

Abstract
Background:
Apical hypertrophic cardiomyopathy (AHCM) is a relatively rare form of hypertrophic cardiomyopathy (HCM), originally described in Japan and later in the West. Limited information is available on this disease in China.
Hypothesis:
This study was designed to describe clinical features and prognoses of patients with AHCM in China.
Methods:
A retrospective study of 208 consecutive patients with AHCM examined at FuWai Hospital was performed. Clinical features, mortality, and cardiovascular morbidity were analyzed.
Results:
The 208 patients with AHCM represented 16.0% of all HCM patients. Among them, 64.4% were pure form and 35.6% were mixed form. Compared with the pure group, the mixed group had a significantly larger left atrial diameter and thicker apical thickness. One hundred ninety‐nine patients had a mean follow‐up of 8.0 ± 3.5 years, cardiovascular mortality was 1.0%, and annual cardiovascular mortality was 0.1%. The 2 cardiovascular deaths were both mixed form. The probability of survival was 97.0 ± 2% at 10 years. Of the patients, 17.8% had 1 or more cardiovascular events. The probability of survival without morbid events at 10 years was 77 ± 4%. Three independent predictors of cardiovascular morbidity were identified: age at diagnosis ≥60 years, left atrial diameter ≥36 mm, and New York Heart Association class ≥III at baseline.
Conclusions:
The prevalence of AHCM is relatively high, and it has a benign prognosis in China. However, 17.8% of patients may develop cardiovascular events. It is important to distinguish the 2 phenotypes of AHCM; the mixed form is less common but more serious than the pure form. © 2011 Wiley Periodicals, Inc.
This work was supported by a grant from the Special Foundation by the Ministry of National Science and Technology. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Introduction
Hypertrophic cardiomyopathy (HCM) is a primary disease of cardiac muscle with heterogeneous morphological, functional, and clinical features, although left ventricular (LV) hypertrophy is the most characteristic feature.1, 2 Apical hypertrophic cardiomyopathy (AHCM) is a relatively rare variant of HCM, in which the hypertrophic myocardium predominantly involves the apex below the papillary muscle. The typical features of AHCM consist of giant negative T waves (GNT) in electrocardiography (ECG), and a spade‐like configuration of the LV cavity at end‐diastole on left ventriculography.3, 4
Previous studies of AHCM in the Japanese population have indicated a benign prognosis.5, 6 It was also associated with a rare occurrence of cardiovascular death and events in Western populations.7, 8 It is not infrequent for AHCM to be associated with arrhythmia and apical infarction with aneurysm.9 However, little is known about the clinical features and prognosis of Chinese patients with AHCM. This study was undertaken to investigate the clinical features and prognostic results in 208 Chinese patients with AHCM.
Methods
Study Population
Patients who were diagnosed with AHCM by echocardiography or magnetic resonance imaging (MRI) between January 1994 and December 2009 at FuWai Hospital were consecutively enrolled in this study. A total of 208 patients were included, and their medical records, ECG, echocardiography, chest radiography, MRI, and angiography from the time of their diagnosis were reviewed retrospectively. The study was approved by the research ethics board of the FuWai Hospital. Data on survival and the clinical status of patients were obtained from a detailed interview or medical records. The primary clinical end points used in this study were death from any cause, heart transplantation, or major therapeutic interventions known to relieve symptoms such as hypertrophic myocardium myectomy and alcohol ablation.
Diagnostic Criteria and Definitions
The diagnosis of AHCM was based on the demonstration by echocardiography or MRI: an asymmetric hypertrophy of left ventricle confined predominantly to the LV apex with maximal apical wall thickness ≥15 mm, and a ratio of maximal apical to posterior wall thickness ≥1.5 in the absence of another cardiac or systemic disease capable of producing a similar degree of hypertrophy.10 AHCM was further subdivided into 2 groups: the pure form was defined as hypertrophy limited to the most distal region at the LV apex below the papillary muscle level, and the mixed form had a coexistent hypertrophy of other segments, such as interventricular septum, which is the most common. The greatest wall thickness was in the apical segment (Figure 1). The definition of cardiovascular death included sudden death as an unexpected sudden collapse in patients who previously had a somewhat stable clinical course, heart failure occurring in the context of cardiac decompensation and a progressive course with limiting symptoms, and stroke as a consequence of embolic events.11 Major cardiovascular events were defined as the occurrence of the following events resulting in hospitalization: syncope, arrhythmia (atrial fibrillation [AF] or atrial flutter, ventricular tachycardia, ventricular fibrillation), progressive heart failure, myocardial infarction unrelated to coronary artery disease, embolic stroke, and apical aneurysm (thin‐walled dyskinesis during systole and akinetic or hypokinetic during diastole of the apex). GNT was defined as a voltage of negative T wave ≥1 mV (≥10 mm) in any lead.
Figure 1.
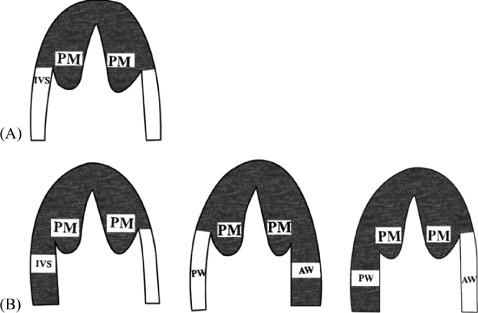
Morphological patterns of the 2 forms in apical hypertrophic cardiomyopathy. (A) Pure form: hypertrophy is limited to the most distal region at the left ventricular apex below the papillary muscle (PM) level. (B) Mixed form: apical hypertrophy extends to other segments, such as the interventricular septum (IVS), left ventricular anterior wall (AW), and left ventricular posterior wall (PW). The greatest wall thickness is in the apical segment.
Echocardiography
Echocardiographic studies were performed with commercially available instruments. Left atrial dimension at end‐systole and standard LV dimensions at end‐diastole and end‐systole, as well as LV wall thickness were obtained from 2‐dimensional images according to the standards of the American Society of Echocardiography.12 The maximal apical wall thickness was evaluated on apical 2‐chamber and apical 4‐chamber view at end‐diastole, and the mean values were measured. The LV outflow tract gradient was calculated from the continuous‐wave Doppler. Peak early‐diastolic mitral flow velocity (VE), peak end‐diastolic mitral flow velocity (VA), and VE/VA ratio were recorded.
Statistics
Continuous variables are expressed as means ± standard deviation, and differences between groups were analyzed by unpaired or paired t tests. Noncontinuous variables were expressed as proportions, and differences between groups were analyzed by χ 2 test or Fisher exact test. Cumulative survival estimates were calculated according to the Kaplan‐Meier method, and the time to the occurrence of fatality or a morbid event was calculated as the difference between the date of diagnosis and the event occurrence date. Risk factors for cardiac death and cardiac events and 95% confidence interval (CI) were determined by multivariate Cox proportional hazards regression models, in which each variable with a P value ≤0.05 in univariate analysis was entered into the mode. The clinical characteristics of the patients at baseline, including sex, age, complications, ECG measurements, and echocardiographic parameters were analyzed by univariate and multivariate Cox regression methods. For all tests, a P value <0.05 was considered a significant difference. Analyses were performed with SPSS software version 17.0 (SPSS, Inc., Chicago, IL)
Results
Baseline
Clinical Characteristics:
The 208 patients with a diagnosis of AHCM represented 16% of all HCM cases (1320) in our hospital. The entire population in this study is Chinese. Clinical variables at baseline are presented in Table 1. The prevalence rate of males is higher than females. There were no differences in age at diagnosis, symptoms, pure or mixed form of AHCM, or the apical thickness between male and female patients. Most patients were symptomatic at the first presentation, and 92.8% of patients were mildly symptomatic (functional New York Heart Association [NYHA] class I/II). The main symptoms were chest discomfort (91.8%), including chest tightness and chest pain, palpitations (30.8%), dyspnea (10.6%), dizziness (9.6%), and presyncope/syncope (7.2%). Although 39.4% of patients had a history of hypertension, blood pressure was well controlled and the mean value was 132 ± 13.5/80 ± 9.8 mm Hg; 22.2% were associated with hyperlipidemia, 15.9% were associated with coronary atherosclerotic heart disease, and 8.7% were associated with diabetes. The management of AHCM patients was mainly based on medication: β‐blocker and calcium channel blocker combination therapy (54.8%), β‐blocker monotherapy (29.8%), and calcium channel blocker monotherapy (10.6%). There were 4.3% of patients who were without medication because of no symptoms or mild symptoms.
Table 1.
Characteristics of the Study Population According to the Pure Form and Mixed Form of Apical Hypertrophic Cardiomyopathy at Baseline
| Features | All Patients (n = 208) | Pure Form (n = 134) | Mixed Form (n = 74) | P Value |
|---|---|---|---|---|
| Male, no. (%) | 153 (73.6) | 97 (72.4) | 56 (75.7) | NS |
| Age at diagnosis, y | 52.2 ± 12.4 | 53.7 ± 11.6 | 49.6 ± 13.4 | 0.020a |
| Family history of HCM, no. (%) | 29 (13.9) | 11 (8.2%) | 18 (24.3%) | 0.013a |
| Hypertension, no. (%) | 82 (39.4) | 58 (43.3) | 24 (32.4) | NS |
| Heart rate, times/min | 69.9 ± 9.7 | 70.0 ± 9.6 | 69.8 ± 10.0 | NS |
| NYHA class ≥III, no. (%) | 15 (7.2) | 4 (3.0) | 11 (14.9) | 0.003a |
| ECG, no. (%) | ||||
| T‐wave inversion | 190 (91.3) | 131 (97.8) | 59 (79.7) | NS |
| GNT | 60 (28.8) | 42 (32.1) | 18 (24.3) | NS |
| LVHV | 125 (60.1) | 84 (62.7) | 41 (55.4) | NS |
| Q waves | 16 (7.7) | 9 (6.7) | 7 (9.5) | NS |
| Echocardiography | ||||
| LVATmax, mm | 40 | 40 | 37 | |
| LVAT, mm | 19.9 ± 4.3 | 19.2 ± 4.1 | 21.3 ± 4.3 | <0.001a |
| IVST, mm | 14.0 ± 6.4 | 11.6 ± 4.6 | 18.4 ± 7.1 | <0.001a |
| LVPWT, mm | 9.9 ± 1.5 | 9.7 ± 1.5 | 10.3 ± 1.4 | 0.001a |
| Ratio LVAT/LVPWT | 2.1 ± 0.8 | 2.03 ± 0.53 | 2.21 ± 1.16 | NS |
| LAd, mm | 37.3 ± 6.0 | 36.5 ± 5.3 | 38.7 ± 6.7 | 0.010a |
| LVEDd, mm | 47.8 ± 5.2 | 48.7 ± 5.1 | 46.3 ± 5.1 | 0.001a |
| LVESd, mm | 30.0 ± 4.7 | 30.4 ± 4.7 | 29.3 ± 4.7 | NS |
| LVEF, % | 66.4 ± 9.3 | 66.6 ± 9.6 | 66.01 ± 8.9 | NS |
| VE, m/s | 0.72 ± 0.19 | 0.71 ± 0.19 | 0.72 ± 0.19 | NS |
| VA, m/s | 0.84 ± 0.23 | 0.82 ± 0.24 | 0.88 ± 0.20 | NS |
| Ratio, VE/VA | 0.91 ± 0.37 | 0.93 ± 0.37 | 0.87 ± 0.36 | NS |
Abbreviations: ECG, electrocardiogram; GNT, giant negative T waves; HCM, hypertrophic cardiomyopathy; IVST, interventricular septum thickness; LAd, left atrial end‐systolic diameter; LVAT, left ventricular apical thickness; LVEDd, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVESd, left ventricular end‐systolic diameter; LVHV, left ventricular high voltage; LVPWT, left ventricular posterior wall thickness; NS, denotes not significant, comparing pure form with mixed form; NYHA, New York Heart Association; VA, peak end‐diastolic mitral flow velocity; VE, peak early‐diastolic mitral flow velocity.
Data are presented as number (%) or mean ± standard deviation.
P < 0.05.
Electrocardiography
In the analysis of initial ECG recordings of all 208 patients, 190 patients (91.3%) presented with T‐wave inversion (including GNT), GNT was observed in 60 patients (28.8%), 125 patients (60.1%) presented with LV high voltage, and 16 patients (7.7%) had Q waves. The proportion of male patients with GNT was higher than that of female patients (P < 0.001). No significant correlation was found between the GNT and the apical thickness, and there was no difference in the diagnosed age or history of hypertension between patients with and without GNT.
Echocardiography, Magnetic Resonance Imaging, and Angiography
All patients received echocardiography at baseline. The echocardiography variables at baseline are presented in Table 1. Only 1 patient, with a mixed form of AHCM, had a mid‐cavity pressure gradient of 32 mm Hg during systole. There were 134 patients (64.4%) with the pure form of AHCM and 35.6% with the mixed form. The clinical characteristics of patients with the pure form and mixed form are summarized in Table 1. The maximum apical thickness was 40 mm in the mixed group and 37 mm in the pure group. There were more patients with a family history of HCM in the mixed group (24.3%) than in the pure group (8.2%). Compared with the pure group, the mixed group demonstrated a significantly worse functional NYHA class, larger left atrial diameter, and smaller LV end‐diastolic diameter (P < 0.05) at baseline. There were no significant differences in gender, history of hypertension, or comorbidities between the 2 forms of AHCM.
One hundred thirty patients had an MRI. There were 31.7% of patients who were not diagnosed with apical hypertrophy by echocardiography initially but were confirmed by MRI eventually. When repeated to look specifically for AHCM, echocardiography confirmed the diagnosis in all of these patients. At baseline, 83 patients received selective coronary and LV angiography, and the spade‐like configuration of the LV cavity at end‐diastole was identified in 27 patients (32.5%). There were 15 patients (18.1%) who presented with myocardial bridging of the left anterior descending coronary, and none had apical aneurysms.
Follow‐Up
One hundred ninety‐one patients (91.8%) were followed up, and the remaining 17 patients (8.2%) were lost, 11 in the pure form and 6 in the mixed form. However, there was no significant difference in the loss rate between the 2 forms, and all 17 patients were mildly symptomatic (functional NYHA class I/II) at baseline. The main reason for loss was an incorrect address and telephone number in the record. The mean follow‐up from the time of diagnosis was 8.0 ± 3.5 years.
Clinical Outcome
Three patients died during the follow‐up period; the overall mortality rate in this study was 1.6%. There was 1 non–cardiac‐related death and 2 cardiovascular deaths, 1 was sudden death and the other caused by heart failure, which was consistent with a cardiovascular mortality of 1.0% and annual cardiovascular mortality of 0.1%. The 2 cardiovascular deaths were both the mixed form of AHCM. The probability of survival at 10 years is 97 ± 2%.
Thirty‐four patients (17.8%) had 1 or more morbid cardiovascular events, of whom 19 (9.9%) had AF, 10 (5.3%) had presyncope/syncope, 5 (2.6%) had ventricular tachycardia, 4 (1.9%) had progressive heart failure with enlarged LV (LV end‐diastolic diameter ≥55 mm), 3 (1.6%) had embolic stroke, 2 (1.0%) had apical myocardial infarction without coronary artery disease, and only 1 patient underwent myectomy because of a progressive obstruction. None had apical aneurysm during follow‐up. The probabilities of event‐free survival at 5 and 10 years were 86 ± 3% and 77 ± 4%, respectively. The multivariate Cox regression analysis identified 3 independent predictors of cardiovascular events: age at diagnosis ≥60 years with a hazard ratio of 2.24 (95% CI: 1.14–8.12, P = 0.020), left atria diameter ≥36 mm with a hazard ratio of 3.14 (95% CI: 1.22–8.12, P = 0.017), NYHA class ≥III with a hazard ratio of 4.86 (95% CI: 1.95–12.12, P = 0.017). There was no significant association between morbid event and gender, history of hypertension, apical wall thickness, GNT, or comorbidities. There was no significant difference in cardiovascular morbidity between the pure and mixed form of AHCM.
Nineteen patients (9.9%) developed AF during follow‐up. A multivariate Cox regression analysis identified 2 predictors of AF: age at diagnosis ≥60 years with a hazard ratio of 6.35 (95% CI: 2.27–17.79, P < 0.001) and left atria diameter ≥36 mm with a hazard ratio of 6.35 (95% CI: 1.34–17.01, P = 0.016). Among the 3 patients with embolic stroke, 2 had AF without anticoagulation.
Echocardiography
The changes in echocardiographic variables are summarized in Table 2. Sixty patients received a second echocardiography examination during a mean follow‐up of 6.5 ± 2.8 years (range, 1.1–13.8 years). There were significant increases in LV end‐systolic diameter (from 37.7 ± 5.5 mm to 39.3 ± 5.0 mm) and left atrial diameter (from 29.9 ± 4.7 mm to 31.4 ± 4.7 mm) in the examined patients.
Table 2.
Changes in Echocardiographic Variables in 60 Patients With Apical Hypertrophic Cardiomyopathy
| Echocardiographic Variable | Baseline | Follow‐Up |
|---|---|---|
| Index of cavity diameter and wall thickness, mm | ||
| LAd | 37.7 ± 5.5 | 39.3 ± 5.0a |
| LVEDd | 48.1 ± 5.1 | 48.8 ± 5.4 |
| LVESd | 29.9 ± 4.7 | 31.4 ± 4.7a |
| LVAT | 18.9 ± 3.4 | 18.6 ± 3.9 |
| IVST | 13.2 ± 3.6 | 13.2 ± 5.1 |
| LVPWT | 10.5 ± 1.6 | 10.7 ± 2.0 |
| Ratio LVAT/LVPWT | 1.86 ± 0.45 | 1.78 ± 0.52 |
| Index of LV's function | ||
| LVEF, % | 67.3 ± 9.1 | 64.6 ± 7.0 |
| VE, m/s | 0.71 ± 0.16 | 0.73 ± 0.18 |
| VA, m/s | 0.83 ± 0.23 | 0.87 ± 0.19 |
| Ratio VE/VA | 0.92 ± 0.33 | 0.85 ± 0.20 |
Data are presented as mean ± SD.
P < 0.05 compared with baseline value in the follow‐up period group by paired t tests.
Abbreviations: IVST, interventricular septum thickness; LAd, left atrial end‐systolic diameter; LV, left ventricular; LVAT, left ventricular apical thickness; LVEDd, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVESd, left ventricular end‐systolic diameter; LVPWT, left ventricular posterior wall thickness; VA, peak end‐diastolic mitral flow velocity; VE, peak early‐diastolic mitral flow velocity.
Discussion
AHCM was originally described in Japan, and there were also some case reports and a few studies about it in China; however, the sample sizes were small and diagnosis criteria were not standardized. In the present study, we characterized clinical features and prognosis in the largest reported Chinese‐patient population with AHCM. These data are likely to influence the diagnosis and management of patients with AHCM.
Clinical Presentation
In this Chinese cohort study, AHCM represented 16% of the entire HCM population, similar to that in Japan (13%–25%)13, 14 and higher than that in Western countries (1%–11%).15 AHCM is more common in Taiwan, with reported cases in 24.5% of all HCM.16 So the incidence of AHCM seems to be higher in Asian countries than Western countries.
GNT in ECG and a spade‐like configuration of the LV cavity at end‐diastole on left ventriculography are typical features of AHCM, although in the present study only 28.8% of patients were found with GNT and 32.5% with the spade‐shaped feature, which may be due to the fact that hypertrophic myocardium is more diffused or limited in a small location. There was no association between GNT and the degree of apical wall thickness, type of AHCM, or morbidity. GNT patterns can disappear,17 and other diseases can also present GNT in ECG, such as raised intracranial pressure, severe myocardial ischemia, and post‐tachycardia syndrome,18 therefore GNT is not the specificity feature of AHCM. Although the mechanism of GNT in AHCM patients remains uncertain, several possibilities have been suggested, including reversal of the sequence of repolarization secondary to severe hypertrophy, myocardial ischemia, and greater metabolic demands due to a more hyperdynamic left ventricle.19
Outcome and Prognosis
Our study showed a benign prognosis in Chinese patients with AHCM in cardiovascular and overall mortality rate. There were 2 cardiovascular deaths during follow‐up, and annual cardiovascular mortality was 0.1%. This was similar to that reported from Japan and the West7, 14 and lower than the annual mortality of 1.4% for the whole HCM group reported by Maron et al.11 In brief, the cardiovascular mortality rate of AHCM patients is low, and prognosis is very optimistic. The most frequent cardiovascular morbid event was AF; among the 3 patients with embolic stroke, 2 had occurrences of AF without anticoagulation. Therefore, the patient should actively use anticoagulants to prevent thrombosis when a complication of AF occurs. In this study, most patients were mildly symptomatic and without obstruction, therefore pharmacological therapy was the initial approach for relieving disabling symptoms. However, implantable cardioverter defibrillators, percutaneous alcohol ablation, and myectomy should be considered when patients experience serious outflow gradients (≥50 mm Hg), frequent presyncope/syncope, malignant arrhythmias, and refractory symptoms.20, 21, 22
Diagnosis
In our research, it is worth noting that echocardiography failed to initially diagnose AHCM in 31.7% of patients, in whom MRI clearly demonstrated hypertrophy of the apex. The possible reasons for missed diagnoses include narrow location of apical hypertrophic myocardium and inadequate visualization of the apex by 2‐dimensional echocardiography. Echocardiography is noninvasive, repeatable, and easy to operate, which remains the first‐line imaging technique for diagnosis of AHCM. Recently, contrast echocardiography has improved the definition of the endocardial border and is also useful in identifying AHCM.23, 24 MRI is emerging as a sensitive and noninvasive examination procedure, which allows better assessment of the degree and extent of apical hypertrophy in AHCM patients than echocardiography.25, 26 Therefore, in patients with suspicion of AHCM and inadequate echocardiography images, the use of contrast echocardiography or MRI should be considered.
Comparison of Pure‐ and Mixed‐Form AHCM
The hypertrophic myocardium is not only limited to the apex of LV, and about 35.6% of patients represented with a mixed form, similar to that reported by Lee et al (32.5%).16 However, the mixed form was more common in Western countries and represented 87.0% of total AHCM.10 Perhaps the location of hypertrophic myocardium is different between Asian and Western countries. The mixed group had a significantly higher ratio of proven familial HCM and worse NYHA class function. Also, the mixed group had a larger left atrial diameter and smaller LV end‐diastolic diameter, which is probably a result of impaired LV diastolic function due to severe hypertrophy.27, 28 There were no cardiovascular deaths in the pure group, whereas 2 cardiovascular deaths occurred in the mixed group during follow‐up. However, no significant differences were found in cardiovascular morbidity between the 2 forms of AHCM, similar to the report of Eriksson et al7; however, a previous investigation of phenotypic spectrum of AHCM has shown that cardiovascular morbidity was significantly higher in patients with the distal‐dominant form.27 The difference in cardiovascular morbidity may be related to the different definitions of mixed form and morbidity and the fact that their study population was older than ours.
Limitations
The major limitation in the present study was that genetic analysis was not performed. Some studies show that gene mutations, such as the troponin T, β‐myosin heavy chain, and α‐cardiac actin are associated with AHCM29, 30, 31; however, the proportion of genotype positive is lower in patients with AHCM than common HCM.32 Genome‐wide association studies and gene expression profiling are needed for a better understanding of the genetic background of the disease. Another potential limitation was the loss of follow‐up. However, the loss rate was similar in the 2 forms, and all of the 17 patients were mildly symptomatic (functional NYHA class I/II).
Conclusion
The prevalence of AHCM is relatively high, and it has a benign prognosis in China. However, 17.8% of patients may develop cardiovascular events, and age at diagnosis of ≥60 years, left atrial diameter ≥36 mm, and NYHA class ≥III at baseline are independent predictors of cardiovascular morbidity. It is important to clinically distinguish the 2 phenotypes of AHCM; the mixed form is less common but more serious. These data are likely to influence the diagnosis and management of patients with AHCM.
References
- 1. Elliott P, McKenna WJ. Hypertrophic cardiomyopathy. Lancet. 2004;363:1881–1891. [DOI] [PubMed] [Google Scholar]
- 2. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology Clinical Expert Consensus Document on Hypertrophic Cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. Eur Heart J. 2003;24:1965–1991. [DOI] [PubMed] [Google Scholar]
- 3. Sakamoto T, Tei C, Murayama M, et al. Giant T wave inversion as a manifestation of asymmetrical apical hypertrophy (AAH) of the left ventricle. Echocardiographic and ultrasono‐cardiotomographic study. Jpn Heart J. 1976;17:611–629. [DOI] [PubMed] [Google Scholar]
- 4. Yamaguchi H, Ishimura T, Nishiyama S, et al. Hypertrophic nonobstructive cardiomyopathy with giant negative T waves (apical hypertrophy): ventriculographic and echocardiographic features in 30 patients. Am J Cardiol. 1979;44:401–412. [DOI] [PubMed] [Google Scholar]
- 5. Sakamoto T, Amano K, Hada Y, et al. Asymmetric apical hypertrophy: ten years experience. Postgrad Med J. 1986;62: 567–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakamoto T, Suzuki J. Apical hypertrophic cardiomyopathy [in Japanese]. Nihon Rinsho. 2000;58:93–101. [PubMed] [Google Scholar]
- 7. Eriksson MJ, Sonnenberg B, Woo A, et al. Long‐term outcome in patients with apical hypertrophic cardiomyopathy. J Am Coll Cardiol. 2002;39:638–645. [DOI] [PubMed] [Google Scholar]
- 8. Partanen J, Kupari M, Heikkila J, et al. Left ventricular aneurysm associated with apical hypertrophic cardiomyopathy. Clin Cardiol. 1991;14:936–939. [DOI] [PubMed] [Google Scholar]
- 9. Furushima H, Chinushi M, Iijima K, et al. Ventricular tachyarrhythmia associated with hypertrophic cardiomyopathy: incidence, prognosis, and relation to type of hypertrophy. J Cardiovasc Electrophysiol. 2010;21:991–999. [DOI] [PubMed] [Google Scholar]
- 10. Louie EK, Maron BJ. Apical hypertrophic cardiomyopathy: clinical and two‐dimensional echocardiographic assessment. Ann Intern Med. 1987;106:663–670. [DOI] [PubMed] [Google Scholar]
- 11. Maron BJ, Olivotto I, Spirito P, et al. Epidemiology of hypertrophic cardiomyopathy‐related death: revisited in a large non‐referral‐based patient population. Circulation. 2000;102: 858–864. [DOI] [PubMed] [Google Scholar]
- 12. Cheitlin MD, Armstrong WF, Aurigemma GP, et al. ACC/AHA/ ASE 2003 guideline update for the clinical application of echocardiography—summary article: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (ACC/AHA/ASE Committee to Update the 1997 Guidelines for the Clinical Application of Echocardiography). J Am Coll Cardiol. 2003;42:954–970. [DOI] [PubMed] [Google Scholar]
- 13. Koga Y, Itaya K, Toshima H. Prognosis in hypertrophic cardiomyopathy. Am Heart J. 1984;108:351–359. [DOI] [PubMed] [Google Scholar]
- 14. Kitaoka H, Doi Y, Casey SA, et al. Comparison of prevalence of apical hypertrophic cardiomyopathy in Japan and the United States. Am J Cardiol. 2003;92:1183–1186. [DOI] [PubMed] [Google Scholar]
- 15. Maron BJ. Hypertrophic cardiomyopathy: a systematic review. JAMA. 2002;287:1308–1320. [DOI] [PubMed] [Google Scholar]
- 16. Lee CH, Liu PY, Lin LJ, et al. Clinical features and outcome of patients with apical hypertrophic cardiomyopathy in Taiwan. Cardiology. 2006;106:29–35. [DOI] [PubMed] [Google Scholar]
- 17. Koga Y, Katoh A, Matsuyama K, et al. Disappearance of giant negative T waves in patients with the Japanese form of apical hypertrophy. J Am Coll Cardiol. 1995;26:1672–1678. [DOI] [PubMed] [Google Scholar]
- 18. Pillarisetti J, Gupta K. Giant inverted T waves in the emergency department: case report and review of differential diagnoses. J Electrocardiol. 2010;43:40–42. [DOI] [PubMed] [Google Scholar]
- 19. Park SY, Park TH, Kim JH, et al. Relationship between giant negative T‐wave and severity of apical hypertrophy in patients with apical hypertrophic cardiomyopathy. Echocardiography. 2010;27: 770–776. [DOI] [PubMed] [Google Scholar]
- 20. Maron BJ. Contemporary insights and strategies for risk stratification and prevention of sudden death in hypertrophic cardiomyopathy. Circulation. 2010;121:445–456. [DOI] [PubMed] [Google Scholar]
- 21. Maron BJ, McKenna WJ, Danielson GK, et al. American College of Cardiology/European Society of Cardiology clinical expert consensus document on hypertrophic cardiomyopathy. A report of the American College of Cardiology Foundation Task Force on Clinical Expert Consensus Documents and the European Society of Cardiology Committee for Practice Guidelines. J Am Coll Cardiol. 2003;42:1687–1713. [DOI] [PubMed] [Google Scholar]
- 22. Schaff HV, Brown ML, Dearani JA, et al. Apical myectomy: a new surgical technique for management of severely symptomatic patients with apical hypertrophic cardiomyopathy. J Thorac Cardiovasc Surg. 2010;139:634–640. [DOI] [PubMed] [Google Scholar]
- 23. Acarturk E, Bozkurt A, Donmez Y. Apical hypertrophic cardiomyopathy: diagnosis with contrast‐enhanced echocardiography—a case report. Angiology. 2003;54:373–376. [DOI] [PubMed] [Google Scholar]
- 24. Moukarbel GV, Alam SE, Abchee AB. Contrast‐enhanced echocardiography for the diagnosis of apical hypertrophic cardiomyopathy. Echocardiography. 2005;22:831–833. [DOI] [PubMed] [Google Scholar]
- 25. Moon JC, Fisher NG, McKenna WJ, et al. Detection of apical hypertrophic cardiomyopathy by cardiovascular magnetic resonance in patients with non‐diagnostic echocardiography. Heart. 2004;90:645–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ates M, Kwong RY, Lipton MJ, et al. Apical hypertrophic cardiomyopathy: diagnosed by cardiac magnetic resonance imaging. Tex Heart Inst J. 2006;33:408–409. [PMC free article] [PubMed] [Google Scholar]
- 27. Kubo T, Kitaoka H, Okawa M, et al. Clinical profiles of hypertrophic cardiomyopathy with apical phenotype—comparison of pure‐apical form and distal‐dominant form. Circ J. 2009;73: 2330–2336. [DOI] [PubMed] [Google Scholar]
- 28. Choi EY, Rim SJ, Ha JW, et al. Phenotypic spectrum and clinical characteristics of apical hypertrophic cardiomyopathy: multicenter echo‐Doppler study. Cardiology. 2008;110:53–61. [DOI] [PubMed] [Google Scholar]
- 29. Arad M, Penas‐Lado M, Monserrat L, et al. Gene mutations in apical hypertrophic cardiomyopathy. Circulation. 2005;112: 2805–2811. [DOI] [PubMed] [Google Scholar]
- 30. Gruner C, Care M, Siminovitch K, et al. Sarcomere protein gene mutations in patients with apical hypertrophic cardiomyopathy. Circ Cardiovasc Genet. 2011;4:288–295. [DOI] [PubMed] [Google Scholar]
- 31. Monserrat L, Hermida‐Prieto M, Fernandez X, et al. Mutation in the alpha‐cardiac actin gene associated with apical hypertrophic cardiomyopathy, left ventricular non‐compaction, and septal defects. Eur Heart J. 2007;28:1953–1961. [DOI] [PubMed] [Google Scholar]
- 32. Seidman CE, Seidman JG. Identifying sarcomere gene mutations in hypertrophic cardiomyopathy: a personal history. Circ Res. 2011; 108: 743–750. [DOI] [PMC free article] [PubMed] [Google Scholar]