

Abstract
Background
Genetic studies have shown that many slow cardiac myosin regulatory light chain 2 (MYL2) gene mutations can cause hypertrophic cardiomyopathy, which is one of the most common causes of heart failure (HF). But until now there has been no pathological or histological evidence that MYL2 may be associated with HF development. Recent microarray studies indicated that myosin heavy chain expression changed in the pathological process of HF. Because MYL2 is a regulatory component of myosin heavy polypeptide, the role of MYL2 protein in HF needs to be studied.
Hypothesis
The level of expression of MYL2 may change in the heart tissue of patients with chronic HF.
Methods
We collected 28 human right auricle samples, 16 from chronic HF patients and 12 from healthy control subjects. Immunohistochemistry was carried out to observe the tissue‐expression pattern of the MYL2 protein and Western blot methods were performed to quantify the relative MYL2 expression level.
Results
In chronic HF patients, the MYL2 protein level decreased significantly compared with normal controls (t test P < 0.05). Among the 16 HF patients, MYL2 expression in the severe HF group (New York Heart Association [NYHA] class III or IV) was even lower than that of the moderate HF group (NYHA class II) (t test P < 0.05).
Conclusions
MYL2 was down‐expressed in HF tissues, and our findings suggested that MYL2 may play a role in the development and progression of chronic HF. Copyright © 2008 Wiley Periodicals, Inc.
The first two authors contributed equally to this work. This study was supported by the National Natural Science Foundation of China (30800457, 30900803) and the Natural Science Program of Hubei Province (2009CDZ013). This study has no relationship with any kind of industry. The authors have no other funding, financial relationships, or conflicts of interest to disclose.
Introduction
Heart failure (HF) is a progressive disorder in which damage to the heart causes weakening of the cardiovascular system. It manifests by fluid congestion or inadequate blood flow to tissues. The prevalence of HF is 2% to 4% in the adult population and increases sharply to > 10% of people age > 65 years.1,2 HF remains one of the most common, costly, disabling, and deadly medical conditions encountered by a wide range of physicians and surgeons in both primary and secondary care.2 Common causes of HF include coronary heart disease, hypertension, valvular heart disease, and cardiomyopathies.1,2 Abnormal cardiac function and cardiomyopathy are major events in the development and progression of the disease.
Over the past few decades, genetic studies have laid a great deal of groundwork in mapping familial cardiomyopathies to chromosomal loci and genes, as well as defining the molecular pathways involved in HF.3, 4, 5 These findings have provided insights into how the structure and function of cardiomyocytes can be compromised by gene defects and how the mutated components of cardiomyocytes can lead to cardiomyopathies. Any of the initial events might trigger the molecular pathways that ultimately result in HF.
MYL2 belongs to the phosphorylatable, regulatory parts of myosin light chains. More than 10 MYL2 gene mutations have been reported to be associated with mid‐left ventricular chamber–type hypertrophic cardiomyopathy (HCM).6, 7, 8, 9, 10, 11, 12 Phosphorylation and Ca2+ binding to the myosin regulatory light chains play an important modulatory role in muscle contraction.13, 14, 15, 16 MYL2 was demonstrated to specifically bind to myosin heavy chains,17 which is one of the most important components of sarcomere filaments. Recent microarray global gene expression studies showed that myosin heavy polypeptides were involved in end‐stage cardiomyopathy.18,19 All these findings give us a clue that a change in MYL2 expression may play a role in the development of cardiac dysfunction and chronic HF. In this study, we measured MYL2 expression levels in myocardium tissue samples from several chronic HF patients and normal controls to test whether MYL2 is involved in the progression of HF, which is one of leading causes of death among cardiomyopathy patients. Furthermore, although a phenotype‐genotype correlation has been established between the MYL2 gene and cardiomyopathies, the underlying molecular mechanism is largely unknown. As MYL2 is a regulatory component of myosin filaments, the direct relationship between the MYL2 protein and HF remains to be studied.
Methods
Study Samples
Tissue samples taken from the right auricle of 28 individuals were studied. Individuals were divided into the non‐HF group (n = 12) and the HF group (n = 16). The 12 samples from nonfailing heart tissue were recruited from organ donors, who had no record of any cardiovascular disorder. The 16 samples from patients with HF were collected during heart valve replacement or heart transplantation surgery. The HF patients were diagnosed by echocardiography according to European Society of Cardiology guidelines.1 Signed informed consent was acquired from all participants or their guardians. This study complied with the Declaration of Helsinki and was approved by the local institution review board.
Immunohistochemistry
Each right auricle specimen was trimmed to measure 5 × 5 × 5 mm, then was fixed in formalin and embedded by paraffin. The paraffin blocks were sliced by a sectioning machine (Leica, Wetzlar, Germany) into 40 consecutive sections, each with a thickness of 5 µm. Of each 40 consecutive sections, 1 was chosen for immunohistochemistry (IHC) staining. For each tissue specimen, a total of 5 stained slides was analyzed. The sections were incubated in 3% hydrogen peroxide (Amresco, Solon, OH) at 37°C for 15 min to remove endogenous peroxidases. Then they were blocked with 3% bovine serum albumin phosphate‐buffered saline (PBS) at 37°C for 20 min. After the excess blocking solution was wiped off, mouse antihuman MYL2 antibody (1:400) (Santa Cruz Biotechnology, Santa Cruz, CA) was added to cover the sections and they were incubated at 4°C overnight. The primary antibody was washed off by PBS and the sections were incubated by goat antimouse interferon globulin (Santa Cruz) at 37°C for 20 min. Thereafter, the antibody binding was stained by the diaminobenzidine (DAB) method using a DAB Kit (Sigma‐Aldrich, St. Louis, MO). The nuclei were shown by counterstaining with hematoxylin (Sigma‐Aldrich) for 15 sec. For negative control, we used PBS instead of primary antibody.
Western Blot
Total tissue protein was extracted from the fixed sample by using the Qproteome FFPE Tissue Kit (Qiagen, Hilden, Germany) and was quantified with a BCA Protein Assay Kit (Thermo Scientific Pierce, Rockford, IL). From each sample, 15 µg protein was loaded and separated by 12% sodium dodecyl sulfate polyacrylamide gel electrophoresis, transferred to nitrocellulose membrane, blocked with 4% milk powder and washed with tris buffered saline Tween (TBST) at room temperature, whereas primary antibody mouse antihuman MYL2 (1:1000) (Santa Cruz) and beta‐actin (1:1000) (Santa Cruz) were allowed to incubate for 4 hours at room temperature, washed with TBST 3 times, then incubated with horseradish peroxidase–labeled secondary antibodies for another 1 hour at room temperature and washed with TBST 3 times. A maximum‐sensitivity ECL kit (Pierce) was used to visualize the protein bands. The molecular weights of MYL2 and beta‐actin were 20 kDa and 42 kDa, respectively.
Image Quantitative Measurement
IHC images were captured on a Nikon Eclipse 80i microscope system (Nikon, Tokyo, Japan). The MYL2 expression level was scaled with positive‐stained integrated optical density (IOD) of each image by Image Pro Plus software, version 6.0 (Media Cybernetics, Silver Spring, MD). The Western blot bands were captured on a ChemiDoc XRS+ Molecular Imager (Bio‐Rad Laboratories, Hercules, CA). The grayscale densities of bands were analyzed by Image Pro Plus software and were quantified by normalizing them with those of beta‐actin.
Statistical Analysis
Data are presented as mean ± SD. Results were assessed by Student t test to determine differences between mean values of the groups, using SPSS v.13.0 (SPSS Inc., Chicago, IL). P values were obtained by 2‐tailed tests and P < 0.05 was considered statistically significant.
Results
Clinical Characteristics of Heart Failure Patients in This Study
Table 1 shows the clinical characteristics of the 16 HF patients. Thirteen of them had valvular heart disease, 1 had dilated cardiomyopathy, and 2 had HCM. They were 8 male and 8 female. The average age of the patients was 42.7 ± 13.7 years. According to the New York Heart Association (NYHA) classification, we defined 7 of them as “moderate HF” (NYHA class II) and the other 9 as “severe HF” (NYHA classes III‐IV). For the 12 patients in the control group, the average age was 51.3 ± 16.8 years, and the male proportion was 50%. Medical records of the 12 control subjects showed that they had no history of any cardiovascular disorders at the time they became involved in this study.
Immunohistochemistry
IHC staining demonstrated MYL2 expression in all 28 right‐auricle tissue samples. In the control group (Figure 1C), the positive brown staining indicated that MYL2 protein was present in the cytoplasm of most myocardial cells, along the sarcomere filament distributions. But in the samples from the HF patients, the MYL2 expression level was lower than in the control group (Figure 1A and 1B). The proportion of positive‐stained cells was smaller, and the positive‐staining areas within cells were also reduced. Optical density analysis showed that in HF patients' samples, the average IOD (33 ± 13 × 106) was significantly lower than that of the control samples (102 ± 33 × 106), t test P = 1.2 × 10−5 (Figure 1D). The average IOD of the severe HF group (27 ± 10 × 106) was even lower than that of the moderate HF group (41 ± 13 × 106), t test P = 0.040 (Figure 1D).
Figure 1.
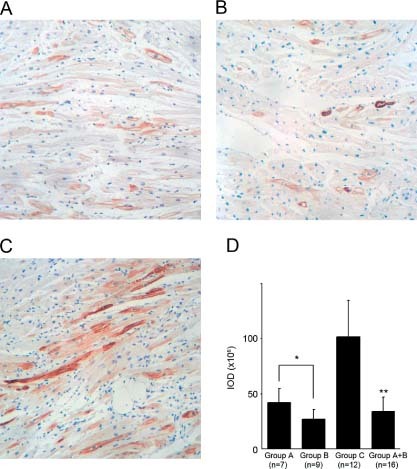
IHC staining. Immunostain of MYL2 in 3 representative samples. View fields were 20× magnified. The brown areas indicate MYL2‐positive staining. The blue areas indicate cell nuclei. (A) Moderate HF patient A10 (NYHA class II); (B) severe HF patient B10 (NYHA class IV); (C) normal control individual C2; (D) IOD measurement of IHC images for each group. The asterisk represents the statistical significance level of P < 0.05 and the double asterisk represents the statistical significance level of P < 0.01. Abbreviations: HF, heart failure; IHC, immunohistochemistry; IOD, integrated optical density; MYL2, myosin regulatory light chain 2; NYHA, New York Heart Association
Western Blot and MYL2 Quantification
Western blot assay was carried out to confirm the IHC findings and to quantify MYL2 expression. Beta‐actin was used as internal control. A specific MYL2 band (about 20 kDa) was observed under the beta‐actin band (about 42 kDa) (Figure 2A). MYL2 expression was weaker in HF patients A10 and B10 compared with control subject C2 (Figure 2A). We quantified the MYL2 expression level of each sample by calculating MYL2/beta‐actin ratios. We normalized the average MYL2 expression level of the control group (Group C) as 100%, and the mean ± SD of the control group was 100% ± 21%. For 16 HF patients, the relative MYL2 level was 69% ± 16%, which was significantly lower than the control group (t test P = 1.6 × 10−4, Figure 2B). MYL2 levels in the severe HF group (Group B) and moderate HF group (Group A) were 62% ± 15% and 79% ± 14%, respectively, and a statistical difference was found (t test P = 0.036, Figure 2B).
Figure 2.
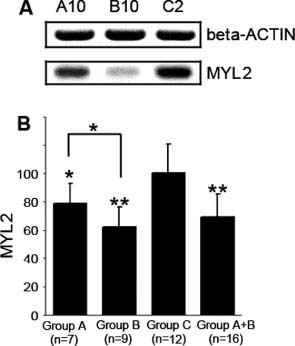
Western blot and quantification of MYL2 protein. MYL2 was down‐expressed in HF tissue. (A) Western blot result of 3 representative samples. A specific ∼20 kDa band was observed for MYL2 protein. For HF patients A10 and B10, MYL2 expression is decreased compared with the C2 normal control. (B) Quantification showed that in moderate HF (group A), severe HF (group B), and total HF patients (group A + B), the relative MYL2 levels were 80%, 60%, and 70%, respectively, of control group C. The asterisk represents the statistical significance level of P < 0.05 and the double asterisk represents the statistical significance level of P < 0.01. The y‐axis is the relative expression level (%). The average expression level of the control group was set as 100%. Abbreviations: HF, heart failure; MYL2, myosin regulatory light chain 2
Discussion
HF can be caused by a variety of cardiovascular diseases, such as cardiomyopathies, hypertension, valvular heart disease, and ischemic heart disease. The end stages of these diseases usually share the common features of cardiac dysfunction and a dilated heart chamber. This suggests that a shared signaling pathway or shared genes may contribute to the development of HF. With the advances of microarray technology, many genes have been associated with the pathophysiology of HF, including several myosin‐related genes encoding myosin light chain kinase 3,18,19 myosin heavy polypeptide 6, and heavy polypeptide 10.20 In this study, we measured MYL2 levels in different human HF tissues using Western blotting and IHC methods. The results showed that MYL2 was down‐regulated in HF tissues, and in severe HF tissues it was even lower. To our knowledge, our in vivo finding is the first report on the possible relationship between myosin regulatory light chain protein level and HF development.
HCM is one of the most common inherited cardiac disorders, with a prevalence in young adults of 0.2%.21 It is one of the most common causes of sudden death in young people.22 Lethal arrhythmia and HF are leading causes of death among HCM patients, increasing the mortality greatly. The correlation between the MYL2 gene and HCM has been previously established, in that > 10 MYL2 mutations have been identified in HCM patients. Many of these mutations affect structure of myosin regulatory light chains, Ca2+ ion binding, and phosophorylation,10 which would impair normal physiological functions of the myocardium and lead to HF. In addition to these genetic findings, our molecular and histological results provided additional evidence that the myosin regulatory light chain may be involved with the failure of myocardial cells. We also extended previously reported microarray findings, in that another myosin family member, MYL2, was associated with not only the onset, but also the development process of HF. Nevertheless, the internal molecular mechanism and pathological process of the role of MYL2 in HF remain unclear. The myosin regulatory light chain may possibly be a “result marker” for failing myocardium, which means it may be passively down‐regulated by other unknown factor in HF development. It could also be an “initial trigger” to lead myocardium dysfunction through an early stage of a certain pathway. More studies should be carried out to elucidate the underlying role of MYL2 in HF development.
Conclusion
We found that MYL2 was down‐expressed in human HF tissues, and the expression level was associated with the severity of the HF. Our findings may provide new insight into the molecular mechanism of HF and suggest a novel latent therapy target.
Acknowledgements
The authors thank all of the participants in this study for their generous donation of their precious tissue samples.
REFERENCES
- 1.Task Force for Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of European Society of Cardiology, Dickstein K, Cohen‐Solal A, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM) [published correction appears in Eur Heart J 2010;12:416]. Eur Heart J 2008; 29: 2388–2442. [DOI] [PubMed] [Google Scholar]
- 2. McMurray JJ, Pfeffer MA. Heart failure. Lancet 2005; 365: 1877–1889. [DOI] [PubMed] [Google Scholar]
- 3. Latronico MV, Elia L, Condorelli G, et al. Heart failure: targeting transcriptional and post‐transcriptional control mechanisms of hypertrophy for treatment. Int J Biochem Cell Biol 2008;40: 1643–1648. [DOI] [PubMed] [Google Scholar]
- 4. Hershberger RE, Cowan J, Morales A, et al. Progress with genetic cardiomyopathies: screening, counseling, and testing in dilated, hypertrophic, and arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Heart Fail 2009; 2: 253–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Oka T, Komuro I. Molecular mechanisms underlying the transition of cardiac hypertrophy to heart failure. Circ J 2008;72(Suppl A): A13–A16. [DOI] [PubMed] [Google Scholar]
- 6. Flavigny J, Richard P, Isnard R, et al. Identification of two novel mutations in the ventricular regulatory myosin light chain gene (MYL2) associated with familial and classical forms of hypertrophic cardiomyopathy. J Mol Med 1998; 76: 208–214. [DOI] [PubMed] [Google Scholar]
- 7. Kabaeva ZT, Perrot A, Wolter B, et al. Systematic analysis of the regulatory and essential myosin light chain genes: genetic variants and mutations in hypertrophic cardiomyopathy. Eur J Hum Genet 2002; 10: 741–748. [DOI] [PubMed] [Google Scholar]
- 8. Richard P, Charron P, Carrier L, et al. Hypertrophic cardiomyopathy: distribution of disease genes, spectrum of mutations, and implications for a molecular diagnosis strategy. Circulation 2003; 107: 2227–2232. [DOI] [PubMed] [Google Scholar]
- 9. Ingles J, Doolan A, Chiu C, et al. Compound and double mutations in patients with hypertrophic cardiomyopathy: implications for genetic testing and counselling. J Med Genet 2005; 42: e59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fokstuen S, Lyle R, Munoz A, et al. A DNA resequencing array for pathogenic mutation detection in hypertrophic cardiomyopathy. Hum Mutat 2008; 29: 879–885. [DOI] [PubMed] [Google Scholar]
- 11. Morita H, Rehm HL, Menesses A, et al. Shared genetic causes of cardiac hypertrophy in children and adults. N Engl J Med 2008; 358: 1899–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Møller DV, Andersen PS, Hedley P, et al. The role of sarcomere gene mutations in patients with idiopathic dilated cardiomyopathy. Eur J Hum Genet 2009; 17: 1241–1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szczesna D, Ghosh D, Li Q, et al. Familial hypertrophic cardiomyopathy mutations in the regulatory light chains of myosin affect their structure, Ca2+ binding, and phosphorylation. J Biol Chem 2001; 276: 7086–7092. [DOI] [PubMed] [Google Scholar]
- 14. Szczesna D, Zhao J, Jones M, et al. Phosphorylation of the regulatory light chains of myosin affects Ca2+ sensitivity of skeletal muscle contraction. J Appl Physiol 2002; 92: 1661–1670. [DOI] [PubMed] [Google Scholar]
- 15. Sherwood JJ, Waller GS, Warshaw DM, et al. A point mutation in the regulatory light chain reduces the step size of skeletal muscle myosin. Proc Natl Acad Sci U S A 2004; 101: 10973–10978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szczesna‐Cordary D, Guzman G, Zhao J, et al. The E22K mutation of myosin RLC that causes familial hypertrophic cardiomyopathy increases calcium sensitivity of force and ATPase in transgenic mice. J Cell Sci 2005; 118: 3675–3683. [DOI] [PubMed] [Google Scholar]
- 17. Wadgaonkar R, Shafiq S, Rajmanickam C, et al. Interaction of a conserved peptide domain in recombinant human ventricular myosin light chain‐2 with myosin heavy chain. Cell Mol Biol Res 1993; 39: 13–26. [PubMed] [Google Scholar]
- 18. Barth AS, Kuner R, Buness A, et al. Identification of a common gene expression signature in dilated cardiomyopathy across independent microarray studies. J Am Coll Cardiol 2006; 48: 1610–1617. [DOI] [PubMed] [Google Scholar]
- 19. Asakura M, Kitakaze M. Global gene expression profiling in the failing myocardium. Circ J 2009; 73: 1568–1576. [DOI] [PubMed] [Google Scholar]
- 20. Min KD, Asakura M, Liao Y, et al. Identification of genes related to heart failure using global gene expression profiling of human failing myocardium. Biochem Biophys Res Commun 2010; 393: 55–60. [DOI] [PubMed] [Google Scholar]
- 21. Maron BJ. Hypertrophic cardiomyopathy: A systemic review. JAMA 2002; 287: 1308–1320. [DOI] [PubMed] [Google Scholar]
- 22. Towbin JA. Hypertrophic cardiomyopathy. Pacing Clin Electrophysiol 2009; 32(Suppl 2): S23–S31. [DOI] [PubMed] [Google Scholar]