

Abstract
Aspirin is integral in the primary and secondary prevention of coronary artery disease and acute coronary syndrome. Given the high clinical importance of aspirin in the management of coronary artery disease, much attention has been directed towards the concept of “aspirin resistance.” Unfortunately, the term aspirin resistance is ill‐defined in the literature, leading to a large variance in the reported prevalence of this phenomenon. In this review, the current understanding of aspirin resistance is discussed. Commonly used functional and diagnostic tests of platelet function, including their strengths and weakness, are reviewed. We next discuss several proposed mechanisms of aspirin resistance and special high‐risk groups at risk for aspirin treatment failure. We then discuss optimal dosing and diagnostic strategies for those populations at risk for aspirin resistance with a focus on tailored aspirin therapy for high‐risk groups. Finally, future topics of interest in the field of aspirin resistance are considered. Clin. Cardiol. 2012 doi: 10.1002/clc.22031
Jonathan Grinstein has no funding, financial relationships, or conflicts of interest to disclose. Christopher P. Cannon receives research grants/support from the following companies: Accumetrics, AstraZeneca, Essentialis, GlaxoSmithKline, Merck, Regeneron, Sanofi, and Takeda. He is also on the advisory board (but funds donated to charity) for Alnylam, Bristol‐Myers Squibb, and Pfizer. He is a Clinical Advisor, equity in Automedics Medical Systems.
Introduction
Aspirin is integral in the primary and secondary prevention of coronary artery disease and acute coronary syndrome (ACS). Aspirin works by irreversibly inhibiting cyclooxygenase‐1 (COX‐1) on platelets, thereby reducing the production of thromboxane A2, a potent vasoconstrictor and platelet activator.1 Among high‐risk patients, antiplatelet agents lead to a 26% reduction in nonfatal myocardial infarction (MI) or death from coronary heart disease2 and a 23% reduction in mortality in patients with ST‐elevation MIs.3 Given the high clinical importance of aspirin in the management of coronary artery disease, much attention has been directed toward the concept of “aspirin resistance.” Unfortunately, the term aspirin resistance is ill‐defined in the literature, leading to a large variance in the reported prevalence of this phenomenon. To complicate the topic further, the term aspirin resistance has been used in the literature both to describe the clinical recurrence of vascular events in patients taking aspirin, also known as aspirin treatment failure, as well as to describe the in vitro and ex vivo resistance of aspirin as determined by an array of platelet function tests.4 Depending on the definition of aspirin resistance used in different studies, the specific testing method used to assess platelet function, and the rate of noncompliance, 2% to 57% of patients have been reported to have suboptimal antiplatelet effects to aspirin therapy.5., 6., 7. Clinically, aspirin resistance is associated with a 3‐fold increase in the risk of death, MI, or cerebrovascular accident (CVA).8
Arguably, the most fundamental definition of aspirin resistance is the lower than normal antiplatelet response to standard doses of aspirin. Near complete inhibition of thromboxane A2 is needed for an effective antiplatelet response by aspirin. Platelet activation occurs with less than 5% of intrinsic thromboxane A2 generation ex vivo9 and when only 2.5% of a given platelet population is aspirin‐free.10 Fortunately, for the great majority of patients, standard doses of aspirin, even low‐dose aspirin, leads to significant and near complete inhibition of COX‐1 and thromboxane A2. Frelinger et al6 followed 700 consecutive aspirin‐treated patients undergoing cardiac catheterization and found that only 1.8% of patients had thromboxane B2 levels, a stable metabolite of thromboxane A2, greater than 10 ng/mL (∼5% of intrinsic activity, as validated above). Platelets from this population also had more arachidonic acid‐induced platelet activation as measured by P‐selectin surface positivity on flow cytometry. In this population of aspirin‐resistant patients, the addition of ex vivo aspirin reduced platelet activation to levels similar to those seen in patients with more complete inhibition of COX‐1, suggesting that the aspirin nonresponders may simply have been underdosed with aspirin. Interestingly, 75% of these aspirin‐resistant patients were receiving low‐dose (81 mg) aspirin,6 suggesting the dose of aspirin may be a factor.
Diagnostic Testing of Platelet Function
As mentioned in the Introduction, the large heterogeneity in experimental design and platelet function assays in the numerous studies investigating aspirin resistance has clouded our understanding of the prevalence and significance of this phenomenon. A comparison of 6 different platelet function assays in 201 patients with stable coronary artery disease receiving daily aspirin showed poor correlation among assays, with anywhere from 2.8% to 59.5% of patients being labeled as aspirin‐resistant depending on the assay used.11., 12. Platelet function assays utilize the tendency of platelets to change shape, aggregate, release metabolic products, or mobilize cell‐surface receptors upon activation.13 In a recent meta‐analysis of 20 studies investigating aspirin resistance, a total of 6 different platelet function assays were used, including studies reflecting both in vivo and ex vivo platelet activity.14 Commonly used platelet function assays, including their strength and limitations, are summarized below (Table 1).
Table 1.
Comparison of Clinical Platelet Function Assays as Compared to Light Transmission Aggregometry as the Standard
| Platelet Function Assay | Sensitivity (CI) | Specificity (CI) | Positive Predictive Value (CI) | Negative Predictive Value (CI) |
|---|---|---|---|---|
| PFA‐100 | 0.75 (0.69–0.81) | 0.40 (0.33–0.47) | 0.05 (0.02–0.08) | 0.97 (0.95–0.99) |
| VerifyNow Aspirin | 0.38 (0.31–0.45) | 0.95 (0.92–0.98) | 0.23 (0.17–0.29) | 0.97 (0.95–0.99) |
| Urinary dTxB2 | 0.13 (0.08–0.18) | 0.77 (0.71–0.83) | 0.02 (0–0.04) | 0.95 (0.92–0.98) |
Abbreviation: CI, confidence interval. Data from Lordkipanidze et al. Eur Heart J. 2007;14:1702–1708.
Cell‐Surface Markers
Flow Cytometric Markers of Platelet Activation:
The 2 most commonly used flow cytometric markers of platelet activation are those directed against the glycoprotein GPIIb–IIIa and the adhesion protein P‐selectin. The PAC1 monoclonal antibody binds to the fibrinogen target of activated GPIIb–IIIa, which is only exposed by activated platelets after a conformational change. This binding site is not present on resting platelets.15 Cross‐bridging of fibrinogen to GPIIb–IIIa is essential for platelet aggregation. P‐selectin is involved in cell adhesion of activated platelets to monocytes and neutrophils. P‐selectin is a component of the α granule of platelets and is mobilized to the platelet surface upon activation. Monoclonal antibodies directed against P‐selectin are only able to bind to activated, degranulated platelets.15 Flow cytometry has the advantage of directly measuring platelet activation as opposed to measuring a surrogate for activation. It is, however, expensive and more labor intensive than many of the other assays.
Platelet Release Products
Soluble P‐Selectin:
As mentioned above, P‐selectin is mobilized to the surface after platelet cell activation. Measurable levels of P‐selectin can be detected in the plasma with enzyme‐linked immunosorbent assay (ELISA) and correlate with platelet activation. Elevated levels are seen in patients with thrombotic disorders, peripheral arterial disease, and ischemic heart disease.16 Soluble P‐selectin is stable and can be stored for several months. However, this test has not been developed into a clinical assay.
Urine or Serum Thromboxane B2:
Arachidonic acid is converted to prostaglandins including thromboxane A2 in a reaction catalyzed by cyclooxygenase. As mentioned above, aspirin inhibits the activation of COX‐1 and subsequently the production of thromboxane A2. Thromboxane B2 is a stable metabolite of thromboxane A2 and is therefore more easily measured. 11‐Dehydrothromboxane B2 can be measured either from the urine or the serum by ELISA relatively inexpensively.17 Urine levels can be variable as they are dependent on both the rate and volume of urine collected. Levels are also associated with cardiovascular risk profiles and clinical events.18
Platelet Aggregation
Light Transmission Aggregometry:
In response to various stimuli, including adenosine diphosphate (ADP), arachidonic acid, and collagen, platelets will aggregate. Transmitted light increases after the aggregation of platelets suspended in plasma, allowing for an indirect measure of platelet function. This assay commonly isolates platelets from other constituents of platelet aggregation found in vivo, namely leukocytes and erythrocytes, although whole‐blood aggregometry may also be utilized. Analysis also occurs in the absence of blood flow and shear stress and therefore is less indicative of physiologic platelet activity when compared to other assays.19 Platelet aggregometry is also quite labor intensive and difficult to standardize across different laboratories.20
VerifyNow:
The VerifyNow system is a point of care system that allows for the rapid detection of platelet aggregation in whole blood. Three different VerifyNow systems are marketed depending on the antiplatelet drug: aspirin, P2Y12 antagonists, and GPIIb–IIIa antagonists. The VerifyNow aspirin system uses disposable cartridges containing an arachidonic acid agonist and fibrinogen‐coated beads. Whole blood is mixed by the movement of electromagnetically‐driven steel balls. Platelets not adequately inhibited by aspirin become activated by arachidonic acid and agglutinate out of the solution. Light absorbance through the sample is measured and an algorithm converts the value into aspirin reaction units (ARUs). An ARU value of >550 designates an aspirin nonresponder, as there is no evidence of an antiplatelet effect of aspirin that is distinguishable from baseline, drug‐free arachidonic acid–induced platelet aggregation.21 Clinically, aspirin resistance as measured using VerifyNow following percutaneous coronary intervention (PCI) is associated with a 3‐fold increase in the composite endpoint of cardiovascular death, MI, angina, and CVA.22 In a comparison of platelet function tests, VerifyNow was associated with the most reproducible test results.12
Platelet Function Analyzer–100:
The Platelet Function Analyzer (PFA)‐100 system measures platelet aggregation using disposable cartridges that simulate injured blood vessels. Whole blood is passed through a small orifice made of collagen and epinephrine or collagen and ADP‐coated membrane under high shear stress. The collagen and epinephrine cartridges are able to detect qualitative platelet defects including aspirin‐induced platelet dysfunction whereas the collagen and ADP cartridges are relatively ineffective at detecting aspirin inhibitory effects.23 Activated platelets aggregate to form a platelet plug. The time needed to close the aperture, or closure time (CT), is measured and used as a surrogate for platelet function.23 There is no agreed upon CT in the literature that defines aspirin resistance, making interpretation difficult. Nonetheless, aspirin resistance as measured using the PFA‐100 is associated with more vascular events than seen in aspirin responders.24
Mechanisms of Aspirin Resistance
There are several proposed mechanisms of aspirin resistance, and true aspirin resistance is likely multifactorial. Aspirin noncompliance clouds our analysis of true aspirin resistance as discussed above and unfortunately is a large contributor to aspirin treatment failure. The rate of aspirin noncompliance is difficult to measure in clinical trials. Schwartz et al25 followed 190 patients after an MI who were thought to be taking daily aspirin and found that 9% had a noninhibited aggregation response by light aggregometry. In these same patients, platelets were inhibited in all but 1 patient 2 hours after observed aspirin ingestion, suggesting that they were aspirin noncompliant.25 Von Pape et al26 found that in 212 patients with a history of MI, 18.4% had a suboptimal antiplatelet response as measured using PFA‐100. These patients underwent counseling and reinforcement and 1 week later, only 10.4% of patients had a suboptimal antiplatelet response. With 4 more weeks of reinforcement and an increase in aspirin dosing from 200 to 300 mg per day only 1.4% of patients were aspirin resistant.26
Even after accounting for noncompliance, a sufficient percentage of patients continue to have suboptimal antiplatelet effects on standard dosing of aspirin. Although the response to aspirin appears to be heritable in certain families with strong family history of coronary heart disease,27 studies investigating genetic polymorphism as an explanation for aspirin resistance have so far been conflicting.28 Interestingly, among high‐risk black and white families with strong family history of heart disease, aspirin response via COX‐1–independent pathways appears to be more heritable than response by COX‐1–dependent pathways.27
Platelet turnover is another mechanism postulated to contribute to aspirin resistance. Aspirin irreversibly inhibits COX‐1 and, as platelets are anucleate, COX‐1 remains inhibited for the 7‐ to 10‐day lifespan of a platelet. Newly formed platelets with intact cyclooxygenase activity can be detected as soon as 4 to 6 hours after aspirin ingestion.10 This platelet turnover is accelerated in certain clinical settings, including diabetes,29 after cardiac surgery,30 and after MI,31., 32. and has been associated with aspirin resistance. Interestingly, a recent analysis of patients with stable coronary artery disease taking once‐daily aspirin showed that up to 25% of patients had suboptimal platelet inhibition with 24‐hour aspirin dosing when analyzed with arachidonic acid‐mediated light transmission aggregometry.33 Diabetes, a condition known to be associated with increased platelet turnover, was correlated with this time‐dependent aspirin resistance. Supporting the theory that aspirin resistance is at least in part secondary to increased platelet turnover, Rocca et al34 examined a group of 100 diabetic patients on chronic aspirin therapy and found that among the group with the fastest recovery of platelet activity, twice‐daily aspirin therapy was associated with significantly more platelet inhibition than once‐daily therapy.
Interacting drugs, most notably nonsteroidal anti‐inflammatory drugs (NSAIDs) such as ibuprofen can reduce the antiplatelet effects of aspirin. Aspirin‐mediated platelet inhibition was reduced when ibuprofen was given 2 hours before aspirin but not when aspirin was administered before ibuprofen. Moreover, the administration of acetaminophen or the COX‐2 antagonist rofecoxib prior to aspirin did not interfere with aspirin's antiplatelet effect.35 It is hypothesized that ibuprofen and other NSAIDs act as competitive inhibitors to aspirin and prevent the ability of aspirin to reach its target on COX‐1.35
Emerging evidence suggests that a significant number of aspirin‐resistant patients are also resistant to P2Y12 antagonists such as clopidogrel. This dual‐antiplatelet resistance likely places these patients at a higher risk of thrombotic complications. Clopidogrel resistance is estimated to occur in 4% and 41% of patients.36., 37. Among patients undergoing elective PCI, in 1 cohort it was estimated that nearly 50% of aspirin‐resistant patients were also resistant to clopidogrel. In patients with dual antiplatelet resistance, there was nearly a 3‐fold increase in the incidence of myonecrosis following PCI compared to dual‐sensitive patients.36 Similarly, among patients who have developed stent thrombosis, 43% of patients were resistant to both aspirin and clopidogrel compared to 14% of controls.38 Aspirin‐resistant patients appear to have platelets that are more sensitive to ADP, which may partially explain the high prevalence of dual‐antiplatelet resistance in this population.39 Furthermore, arachidonic acid appears to be able to incompletely activate platelets through an ADP‐dependent and COX‐independent pathway in COX‐inhibited cells, suggesting some overlap in these pathways.6
High‐Risk Groups
The ideal dosing strategy for aspirin would involve prescribing the lowest possible dose of aspirin needed to achieve the desired antiplatelet effect and minimize side effects while at the same time identify those at high risk for aspirin resistance and modifying their dosing as needed to achieve effective platelet inhibition. Several groups of patients have been identified as being at a higher risk of aspirin resistance (Table 2).
Table 2.
Proposed Mechanisms of Aspirin Resistance in High‐Risk Groups
| Group | Proposed Mechanisms of Resistance |
|---|---|
| Diabetes mellitus | Increased platelet turnover; decreased aspirin‐mediated acetylation |
| Obesity | Increased baseline platelet reactivity; increased volume of distribution; prothrombotic effects of leptin |
| Women | Increased COX‐1–independent signaling; increased baseline platelet reactivity |
| Post‐ACS | Increased platelet turnover; increased inflammation and platelet aggregation |
| History of stent thrombosis | Increased platelet turnover; increased peri‐procedural inflammation |
| Post‐CABG | Increased platelet turnover; platelet activation by bypass pump |
ACS, acute coronary syndrome; CABG, coronary artery bypass graft; COX‐1, cyclooxygenase‐1.
Diabetes Mellitus
With the Third Report of the National Cholesterol Education Program (Adult Treatment Panel III), diabetes mellitus was upgraded to a coronary heart disease risk equivalent.40 In diabetic patients, aspirin therapy is recommended for the secondary prevention of coronary heart disease and for primary prevention in patients at increased cardiovascular risk including those over the age of 40 years or those with additional risk factors.41 There are several proposed mechanisms for the increased rate of aspirin resistance that has been observed in the diabetic population. As mentioned above in the Mechanisms of Aspirin Resistance section, diabetes is associated with increased platelet turnover,29 suggesting that once‐daily aspirin therapy may be insufficient in this population.34 Increased protein glycosylation in poorly controlled diabetes has been reported to reduce aspirin‐mediated acetylation and inhibition of COX‐1, possibly through a competitive mechanism.42 This mechanism may, in part, explain why dual‐antiplatelet therapy with aspirin and clopidogrel was more effective at reducing platelet activation than low‐dose43., 44. or high‐dose44 aspirin monotherapy in diabetic patients with coronary artery disease. Improved glycemic control reduces COX‐1–mediated platelet activation as measured by thromboxane B2 levels independent of aspirin therapy, suggesting that poor glycemic control also increases platelet activation through independent mechanisms.45
Obesity
Obesity is commonly associated with other coronary artery disease risk factors including hyperlipidemia, hypertension, and diabetes. Accordingly, many obese patients are on aspirin for the primary and secondary prevention of coronary artery disease. Emerging evidence suggests that obesity, independent of its associated comorbid conditions, may be associated with aspirin resistance. Obesity is associated with a modest yet significant increase in baseline platelet reactivity. After the addition of low‐dose (50–81 mg) aspirin, platelets in obese patients have more incomplete inhibition of aggregation to arachidonic acid46., 47. and higher levels of thromboxane B2.47 These changes remained significant after accounting for potentially confounding variables including hypertension, fasting blood sugar, and hyperlipidemia. It has been proposed that the increased volume of distribution of aspirin in obese individuals may, in part, explain the higher prevalence of aspirin resistance in this population. Bordeaux et al47 attempted to address this hypothesis by comparing aspirin resistance in obese participants in response to low‐dose and high‐dose aspirin. Their results were not powered for this analysis although they did show lower, although nonsignificant, aspirin resistance in obese patients with the addition of high‐dose aspirin compared to low‐dose aspirin.47 Additional studies are needed to further address this mechanism of resistance in this population. Additionally, leptin, a hormone that is elevated in obesity, appears to have prothrombotic effects. Leptin receptors have been identified on the surface of platelets. At concentrations typically found in obesity but not in non‐obese individuals, leptin markedly enhanced platelet aggregation48 and in mouse models was associated with heightened thrombosis.49
Women
While aspirin has proven benefit in the primary prevention of cardiovascular events in men, its benefit in women is less clear. The Women's Health Study included nearly 40,000 women without known coronary artery disease who were randomly assigned to either 100 mg of aspirin on alternating days or placebo and followed for 10 years. While the study showed a 24% risk reduction in ischemic strokes, there was no reduction in the rate of MIs or death from cardiovascular causes.50 Subgroup analysis of the HOT trial also showed a gender bias of aspirin's therapeutic effect because aspirin was associated with a 42% reduction in the rate of MI in male patients with diastolic hypertension but was associated with a nonsignificant 19% reduction of infarction in hypertensive women.51 Of note, the Women's Health Study used mean aspirin doses less than the standard mean dose of 81 mg daily most commonly used in the United States. In a direct comparison of aspirin 81 mg daily versus 100 mg every other day in women who otherwise would have met inclusion criteria for The Women's Health Study, daily aspirin was associated with less aspirin resistance than every other day aspirin, suggesting that the results of the Women's Health Study may have underestimated the cardioprotective effects of aspirin.52 In addition, the Antithrombotic Trialists' Collaboration found in both men and women that doses of aspirin less than 75 mg per day had a nonsignificant decrease in vascular events.2 Thus, in the Women's Health Study, the total dose of aspirin may simply have been too low, possibly accounting for the observed ineffectiveness in women. Additional studies of platelet function in women suggest that aspirin resistance might be mediated by COX‐1–independent pathways. Men and women treated with aspirin have similar inhibition of arachidonic acid–mediated aggregation; however, when COX‐1–independent pathways were studied, such as ADP‐ and collagen‐mediated aggregation, women have more residual platelet activity than men after low‐dose53., 54. and‐high dose54 aspirin. Women have more reactive platelets than men at baseline prior to aspirin administration and this appears to be the greatest contributor to the gender differences seen in response to aspirin.53., 54. Thus it is biologically possible, but not proven that women exhibit more aspirin resistance than men.
Post–ACS
ACS, defined as unstable angina, non–ST‐segment elevation MI (NSTEMI), and ST‐segment elevation MI (STEMI), is associated with an increased incidence of aspirin resistance both during the event and during the post‐ACS period. Among patients admitted to the emergency room with chest pain, Aydinalp et al55 showed nearly a 2‐fold increase in the prevalence of aspirin resistance, using the PFA‐100 assay, in those patients with a confirmed diagnosis of ACS compared to those who had been ruled out for ACS (40.7% vs 17.2%). Similarly, patients with acute MI (NSTEMI or STEMI) are more likely to be aspirin resistant than in those without acute infarction.56 Unfortunately, neither of these studies took into account the potential for patient noncompliance. The increased rates of aspirin resistance during acute MIs may be a transient phenomenon secondary to the heightened inflammatory and prothrombotic state during the peri‐ACS period. In a study of STEMI patients admitted to the intensive care unit (ICU), there were significantly more aspirin nonresponders, as measured by thromboxane B2 levels and arachidonic acid–mediated platelet aggregation, compared to controls upon first evaluation in the ICU. Interestingly, among these nonresponders, reevaluation of platelet function 24 to 48 hours after the initial evaluation showed that the majority of patients had optimal platelet inhibition by aspirin.57 As mentioned above, platelet turnover appears to be more rapid in patients with acute MI and may partially explain the higher rates of aspirin resistance.31., 32. There is a reduction of large platelets at the time of admission but not during recovery in patients with an acute MI, suggesting that there may be a consumption of large platelets at the time of thrombus formation.58 ADP levels have been shown to be higher during the acute infarct setting in patients with STEMI, and higher levels correlate with increased platelet aggregation. Likewise, aspirin‐resistant patients have higher ADP levels than aspirin responders.59 These findings suggest that higher ADP levels in ACS lead to more platelet aggregation that may need higher doses of aspirin to suppress in some patients. However, despite the heightened platelet activity seen in the peri‐ACS period, the CURRENT‐OASIS 7 investigators found no difference between low‐dose (75 mg to 100 mg) and high‐dose (300 mg to 325 mg) aspirin in the rate of cardiovascular death, MI, or stroke at 30 days in patients with ACS undergoing PCI.60
History of Stent Thrombosis
A history of stent thrombosis after PCI has similarly been associated with increased rates of aspirin resistance. Stent thrombosis is a rare but severe complication following PCI, with an estimated occurrence of 0.6% within 15 months of follow‐up.61 Dual resistance to aspirin and clopidogrel at the time of drug‐eluting stent implantation is associated with an 11% risk of definite or probable stent thrombosis at 6 months compared to only 2% in responders.62 Würtz et al63 examined the antiplatelet effects of aspirin in patients with a confirmed history of stent thrombosis compared to those without such a history and found increased platelet aggregation in patients with a history of thrombosis. Those with a history of stent thrombosis had a higher but nonsignificant fraction of immature platelet cells, suggesting that rapid platelet turnover may, in part, contribute to the heightened aspirin resistance in this population. Pinto Slottow et al38 found that, among patients with a history of stent thrombosis, 23% were aspirin resistant compared to 5% of controls and 43% of patients were resistant to both aspirin and clopidogrel compared to 14% of controls. Interestingly, aspirin resistance in patients with a history of stent thrombosis appears to be more prevalent in early stent thrombosis (<30 days) than late stent thrombosis (>30 days).38 This may be related to heightened inflammation and periprocedural changes to the coronary anatomy after stent implantation, which increases the prothrombotic risk.
Post–Coronary Artery Bypass Graft Surgery
Early thrombosis is a major cause of vein graft occlusion in the first month following coronary artery bypass graft (CABG) surgery, occurring in 8% to 18% of all vein grafts.64 Aspirin has been shown to have beneficial effects on vein graft patency for the first year following CABG.65 Up to two‐thirds of patients are aspirin resistant, as assessed by arachidonic acid–mediated platelet aggregation, following CABG, especially within the first 10 days following surgery.64 Traditional, on‐pump cardiopulmonary bypass leads to a decline in circulating platelet count by as much as 30% to 50%. This leads to increased platelet turnover and the counts often recover or surpass preoperative levels by postoperative day 10.66 As mentioned above, this increased platelet turnover partially explains the high degree of aspirin resistance seen in post‐CABG patients. Platelets also appear to be activated by cardiopulmonary bypass, undergoing morphological changes and release of alpha granules.66 Off‐pump coronary artery bypass surgery is associated with less platelet activation and aggregation than on‐pump surgery.67 As well, platelets are more responsive to aspirin after off‐pump coronary surgery compared to on‐pump surgery although aspirin resistance has been described in the off‐pump surgical population.64 Wang et al68 found that 29.7% of their cohort of off‐pump surgery patients was aspirin resistant, using arachidonic acid–mediated platelet aggregometry, on postoperative day 1 and only 4.5% remained resistant on postoperative day 10. All patients were aspirin sensitive by 6 months of follow up.68 A large meta‐analysis comparing lower doses of aspirin (75–325 mg) compared to higher doses of aspirin (500–1500 mg) failed to show any statistical significance in occlusion risks following CABG but did show more adverse reactions in the high‐dose aspirin group.69 Likewise, more frequent aspirin dosing was no more efficacious than once‐daily dosing.64
Clinical Implications and Choice of Optimal Dosing Strategy
Multiple well‐designed trials have shown that low‐dose aspirin is equally if not more effective than high‐dose aspirin as an antithrombotic agent. For the majority of patients, aspirin appears to lack dose‐responsiveness in its antithrombotic effects but does demonstrate dose‐dependence for its side effects, notably gastrointestinal bleeding.1 Aspirin at a dose of 75 mg per day is equally as effective at reducing the risk of MI or death in patients with unstable angina compared to doses as high as 1300 mg per day.70 Similarly, 100 mg of aspirin had a similar antithrombotic profile as 1200 mg of aspirin in preventing the incidence of early graft occlusion following CABG.71 Even aspirin doses as low as 50 mg per day are equally as effective as 100 mg per day at preventing cardiovascular events in patients with coronary heart disease.72 Among high‐risk patients, there is some evidence that lower doses of aspirin may in fact be superior to high‐dose aspirin at reducing serious vascular events. The Antithrombotic Trialists' Collaboration conducted a large meta‐analysis investigating various antiplatelet regimens involving patients with acute or previous vascular disease as well as those with predisposing conditions for vascular disease. They found that in an indirect comparison among different aspirin doses, low‐dose (75–150 mg) aspirin was associated with a larger proportional reduction in vascular events compared to medium‐dose (160–325 mg) aspirin or high‐dose (500–1500 mg) aspirin (32%, 26%, and 19%, respectively).2
While once‐daily low‐dose aspirin appears to be the optimal dosing strategy for the majority of patients, as described above in the High‐Risk Groups section, a significant percentage of patients have suboptimal antiplatelet response to standard aspirin dosing. In a large systemic review, Hovens et al73 found that the prevalence of aspirin resistance was less among patients receiving greater than 300 mg of aspirin per day (19%) compared to those receiving less than 100 mg of aspirin per day (36%). It appears that the dose‐dependent effect of aspirin in aspirin‐resistant patients may be mediated by non–COX‐1 pathways. Gurbel et al74 showed that aspirin at doses of 81 mg, 162 mg, and 325 mg all had near total inhibition of arachidonic acid–induced platelet aggregation with no difference seen among dose. Conversely, there was a dose‐dependent inhibition of platelet function as assessed by non–COX‐1 pathways because aspirin at doses of 162 mg and 325 mg was more effective at reducing ADP‐ and collagen‐mediated aggregation as well at prolonging CT as measured by PFA‐100 than 81 mg of aspirin.74 This interplay between COX‐1–dependent and ADP‐dependent pathways was also described by Frelinger et al,6 who showed that residual arachidonic acid–induced platelet activation was less in patients taking clopidogrel, an ADP receptor antagonist, compared to controls. Adjunctive clopidogrel, in addition to low‐dose aspirin, has been shown to be more effective at inhibiting platelet aggregation to ADP or collagen than high‐dose aspirin (300 mg) monotherapy in diabetic patients with coronary artery disease who were resistant to low‐dose aspirin.44 Moreover, as described above in the Mechanisms of Aspirin Resistance section, emerging evidence suggests that in certain patient populations with rapid platelet turnover, twice‐daily aspirin may be more effective than once‐daily aspirin.34
Based on the above data, it has been proposed that certain high‐risk patient populations might benefit from diagnostic screening for tailored antiplatelet therapy. In a small study conducted by Capodanno et al,75 30 patients with type 2 diabetes mellitus and coronary artery disease underwent a weekly aspirin dose and/or frequency escalation scheme followed by platelet functional testing using the VerifyNow assay. Platelet function was tested in each patient after 1 week of either daily aspirin at a dose of 81 mg, 162 mg, or 325 mg, or twice‐daily aspirin at a dose of 81 mg or 162 mg. Similar to the findings of Rocca et al,34 twice‐daily aspirin was associated with more platelet inhibition than once‐daily aspirin.75 Neubauer et al76 recently studied a dose escalation and subsequent therapeutic substitution algorithm in aspirin and clopidogrel nonresponders in patients undergoing PCI for ACS or stable angina. Patients underwent whole‐blood aggregometry to both ADP and arachidonic acid. Aspirin nonresponders were treated with dose escalation from 100 mg daily to 300 mg daily and if needed to 500 mg daily, with adequate platelet response seen in all patients after dose escalation. Initial clopidogrel nonresponders to 75 mg of clopidogrel were treated initially with 150 mg of clopidogrel daily. For those who continued to have suboptimal antiplatelet response, ADP antagonism was substituted to either ticlodipine or prasugrel. With this algorithm (Figure 1), the initial clopidogrel nonresponder rate was reduced by nearly 87%.76
Figure 1.
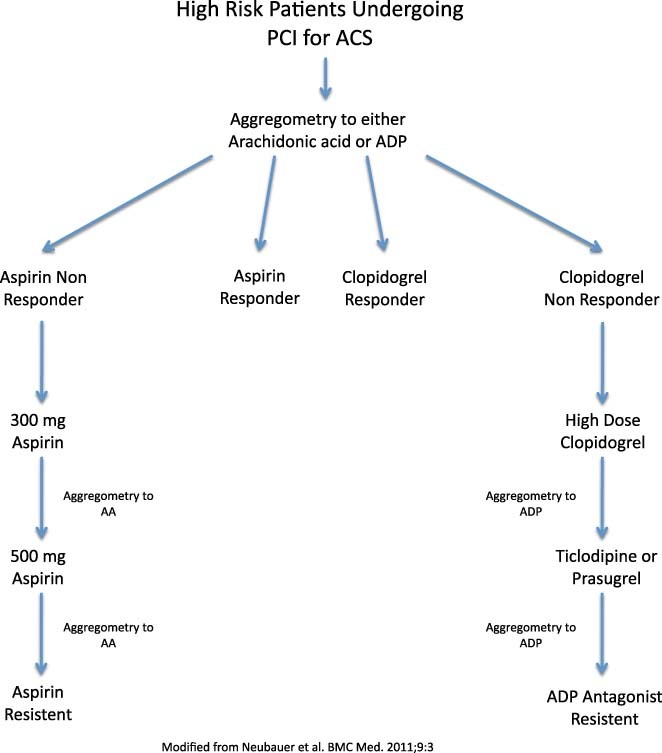
Dose escalation and therapeutic substitution algorithm for patients at high risk for aspirin and/or clopidogrel resistance after PCI. Abbreviation: PCI, percutaneous coronary intervention.
While the algorithm proposed by Neubauer et al76 may have been effective in their particular patient population of patients undergoing PCI, simple aspirin dose escalation is unlikely to be effective in all cases of aspirin resistance, as described above in the Clinical Implications and Choice of Optimal Dosing Strategy section. However, the concept of tailored therapy on an individual or perhaps population level is intriguing for high‐risk patient populations. In an ambulatory setting, a point of care system of testing such as VerifyNow or PFA‐100 is the most practical method of analysis. Patients at high risk for aspirin resistance such as diabetics, women, or the obese, particularly those with recurrent vascular events, or those with a history of stent thrombosis, might undergo platelet function testing in the future. To maximize the antiplatelet response, both the COX‐1–dependent and ‐independent pathways should be assayed, both before and after tailored antiplatelet therapy, and medication adjustments could be made accordingly with either dose or frequency escalation of aspirin and/or addition of an ADP antagonist.
Future Exploration
It is apparent that, although intellectually promising, additional studies validating tailored antiplatelet therapy, including a cost analysis, are needed before such a system is likely to be implemented in large scale. Additionally, studies directly comparing aspirin dose and frequency escalation with dual antiplatelet therapy in high‐risk patient populations is still lacking. Finally, additional testing on the optimal aspirin dose in patients on alternative ADP receptor antagonists is needed given the growing evidence of superiority of the novel ADP receptor antagonists, prasugrel77 and ticagrelor,78 compared to clopidogrel. Of particular interest is the interaction between aspirin and ticagrelor at higher aspirin doses. The PLATO trial showed superiority of ticagrelor compared to clopidogrel overall across the international trial but failed to show superiority within North America, where aspirin doses in patients with ACS tend to be higher.78 Accordingly, the U.S. Food and Drug Administration has recommended that aspirin doses not exceed 100 mg per day in patients receiving concomitant ticagrelor because higher doses may decrease the effectiveness of the drug.79 Additional studies investigating the interactions of aspirin and ticagrelor are needed in order to better understand optimal aspirin dosing in this population.
References
- 1. Patrono C, Coller B, Fitzgerald GA, et al. Platelet active drugs: the relationships among dose, effectiveness, and side‐effects. Chest. 2004;126:234S–264S. [DOI] [PubMed] [Google Scholar]
- 2. Antithrombotic Trialists' Collaboration . Collaborative meta‐analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ. 2002;324:71–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. dummy Randomised trial of intravenous streptokinase, oral aspirin, both, or neither among 17,187 cases of suspected acute myocardial infarction: ISIS‐2. ISIS‐2 (Second International Study of Infarct Survival) Collaborative Group. Lancet. 1988;2:349–360. [PubMed] [Google Scholar]
- 4. Sanderson S, Emery J, Baglin T, et al. Narrative review: aspirin resistance and its clinical implications. Ann Intern Med. 2005;142:370–380. [DOI] [PubMed] [Google Scholar]
- 5. Sane DC, McKee SA, Malinin AI, et al. Frequency of aspirin resistance in patients with congestive heart failure treated with antecedent aspirin. Am J Cardiol. 2002;90:893–895. [DOI] [PubMed] [Google Scholar]
- 6. Frelinger AL, Furman MI, Linden MD, et al. Residual arachidonic acid‐induced platelet activation via an adenosine diphosphate‐dependent but cyclooxygenase‐1 and cyclooxygenase 2‐independent pathway. A 700‐patient study of aspirin resistance. Circulation. 2006;113:2888–2896. [DOI] [PubMed] [Google Scholar]
- 7. FitzGerald R, Pirmohamed M. Aspirin resistance: effect of clinical, biochemical and genetic factors. Pharmacol Ther. 2011;130: 213–225. [DOI] [PubMed] [Google Scholar]
- 8. Gum PA, Kottke‐Marchant K, Welsh PA, et al. A prospective, blinder determination of the natural history of aspirin resistance among stable patients with cardiovascular disease. J Am Coll Cardiol. 2003;41:961–965. [DOI] [PubMed] [Google Scholar]
- 9. Reilly IAG, Fitzgerald GA. Inhibition of thromboxane formation in vivo and ex vivo: implications for therapy with platelet inhibitory drugs. Blood. 1987;69:180–186. [PubMed] [Google Scholar]
- 10. Di Minno G, Silver MJ, Murphy S. Monitoring the entry of new platelets into the circulation after ingestion of aspirin. Blood. 1983;61:1081–1085. [PubMed] [Google Scholar]
- 11. Lordkipanidze M, Pharand C, Schampaert E, et al. A comparison of six major platelet function tests to determine the prevalence of aspirin resistance in patients with stable coronary artery disease. Eur Heart J. 2007;14:1702–1708. [DOI] [PubMed] [Google Scholar]
- 12. Grove EL, Hvas AM, Johnsen HL, et al. A comparison of platelet function tests and thromboxane metabolites to evaluate aspirin response in health individuals and patients with coronary artery disease. Thromb Haemost. 2010;103:1245–1253. [DOI] [PubMed] [Google Scholar]
- 13. Kamath S, Blann AD, Lip GY. Platelet activation: assessment and quantification. Eur Heart J. 2001;22:1561–1571. [DOI] [PubMed] [Google Scholar]
- 14. Krasopoulos G, Brister SJ, Beattie WS, et al. Aspirin “resistance” and risk of cardiovascular morbidity: a systematic review and metaanalysis. BMJ. 2008;336:195–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Michelson AD, Barnard MR, Krueger LA, et al. Evaluation of platelet function by flow cytometry. Methods. 2000;21:259–270. [DOI] [PubMed] [Google Scholar]
- 16. Blann AD, Lip GY. Hypothesis: is soluble P‐selectin a new marker of platelet activation? Atherosclerosis. 1997;128:135–138. [DOI] [PubMed] [Google Scholar]
- 17. Perneby C, Granstrom E, Beck O, et al. Optimization of an enzyme immunoassay for 11‐dehydro‐thromboxane B(2) in urine: comparison with GC‐MS. Thromb Res. 1999;96:427–436. [DOI] [PubMed] [Google Scholar]
- 18. Faraday N, Becker DM, Yanek LR, et al. Relation between atherosclerosis risk factors and aspirin resistance in a primary prevention population. Am J Cardiol. 2006;98:774–779. [DOI] [PubMed] [Google Scholar]
- 19. Hankey GJ, Eikelboom JW. Aspirin resistance. Lancet. 2006;367: 606–616. [DOI] [PubMed] [Google Scholar]
- 20. Michelson AD. Platelet function testing in cardiovascular diseases. Circulation. 2004;110:e489–e493. [DOI] [PubMed] [Google Scholar]
- 21. Van Werkum JW, Harmsze AM, Elsenberg HA, et al. The use of the VerifyNow system to monitor antiplatelet therapy: a review of the current evidence. Platelets. 2008;19:479–488. [DOI] [PubMed] [Google Scholar]
- 22. Lee PY, Chen WH, Ng W, et al. Low‐dose aspirin increases aspirin resistance in patients with coronary artery disease. Am J Med. 2005;118:723–727. [DOI] [PubMed] [Google Scholar]
- 23. Mammen EF, Comp PC, Gosselin R. PFA‐100TM system: a new method for assessment of platelet dysfunction. Semin Thromb Hemost. 1998;24:195–202. [DOI] [PubMed] [Google Scholar]
- 24. Crescente M, Di Castelnuovo A, Iacoviello L, et al. Response variability to aspirin as assessed by the platelet function analyzer (PFA)‐100. A systematic review. Thromb Haemost. 2008;99: 14–26. [DOI] [PubMed] [Google Scholar]
- 25. Schwartz KA, Schwartz DE, Ghosheh K, et al. Compliance as a critical consideration in patients who appear to be resistant to aspirin after healing of myocardial infarction. Am J Cardiol. 2005;95:973–975. [DOI] [PubMed] [Google Scholar]
- 26. Von Pape KW, Strupp G, Banzel T, et al. Effect of compliance and dosage adaption of long term aspirin on platelet function with PFA‐100ˆ® in patients after myocardial infarction. Thromb Haemost. 2005;94:889–891. [DOI] [PubMed] [Google Scholar]
- 27. Faraday N, Yanek LR, Mathias R, et al. Heritability of platelet responsiveness to aspirin in activation pathways directly and indirectly related to cyclooxygenase‐1. Circulation. 2007;115: 2490–2496. [DOI] [PubMed] [Google Scholar]
- 28. Goodman T, Ferro A, Sharma P. Pharmacogenetics of aspirin resistance: a comprehensive systematic review. Br J Clin Pharmacol. 2008;66:222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Winocour PD. Platelet turnover in advanced diabetes. Eur J Clin Invest. 1994;24(Suppl 1):34–37. [DOI] [PubMed] [Google Scholar]
- 30. Arazi HC, Doiny DG, Torcivia RS, et al. Impaired antiplatelet effect of aspirin, inflammation and platelet turnover in cardiac surgery. Interact Cardiovasc Thorac Surg. 2010;10:863–867. [DOI] [PubMed] [Google Scholar]
- 31. Hamsten A, Svensson J, Waldius G, et al. Shortened megakaryocyte‐platelet regeneration time in young survivors of myocardial infarction. Am Heart J. 1985;110:1154–1160. [DOI] [PubMed] [Google Scholar]
- 32. Grove EL, Hvas AM, Kristensen SD. Immature platelets in patients with acute coronary syndromes. Thromb Haemost. 2009; 101:151–156. [PubMed] [Google Scholar]
- 33. Henry P, Vermillet A, Boval B, et al. 24‐hour time dependent aspirin efficacy in patients with stable coronary artery disease. Thromb Haemost. 2010;105:336–344. [DOI] [PubMed] [Google Scholar]
- 34. Rocca B, Santilli F, Pitocco D, et al. Variability in the recovery rate of platelet cyclooxygenase activity during chronic therapy with low‐dose aspirin in type 2 diabetes [Abstract]. Circulation. 2010; 122:A12233. [Google Scholar]
- 35. Catella‐Lawson F, Reilly MP, Kapoor SC, et al. Cyclooxygenase inhibitors and the antiplatelet effects of aspirin. N Engl J Med. 2001;345:1809–1817. [DOI] [PubMed] [Google Scholar]
- 36. Lev EI, Patel RT, Maresh KJ, et al. Aspirin and clopidogrel drug response in patients undergoing percutaneous coronary intervention. The role of dual drug resistance. J Am Coll Cardiol. 2006; 47:27–33. [DOI] [PubMed] [Google Scholar]
- 37. Price MJ, Berger PB, Teirstein PS, et al. Standard‐ vs high‐dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA. 2011;305:1097–1105. [DOI] [PubMed] [Google Scholar]
- 38. Pinto Slottow TL, Bonello L, Gavini R, et al. Prevalence of aspirin and clopidogrel resistance among patients with and without drug‐eluting stent thrombosis. Am J Cardiol. 2009;104: 525–530. [DOI] [PubMed] [Google Scholar]
- 39. Macchi L, Christiaens L, Brabant S, et al. Resistance to aspirin in vitro is associated with increased platelet sensitivity to adenosine diphosphate. Thromb Res. 2002;107:45–49. [DOI] [PubMed] [Google Scholar]
- 40. Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation. 2002;106:3143–3421. [PubMed] [Google Scholar]
- 41. Buse JB, Ginsberg HN, Bakris GL, et al.; for the American Heart Association and American Diabetes Association. Primary prevention of cardiovascular diseases in people with diabetes mellitus: a scientific statement from the American Heart Association and the American Diabetes Association. Circulation. 2007;115: 114–126. [DOI] [PubMed] [Google Scholar]
- 42. Watala C, Pluta J, Golanski J, et al. Increased protein glycation in diabetes mellitus is associated with decreased aspirin‐mediated protein acetylation and reduced sensitivity of blood platelets to aspirin. J Mol Med. 2005;83:148–158. [DOI] [PubMed] [Google Scholar]
- 43. Serebruany VL, Malinin AI, Pokov A, et al. Effects of clopidogrel and aspirin in combination versus aspirin alone on platelet activation and major receptor expression in diabetic patients: the Plavix Use for Treatment of Diabetes (PLUTO‐Diabetes) trial. Am Heart J. 2008;155:93.e1–93.e7. [DOI] [PubMed] [Google Scholar]
- 44. Duzenli MA, Ozdemir K, Aygul N, et al. Comparison of increased aspirin dose versus combined aspirin plus clopidogrel therapy in patients with diabetes mellitus and coronary heart disease and impaired antiplatelet response to low‐dose aspirin. Am J Cardiol. 2008;102:396–400. [DOI] [PubMed] [Google Scholar]
- 45. Santilli F, Davì G, Consoli A, et al. Thromboxane‐dependent CD40 ligand release in type 2 diabetes mellitus. J Am Coll Cardiol. 2006; 47:391–397. [DOI] [PubMed] [Google Scholar]
- 46. Tamminen M, Lassila R, Westerbacka J, et al. Obesity is associated with impaired platelet‐inhibitory effect of acetylsalicylic acid in nondiabetic subjects. Int J Obes Relat Metab Disord. 2003; 27:907–911. [DOI] [PubMed] [Google Scholar]
- 47. Bordeaux BC, Qayyum R, Yanek LR, et al. Effect of obesity on platelet reactivity and response to low‐dose aspirin. Prev Cardiol. 2010;13:56–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Nakata M, Yada T, Soejima N, et al. Leptin promotes aggregation of human platelets via the long form of its receptor. Diabetes. 1999;48:426–429. [DOI] [PubMed] [Google Scholar]
- 49. Konstantinides S, Schafer K, Koschnick S, et al. Leptin‐dependent platelet aggregation and arterial thrombosis suggests a mechanism for atherothrombotic disease in obesity. J Clin Invest. 2001;108:1533–1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ridker PM, Cook NR, Lee IM, et al. A randomized trial of low‐dose aspirin in the primary prevention of cardiovascular disease in women. N Engl J Med. 2005;352:1293–1304. [DOI] [PubMed] [Google Scholar]
- 51. Kjeldsen SE, Kolloch RE, Leonetti G, et al. Influence of gender and age on preventing cardiovascular disease by antihypertensive treatment and acetylsalicylic acid. The HOT study. J Hypertens. 2000;18:629–642. [DOI] [PubMed] [Google Scholar]
- 52. Swain L, Hillman RS. Aspirin administered to women at 100 mg every other day produces less platelet inhibition than aspirin administered at 81 mg per day: implications for interpreting the Women's Health Study. J Thromb Thrombolysis. 2009;28: 94–100. [DOI] [PubMed] [Google Scholar]
- 53. Becker DM, Segal J, Vaidya D, et al. Sex differences in platelet reactivity and response to low‐dose aspirin therapy. JAMA. 2006;295:1420–1427. [DOI] [PubMed] [Google Scholar]
- 54. Qayyum R, Becker DM, Yanek LR, et al. Platelet Inhibition by aspirin 81 and 325mg/day in men versus women without clinically apparent cardiovascular disease. Am J Cardiol. 2008;101: 1359–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Aydinalp A, Atar I, Gulmez O, et al. The clinical significance of aspirin resistance in patients with chest pain. Clin Cardiol. 2010; 33:E1–E7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Poulsen TS, Jørgensen B, Korsholm L, et al. Prevalence of aspirin resistance in patients with an evolving acute myocardial infarction. Thromb Res. 2007;119:555–562. [DOI] [PubMed] [Google Scholar]
- 57. Valles J, Santos MT, Fuset MP, et al. Partial inhibition of platelet thromboxane A2 synthesis by aspirin is associated with myonecrosis in patients with ST‐segment elevation myocardial infarction. Am J Cardiol. 2007;99:19–25. [DOI] [PubMed] [Google Scholar]
- 58. Erne P, Wardle J, Sanders K, et al. Mean platelet volume and size distribution and their sensitivity to agonists in patients with coronary artery disease and congestive heart failure. Thromb Haemost. 1988;59:259–263. [PubMed] [Google Scholar]
- 59. Borna C, Lazarowski E, van Heusden C, et al. Resistance to aspirin is increased by ST‐elevation myocardial infarction and correlates with adenosine diphosphate levels. Thromb J. 2005;3:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. The CURRENT‐OASIS Investigators. Dose comparisons of clopidogrel and aspirin in acute coronary syndromes. N Engl J Med. 2010;363:930–942. [DOI] [PubMed] [Google Scholar]
- 61. Kaltoft A, Jensen LO, Maeng M, et al. 2‐year clinical outcomes after implantation of sirolimus‐eluting, paclitaxel‐eluting, and bare‐metal coronary stents: results from the WDHR (Western Denmark Heart Registry). J Am Coll Cardiol. 2009;53: 658–664. [DOI] [PubMed] [Google Scholar]
- 62. Gori AM, Marcucci R, Migliorini A, et al. Incidence and clinical impact of dual nonresponsiveness to aspirin and clopidogrel in patients with drug‐eluting stents. J Am Coll Cardiol. 2008;52:734–739. [DOI] [PubMed] [Google Scholar]
- 63. Würtz M, Grove EL, Wulff LN , et al. Patients with previous definite stent thrombosis have a reduced antiplatelet effect of aspirin and a larger fraction of immature platelets. JACC Cardiovasc Interv. 2010;3:828–835. [DOI] [PubMed] [Google Scholar]
- 64. Zimmermann N, Gams E, Hohlfeld T. Aspirin in coronary artery bypass surgery: new aspects of and alternatives for an old antithrombotic agent. Eur J Cardiothorac Surg. 2008;34: 93–108. [DOI] [PubMed] [Google Scholar]
- 65. Goldman S, Copeland J, Moritz T, et al. Saphenous vein graft patency 1 year after coronary artery bypass surgery and effects of antiplatelet therapy. Results of a Veterans Administration Cooperative Study. Circulation. 1989;80:1190–1197. [DOI] [PubMed] [Google Scholar]
- 66. Hyde JA, Chin JA, Graham TR. Platelets and cardiopulmonary bypass. Perfusion. 1998;13:389–409. [DOI] [PubMed] [Google Scholar]
- 67. Ballotta A, Saleh HZ, El Baghdady HW, et al. Comparison of early platelet activation in patients undergoing on‐pump versus off‐pump coronary artery bypass surgery. J Thorac Cardiovasc Surg. 2007;134:132–138. [DOI] [PubMed] [Google Scholar]
- 68. Wang Z, Gao F, Men J, et al. Aspirin resistance in off‐pump coronary artery bypass grafting. Eur J Cardiothorac Surg. 2012;41:108–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Antiplatelet Trialists' Collaboration. Collaborative overview of randomized trials of antiplatelet therapy‐I: Prevention of death, myocardial infarction, and stroke by prolonged antiplatelet therapy in various categories of patients. Br Med J. 1994;308:81–106. [PMC free article] [PubMed] [Google Scholar]
- 70. Cairns JA, Gent M, Singer J, et al. Aspirin, sulfinpyrazone, or both in unstable angina. N Engl J Med. 1985;313:1369–1375. [DOI] [PubMed] [Google Scholar]
- 71. Brown BG, Cukingnan RA, DeRouen T, et al. Improved graft patency in patients treated with platelet‐inhibiting therapy after coronary bypass surgery. Circulation. 1985;72:138–146. [DOI] [PubMed] [Google Scholar]
- 72. Barent R, Auer J, Franklin B, et al. Platelet response to aspirin 50 and 100 mg in patients with coronary heart disease over a five‐year period. Am J Cardiol. 2011;108:644–650. [DOI] [PubMed] [Google Scholar]
- 73. Hovens MMC, Snoep JD, Eikenboom JCJ, et al. Prevalence of persistent platelet reactivity despite use of aspirin: a systematic review. Am Heart J. 2007;153:175–181. [DOI] [PubMed] [Google Scholar]
- 74. Gurbel PA, Bliden KP, DiChiara J, et al. Evaluation of dose‐related effects of aspirin on platelet function: results from the aspirin0induced platelet effect (ASPECT) study. Circulation. 2007;115:3156–3164. [DOI] [PubMed] [Google Scholar]
- 75. Capodanno D, Patel A, Dharmashankar K, et al. Pharmacodynamic effects of different aspirin dosing regimens in type 2 diabetes mellitus patients with coronary artery disease. Circ Cardiovasc Interv. 2011:4:180–187. [DOI] [PubMed] [Google Scholar]
- 76. Neubauer H, Kaiser AF, Endres HG, et al. Tailored antiplatelet therapy can overcome clopidogrel and aspirin resistance ‐ the Bochum clopidogrel and aspirin plan (BOCLA‐Plan) to improve antiplatelet therapy. BMC Med. 2011;9:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001–2015. [DOI] [PubMed] [Google Scholar]
- 78. Wallentin L, Becker RC, Budaj A, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361:1045–1057. [DOI] [PubMed] [Google Scholar]
- 79. Gaglia MA, Waksman R. Overview of the 2010 Food and Drug Administration Cardiovascular and Renal Drugs Advisory Committee Meeting Regarding Ticagrelor. Circulation. 2011;123: 451–456. [DOI] [PubMed] [Google Scholar]