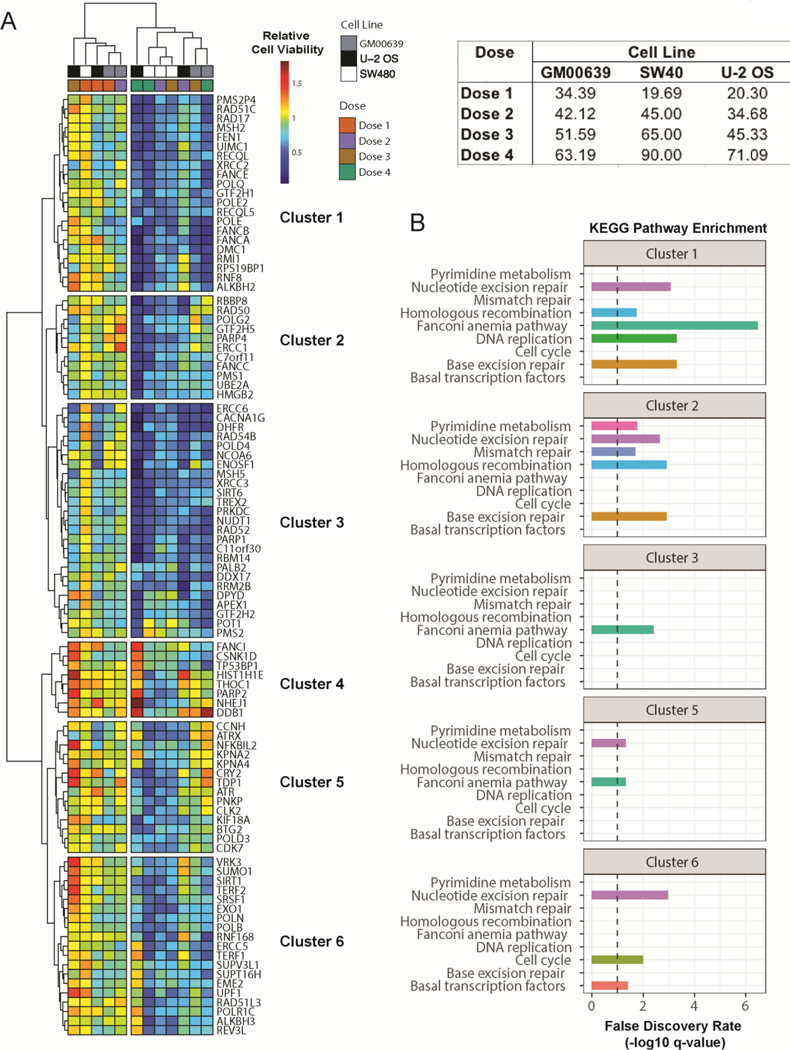
(A) Heatmap of relative cell proliferation suppression for a given formaldehyde dose across cell lines. Each map cell represents proliferation relative to an untreated control for a given gene and cell line. Hierarchical analyses of genes (rows) identified six clusters. Cell-line specific siRNA doses are shown in the table (top-right). (B) KEGG Pathway enrichment for gene clusters presented in A (FDR corrected p-values). Dotted lines represent an FDR threshold of 10%.
DNA damage repair pathways are highly conserved at both the functional and protein levels (56,57). Thus, we asked how well our human cell line results corresponded to our comparable essential gene yeast screen to identify genes that modulate cellular responses to formaldehyde (20). Among the 23 sensitizing genes we identified in all three human cell lines, 17 have a yeast homolog, including one which is essential and thus was not represented in our yeast deletion strain library. Of the 16 remaining genes from the human DDR screen, 9 also conferred formaldehyde sensitivity in our yeast deletion strain screen and mapped to comparable functional pathways: HR, DSB repair, DNA replication, DNA damage checkpoints, and cell cycle regulation (Table 1). Although deletion of RAD57 did not sensitize cells to formaldehyde in our yeast screen, a comparable screen done in diploid yeast demonstrated that RAD57 was required for formaldehyde tolerance (22). Together, these results identify several important functional pathways that strongly modulate formaldehyde toxicity across different functional pathways and eukaryotic species.
Table 1. Conserved genes and pathways that mitigate proliferation suppression following formaldehyde exposure.
Human and S. cerevisiae gene names, along with the yeast systemic names are provided. ‘Sensitivity’ indicates the level of sensitivity to formaldehyde noted in our previously published yeast screen (20), whereas ‘Description’ provides brief annotations of the human gene to specific DDR or DNA metabolic pathways (69,70)
| Human Gene | S. cerevisiae Gene | Systematic Name | Sensitivity | Description |
|---|---|---|---|---|
| CACNA1G | CCH1 | YGR217W | Not Sensitive | Calcium Voltage Channel |
| DMC1 | ECM30 | YLR436C | Sensitive | HR/DSB Repair/Mitosis |
| ERCC6 | RAD26 | YJR035W | Not Sensitive | NER/BER |
| FANCB | - | - | - | Fanconi Anemia Pathway |
| FANCE | - | - | - | Fanconi Anemia Pathway |
| FEN1 | RAD27 | YKL113C | Moderately | BER/DNA Replication |
| MPLKIP | CDC5P | YMR001C | Essential | Cell Cycle/ Mitosis |
| MSH2 | MSH2 | YOL090W | Moderately | Mismatch Repair |
| MSH5 | MSH5 | YDL154W | Not Sensitive | Mismatch Repair |
| NUDT1 | NPY1 | YGL067W | Not Sensitive | BER |
| PMS2P4 | PMS1 | YNL082W | Not Sensitive | Mismatch Repair |
| PRKDC | - | - | - | NHEJ/DSB Repair |
| RAD17 | RAD24 | YER173W | Sensitive | Cell Cycle/DNA Damage Checkpoint |
| RAD52 | RAD52 | YML032C | Sensitive | HR/DSB Repair/DNA Replication |
| RBBP8 | SAE2 | YGL175C | Not Sensitive | Cell Cycle/DNA Damage Checkpoint |
| RBM14 | PSP2 | YML017W | Sensitive | Mitochondrial mRNA Splicing |
| RECQL | SGS1 | YMR190C | Sensitive | HR/DSB Repair/Replication |
| RPS19BP1 | - | - | - | Ribosomal Subunit |
| RRM2B | RNR4 | YGR180C | Sensitive | DNA Damage Checkpoint/Replication |
| SIRT6 | SIR2 | YDL042C | Moderately | Chromatin Modification/DNA Replication |
| TREX2 | - | - | - | HR/DSB Repair |
| UIMC1 | - | - | - | HR/DSB Repair/Chromatin Modification |
| XRCC3 | RAD57 | YDR004W | Not Sensitive | HR/DSB Repair |
4. Discussion
Our analyses of genetic determinants of formaldehyde toxicity in human cells identified 23 genes and four functional pathways that modulate the response to chronic formaldehyde exposure in three unrelated human cell lines. We also identified 94 additional genes that conferred sensitivity when depleted in two cell lines, and an additional 118 genes that conferred sensitivity when depleted in a single cell line (Figure 2). The high fraction of siRNAs that modified the response of one or more cell lines to formaldehyde exposure likely reflects the design of the 320 DDR siRNA library, which was focused on genes associated with DNA damage repair and related processes such as replication and mitotic cell division (54). Among these we identified four functional pathways that when perturbed were strongly sensitizing to formaldehyde: HR, DSB repair, DNA replication, and IR response. These pathways detect and respond to different types of macromolecular damage mediated by formaldehyde, and are functionally interrelated in part due to the sharing of key proteins (Figure 4, Table S4).
One surprise in our screen data was that the Fanconi anemia (FA) pathway was not identified as a consistent contributor to mitigation of formaldehyde toxicity. However, siRNA-mediated depletion of 11 of the 22 FA complementation group genes included in the 320 DDR library did sensitize one or more cell lines to formaldehyde exposure (Figure 2, Table S1). This apparent lack of consistent Fanconi pathway-specific modulation across all three cell lines may reflect the redundant annotation of FANC proteins to additional pathways that mitigate formaldehyde toxicity, and the inclusion of only half of the currently recognized FANC genes as targets in our 320 DDR siRNA library (58)(Table S4). Genetic modifiers of FA pathway function may influence dose-dependent formaldehyde suppression of proliferation in individual cell lines. Two groups of genetic modifiers with the potential to modify FANC gene function include the alcohol and aldehyde dehydrogenase (ALDH and ADH) gene families that catabolize both endogenous and exogenous formaldehyde and other aldehydes. Individual members of these gene families can strongly sensitize cells to aldehyde exposure and can promote disease progression in FA patients (29,59,60).
As part of our screen, we also identified several genes that conferred resistance to formaldehyde exposure in a specific cell line. Depletion of ATM was strongly protective in SW480 (Supplementary Table S1). This is reminiscent of other studies in which attenuation of the DNA damage response where ATM plays a key role blunted the toxicity of DNA damaging agents including IR (61,62). We also identified a protective effect of DDB1 depletion in GM00639 and U-2 OS cells (Supplementary Table S1 and Supplementary Figure S1). DDB1 is the large subunit of the UV-damage DNA-binding protein complex (the UV-DDB complex) that participates in NER. It also participates in DCX (DDB1-CUL4-X-box) E3 ubiquitin-protein ligase complexes that may promote DNA repair by modifying individual proteins and chromatin (63). Of note, the depletion of DDB2, the protein heterodimer partner of DDB1, did not confer a similar protective effect (Supplementary Table S1), indicating that this protective effect may not depend directly on the UV-DDB complex.
In summary, our results demonstrate the ability of systematic genetic screens to identify functionally important pathways that modulate the response of human cells to formaldehyde exposure, and the use of these pathways to mitigate the effects of formaldehyde. Our results also highlight additional variables that need to be better defined before we can confidently extrapolate from cell-based screening data to tissue or organ-level effects. These toxicity determinants include: target tissue or organ cell types and their cycling or mitotic activity, tissue-level detoxification/quenching pathways, and the differential expression of genes that modify cellular responses following formaldehyde exposure. For example, the DNA damage response can vary greatly across tissue types and species as a function of tissue-specific gene expression and germline or somatic genetic variation (66–68). Our results also highlight the importance of performing studies in cell types relevant to the mode of exposure, and caution against making summary gene-specific statements in toxicity studies as gene expression levels, cell and tissue types and germline and acquired mutations can all modulate cellular responses. At the organismal level, both formaldehyde dose and route(s) of exposure will further strongly determine both the response and toxicity. Once better understood, these determinants of variability will provide a sound basis for performing genotoxic studies or to improve exposure and occupational hazard guidelines.
Supplementary Material
Highlights.
A DNA Damage Response siRNA screen identified cellular responses to formaldehyde.
23 DNA Damage Response genes were identified to mitigate formaldehyde sensitivity.
Four common pathways mitigate formaldehyde sensitivity in three human cell lines.
Genetic and phenotypic heterogeneity in cell lines affect response to formaldehyde.
Acknowledgements
The authors are grateful to members of the McCullough lab, R. Stephen Lloyd, and Lloyd lab members for helpful discussions, James Annis of the Quellos HTS Facility for assistance in conducting the siRNA screen, and Dr. Laura Heiser for valuable insights regarding the data analyses.
Funding
This work was supported by National Institutes of Health awards R01 CA106858 and a Diversity Supplement (PA-16–288) to R01CA106858 to AKM, and P01 CA077852 to NC and RJM Jr., and Department of Defense Bone Marrow Failure Program Award BM130174 to RJM Jr. Funding for open access charge: Oregon Health & Science University.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data availability
Raw exome sequencing data generated from the cell line GM00639 (CC1 clonal derivative) have been deposited in the Sequencing Read Archive SRA Accession: SRP131620.
Supplementary data
Supplementary Data are available online.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
References
- 1.Swenberg JA, Moeller BC, Lu K, Rager JE, Fry RC and Starr TB (2013) Formaldehyde carcinogenicity research: 30 years and counting for mode of action, epidemiology, and cancer risk assessment. Toxicol Pathol, 41, 181–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hauptmann M, Stewart PA, Lubin JH, Beane Freeman LE, Hornung RW, Herrick RF, Hoover RN, Fraumeni JF Jr., Blair A and Hayes RB (2009) Mortality from lymphohematopoietic malignancies and brain cancer among embalmers exposed to formaldehyde. J Natl Cancer Inst, 101, 1696–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hauptmann M, Lubin JH, Stewart PA, Hayes RB and Blair A (2004) Mortality from solid cancers among workers in formaldehyde industries. Am J Epidemiol, 159, 1117–1130. [DOI] [PubMed] [Google Scholar]
- 4.Hauptmann M, Lubin JH, Stewart PA, Hayes RB and Blair A (2003) Mortality from lymphohematopoietic malignancies among workers in formaldehyde industries. J Natl Cancer Inst, 95, 1615–1623. [DOI] [PubMed] [Google Scholar]
- 5.Heck HD, Casanova-Schmitz M, Dodd PB, Schachter EN, Witek TJ and Tosun T (1985) Formaldehyde (ch2o) concentrations in the blood of humans and fischer-344 rats exposed to ch2o under controlled conditions. Am Ind Hyg Assoc J, 46, 1–3. [DOI] [PubMed] [Google Scholar]
- 6.Luo W, Li H, Zhang Y and Ang CY (2001) Determination of formaldehyde in blood plasma by high-performance liquid chromatography with fluorescence detection. J Chromatogr B Biomed Sci Appl, 753, 253–257. [DOI] [PubMed] [Google Scholar]
- 7.Nagy K, Pollreisz F, Takats Z and Vekey K (2004) Atmospheric pressure chemical ionization mass spectrometry of aldehydes in biological matrices. Rapid Commun Mass Spectrom, 18, 2473–2478. [DOI] [PubMed] [Google Scholar]
- 8.Regazzoni LG, Grigoryan H, Ji Z, Chen X, Daniels SI, Huang D, Sanchez S, Tang N, Sille FC, Iavarone AT et al. (2017) Using lysine adducts of human serum albumin to investigate the disposition of exogenous formaldehyde in human blood. Toxicol Lett, 268, 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA and Shi Y (2004) Histone demethylation mediated by the nuclear amine oxidase homolog lsd1. Cell, 119, 941–953. [DOI] [PubMed] [Google Scholar]
- 10.Walport LJ, Hopkinson RJ and Schofield CJ (2012) Mechanisms of human histone and nucleic acid demethylases. Curr Opin Chem Biol, 16, 525–534. [DOI] [PubMed] [Google Scholar]
- 11.Kato S, Burke PJ, Fenick DJ, Taatjes DJ, Bierbaum VM and Koch TH (2000) Mass spectrometric measurement of formaldehyde generated in breast cancer cells upon treatment with anthracycline antitumor drugs. Chem Res Toxicol, 13, 509–516. [DOI] [PubMed] [Google Scholar]
- 12.Kato S, Burke PJ, Koch TH and Bierbaum VM (2001) Formaldehyde in human cancer cells: Detection by preconcentration-chemical ionization mass spectrometry. Anal Chem, 73, 2992–2997. [DOI] [PubMed] [Google Scholar]
- 13.Costa S, Carvalho S, Costa C, Coelho P, Silva S, Santos LS, Gaspar JF, Porto B, Laffon B and Teixeira JP (2015) Increased levels of chromosomal aberrations and DNA damage in a group of workers exposed to formaldehyde. Mutagenesis, 30, 463–473. [DOI] [PubMed] [Google Scholar]
- 14.National Toxicology Program. (2014) Formaldehyde. Rep Carcinog. Thirteen, 13. [Google Scholar]
- 15.National Institute of Environmental Health Sciences. (2014) Review of the formaldehyde assessment in the national toxicology program 12th report on carcinogens [PubMed]
- 16.U.S. Department of Health and Human Services. (2011) Report on carcinogens, twelfth edition. Natl. Toxicol. Program, 12, 53. [Google Scholar]
- 17.Zhang L, Steinmaus C, Eastmond DA, Xin XK and Smith MT (2009) Formaldehyde exposure and leukemia: A new meta-analysis and potential mechanisms. Mutat Res, 681, 150–168. [DOI] [PubMed] [Google Scholar]
- 18.Jensen RP, Luo W, Pankow JF, Strongin RM and Peyton DH (2015) Hidden formaldehyde in e-cigarette aerosols. N Engl J Med, 372, 392–394. [DOI] [PubMed] [Google Scholar]
- 19.Peteffi GP, Antunes MV, Carrer C, Valandro ET, Santos S, Glaeser J, Mattos L, da Silva LB and Linden R (2016) Environmental and biological monitoring of occupational formaldehyde exposure resulting from the use of products for hair straightening. Environ Sci Pollut Res Int, 23, 908–917. [DOI] [PubMed] [Google Scholar]
- 20.de Graaf B, Clore A and McCullough AK (2009) Cellular pathways for DNA repair and damage tolerance of formaldehyde-induced DNA-protein crosslinks. DNA Repair (Amst), 8, 1207–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kumari A, Lim YX, Newell AH, Olson SB and McCullough AK (2012) Formaldehyde-induced genome instability is suppressed by an xpf-dependent pathway. DNA Repair (Amst), 11, 236–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.North M, Gaytan BD, Romero C Jr., De La Rosa VY, Loguinov A, Smith MT, Zhang L and Vulpe CD (2016) Functional toxicogenomic profiling expands insight into modulators of formaldehyde toxicity in yeast. Front Genet, 7, 200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Quievryn G and Zhitkovich A (2000) Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis, 21, 1573–1580. [PubMed] [Google Scholar]
- 24.Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA et al. (2016) Mechanism and regulation of DNA-protein crosslink repair by the DNA-dependent metalloprotease sprtn. Mol Cell, 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stingele J, Schwarz MS, Bloemeke N, Wolf PG and Jentsch S (2014) A DNA-dependent protease involved in DNA-protein crosslink repair. Cell, 158, 327–338. [DOI] [PubMed] [Google Scholar]
- 26.Langevin F, Crossan GP, Rosado IV, Arends MJ and Patel KJ (2011) Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature, 475, 53–58. [DOI] [PubMed] [Google Scholar]
- 27.Ren X, Ji Z, McHale CM, Yuh J, Bersonda J, Tang M, Smith MT and Zhang L (2013) The impact of fancd2 deficiency on formaldehyde-induced toxicity in human lymphoblastoid cell lines. Arch Toxicol, 87, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rosado IV, Langevin F, Crossan GP, Takata M and Patel KJ (2011) Formaldehyde catabolism is essential in cells deficient for the fanconi anemia DNA-repair pathway. Nat Struct Mol Biol, 18, 1432–1434. [DOI] [PubMed] [Google Scholar]
- 29.Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L et al. (2015) Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol Cell, 60, 177–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kumari A, Owen N, Juarez E and McCullough AK (2015) Blm protein mitigates formaldehyde-induced genomic instability. DNA Repair (Amst), 28, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakano T, Katafuchi A, Matsubara M, Terato H, Tsuboi T, Masuda T, Tatsumoto T, Pack SP, Makino K, Croteau DL et al. (2009) Homologous recombination but not nucleotide excision repair plays a pivotal role in tolerance of DNA-protein cross-links in mammalian cells. J Biol Chem, 284, 27065–27076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chanet R, Izard C and Moustacchi E (1975) Genetic effects of formaldehyde in yeast. I. Influence of the growth stages on killing and recombination. Mutat Res, 33, 179–186. [DOI] [PubMed] [Google Scholar]
- 33.Chanet R, Izard C and Moustacchi E (1976) Genetic effects of formaldehyde in yeast. Ii. Influence of ploidly and of mutations affecting radiosensitivity on its lethal effect. Mutat Res, 35, 29–38. [DOI] [PubMed] [Google Scholar]
- 34.Chanet R and von Borstel RC (1979) Genetic effects of formaldehyde in yeast. Iii. Nuclear and cytoplasmic mutagenic effects. Mutat Res, 62, 239–253. [DOI] [PubMed] [Google Scholar]
- 35.Yasokawa D, Murata S, Iwahashi Y, Kitagawa E, Nakagawa R, Hashido T and Iwahashi H (2010) Toxicity of methanol and formaldehyde towards saccharomyces cerevisiae as assessed by DNA microarray analysis. Appl Biochem Biotechnol, 160, 1685–1698. [DOI] [PubMed] [Google Scholar]
- 36.Ho YC, Huang FM and Chang YC (2007) Cytotoxicity of formaldehyde on human osteoblastic cells is related to intracellular glutathione levels. J Biomed Mater Res B Appl Biomater, 83, 340–344. [DOI] [PubMed] [Google Scholar]
- 37.Khan M, Naqvi AH and Ahmad M (2015) Comparative study of the cytotoxic and genotoxic potentials of zinc oxide and titanium dioxide nanoparticles. Toxicol Rep, 2, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidorova JM, Kehrli K, Mao F and Monnat R Jr. (2013) Distinct functions of human recq helicases wrn and blm in replication fork recovery and progression after hydroxyurea-induced stalling. DNA Repair (Amst), 12, 128–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Swanson C, Saintigny Y, Emond MJ and Monnat RJ Jr. (2004) The werner syndrome protein has separable recombination and survival functions. DNA Repair (Amst), 3, 475–482. [DOI] [PubMed] [Google Scholar]
- 40.Leibovitz A, Stinson JC, McCombs WB 3rd, McCoy CE, Mazur KC and Mabry ND (1976) Classification of human colorectal adenocarcinoma cell lines. Cancer Res, 36, 4562–4569. [PubMed] [Google Scholar]
- 41.Ponten J and Saksela E (1967) Two established in vitro cell lines from human mesenchymal tumours. Int J Cancer, 2, 434–447. [DOI] [PubMed] [Google Scholar]
- 42.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D et al. (2012) The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature, 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM et al. (2017) Defining a cancer dependency map. Cell, 170, 564–576 e516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saintigny Y, Makienko K, Swanson C, Emond MJ and Monnat RJ Jr. (2002) Homologous recombination resolution defect in werner syndrome. Mol Cell Biol, 22, 6971–6978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H and Durbin R (2009) Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M et al. (2010) The genome analysis toolkit: A mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res, 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.DePristo MA, Banks E, Poplin R, Garimella KV, Maguire JR, Hartl C, Philippakis AA, del Angel G, Rivas MA, Hanna M et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat Genet, 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J et al. (2013) From fastq data to high confidence variant calls: The genome analysis toolkit best practices pipeline. Curr Protoc Bioinformatics, 43, 11 10 11–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang K, Li M and Hakonarson H (2010) Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res, 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA and Abecasis GR (2015) Genomes project consortium:A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB et al. (2016) Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536, 285–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M and Getz G (2015) Oncotator: Cancer variant annotation tool. Hum Mutat, 36, E2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim JW, Botvinnik OB, Abudayyeh O, Birger C, Rosenbluh J, Shrestha Y, Abazeed ME, Hammerman PS, DiCara D, Konieczkowski DJ et al. (2016) Characterizing genomic alterations in cancer by complementary functional associations. Nat Biotechnol, 34, 539–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kehrli K, Phelps M, Lazarchuk P, Chen E, Monnat R Jr. and Sidorova JM (2016) Class i histone deacetylase hdac1 and wrn recq helicase contribute additively to protect replication forks upon hydroxyurea-induced arrest. J Biol Chem, 291, 24487–24503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP et al. (2015) String v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res, 43, D447–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aravind L, Walker DR and Koonin EV (1999) Conserved domains in DNA repair proteins and evolution of repair systems. Nucleic Acids Res, 27, 1223–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Eisen JA and Hanawalt PC (1999) A phylogenomic study of DNA repair genes, proteins, and processes. Mutat Res, 435, 171–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mamrak NE, Shimamura A and Howlett NG (2017) Recent discoveries in the molecular pathogenesis of the inherited bone marrow failure syndrome fanconi anemia. Blood Rev, 31, 93–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hira A, Yabe H, Yoshida K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Nakamura J, Kojima S et al. (2013) Variant aldh2 is associated with accelerated progression of bone marrow failure in japanese fanconi anemia patients. Blood, 122, 3206–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Garaycoechea JI, Crossan GP, Langevin F, Daly M, Arends MJ and Patel KJ (2012) Genotoxic consequences of endogenous aldehydes on mouse haematopoietic stem cell function. Nature, 489, 571–575. [DOI] [PubMed] [Google Scholar]
- 61.Blackford AN and Jackson SP (2017) Atm, atr, and DNA-pk: The trinity at the heart of the DNA damage response. Mol Cell, 66, 801–817. [DOI] [PubMed] [Google Scholar]
- 62.Ceccaldi R, Parmar K, Mouly E, Delord M, Kim JM, Regairaz M, Pla M, Vasquez N, Zhang QS, Pondarre C et al. (2012) Bone marrow failure in fanconi anemia is triggered by an exacerbated p53/p21 DNA damage response that impairs hematopoietic stem and progenitor cells. Cell Stem Cell, 11, 36–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Iovine B, Iannella ML and Bevilacqua MA (2011) Damage-specific DNA binding protein 1 (ddb1): A protein with a wide range of functions. Int J Biochem Cell Biol, 43, 1664–1667. [DOI] [PubMed] [Google Scholar]
- 64.Fu W, Ligabue A, Rogers KJ, Akey JM and Monnat RJ Jr. (2017) Human recq helicase pathogenic variants, population variation and “missing” diseases. Hum Mutat, 38, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rogers KJ, Fu W, Akey JM and Monnat RJ Jr. (2014) Global and disease-associated genetic variation in the human fanconi anemia gene family. Hum Mol Genet, 23, 6815–6825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Blanpain C, Mohrin M, Sotiropoulou PA and Passegue E (2011) DNA-damage response in tissue-specific and cancer stem cells. Cell Stem Cell, 8, 16–29. [DOI] [PubMed] [Google Scholar]
- 67.Lans H and Vermeulen W (2015) Tissue specific response to DNA damage: C. Elegans as role model. DNA Repair (Amst), 32, 141–148. [DOI] [PubMed] [Google Scholar]
- 68.Moser R, Xu C, Kao M, Annis J, Lerma LA, Schaupp CM, Gurley KE, Jang IS, Biktasova A, Yarbrough WG et al. (2014) Functional kinomics identifies candidate therapeutic targets in head and neck cancer. Clin Cancer Res, 20, 4274–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT et al. (2000) Gene ontology: Tool for the unification of biology. The gene ontology consortium. Nat Genet, 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.The Gene Ontology, C. (2017) Expansion of the gene ontology knowledgebase and resources. Nucleic Acids Res, 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Exome sequencing data are deposited in the Sequence Read Archive (SRA), accession number SRP131620.