

Abstract
Subacute and chronic cardiac adaptations to marathon running may increase risk for sudden death. Herein, it is proposed that cardiac arrhythmogenic remodeling resulting from prolonged strenuous exertion may also have a systemic vascular component. Marathon running reduces coronary perfusion pressure and causes acute endothelial damage, possibly via altering concentrations of circulating angiogenic growth factors with novel vasoregulatory properties. Marathon runners have increased arterial stiffness and augmented pressure from wave reflections contributing to a widening of pulse pressure. Pulsatile hemodynamics may contribute to target organ damage. Moreover, each of these vascular maladaptations (increased arterial stiffness, augmented pressure from wave reflections, and widened pulse pressure) has been associated with atrial fibrillation and may provide a substrate for lethal arrhythmogenesis in the marathon runner. Clin. Cardiol. 2012 DOI: 10.1002/clc.21009
The author has no funding, financial relationships, or conflicts of interest to disclose.
This article first published online 28 November 2011. It has since changed. The term “coronary pulse pressure” has been changed to “coronary perfusion pressure.”
With the Persian fleet descending on the Greek city of Marathon in 490 BC, a young runner by the name of Phidippides was sent as a messenger to enlist the support of the Spartan army. Upon return to Marathon, his running skills were once again employed, this time to speed word of the recent Athenian victory over the Persians. Running over 250 km in a short period of time (3 days), Phidippides arrived in Athens and quickly succumbed to his exhaustion. With 1 final breath it is purported that he exalted “Nike!”—paying homage to the Greek goddess of victory—and died.
Phidippides cardiomyopathy has been recently proposed to account for the small percentage of highly exercise‐trained individuals who suffer sudden cardiac death (SCD) without clinical manifestations of coronary disease, valvular disease, or congenital cardiac disease such as hypertrophic cardiomyopathy. Given the wealth of information on myocardial damage and ventricular dysfunction with long‐distance running, it is extremely attractive to focus on the cardiac phenotype of this new cardiomyopathy. However, an equally important question exists: how healthy is the systemic vasculature of the long‐distance/marathon runner?
With advancing age, there is pervasive vascular dysfunction that manifests as stiffening of the great vessels (ie, central arteries such as the carotid and aorta) and augmented pressure from wave reflections. Increased pressure pulsatility stemming from arterial stiffness and wave reflections contributes to subclinical target organ damage (impaired vascular reactivity due to endothelial damage and increased left ventricular (LV) pressure effort/afterload contributing to cardiac hypertrophy). Moreover, each of the aforementioned vascular maladaptations is an independent predictor of future cardiovascular events.1., 2. Although habitual physical activity and moderate‐intensity aerobic/endurance exercise training are known to have favorable effects on systemic vascular structure and function (ie, reduced arterial stiffness, attenuated pressure from wave reflections, improved endothelial function), higher‐intensity exercise may paradoxically have the opposite effect. Thus, there appears to be a U curve with respect to exercise and vascular function.3 Inactivity is detrimental, but excessive activity may be equally detrimental.
Cross‐sectional studies note that marathon runners have increased central artery stiffness.3 It has been suggested that exercise training‐induced bradycardia results in a compensatory increase in stroke volume (commensurate with cardiac chamber enlargement) to maintain cardiac output.3 Increased load on the vasculature over time may result in elastin fatigue and fragmentation, contributing to a loss of arterial compliance. Marathon runners may also have increased pressure from wave reflections and increased central pulse pressure.4., 5. Interestingly, the reasons for this may also reside in exercise training‐induced bradycardia and increased stroke volume. With each cardiac ejection, a pressure wave is generated that traverses the aorta. Arriving at areas of impedance mismatch (arterial branch points, smaller arteries/arterioles), this pressure wave is reflected back toward the heart, adjoining a newly generated pressure wave. Thus, the contour of the central blood pressure waveform and overall pulse pressure is due to the combination of a forward and reflected wave. With a reduction in heart rate as is seen with chronic exercise training, systolic ejection duration is increased, altering pressure wave temporal associations such that the reflected pressure wave has more time to arrive during systole than during diastolic decay.6 This in turn will increase overall pulse pressure. Recently, Papaioannou et al have shown that the correlation of augmentation index (a proxy of wave reflection) with heart rate is higher in subjects with higher levels of aortic stiffness.7 That is, the same reduction in heart rate induces a greater increase in pressure from wave reflections in persons with stiffer vessels compared to those with more compliant vessels,7 of particular concern to the bradycardic marathon runner with stiff arteries.
Increased pulse pressure may also stem from a larger outgoing/forward pressure wave.8 Recently, it has been demonstrated that central pulse pressure is highly correlated with stroke volume in endurance exercise‐trained individuals.5 Changes in central pulse pressure may be load dependent. An increase in stroke volume secondary to transient increases in preload has been shown to augment forward wave pressure, increasing central pulse pressure.9 Thus, exercise training‐induced bradycardia, coupled with increased stroke volume from enhanced LV contractility and enlarged chamber volume, will increase both forward and reflected pressure waves, increasing pulse pressure in the marathon runner.
Increased arterial stiffness,10 augmented pressure from wave reflections,11 and increased pulse pressure12 have each been associated with atrial fibrillation, offering a mechanistic link among large artery dysfunction, pulsatile hemodynamics, and SCD (Figure 1). Increased arterial stiffness and pressure from wave reflections increases afterload, thereby promoting ventricular hypertrophy and impaired LV relaxation. This in turn may increase left atrial size. Left atrial size, a load‐sensitive manifestation of left atrial volumetric remodeling, has been proposed as a nonspecific end organ marker of chronic LV diastolic burden.13 Elevated left atrial size and impaired LV diastolic function may lead to fibrosis and electrical remodeling in the atrium, providing a substrate for the development of atrial fibrillation.12 Parenthetically, although chronic exercise training has been shown to increase systolic function, it does not appear to be able to modulate age‐associated reductions in diastolic relaxation properties,14 and some studies even note a possible reduction in early LV diastolic filling properties in older master athletes.15 Increased pressure from wave reflections subsequently increasing LV pressure effort/afterload in highly endurance‐trained athletes/marathon runners may offer insight into these observations.4
Figure 1.
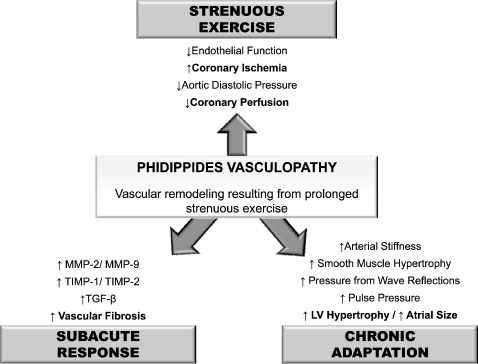
Acute and chronic changes in systemic vascular structure and function with long‐distance exercise/marathon training. Abbreviations: MMP, matrix metalloproteinase; TGF, transforming growth factor; TIMP, tissue inhibitor of matrix metalloprotease.
High‐intensity exercise increases arterial stiffness.16 Following completion of a marathon, there are large reductions in pressure from wave reflections, aortic pulse pressure, and aortic diastolic pressure.3 This is clinically relevant, as aortic diastolic pressure is a significant determinant of coronary perfusion pressure and subendocardial viability. Reduced central pulse pressure may be due to reduced forward wave genesis owing to reduced venous return and preload due to peripheral venous pooling. Reduced central pulse pressure may also be due to reduced pressure from wave reflections owing to peripheral vasodilation altering reflection sites (ie, impedance matching allowing the outgoing pressure wave to move further into the periphery before being reflected, increasing round‐trip travel time). Myocardial ischemia from reduced coronary and subendocardial perfusion is a prime arrhythmogenic substrate increasing the risk for SCD (Figure 1).
Running a marathon reduces femoral artery flow‐mediated dilation, suggesting endothelial damage.17 Endothelial dysfunction is also associated with atrial fibrillation18 and has been linked to SCD.19 The mechanisms responsible for endothelial dysfunction following a marathon have not been specifically examined. It is possible that increased pressure pulsatility related to arterial stiffness and wave reflection causes downstream endothelial damage. Studies have noted associations between arterial stiffness/pulse pressure and flow‐mediated dilation.20 Marathon running is also associated with an activated host response, and inflammation may acutely impair endothelial function.21 Finally, marathon running causes release of novel angiogenic peptides that have systemic endothelial regulatory actions, such as soluble fms‐like tyrosine kinase‐1 (sFlt‐1)22 and endoglin.23 Coronary ischemia has been shown to cause release of sFlt‐1 from coronary endothelial cells into the systemic circulation.24 sFlt‐1 impairs endothelial function25 by reducing nitric oxide (NO) bioavailability and increasing endothelial cell apoptosis,26 negatively regulating endothelial cell proliferation27 and sensitizing endothelial cells to proinflammatory factors.28 Endoglin is a transforming growth factor‐β coreceptor highly expressed in endothelial cells and is associated with LV filling pressures and LV dysfunction.29 Like sFlt‐1, endoglin has been shown to regulate the expression and activity of endothelial NO synthase,30 reducing NO production31 and making it an important modulator of endothelial vasomotor tone.32 Endothelial dysfunction in the acute setting may also contribute to coronary ischemia and subsequent arrhythmia.
Animal models have demonstrated that intense exercise increases factors that modulate extracellular matrix turnover (ie, matrix metalloproteinase (MMP)‐2, MMP‐9, tissue inhibitor of metalloproteinase (TIMP)‐1, TIMP‐2),33., 34. which has been seen in humans in vivo.35., 36. In addition to contributing to cardiac stiffness, these factors also increase arterial stiffness,37 and the process of cardiac fibrosis and vascular fibrosis is certainly intertwined.38 The subacute effects may persist upward of 2 weeks following exercise,36 and exercise‐induced muscle damage/inflammation is associated with subacute increases in arterial stiffness.39
In conclusion, akin to what has been postulated with cardiac maladaptation, it is proposed herein that repetitive, sustained elevations of cardiac output for several hours in predisposed individuals causes fatigue fracture of elastin fibers and stimulates resident macrophages, pericytes, and fibroblasts, resulting in the deposition of collagen and vascular fibrosis, and an increase in arterial stiffness. This, coupled with bradycardia‐induced augmented pressure from wave reflections and load‐dependent increases in forward wave pressure, will increase pressure pulsatility, contributing to downstream endothelial damage, target organ damage (ie, LV hypertrophy and atrial enlargement), coronary ischemia, atrial fibrillation, and possible SCD. Thus, cardiac arrhythmogenic remodeling resulting from prolonged strenuous exertion may have a vascular component. Phidippides cardiomyopathy may extend beyond to cardiac chamber walls and should rightfully be considered a cardiovascular disease.
References
- 1. Vlachopoulos C, Aznaouridis K, Stefanadis C. Prediction of cardiovascular events and all‐cause mortality with arterial stiffness: a systematic review and meta‐analysis. J Am Coll Cardiol. 2010; 55:1318–1327. [DOI] [PubMed] [Google Scholar]
- 2. Yeboah J, Folsom AR, Burke GL, et al. Predictive value of brachial flow‐mediated dilation for incident cardiovascular events in a population‐based study: the multi‐ethnic study of atherosclerosis. Circulation. 2009;120:502–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Vlachopoulos C, Kardara D, Anastasakis A, et al. Arterial stiffness and wave reflections in marathon runners. Am J Hypertens. 2010; 23:974–979. [DOI] [PubMed] [Google Scholar]
- 4. Heffernan KS, Jae SY, Tomayko E, et al. Influence of arterial wave reflection on carotid blood pressure and intima‐media thickness in older endurance trained men and women with pre‐hypertension. Clin Physiol Funct Imaging. 2009;29:193–200. [DOI] [PubMed] [Google Scholar]
- 5. Laurent P, Marenco P, Castagna O, et al. Differences in central systolic blood pressure and aortic stiffness between aerobically trained and sedentary individuals. J Am Soc Hypertens. 2011;5: 85–93. [DOI] [PubMed] [Google Scholar]
- 6. Wilkinson IB, Mohammad NH, Tyrrell S, et al. Heart rate dependency of pulse pressure amplification and arterial stiffness. Am J Hypertens. 2002;15:24–30. [DOI] [PubMed] [Google Scholar]
- 7. Papaioannou TG, Vlachopoulos CV, Alexopoulos NA, et al. The effect of heart rate on wave reflections may be determined by the level of aortic stiffness: clinical and technical implications. Am J Hypertens. 2008;21:334–340. [DOI] [PubMed] [Google Scholar]
- 8. Mitchell GF, Wang N, Palmisano JN, et al. Hemodynamic correlates of blood pressure across the adult age spectrum: noninvasive evaluation in the Framingham Heart Study. Circulation. 2010; 122:1379–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Heffernan KS, Sharman JE, Yoon ES, et al. Effect of increased preload on the synthesized aortic blood pressure waveform. J Appl Physiol. 2010;109:484–490. [DOI] [PubMed] [Google Scholar]
- 10. Drager LF, Bortolotto LA, Pedrosa RP, et al. Left atrial diameter is independently associated with arterial stiffness in patients with obstructive sleep apnea: potential implications for atrial fibrillation. Int J Cardiol. 2010;144:257–259. [DOI] [PubMed] [Google Scholar]
- 11. Doi M, Miyoshi T, Hirohata S, et al. Increased augmentation index of the radial pressure waveform in patients with paroxysmal atrial fibrillation. Cardiology. 2009;113:138–145. [DOI] [PubMed] [Google Scholar]
- 12. Mitchell GF, Vasan RS, Keyes MJ, et al. Pulse pressure and risk of new‐onset atrial fibrillation. JAMA. 2007;297:709–715. [DOI] [PubMed] [Google Scholar]
- 13. Abhayaratna WP, Seward JB, Appleton CP, et al. Left atrial size: physiologic determinants and clinical applications. J Am Coll Cardiol. 2006;47:2357–2363. [DOI] [PubMed] [Google Scholar]
- 14. Nottin S, Nguyen LD, Terbah M, et al. Long‐term endurance training does not prevent the age‐related decrease in left ventricular relaxation properties. Acta Physiol Scand. 2004;181: 209–215. [DOI] [PubMed] [Google Scholar]
- 15. Fleg JL, Shapiro EP, O'Connor F, et al. Left ventricular diastolic filling performance in older male athletes. JAMA. 1995;273: 1371–1375. [PubMed] [Google Scholar]
- 16. Rossow L, Fahs CA, Guerra M, et al. Acute effects of supramaximal exercise on carotid artery compliance and pulse pressure in young men and women. Eur J Appl Physiol. 2010;110: 729–737. [DOI] [PubMed] [Google Scholar]
- 17. Dawson EA, Whyte GP, Black MA, et al. Changes in vascular and cardiac function after prolonged strenuous exercise in humans. J Appl Physiol. 2008;105:1562–1568. [DOI] [PubMed] [Google Scholar]
- 18. Guazzi M, Arena R. Endothelial dysfunction and pathophysiological correlates in atrial fibrillation. Heart. 2009;95:102–106. [DOI] [PubMed] [Google Scholar]
- 19. Lerman A, Zeiher AM. Endothelial function: cardiac events. Circulation. 2005;111:363–368. [DOI] [PubMed] [Google Scholar]
- 20. Kuvin JT, Sidhu M, Patel AR, et al. Pulse pressure and peripheral arterial vasoreactivity. J Hum Hypertens. 2005;19:501–502. [DOI] [PubMed] [Google Scholar]
- 21. Adams V, Linke A, Breuckmann F, et al. Circulating progenitor cells decrease immediately after marathon race in advanced‐age marathon runners. Eur J Cardiovasc Prev Rehabil. 2008;15: 602–607. [DOI] [PubMed] [Google Scholar]
- 22. Weissgerber TL, Davies GA, Roberts JM, Modification of angiogenic factors by regular and acute exercise during pregnancy. J Appl Physiol. 2010;108:1217–1223. [DOI] [PubMed] [Google Scholar]
- 23. Tchou I, Margeli A, Tsironi M, et al. Growth‐differentiation factor‐15, endoglin and N‐terminal pro‐brain natriuretic peptide induction in athletes participating in an ultramarathon foot race. Biomarkers. 2009;14:418–422. [DOI] [PubMed] [Google Scholar]
- 24. Kapur NK, Heffernan KS, Yunis AA, et al. Elevated soluble fms‐like tyrosine kinase‐1 levels in acute coronary occlusion. Arterioscler Thromb Vasc Biol. 2011;31:443–450. [DOI] [PubMed] [Google Scholar]
- 25. Maynard SE, Min JY, Merchan J, et al. Excess placental soluble fms‐like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest. 2003;111:649–658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Di Marco GS, Reuter S, Hillebrand U, et al. The soluble VEGF receptor sFlt1 contributes to endothelial dysfunction in CKD. J Am Soc Nephrol. 2009;20:2235–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fong GH, Rossant J, Gertsenstein M, et al. Role of the Flt‐1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature. 1995;376:66–70. [DOI] [PubMed] [Google Scholar]
- 28. Cindrova‐Davies T, Sanders DA, Burton GJ, et al. Soluble FLT1 sensitizes endothelial cells to inflammatory cytokines by antagonizing VEGF receptor‐mediated signalling. Cardiovasc Res. 2011; 89:671–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kapur NK, Heffernan KS, Yunis AA, et al. Usefulness of soluble endoglin as a noninvasive measure of left ventricular filling pressure in heart failure. Am J Cardiol. 2010;106:1770–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Belik J, Jerkic M, McIntyre BA, et al. Age‐dependent endothelial nitric oxide synthase uncoupling in pulmonary arteries of endoglin heterozygous mice. Am J Physiol Lung Cell Mol Physiol. 2009;297: L1170–L1178. [DOI] [PubMed] [Google Scholar]
- 31. Sandrim VC, Palei AC, Metzger IF, et al. Nitric oxide formation is inversely related to serum levels of antiangiogenic factors soluble fms‐like tyrosine kinase‐1 and soluble endogline in preeclampsia. Hypertension. 2008;52:402–407. [DOI] [PubMed] [Google Scholar]
- 32. Jerkic M, Rivas‐Elena JV, Prieto M, et al. Endoglin regulates nitric oxide‐dependent vasodilatation. FASEB J. 2004;18:609–611. [DOI] [PubMed] [Google Scholar]
- 33. Koskinen SO, Ahtikoski AM, Komulainen J, et al. Short‐term effects of forced eccentric contractions on collagen synthesis and degradation in rat skeletal muscle. Pflugers Arch. 2002;444: 59–72. [DOI] [PubMed] [Google Scholar]
- 34. Koskinen SO, Wang W, Ahtikoski AM, et al. Acute exercise induced changes in rat skeletal muscle mRNAs and proteins regulating type IV collagen content. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1292–R1300. [DOI] [PubMed] [Google Scholar]
- 35. Koskinen SO, Hoyhtya M, Turpeenniemi‐Hujanen T, et al. Serum concentrations of collagen degrading enzymes and their inhibitors after downhill running. Scand J Med Sci Sports. 2001;11: 9–15. [DOI] [PubMed] [Google Scholar]
- 36. Mackey AL, Donnelly AE, Turpeenniemi‐Hujanen T, et al. Skeletal muscle collagen content in humans after high‐force eccentric contractions. J Appl Physiol. 2004;97:197–203. [DOI] [PubMed] [Google Scholar]
- 37. Tan J, Hua Q, Xing X, et al. Impact of the metalloproteinase‐9/ tissue inhibitor of metalloproteinase‐1 system on large arterial stiffness in patients with essential hypertension. Hypertens Res. 2007; 30:959–963. [DOI] [PubMed] [Google Scholar]
- 38. Nehme J, Mercier N, Labat C, et al. Differences between cardiac and arterial fibrosis and stiffness in aldosterone‐salt rats: effect of eplerenone. J Renin Angiotensin Aldosterone Syst. 2006;7: 31–39. [DOI] [PubMed] [Google Scholar]
- 39. Barnes JN, Trombold JR, Dhindsa M, et al. Arterial stiffening following eccentric exercise‐induced muscle damage. J Appl Physiol. 2010;109:1102–1108. [DOI] [PubMed] [Google Scholar]