

Gas gangrene, or clostridial myonecrosis, is usually caused by Clostridium perfringens and may occur spontaneously in association with diabetes mellitus, peripheral vascular disease, or some malignancies but more often after contamination of a deep surgical or traumatic lesion. If not controlled, clostridial myonecrosis results in multiorgan failure, shock, and death, but very little is known about the muscle regeneration process that follows myonecrosis when the infection is controlled.
KEYWORDS: Clostridium perfringens, clostridial myonecrosis, gas gangrene, host-pathogen interactions, innate immunity, muscle regeneration
ABSTRACT
Gas gangrene, or clostridial myonecrosis, is usually caused by Clostridium perfringens and may occur spontaneously in association with diabetes mellitus, peripheral vascular disease, or some malignancies but more often after contamination of a deep surgical or traumatic lesion. If not controlled, clostridial myonecrosis results in multiorgan failure, shock, and death, but very little is known about the muscle regeneration process that follows myonecrosis when the infection is controlled. In this study, we characterized the muscle regeneration process after myonecrosis caused in a murine experimental infection with a sublethal inoculum of C. perfringens vegetative cells. The results show that myonecrosis occurs concomitantly with significant vascular injury, which limits the migration of inflammatory cells. A significant increase in cytokines that promote inflammation explains the presence of an inflammatory infiltrate; however, impaired interferon gamma (IFN-γ) expression, a reduced number of M1 macrophages, deficient phagocytic activity, and a prolongation of the permanence of inflammatory cells lead to deficient muscle regeneration. The expression of transforming growth factor β1 (TGF-β1) agrees with the consequent accumulation of collagen in the muscle, i.e., fibrosis observed 30 days after infection. These results provide new information on the pathogenesis of gas gangrene caused by C. perfringens, shed light on the basis of the deficient muscle regenerative activity, and may open new perspectives for the development of novel therapies for patients suffering from this disease.
INTRODUCTION
Clostridium perfringens, a Gram-positive, anaerobic, spore-forming rod widely distributed in nature, is the most frequent cause of gas gangrene or clostridial myonecrosis. This life-threatening acute soft tissue infection is characterized by fever, sudden onset of prominent pain, gas accumulation at the infection site, massive local edema, and extensive muscular necrosis (1).
Gas gangrene frequently occurs after the introduction of the bacteria in a deep lesion or in a surgical wound (1). Infection is established under an anaerobic environment adequate for clostridial growth, and myonecrosis spreads within a few hours (1). In a murine model of gas gangrene, myonecrosis occurs before the appearance of significant clinical signs and the transcription of several cytokines and chemokines, and it is induced after infection with a lethal inoculum of C. perfringens (2). However, very little is known about the myonecrosis and muscle regeneration process that follow an experimental infection with a nonlethal C. perfringens inoculum.
Muscle regeneration after myonecrosis occurs in three sequential and interrelated phases: inflammation, regeneration, and remodeling (3). Initially, myofiber injury is associated with the entry of extracellular calcium, which induces a series of degenerative events, including hypercontraction, mitochondrial alterations, and the activation of calcium-dependent proteases, leading to necrosis of the myofibers (3, 4). Moreover, the disruption of the sarcolemma results in an increase in the serum level of creatine kinase (CK), an enzyme normally restricted to the myofiber cytosol (5). The presence of necrotic myofibers activates the inflammatory response, and an influx of cells from the immune system then occurs in the necrotic muscle (3). Inflammation is a critical phase of the regenerative process (3). Myofiber necrosis activates the synthesis and release of a plethora of signaling molecules into the extracellular space, and these mediators induce the sequential attraction and activation of diverse cell populations that promote inflammation and muscle regeneration (5, 6). The vascular network has an important role in this process, as it has an impact on the distribution of inflammatory cells, growth factors, cytokines, chemokines, and nutrients. Therefore, alterations in vascular integrity can affect the regenerative process (7). The regeneration phase begins with the activation of satellite cells (SC) that reside on the surface of the myofibers (8). Following myonecrosis, some SC proliferate and differentiate, whereas others return to quiescence as a reserve population of myogenic cells (8). Postmitotic precursor cells derived from activated SC then form multinucleated myotubes and proceed through a stage of regeneration that is dominated by terminal differentiation and growth (8). When the formation of contractile myofibers is complete, the size of the newly formed fibers increases, and the nucleus is displaced to the periphery of the fiber (5).
The final phase of the regenerative process includes the remodeling of connective tissue, angiogenesis, and functional recovery of the regenerated myofibers (3). After muscle injury, the extracellular matrix is remodeled, resulting in the overproduction of several collagen types (3); however, the overproduction of collagens within the necrotic area could lead to excessive scarring and loss of muscle function (3). Transforming growth factor β1 (TGF-β1) has been identified as a key factor in the activation of the fibrosis cascade in injured skeletal muscle (3). The processes of neovascularization and reinnervation play a critical role in determining the regeneration potential of the injured muscle (4).
The influx of inflammatory cells to the site of myonecrosis is paramount for efficient regeneration (9). The inflammatory response during the early stages of muscle regeneration is temporally and spatially coupled to the initial stages of myogenesis, when SC are activated and initiate their proliferation and differentiation (8). Neutrophils positive for the lymphocyte antigen 6 complex (Ly6C) are the first inflammatory cells to invade the necrotic muscle. The intramuscular density of these cells increases in the first 6 h after myonecrosis, reaching a peak 24 h after injury and then gradually returning to normal (5, 6, 8). Resident tissue macrophages, which are positive for F4/80 (a specific marker of murine macrophages) and Ly6C, promote a marked influx of neutrophils through the release of chemoattractants, such as murine chemokine keratinocyte chemoattractant (KC or CXCL1) and macrophage inflammatory protein 2 (MIP2 or CXCL2). Neutrophils initiate the process of removal of necrotic myofibers and cellular debris by phagocytosis and by the rapid release of high concentrations of free radicals and proteases (3, 4). In addition, neutrophils secrete proinflammatory cytokines that stimulate the arrival of macrophages, further promoting tissue inflammation and mediating the regeneration process (3, 4, 10).
Two populations of macrophages sequentially invade the necrotic muscle tissue. A population of CD68+ CD163− F4/80+ macrophages (M1 macrophages) closely follows the invasion of neutrophils, reaching a maximum concentration approximately 2 days after injury and subsequently decreasing in number (6). The Th1 response is characterized by the presence of interleukin-1 (IL-1), IL-2, interferon gamma (IFN-γ), and tumor necrosis factor alpha (TNF-α) (11). In turn, M1 macrophages are capable of damaging the host tissue by releasing free radicals that can disrupt cell membranes. They also phagocytose the necrotic muscle and promote the proliferation of SC (3, 4). The Th2 response involves high levels of IL-4, IL-5, IL-6, IL-10, and IL-13, which have anti-inflammatory effects and deactivate M1 macrophages (11). Nonphagocytic CD68− CD163+ F4/80+ 206+ macrophages, known as M2 macrophages, invade the muscle and reach their maximum peak approximately 4 days after injury, but the number of these cells remains elevated in the necrotic muscle by periods of up to 2 weeks (6). These tissue-remodeling macrophages decrease the inflammatory response and promote angiogenesis as well as myoblast proliferation, growth, and differentiation (3, 4).
Stimulation by IFN-γ is essential for the classic activation of the Th1 phenotype (11). IFN-γ is also a powerful activator of neutrophils and M1 macrophages (8). Although IFN-γ is usually a product of “natural killer” cells and T cells, it could also be expressed by M1 macrophages having an autocrine role in their activation (6, 8). Furthermore, the stimulation of IFN-γ can increase the response of neutrophils to chemotactic cytokines, potentially increasing their invasion to the sites of injury (6).
TNF-α is another Th1 cytokine highly expressed by M1 macrophages. The level of TNF-α in the muscle reaches its peak approximately 24 h after the onset of the injury, which coincides with the invasion of neutrophils and M1 macrophages and with the increase in secondary myonecrosis generated by myeloid cells (6). Part of the myeloid cell-mediated injury to myofibers is caused by nitric oxide (NO) derived from inducible nitric oxide synthase (iNOS), and TNF-α can stimulate M1 macrophages to elevate iNOS expression, promoting further damage (6). The potential of TNF-α to promote muscle repair and regeneration lies in its direct action on muscle cells (8).
Another important factor that influences the muscle regeneration process is TGF-β1, which has been recognized as a modulator of myoblast activity (12). In general, TGF-β1 plays a negative role in the regulation of myogenesis; it is highly expressed in quiescent SC and represses the progress of the cell cycle in these cells, repressing the expression of MyoD and myogenin (13).
In this study, we characterize the muscle regeneration process after myonecrosis caused by a limited sublethal infection induced in mice by the intramuscular injection of 106 wild-type C. perfringens vegetative cells.
RESULTS AND DISCUSSION
A sublethal inoculum of C. perfringens induces myonecrosis.
C. perfringens is an anaerobic bacterium that induces gas gangrene, a devastating disease characterized by severe myonecrosis. We have previously shown that an intramuscular inoculum of 6 × 108 CFU of C. perfringens induces severe myonecrosis, as evidenced by a rapid and sustained release of CK into the circulation (14). In this work, it was found that an inoculum of 1 × 106 CFU of this bacterium also induces myonecrosis, as evidenced by a significant increase in plasma CK 5 h after infection (P < 0.01). However, with this inoculum, at 24 h postinfection, plasma CK activity showed no significant difference compared to controls (Fig. 1A), indicating that the infection is controlled by the immune system and that myonecrosis becomes limited. Histological analysis of infected muscle showed areas of myonecrosis characterized by myofibrillar hypercontraction of myofibers from 5 h postinfection (Fig. 1B). Bacterial aggregates were evident between the myofibers, whereas the inflammatory infiltrate was distributed in a nonhomogeneous way in the muscle, without accumulation of neutrophils inside the venules (Fig. 1B). Accordingly, it was previously reported that inhibition of chemotaxis at the site of infection by C. perfringens depends on the size of the inoculum, and thus, a sublethal inoculum does not prevent the inflow of the inflammatory infiltrate into the infected muscle; hence, the immune system controls the infection and inhibits the establishment of the bacteria (15). Thus, our model of a sublethal inoculum of the bacteria allowed us to study the development of the muscle regenerative response.
FIG 1.

Myonecrosis induced by infection with a sublethal inoculum of C. perfringens. Groups of 6 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens. (A) CK activity was determined in plasma 5 and 24 h after infection. Results show means ± standard errors (SE). *, P < 0.01 for samples that differ statistically from the control. (B) Sections of injected muscles were taken 5 h, 1 day, and 3 days after infection and stained with hematoxylin and eosin (HE). Abundant necrotic muscle fibers are observed by as early as 5 h after infection. Bacteria and an inflammatory infiltrate are evident in the necrotic muscle (arrowheads and asterisks, respectively). Myonecrosis is evident by hypercontraction bands (arrows). Notice the presence of bacteria near a necrotic fiber 3 days after infection (inset). Control muscle injected with PBS shows a normal histological pattern. Bars, 100 μm.
C. perfringens persists in infected muscle.
Histological analysis showed that C. perfringens persisted in the infected tissue until 3 days postinfection. Further counts of CFU revealed that viable bacteria persisted in the muscle up to day 9 postinfection (see Fig. S1 in the supplemental material). The CFU counts were 3 × 104 CFU (3% of the initial inoculum) at 24 h, 8 × 102 CFU at 5 days, and 4 CFU at 9 days.
Although C. perfringens infection is controlled before 24 h and an inflammatory infiltrate is present, the bacteria persist in the muscular tissue. Previously, O’Brien and Melville (16) found viable bacteria in 5% of a sublethal inoculum of C. perfringens in the muscle tissue of mice 24 h after infection and even detected bacteria at 72 h postinfection. In the present study, 3% of a sublethal inoculum was recovered similarly 24 h after infection, but the most relevant finding was the presence of viable bacteria even 5 days and 9 days after infection. The persistence of C. perfringens for such a period is relevant at the clinical level, because some cases of gangrene develop several days after trauma or surgery (16).
A sublethal inoculum of C. perfringens impairs the muscle regeneration process.
To characterize the process of muscle regeneration, a histological analysis of the infected muscles was performed at 7 days postinfection. At this time, the presence of regenerative myofibers, characterized by central nuclei (Fig. 2) near or within a fibrous matrix, was observed. In addition, cellular detritus from necrotic myofibers had not been removed despite the presence of an inflammatory infiltrate even at 14 and 30 days postinfection (Fig. 2).
FIG 2.

Alterations in muscle regeneration after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and muscle sections from samples collected at 7, 14, and 30 days were stained with HE. The inset at 7 days shows regenerative cells with central nuclei; asterisks indicate the presence of cellular debris embedded in a fibrous matrix at different times. Bars, 100 μm.
Due to the presence of regenerative cells, cell debris, and fibrotic areas, regeneration and necrosis areas were quantified 14 and 30 days after muscle injury (Fig. 3A). At 24 h postinfection, the area of myonecrosis was 48.3% ± 6.5% of the total area of the muscle (initial lesion), and at 14 days, the percentage of the area corresponding to nonregenerated muscle was 22.1% ± 4.5%, while only 27.9% ± 5.3% of the muscle was occupied by regenerating myofibers (Fig. 3A), hence evidencing a deficient muscular regeneration process.
FIG 3.
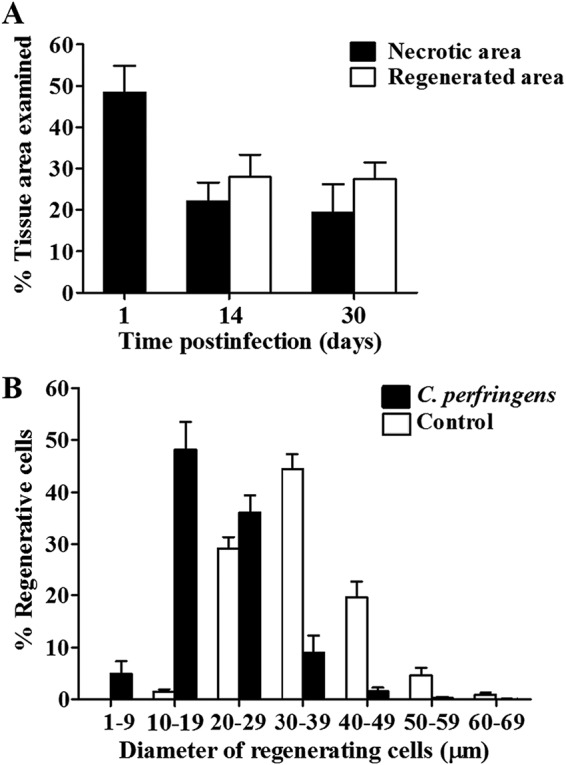
Muscle regeneration efficiency and regenerating myofiber sizes after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and muscle sections from samples collected at 1, 14, and 30 days were stained with HE. (A) The extent of myonecrosis was determined 1 day after infection as the percentage of the examined area corresponding to necrotic myofibers. The percentage of the necrotic area at 14 and 30 days corresponds to nonregenerated muscle, including cellular debris and fibrotic zones, while the percentage of the regenerated area was determined at 14 and 30 days postinfection as the area encompassing regenerative myofibers. (B) Quantification of regenerative myofibers according to their diameter at 30 days postinfection and comparison with controls injected with sterile PBS. Results show means ± SE.
Although a portion of the necrotic muscle was regenerated, when determining the size of the regenerative myofibers, it was observed that they differed significantly from the size of the fibers in control muscles at 30 days (Fig. 3B). While in the controls, 44.5% ± 2.8% of the myofibers had a diameter of between 30 and 39 μm, in the muscles infected with C. perfringens, 48.1% ± 5.4% of the myofibers had a diameter of between 10 and 19 μm. Moreover, 5.0% ± 2.4% of the regenerative cells in the infected muscles corresponded to myofibers with a small diameter of between 1 and 9 μm, indicating deficient regeneration (Fig. 3B).
Another characteristic of deficient regeneration is the replacement of myofibers by fibrous tissue. A stain specific for collagen fibers was made with Sirius red. Red areas (indicative of collagen deposition) around small regenerative cells were evident at 7 days postinfection in the necrotic muscle. These areas remained over time and showed greater intensity 14 days and 30 days (Fig. 4A) after muscle injury. When a quantitative analysis was performed at 30 days, the control showed only 3.1% ± 0.5% collagenous material in the total area, while in the muscles infected with a sublethal inoculum of C. perfringens, 23.5% ± 5.6% of the muscle corresponded to collagen deposition, which represents a significant difference between both groups (P < 0.05) (Fig. 4B). Thus, the deficient muscle regenerative outcome in this model correlates with increased collagen deposition, underscoring the replacement of myofibers by a fibrotic matrix.
FIG 4.

Collagen deposition in mouse gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens. (A) Sections from muscle samples collected 7, 14, and 30 days after injection were stained with Sirius red and Fast green. Red areas correspond to collagen fibers, while green areas correspond to other proteins. Controls were injected with sterile PBS. Bars, 100 μm. (B) Fibrotic muscle was quantified at 30 days as the percentage of the examined area corresponding to collagen. Results show means ± SE. *, P < 0.05 for samples with a statistically significant difference compared with the control.
A sublethal inoculum of C. perfringens alters capillary vessels and nerves in infected muscle.
To determine whether infection with C. perfringens causes injury to the vasculature, immunostaining was performed with antibodies specific for the endothelial growth factor receptor Flk-1. Six hours after infection with a sublethal inoculum of C. perfringens, a decrease in the number of capillary vessels was evident, and approximately 75% of the vessels disappeared at 24 h postinfection (Fig. 5A). The lack of capillary vessels was observed mainly in areas of myonecrosis at 6 h and 1 day postinfection; in addition, the structures that showed a positive staining signal in these areas were smaller than those of the control samples, highlighting that they were probably either nonfunctional capillary vessels or endothelial cell debris (Fig. 5A).
FIG 5.

Capillary vessels in gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens. (A) Sections from muscle samples collected 6 h, 1 day, and 30 days after injection were stained with anti-Flk-1 antibodies for detection by immunofluorescence. Arrowheads indicate areas without capillaries at 6 h and 1 day postinfection, while arrows point to the presence of capillaries in fibrotic areas at 30 days postinfection. Bars, 100 μm. (B and C) The numbers of capillaries per area (B) and per myofiber (C) were determined at different times after infection. Controls were injected with sterile PBS. Results show means ± SE. *, P < 0.05 for samples with a statistically significant difference compared with the control.
When the capillary vessels were quantified by area of tissue, the control showed an average of 805.3 ± 43.8 capillaries per mm2, while in the muscles infected with C. perfringens, the number of capillaries per square millimeter decreased significantly to 201.3 ± 51.5 at 6 h (P < 0.05) and 203 ± 52.9 at 24 h postinfection (P < 0.05) (Fig. 5B). Similar results were obtained for the capillary/myofiber ratio: while the control showed an average of 1.1 ± 0.1 capillaries per myofiber, in the infected muscles, the average significantly dropped to 0.4 ± 0.1 capillaries per myofiber at 6 h (P < 0.05) and to 0.30 ± 0.05 capillaries per myofiber at 24 h postinfection (P < 0.05) (Fig. 5C).
Despite the significant decrease in the density of capillary vessels at early times after infection, at 30 days, there were no significant differences in the numbers of capillaries per area or muscle tissue compared to the controls (Fig. 5B and C), and they were evident even in the fibrotic tissue (Fig. 5A), indicating that a revascularization process took place. However, it is likely that the early disruption of the microvascular network in the infected muscle affects the process of regeneration, since key steps in muscle regeneration require that an intact blood supply is present within the first hours after myonecrosis. Muscle healing is critically affected by ischemia associated with a deficient blood supply (17). The most critical consequence of ischemia is a decrease in the cellular energy supply (17), as energy is required for every aspect of the wound-healing process, such as protein synthesis, cell migration and proliferation, membrane transport, and growth factor production (17). Under these circumstances, the observed revascularization process may have occurred at a time when the muscle regenerative process had already been impaired.
Histological examination of the nerves in muscle sections at 24 h postinfection revealed damage to axons in intramuscular nerves, evidenced by a loss of intra-axonal material and the presence of digestion chambers in nerves. In some axons, there was a loss of myelin sheaths, but no damage was observed in the perineurium. The antibody used against the neurofilament of the axons allowed their detection by immunofluorescence (Fig. 6A). A decreased number of nerves was also observed 3 days after infection with C. perfringens (1.7 ± 0.3 nerves per mm2), compared with the control (3.2 ± 0.1 nerves per mm2) (P < 0.05) (Fig. 6B). In addition, a decreased number of axons within the nerves was observed (Fig. 6A): while in the control muscle, the average number of axons was 17.0 ± 1.3 axons per 1,000 μm2, in the affected muscles, it decreased significantly to 6.7 ± 1.1 axons per 1,000 μm2 (P < 0.05) (Fig. 6C). Despite the decrease in the number of axons observed at 3 days postinfection, when the samples were analyzed 30 days after muscle injury, no significant differences were found in relation to the controls for the number of nerves per area or for the number of axons per 1,000 μm2 (Fig. 6C). Hence, a reinnervation process ensued in muscle after myonecrosis.
FIG 6.

Innervation in mouse gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens. (A) Sections from muscle samples collected at 3 and 30 days were stained with anti-heavy neurofilament protein antibodies for detection by immunofluorescence. The arrow indicates nerve alterations at 3 days postinfection. Bars, 100 μm. (B and C) The number of nerves per area (B) and the number of axons inside nerves (C) were determined at different times after infection. Controls were injected with sterile PBS. Results show means ± SE. *, P < 0.05 for samples with a statistically significant difference compared with the control.
A sublethal inoculum of C. perfringens increases the expression of mediators of the inflammatory response and fibrosis in infected muscle.
To evaluate the immune response after infection with 1 × 106 CFU of C. perfringens, the expression levels of proinflammatory cytokines (IL-1β, IL-6, TNF-α, and IFN-γ) and anti-inflammatory cytokines (IL-4, IL-10, and IL-13) were determined by reverse transcription-PCR (RT-PCR) and enzyme-linked immunosorbent assays (ELISAs) (Fig. 7). Furthermore, the expression levels of neutrophil (MIP2 and KC) and macrophage (monocyte chemoattractant protein 1 [MCP-1]) chemoattractant cytokines were also analyzed (Fig. 8).
FIG 7.

Cytokine expression in mouse gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 4 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and the expression of cytokines in infected muscles was measured by RT-PCR (A) and by ELISAs (B) at different times. The results obtained with three normalization genes (GADPH, RPL13A, and RNSP1) were incorporated in panel A, based on 2−ΔΔCT calculation. Controls were injected with sterile PBS. Results show means ± SE. *, P < 0.01; **, P < 0.05 (for samples with a statistically significant difference compared with controls). ND, not determined.
FIG 8.

Chemoattractant expression in mouse gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 4 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and the expression of chemoattractants in infected muscles was measured by RT-PCR at different times. The results obtained with three normalization genes (GADPH, RPL13A, and RNSP1) were incorporated, based on 2−ΔΔCT calculation. Controls were injected with sterile PBS. Results show means ± SE. *, P < 0.01; **, P < 0.05 (for samples with a statistically significant difference compared with controls).
Significantly increased expression levels of mRNAs for IL-1β and IL-6 were observed at 6 h postinfection compared to controls, which lasted up to at least 48 h (P < 0.01) (Fig. 7A). The expression of TNF-α also showed a significant increase from 6 h on (P < 0.05) and remained elevated for at least 48 h postinfection (P < 0.01) (Fig. 7A). Furthermore, a significant increase in the expression of IL-10 was observed in comparison to controls from 1 day (P < 0.01) up to 6 days (P < 0.05), reaching a maximum peak 24 h after infection with the bacteria (P < 0.01) (Fig. 7A). When immunoassays were carried out to quantify the levels of various cytokines in the muscle, confirmatory results were obtained, since the level of IL-1β remained high for at least 48 h postinfection, and that of IL-6 increased significantly from 6 to 48 h postinfection (P < 0.01), reaching a maximum level at 6 h, while TNF-α reached a maximum level at 48 h postinfection (P < 0.01). Similarly, in a lethal experimental infection, the transcription of TNF-α, IL-1β, and IL-6 was induced 1.5 h after inoculation of 109 C. perfringens vegetative cells (2). In contrast, IFN-γ showed a significant increase only at the transcriptional level at 6 h postinfection (P < 0.01) but not at the protein level (Fig. 7). Interestingly, in a characterization of the intestinal immune response to C. perfringens infection in broiler chickens, it was reported that the production of IFN-γ does not increase either (18, 19).
There was not a significant difference in the levels of expression of IL-4 between infected mice and controls, whereas for IL-13, there was a significant difference only at 6 days postinfection (P < 0.05) (Fig. S2 and S3). On the other hand, IL-10 showed a significant increase 2 days after infection (P < 0.05), and the amount of protein remained slightly larger than in controls until day 6 (Fig. 7B).
The chemokines MIP2 and KC showed a significant increase in expression from 6 h to 48 h after infection with a sublethal inoculum of C. perfringens in comparison with controls (P < 0.01) (Fig. 8). The highest KC expression level occurred at 6 h postinfection (maximum peak) and remained high even at 48 h. The highest MCP-1 expression level occurred at 6 h postinfection, and although it decreased at 24 h postinfection and increased again at 48 h, it was significantly higher than that observed in the controls at all evaluated times (Fig. 8).
Because TGF-β1 has been associated with fibrosis in the process of muscle regeneration, its gene expression and protein concentration were analyzed. A bimodal behavior was observed for TGF-β1 at both the transcriptional and protein levels. When the relative mRNA expression level was analyzed, a significant increase was observed 1 day after infection (P < 0.05) in relation to the control, and expression decreased at 2 days but increased again at 3 days postinfection (P < 0.01), remaining elevated at least until 6 days postinfection (P < 0.05) (Fig. 9A).
FIG 9.

TGFB1 expression in mouse gastrocnemius after experimental infection with a sublethal inoculum of C. perfringens. Groups of 4 to 6 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and the expression of TGF-β1 in infected muscles was measured by RT-PCR (A) and by ELISAs (B) at different times. Controls were injected with sterile PBS. Results show means ± SE. *, P < 0.01; **, P < 0.05 (for samples with a statistically significant difference compared with the control).
At the protein level, a significantly larger amount of TGF-β1 was detected in the infected muscle than in the controls 1 day after infection (P < 0.01), and although there was a decrease at 3 days, the protein concentration remained elevated at 6 days postinfection in infected muscles, compared to the controls (P < 0.05) (Fig. 9B). Thus, the increase in TGF-β1 correlates with the deficient regeneration observed after infection with a sublethal dose of C. perfringens. This fits with the known role of this mediator, which favors collagen deposition, i.e., fibrosis, and inhibits myogenic cell differentiation.
A sublethal inoculum of C. perfringens alters the influx of neutrophils and macrophages to infected muscle.
Neutrophils are the first cells that reach the muscle after myonecrosis and are followed by M1 and M2 macrophages before the resolution of myonecrosis (8). In injuries induced by a lethal inoculum of C. perfringens, the absence of inflammatory cells in the infected muscle and the presence of neutrophils attached to the endothelium due to the overexpression of adhesion molecules have been reported (1). However, when a sublethal inoculum of this bacterium was used, an inflammatory infiltrate was observed in the muscle, with the presence of neutrophils, both aggregated and dispersed, in the necrotic muscle (Fig. 10A). The presence of neutrophils was evident 5 h after infection (Fig. 10A), when they reached a number of 505.2 ± 38 cells per mm2 (Fig. 10B). At 24 h, their number was 548.4 ± 56.4 cells per mm2; at 3 days, it was 342.2 ± 36.6 cells per mm2; and at 5 days after infection, numbers dropped to 81.7 ± 9.9 cells per mm2 (Fig. 10A and B). These observations agree with the above-described pattern of early neutrophil influx as the first wave of inflammatory cells in injured tissues.
FIG 10.

Recruitment of neutrophils at the site of injury after experimental infection with a sublethal inoculum of C. perfringens. (A) Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and the presence of neutrophils was determined using antielastase antibodies at the indicated times. (B) For each time, the number of cells corresponding to neutrophils was determined. PMNs, polymorphonuclear leukocytes. Bars, 50 μm.
M1 macrophages immunostained with anti-iNOS were detected in small amounts during the study period. Their density peaked at 2 to 3 days postinfection (142.6 ± 18.1 cells per mm2) (Fig. 11A and B) and declined further until 5 days postinfection (25.4 ± 8.5 cells per mm2) (Fig. 11B). M2 macrophages immunostained with antiarginase antibodies reached a maximum density at 7 days postinfection (616.2 ± 179.4 cells per mm2) (Fig. 11A and B) and were detected even at 14 days postinfection (41.8 ± 18.1 cells per mm2) (Fig. 11B).
FIG 11.

Recruitment of M1 and M2 macrophages to the site of injury after experimental infection with a sublethal inoculum of C. perfringens. Groups of 3 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens, and the presence of M1 and M2 macrophages was determined using anti-iNOS and antiarginase antibodies, respectively, at the indicated times. (A) The highest intensity observed was related to the maximum number of cells in the muscle. (B) For each time, the number of cells corresponding to M1 and M1 macrophages was determined. Bars, 50 μm.
Little influx of M1 macrophages (iNOS positive [iNOS+]) in the muscle of infected animals was evident (Fig. 11A and B), which suggests alterations in this population of inflammatory cells after infection with a sublethal inoculum of C. perfringens. This could be due to the observed absence of IFN-γ. Accordingly, in experiments in which IFN-γ signaling has been blocked in injured muscles, there is a reduction in the expression in macrophages of transcripts that indicate the activation of the M1 phenotype, such as iNOS (20). The classical inflammation response after tissue injury occurs within the following 5 days (21). Normal remodeling of the muscle is very dependent on the timing of the M1 and M2 macrophage response (22, 23). The transition to M2 macrophages is critical for muscle regeneration, and prolonged inflammation results in fibrosis (22, 23).
Macrophages play a central role in wound healing directing T-cell activation, promoting stem cell and progenitor cell migration, activating angiogenic responses, and guiding extracellular matrix remodeling (24). They phagocytose cellular debris generated during tissue remodeling, recycling important molecular components to be reused (25). The phenotype of macrophages can highly influence the progression of a disease or injury (25). The same macrophages can switch between proinflammatory and prohealing states depending on the surrounding environmental cues, and the switch from the M1 to the M2 phenotype occurs in stages in response to the upregulation of IL-4 and IL-13 (25). M2 macrophages express high levels of IL-10, low levels of IL-12 (3), and prohealing states depending on the environment (25). IL-10, IL-4, and IL-13 have defined roles in the activation of M2 macrophages, and they comprise several subpopulations that determine the outcome of the regenerative process (25–27). M2a macrophages are activated by IL-4 and IL-13, secrete IL-10, express arginase 1, and promote wound healing and muscle regeneration, while M2b macrophages are activated by immune complexes or Toll-like receptors and release anti-inflammatory cytokines associated with the Th2 response (27, 28). M2c macrophages are activated by IL-10, release cytokines that deactivate the M1 phenotype, and promote the proliferation of nonmyeloid cells and the deposition of the extracellular matrix (8, 27).
Recruited monocytes fail to differentiate adequately in tissue remodeling in muscular dystrophy (29), insulin resistance (30), and advanced age (31), leading to unsuccessful remodeling and tissue repair (32–34). Similar to muscular dystrophies, muscle loss due to aging is likely caused by a prolonged inflammatory response, characterized by a higher expression level of IL-1β. If the M1 response lasts too long, the new tissue is highly fibrotic, leading to decreased function. In myonecrosis induced by a sublethal inoculum of C. perfringens, altered muscle regeneration was observed in association with both a delayed recruitment of macrophages to the site of infection and altered production of cytokines.
After infection with a sublethal inoculum of C. perfringens, a significant increase in the gene expression level of IL-10 in the muscle was observed from 1 day (maximum peak) to 6 days after infection, and a significant increase at the protein level was observed from 2 days after infection (maximum peak) (Fig. 7A and B). Although no significant changes were detected in the expression of IL-4, for IL-13, an increase in the gene expression level was observed after 6 days (Fig. S2 and S3). The influx of both M1 and M2 macrophages into the infected muscle was observed from the first day postinfection. The presence of M1 macrophages in the infected muscle was scarce, having its maximum peak by 3 days, whereas M2 macrophages were more abundant than M1 macrophages in the infected muscle, having a maximum peak by 7 days and remaining high even after 14 days (Fig. 11B). The altered arrival of M1 and M2 macrophages into the infected muscle after infection in our model is likely to alter the muscle regeneration process.
When analyzing the arrival and permanence of the different cell populations using a sublethal dose of C. perfringens vis-à-vis the typical inflammatory response reported in the literature (Fig. 12), there is a prolongation of the inflammatory response in the C. perfringens model. The neutrophils remain in the infected muscle for up to 5 days, which is complemented with high expression levels of proinflammatory cytokines for up to at least 2 days. In addition, there is a limited arrival of M1 macrophages, which remain within the infected muscle for a longer time span than expected. We proposed that given the absence of IFN-γ in the acute phase of infection, the number and activity of M1 macrophages were affected. One possible cause of this could be an altered function of neutrophils due to the direct effect of C. perfringens toxins on these cells. It has been reported that perfringolysin O (PFO) induces cytotoxicity in neutrophils (35), and C. perfringens phospholipase C (CpPLC) interferes with the replacement of mature neutrophils in the peripheral circulation, inhibiting their maturation (36). Additionally, vascular injury caused by C. perfringens infection could also contribute to the prevention of the influx of cells of the inflammatory response. Thus, the direct and indirect effects of infection with C. perfringens on inflammatory cells could contribute to inducing a deficient regenerative response.
FIG 12.

Time course of changes in cytokine expression and intramuscular myeloid cell populations after acute muscle injury, compared with those after experimental infection with a sublethal inoculum of C. perfringens. (A) Expected cytokine expression and changes of myeloid cell populations after acute muscle injury. (Adapted from reference 27 with permission of the publisher.) (B) Cytokine expression and changes of myeloid cell populations after intramuscular infection with a sublethal inoculum of C. perfringens. Asterisks indicate critical points for muscle regeneration that were affected in this model.
After muscle injury, the regeneration process is mediated by a specific type of stem cells, SC, but it also involves the interaction of these myogenic cells with other resident cells, inflammatory cells, blood vessels, nerves, and the extracellular matrix (14, 37). There are at least three basic requirements for the process of muscle regeneration to occur: (i) adequate blood flow in the regenerative muscle, (ii) innervation of regenerative cells, and (iii) permanence of the basal lamina around the necrotic myofibers, which serves as a scaffold and substrate for regeneration (7). Impairment of any of these contributory factors results in defective muscle regeneration.
The importance of blood flow lies, on one hand, in the inflow of the inflammatory infiltrate to the site of the lesion and, in addition, the provision of oxygen, nutrients, and ATP to the regenerating muscle. When vascular density was analyzed after infection with a sublethal inoculum of C. perfringens, it was observed that the microvasculature is affected within the first hours after infection, a time that coincides with the arrival of neutrophils, the first inflammatory cells present in the infected muscle. Although the presence of an inflammatory infiltrate is evident, vascular injury and the direct effects of CpPLC and PFO on cells of the immune system can affect the migration of inflammatory cells to the myonecrotic area, which results in deficient debris removal. Thus, the combination of a direct inhibitory action of C. perfringens toxins and a disruption of the microvascular network is likely to affect the timely arrival of inflammatory cells. It has been reported that disturbances that affect the removal of dead cells delay the process of muscle regeneration (8, 38). This may be due to the fact that the persistence of cellular debris becomes a physical obstacle for muscle regeneration.
Another consequence of injury to the vascular system in the regenerative process is a limitation in the available oxygen, which is critical for muscle healing. Hypoxia can also promote the proliferation of bacteria and the further development of gas gangrene, hence generating a vicious cycle (39). In studies carried out with snake venoms, alteration of the microvasculature affects the regenerative process, favoring the replacement of muscle by fibrotic muscle; moreover, regenerative myofibers have small diameters (14, 40, 41). Although our findings showed that the capillary density is restored at 30 day, it is possible that the severe injury to blood vessels induced by C. perfringens in the first hours after infection (Fig. 5) could be one of the causes behind the impaired regenerative process.
Innervation is another requirement for a successful muscle regeneration process. The proliferation and fusion of myogenic cells occur in muscles that regenerate in the absence of nerves and in those in which the nerves are intact (42). However, although the initial events of the muscle regeneration process can occur in the absence of innervation, the latter is required for growth and recovery of muscle function (42, 43). In the absence of innervation, regenerating cells do not reach their maturity. In this model, although the density and structure of the nerves were affected with a sublethal inoculum of C. perfringens 3 days after infection, innervation was recovered at 30 days (Fig. 6B). Consequently, innervation does not appear to be a cause of deficient regeneration after C. perfringens-induced myonecrosis. However, the question remains as to whether C. perfringens toxins induce injury at the synaptic level and neuromuscular transmission, as has been reported for the lethal toxin of Clostridium sordellii (44).
For a successful muscle regeneration process, scaffolding is required to maintain the position of the myofibers. The processes of muscle necrosis and regeneration involve a complex turnover of the extracellular matrix, which is a determinant of an effective regenerative response. The process of matrix deposition after myonecrosis, although initially beneficial, affects the regenerative process if it continues without control, resulting in the permanent accumulation of collagen around the myofibers, which may even lead to muscle replacement by fibrous tissue (45). It has been reported that MCP-1 is a profibrotic mediator whose neutralization reduces the extent of fibrosis (46, 47). Fibroblasts contribute to the formation of fibrous muscle by the production and accumulation of components of the extracellular matrix, such as hyaluronic acid, fibronectin, proteoglycans, and interstitial collagens (45). When histological sections of animals infected with a sublethal inoculum of C. perfringens were analyzed, it was observed that at 30 days, almost 25% of the muscle area corresponded to collagen that accumulated in areas where the regenerative process was deficient (Fig. 4). This may be associated with the overexpression of some mitogenic chemokines and cytokines, produced by neutrophils and macrophages, which are also involved in the mechanism of fibrogenesis.
Experimental models of fibrosis have documented potent antifibrotic properties for cytokines associated with the Th1 response, such as IFN-γ (47); thus, the absence of this cytokine in our study model could be associated with the overproduction of extracellular matrix. On the other hand, IL-10 and TGF-β activate a subpopulation of M2 macrophages that promotes the deposition of extracellular matrix, leading to fibrosis under different pathogenic conditions (8, 45). TGF-β has been associated with the development of fibrosis in a number of diseases, as it is one of the main activators of macrophages and fibroblasts; of the three isotypes of TGF-β in mammals, muscle fibrosis is mainly attributed to the TGF-β1 isoform (47, 48). In this model, an increase in the level of TGF-β1 was evidenced in both early and late time intervals (Fig. 9) and hence may be related to the collagen accumulation evidenced from 7 days after infection.
IFN-γ has a relevant function in muscle regeneration, since this molecule directly regulates myogenic cells in their differentiation process (8, 20). Therefore, the alterations in the regenerative response observed in this study could also be a consequence of the absence of IFN-γ. In general, infection with a sublethal inoculum of C. perfringens stimulates the expression of chemoattractants and cytokines that stimulate the arrival of different inflammatory cells to the site of the lesion. However, it has been previously reported that the bacterium generates toxins capable of directly affecting neutrophils and macrophages, and as a consequence, their function is altered. Additionally, the recruitment of M1 macrophages to the infection site was affected by the lack of IFN-γ. On the other hand, the bacterium causes injury to the microvasculature, which affects the migration of phagocytes into the necrotic muscle. The alteration of the influx of different inflammatory cells leads to deficient cell debris removal. The above-described observations, coupled with the release of factors that stimulate the overproduction of collagen, such as TGF-β1, could explain the deficient muscle regeneration characterized by fibrosis and small regenerative myofibers.
Although muscle regeneration is highly efficient in many clinical and experimental models, provided that basic requirements are fulfilled, muscle regeneration after myonecrosis induced by a sublethal inoculum of C. perfringens is deficient, probably due to altered events in the initial phases following injury, which are critical and influence the overall outcome of the regeneration process. The inflammatory response induced by C. perfringens is characterized by alterations in the early influx of inflammatory cells, mainly neutrophils, and M1 and M2 macrophages to the site of infection. These cells remain in the muscle for prolonged periods of time and are likely to be functionally impaired. In the case of M1 macrophages, their limited recruitment is possibly due to the low levels of IFN-γ produced. Our findings highlight several aspects of the regenerative muscle response which are affected in the experimental model of gas gangrene used. Understanding the mechanisms of the inflammatory and regenerative responses in the muscle after infection by C. perfringens could be crucial for understanding the reasons behind such deficient regenerative responses and for devising innovative therapeutic strategies for this severe muscular pathology.
MATERIALS AND METHODS
Bacterial culture.
C. perfringens (strain JIR325) was grown on brain heart infusion (BHI) broth in an anaerobic chamber until an optical density at 600 (OD600) of 0.47 was reached. The culture was centrifuged and washed two times with 0.12 M NaCl–0.04 M phosphates (pH 7.2) (phosphate-buffered saline [PBS]). The number of CFU per 100 μl was determined by plating serial 10-fold dilutions on BHI agar plates supplemented with yolk.
Experimental infection.
CD-1 mice with a body weight of 18 to 20 g were injected in the left gastrocnemius with 1 × 106 CFU of C. perfringens JIR325 diluted in 100 μl of PBS. All the procedures involving the use of animals in this study were approved by the Institutional Committee for the Care and Use of Laboratory Animals (CICUA) of the Universidad de Costa Rica (approval number CICUA-098-17) and met Animal Research Reporting In Vivo Experiments (ARRIVE) guidelines (59) and the International Guiding Principles for Biomedical Research Involving Animals of the Council of International Organizations of Medical Sciences (CIOMS) (60).
CK activity assay.
The plasma activity of creatine kinase (CK), an intracellular enzyme present in high concentrations in skeletal muscle, was used as a myotoxicity indicator. Mice were anesthetized and bled from the ocular plexus at 5 and 24 h postinfection, and blood samples were collected into heparinized capillary tubes. The CK activity in plasma was determined using the CK-NAC UV Unitest enzyme assay (Wiener Lab, Argentina), according to the manufacturer’s instructions.
Determination of the persistence of C. perfringens in muscle.
Groups of 4 CD-1 mice were injected intramuscularly with 1 × 106 CFU of C. perfringens and sacrificed at 1, 5, and 9 days postinfection. Controls were injected with sterile PBS. Gastrocnemius muscles were dissected under aseptic conditions, weighed, finely cut in a petri dish with sterile knives, suspended in sterile PBS, and shaken with a vortex; serial dilutions were made; 100 μl of each dilution was plated in duplicate on BHI agar plates supplemented with egg yolk; and the number of CFU per milligram of infected tissue was determined after 24 h.
Histological analysis.
For histological analysis, mice were injected with 1 × 106 CFU of C. perfringens JIR325 diluted in 100 μl of PBS or with sterile PBS. Animals were sacrificed in the phase of muscle necrosis at 1, 5, and 24 h postinfection and in times that cover the different phases of the process of muscle regeneration, i.e., 3, 5, 7, 14, and 30 days postinfection. The injected gastrocnemius muscles were dissected and placed in a zinc fixative solution (calcium acetate at 3 mM, zinc acetate at 27 mM, zinc chloride at 36 mM, and Tris buffer at 0.1 M [pH 7.4]) for at least 48 h at 4°C. The dissected muscles were dehydrated in ethanol, placed in xylene, and embedded in paraffin. Three nonconsecutive, 4-μm-thick (at least 40 μm apart) sections were used to analyze different points from the midregion of each muscle and placed on glass slides. Sections were deparaffinized in xylene, hydrated in decreasing concentrations of ethanol and distilled water, and stained with hematoxylin and eosin (HE) for microscopic evaluation. Images of total muscle were captured from each section using an Evolution MP camera (Media Cybernetics, USA) and analyzed using Image Pro 6.3 image analysis software (Media Cybernetics, USA). The necrotic area was estimated in samples collected at 24 h postinfection, considering the percentage of the area observed corresponding to hypercontracted necrotic myofibers. The average necrotic area in three nonconsecutive sections was calculated per muscle. Areas of regeneration and of a lack of regeneration were estimated in samples collected at 14 and 30 days postinfection; areas of regeneration corresponded to the percentage of the examined area characterized by the presence of regenerating myofibers (with centrally located nuclei), while nonregenerative areas were defined as the percentage of the examined area corresponding to cell debris and fibrotic muscle. The diameters of regenerating myofibers were also determined. The average diameter of the regenerative fibers in three nonconsecutive sections was calculated per muscle.
Collagen staining.
Groups of 3 mice were injected with C. perfringens JIR325 or with sterile PBS and sacrificed at 7, 14, and 30 days postinfection. Increments of collagen in the muscle were detected by staining with Direct Red 80 (Sigma-Aldrich, USA) (0.1% in a saturated picric acid solution), which stains collagen, and Fast green FCF (0.1%) (Sigma-Aldrich, USA), which stains other proteins, for 1 h at room temperature, according to a previously described procedure (49). Slides were washed with acidified water (5 ml of glacial acetic acid per liter), dehydrated, and cleared in xylene, and microscopic evaluation was performed. Images of three sections were captured per muscle using an Evolution MP camera (Media Cybernetics, USA) and analyzed using Image Pro 6.3 image analysis software (Media Cybernetics, USA). The percentage of fibrosis (collagen deposition) in total muscle was quantified at 30 days postinfection, using ImageJ 1.51K image analysis software (National Institutes of Health, USA).
Quantitative PCR.
Relative expression levels of transcripts coding for IL-1β, IL-6, TNF-α, IFN-γ, TGF-β1, IL-13, IL-10, MIP2, KC, and MCP-1 were determined at time points similar to those reported previously for other muscle injury models (6, 8, 11). Groups of six mice were injected with C. perfringens JIR325 or with sterile PBS in the left gastrocnemius and sacrificed at 6 h and 1, 2, 3, and 6 days postinfection. The left gastrocnemius muscles were rapidly dissected and ground under sterile conditions. Total RNA was extracted using TRIzol reagent (Ambion, Invitrogen), according to the manufacturer’s instructions, and quantified using a NanoDrop 2000c spectrophotometer (Thermo Scientific, USA). RNA was retrotranscribed to cDNA with a RevertAid H Minus first-strand cDNA synthesis kit (Fermentas, Thermo Fisher Scientific) in a 2720 thermal cycler (Applied Biosystems) using 4 μg of total RNA and random-hexamer primers. A total of 160 ng of cDNA was used for each reaction for quantitative real-time PCR (qPCR) using LightCycler 480 SYBR green I master (Roche Diagnostics) and a LightCycler 480 real-time PCR device (Roche Diagnostics). The selected primers used to assess inflammatory-response-specific genes by PCR are listed in Table 1. Genes used as reference genes were the housekeeping genes glyceraldehyde-3-phosphate dehydrogenase (GAPDH), RNA-binding protein S1 (RNSP1), and ribosomal protein L13A (RPL13A) (50). The cycle number at which the reaction crossed an arbitrarily placed threshold (CT) was determined, and the relative expression level of each gene regarding the mean expression levels of control genes was determined using the 2−ΔΔCT method, where ΔCT = CTgene − CTcontrol genes (mean) and ΔΔCT = ΔCTmice + C. perfringens − ΔCTcontrol mice (mean) (51). Left gastrocnemius muscles of healthy mice injected with sterile PBS were used as controls.
TABLE 1.
Primers used to assess the expression levels for inflammatory mediators
| Gene | Primer directionb | Primer sequence (5′–3′) | Amplicon size (bp) | Reference |
|---|---|---|---|---|
| MIP2 | Fwd | AGGGCGGTCAAAAAGTTTGC | 194 | 52 |
| Rev | CGAGGCACATCAGGTACGAT | |||
| KC | Fwd | GCTGGGATTCACCTCAAGAA | 180 | 53 |
| Rev | TCTCCGTTACTTGGGGACAC | |||
| MCP-1 | Fwd | ATGCAGTTAATGCCCCACTC | 167 | 54 |
| Rev | TTCCTTATTGGGGTCAGCAC | |||
| IL-6 | Fwd | GAACAACGATGATGCACTTGC | 154 | 37 |
| Rev | CTTCATGTACTCCAGGTAGCTATGGT | |||
| IL-10 | Fwd | CAAGGAGCATTTGAATTCCC | 157 | 36 |
| Rev | GGCCTTGTAGACACCTTGGTC | |||
| TNF-α | Fwd | CTTCTGTCTACTGAACTTCGGG | 163 | 37 |
| Rev | CACTTGGTGGTTTGCTACGAC | |||
| IFN-γ | Fwd | TGCTGATGGGAGGAGATGTCT | 101 | 55 |
| Rev | TTTCTTTCAGGGACAGCCTGTT | |||
| IL-1β | Fwd | TGACGTTCCCATTAGACAACTG | 231 | 23 |
| Rev | CCGTCTTTCATTACACAGGACA | |||
| TGF-β1 | Fwd | GAGACGGAATACAGGGCTTTC | 240 | 56 |
| Rev | TCTCTGTGGAGCTGAAGCAAT | |||
| IL-13 | Fwd | TCTTGCTTGCCTTGGTGGTCTCGC | 220 | 57 |
| Rev | GATGGCATTGCAATTGGAGATGTTG | |||
| GAPDHa | Fwd | AACCTGCCAAGTATGATGAC | 191 | 58 |
| Rev | ATACCAGGAAATGAGCTTGA | |||
| RPL13Aa | Fwd | CCTGCTGCTCTCAAGGTTGTT | 146 | 36 |
| Rev | CGATAGTGCATCTTGGCCTTT | |||
| RNSP1a | Fwd | AGGCTCACCAGGAATGTGAC | 196 | 36 |
| Rev | CTTGGCCATCAATTTGTCCT | |||
Reference gene used for data normalization.
Fwd, forward; Rev, reverse.
ELISAs.
The IL-1β, IL-6, TNF-α, IFN-γ, TGF-β1, IL-4, and IL-10 protein contents in the muscle were determined by a capture ELISA at time points at which their expression had been reported in other studies of muscle injuries (6, 8, 11). Groups of 6 mice were injected intramuscularly in the left gastrocnemius with C. perfringens JIR325 or with sterile PBS. At various time intervals (6 h and 1, 2, 3, and 6 days), mice were killed, and the injected gastrocnemius muscles were dissected, frozen with liquid nitrogen, and homogenized in a sterile pyrogen-free saline solution with Complete EDTA-free protease inhibitor (Roche Diagnostics). Muscle homogenates were centrifuged, and the supernatants were collected and stored at −70°C. IL-1β, TNF-α, IFN-γ, IL-4, and IL-10 levels were quantitated using ELISA kits from R&D Systems (USA), while IL-6 and TGF-β1 levels were quantitated using ELISA kits from eBioscience (San Diego, CA, USA), according to the manufacturers’ instructions. Left gastrocnemius muscles of healthy mice injected with sterile PBS were used as controls.
Quantification of inflammatory cells.
In order to quantify the numbers of different inflammatory cells at different time points during the regenerative process, groups of three mice were injected with C. perfringens JIR325 or with sterile PBS (negative controls) and sacrificed during the acute phase (1, 3, 5, and 24 h postinfection) and the chronic phase (3, 5, 7, and 14 days postinfection). Injected muscles were dissected and embedded in paraffin, as described above. For each muscle, three nonconsecutive, 4-μm-thick (at least 40 μm apart) sections were used to analyze different points from the midregion of each muscle. The sections were placed on positively charged glass slides, deparaffinated in xylene, and hydrated. Fluorescence immunohistochemistry (IHC) for neutrophils was performed using anti-neutrophil elastase rabbit polyclonal antibody (pAb) (EMD Millipore), antigen retrieval was carried out by placing the slides in citrate buffer (pH 6) at 50°C for 10 min, and blockage steps were performed with the Dako Cytomation biotin blocking system (Dako, USA) as well as with serum-free protein block (Dako, Denmark), according to the manufacturers’ instructions. Sections were incubated overnight with anti-neutrophil elastase antibody (diluted 1:10) at 4°C in a wet chamber. Next, sections were washed with PBS and incubated with biotinylated polyclonal goat anti-rabbit immunoglobulins (Dako, Denmark) (diluted 1:200) for 1 h at room temperature. After washing with PBS, sections were incubated with streptavidin-Alexa Fluor 488 (Invitrogen) (diluted 1:200) for 30 min at room temperature. Finally, nuclear staining was performed with bis-benzamide Hoechst stain (Sigma, USA) in a final concentration of 0.5 μg/ml. For M1 macrophage immunohistochemical staining, the protocol was similar to the one used for neutrophils, with the following modifications: antigen retrieval was carried out using 0.6 U/ml of proteinase K (Fermentas, Thermo Fisher Scientific) for 5 min at room temperature, and the primary antibody used was rabbit polyclonal anti-iNOS (Abcam, USA) (diluted 1:75). M2 macrophages were stained with a goat polyclonal antiarginase antibody (Abcam, USA). Briefly, antigen retrieval was carried out using proteinase K (Fermentas, Thermo Fisher Scientific), the blockage step was performed with serum-free protein block (Dako, Denmark) for 10 min at room temperature, and sections were then incubated overnight with the primary antibody (diluted 1:50) at 4°C in a wet chamber. Donkey F(ab′)2 anti-goat IgG(H+L) (Alexa Fluor 488) (preadsorbed; Abcam, USA) (diluted 1:200) was used as a secondary antibody, sections were incubated for 1 h at room temperature, nuclear staining was then performed, and three sections per muscle were evaluated microscopically. Images were captured using an Evolution MP camera (Media Cybernetics, USA), and the number of cells per square millimeter of muscle was determined with Image Pro 6.3 software (Media Cybernetics, USA).
Quantification of capillaries in muscle tissue.
Groups of three mice were injected with C. perfringens JIR325 or with sterile PBS (negative controls) and sacrificed at times covering the degenerative phase (6 and 24 h) and the regenerative phase (30 days). Injected muscles were dissected and embedded in paraffin, as described above. From the midregion of each muscle, three nonconsecutive sections of a 4-μm thickness (at least 40 μm apart) were obtained and placed on positively charged glass slides, and sections were then deparaffinized in xylene and hydrated. Fluorescence immunohistochemistry was carried out in order to detect capillary vessels. For antigen retrieval, proteinase K (Fermentas, Thermo Fisher Scientific) was used for 5 min at room temperature, and blockage steps were then performed with H2O2 (3%) for 30 min, the Dako Cytomation biotin blocking system (Dako, USA) for 10 min each, and serum-free protein block (Dako, Denmark) for 1 h. Sections were incubated overnight with purified anti-mouse Flk-1 (IHC), a receptor for vascular endothelial growth factor (VEGF) (BD Pharmingen) (diluted 1:50), at 4°C in a wet chamber. Next, sections were washed with PBS and incubated with polyclonal goat anti-rabbit immunoglobulins (biotinylated) (Dako, Denmark) (diluted 1:200) for 1 h at room temperature. In order to amplify the signal, the Biotin-XX Tyramide SuperBoost kit (Invitrogen) was used according to the manufacturer’s instructions. Streptavidin-Alexa Fluor 488 (Invitrogen) (diluted 1:300) was used as the final fluorophore for 30 min at room temperature. Nuclear staining was performed with bis-benzamide Hoechst stain (Sigma, USA) in a final concentration of 0.5 μg/ml. Samples were analyzed microscopically, and images were captured using an Evolution MP camera (Media Cybernetics, USA). Capillaries were identified as immunostained ring structures with diameters of 5 to 10 μm with a central lumen. The antibody used is specific for endothelial cells. The number of capillary vessels was determined manually, and the average from three nonconsecutive sections was calculated per muscle. Ratios of capillary vessels per square millimeter and numbers of capillary vessels/numbers of myofibers were estimated. Sizes and areas were calculated using Image Pro 6.3 software (Media Cybernetics, USA).
Quantification of nerves.
Groups of three mice were injected with C. perfringens JIR325 or with sterile PBS (negative controls) and sacrificed at 3 and 30 days postinfection, to have a view of the end of the degenerative and the regenerative phases. Muscles were processed as described above and embedded in paraffin. For each muscle, three nonconsecutive, 4-μm-thick (at least 40 μm apart) sections were obtained and placed on positively charged glass slides, deparaffinized in xylene, and hydrated. Antigen retrieval was performed using proteinase K (Fermentas, Thermo Fisher Scientific) for 5 min at room temperature. For blockage steps, the Dako Cytomation biotin blocking system and serum-free protein block (Dako, Denmark) were used according to the manufacturers’ instructions. Sections were incubated overnight with an anti-200-kDa neurofilament heavy antibody neuronal marker (Abcam, USA) (diluted 1:1,000) at 4°C in a wet chamber. Next, sections were washed with PBS and incubated with polyclonal goat anti-rabbit immunoglobulins (biotinylated) (Dako, Denmark) (diluted 1:200) for 1 h at room temperature. After washing with PBS, sections were incubated with streptavidin-Alexa Fluor 488 (Invitrogen) (diluted 1:200) for 30 min at room temperature. Finally, nuclear staining was performed with bis-benzamide Hoechst stain (Sigma, USA) in a final concentration of 0.5 μg/ml. Microscopic images of the complete histological sections were captured using an Evolution MP camera (Media Cybernetics, USA). Positively stained structures with areas of between 50 and 8,000 μm2 were considered. The number of nerves per square millimeter and the number of axons per square micrometer were determined manually, and the average number of nerves in three nonconsecutive sections was calculated per muscle. Sizes and areas were calculated with Image Pro 6.3 software (Media Cybernetics, USA).
Statistical analysis.
Data were analyzed by using IBM SPSS statistics software or GraphPad Prism version 5.00 for Windows. CK, RT-PCR, and ELISA data were analyzed using a Kruskal-Wallis test and a Dunn test for post hoc analysis. The Mann-Whitney U test was used for analysis related to muscle regeneration processes, area, myofibers, capillary vessels, and nerve quantification.
Supplementary Material
ACKNOWLEDGMENTS
This study was funded by the Vicerrectoría de Investigación, Universidad de Costa Rica (project no. 741-B8-135 to M.F.-D.). We also acknowledge support from the Ministerio de Ciencia, Tecnología y Telecomunicaciones (MICITT). The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. We declare that no competing interests exist.
M.F.-D. conceptualized, supervised, and obtained funds for the project; M.F.-D., A.A.-G., A.M.Z.-P., and C.S. conceived and designed experiments; A.M.Z.-P., M.F.-D., and C.S. conducted experiments; M.F.-D., A.A.-G., A.M.Z.-P., C.S., and J.M.G. analyzed data; M.F.-D., A.A.-G., and A.M.Z.-P. wrote the paper; and M.F.-D., A.A.-G., A.M.Z.-P., J.M.G., and C.S. edited the paper.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00200-19.
REFERENCES
- 1.Stevens DL, Bryant AE. 2017. Necrotizing soft-tissue infections. N Engl J Med 377:2253–2265. doi: 10.1056/NEJMra1600673. [DOI] [PubMed] [Google Scholar]
- 2.Low LY, Harrison PF, Gould J, Powell DR, Choo JM, Forster SC, Chapman R, Gearing LJ, Cheung JK, Hertzog P, Rood JI. 2018. Concurrent host-pathogen transcriptional responses in a Clostridium perfringens murine myonecrosis infection. mBio 9:e00473-18. doi: 10.1128/mBio.00473-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Carosio S, Berardinelli M, Aucello M, Musaró A. 2011. Impact of ageing on muscle cell regeneration. Ageing Res Rev 10:35–42. doi: 10.1016/j.arr.2009.08.001. [DOI] [PubMed] [Google Scholar]
- 4.Turner N, Badylak S. 2012. Regeneration of skeletal muscle. Cell Tissue Res 347:759–774. doi: 10.1007/s00441-011-1185-7. [DOI] [PubMed] [Google Scholar]
- 5.Karalaki M, Fili S, Philippou A, Koutsilieris M. 2009. Muscle regeneration: cellular and molecular events. In Vivo 23:779–796. [PubMed] [Google Scholar]
- 6.Tidball J. 2011. Mechanisms of muscle injury, repair, and regeneration. Compr Physiol 1:2029–2062. doi: 10.1002/cphy.c100092. [DOI] [PubMed] [Google Scholar]
- 7.Gutiérrez JM, Escalante T, Hernández R, Gastaldello S, Saravia-Otten P, Rucavado A. 2018. Why is skeletal muscle regeneration impaired after myonecrosis induced by viperid snake venoms? Toxins (Basel) 10:E182. doi: 10.3390/toxins10050182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tidball J. 2017. Regulation of muscle growth and regeneration by the immune system. Nat Rev Immunol 17:165–177. doi: 10.1038/nri.2016.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chazaud B, Sonnet C, Lafuste P, Bassez G, Rimaniol AC, Poron F, Authier FJ, Dreyfus PA, Gherardi RK. 2003. Satellite cells attract monocytes and use macrophages as a support to escape apoptosis and enhance muscle growth. J Cell Biol 163:1133–1143. doi: 10.1083/jcb.200212046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Filippo K, Henderson R, Laschinger M, Hogg N. 2008. Neutrophil chemokines KC and macrophage-inflammatory protein-2 are newly synthesized by tissue macrophages using distinct TLR signaling pathways. J Immunol 180:4308–4315. doi: 10.4049/jimmunol.180.6.4308. [DOI] [PubMed] [Google Scholar]
- 11.Tidball JG. 2008. Inflammation in skeletal muscle regeneration, p 243–268. In Schiaffino S, Partridge T (ed), Skeletal muscle repair and regeneration. Springer, Dordrecht, Netherlands. [Google Scholar]
- 12.Chargé S, Rudnicki M. 2004. Cellular and molecular regulation of muscle regeneration. Physiol Rev 84:209–238. doi: 10.1152/physrev.00019.2003. [DOI] [PubMed] [Google Scholar]
- 13.Fu X, Wang H, Hu P. 2015. Stem cell activation in skeletal muscle regeneration. Cell Mol Life Sci 72:1663–1677. doi: 10.1007/s00018-014-1819-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Monturiol-Gross L, Flores-Díaz M, Araya-Castillo C, Pineda-Padilla MJ, Clark GC, Titball RW, Alape-Girón A. 2012. Reactive oxygen species and the MEK/ERK pathway are involved in the toxicity of Clostridium perfringens α-toxin, a prototype bacterial phospholipase C. J Infect Dis 206:1218–1226. doi: 10.1093/infdis/jis496. [DOI] [PubMed] [Google Scholar]
- 15.O’Brien DK, Therit BH, Woodman ME, Melville SB. 2007. The role of neutrophils and monocytic cells in controlling the initiation of Clostridium perfringens gas gangrene. FEMS Immunol Med Microbiol 50:86–93. doi: 10.1111/j.1574-695X.2007.00235.x. [DOI] [PubMed] [Google Scholar]
- 16.O’Brien D, Melville S. 2004. Effects of Clostridium perfringens alpha-toxin (PLC) and perfringolysin O (PFO) on cytotoxicity to macrophages, on escape from the phagosomes to macrophages, and on persistence of C. perfringens in host tissues. Infect Immun 72:5204–5215. doi: 10.1128/IAI.72.9.5204-5215.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kotwal GJ, Chien S. 2017. Macrophage differentiation in normal and accelerated wound healing, p 353–364. In Kloc M. (ed), Macrophages: origin, functions and biointervention. Springer, Cham, Switzerland. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fasina YO, Lillehoj HS. 2019. Characterization of intestinal immune response to Clostridium perfringens infection in broiler chickens. Poult Sci 98:188–198. doi: 10.3382/ps/pey390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alizadeh M, Rogiewicz A, McMillan E, Rodriguez-Lecompte JC, Patterson R, Slominski BA. 2016. Effect of yeast-derived products and distillers dried grains with solubles (DDGS) on growth performance and local innate immune response of broiler chickens challenged with Clostridium perfringens. Avian Pathol 45:334–345. doi: 10.1080/03079457.2016.1155693. [DOI] [PubMed] [Google Scholar]
- 20.Cheng M, Nguyen M, Fantuzzi G, Koh T. 2008. Endogenous interferon-γ is required for efficient skeletal muscle regeneration. Am J Physiol Cell Physiol 294:C1183–C1191. doi: 10.1152/ajpcell.00568.2007. [DOI] [PubMed] [Google Scholar]
- 21.Novak ML, Koh TJ. 2013. Phenotypic transitions of macrophages orchestrate tissue repair. Am J Pathol 183:1352–1363. doi: 10.1016/j.ajpath.2013.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chazaud B. 2016. Inflammation during skeletal muscle regeneration and tissue remodeling: application to exercise-induced muscle damage management. Immunol Cell Biol 94:140–145. doi: 10.1038/icb.2015.97. [DOI] [PubMed] [Google Scholar]
- 23.Xiao W, Liu Y, Chen P. 2016. Macrophage depletion impairs skeletal muscle regeneration: the roles of pro-fibrotic factors, inflammation, and oxidative stress. Inflammation 39:2016–2028. doi: 10.1007/s10753-016-0438-8. [DOI] [PubMed] [Google Scholar]
- 24.Castiglioni A, Corna G, Rigamonti E, Basso V, Vezzoli M, Monno A, Almada AE, Mondino A, Wagers AJ, Manfredi AA, Rovere-Querini P. 2015. FOXP3+ T cells recruited to sites of sterile skeletal muscle injury regulate the fate of satellite cells and guide effective tissue regeneration. PLoS One 10:e0128094. doi: 10.1371/journal.pone.0128094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Juban G, Chazaud B. 2017. Metabolic regulation of macrophages during tissue repair: insights from skeletal muscle regeneration. FEBS Lett 591:3007–3021. doi: 10.1002/1873-3468.12703. [DOI] [PubMed] [Google Scholar]
- 26.Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. 2004. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol 25:677–686. doi: 10.1016/j.it.2004.09.015. [DOI] [PubMed] [Google Scholar]
- 27.Tidball J, Villalta A. 2010. Regulatory interactions between muscle and the immune system during muscle regeneration. Am J Physiol Regul Integr Comp Physiol 298:R1173–R1187. doi: 10.1152/ajpregu.00735.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rigamonti E, Touvier T, Clementi E, Manfredi AA, Brunelli S, Rovere-Querini P. 2013. Requirement of inducible nitric oxide synthase for skeletal muscle regeneration after acute damage. J Immunol 190:1767–1777. doi: 10.4049/jimmunol.1202903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villalta SA, Nguyen HX, Deng B, Gotoh T, Tidball JG. 2009. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum Mol Genet 18:482–496. doi: 10.1093/hmg/ddn376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Olefsky JM, Glass CK. 2010. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 72:219–246. doi: 10.1146/annurev-physiol-021909-135846. [DOI] [PubMed] [Google Scholar]
- 31.Mahbub S, Deburghgraeve CR, Kovacs EJ. 2012. Advanced age impairs macrophage polarization. J Interferon Cytokine Res 32:18–26. doi: 10.1089/jir.2011.0058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Carvalho SC, Apolinário LM, Matheus SM, Santo Neto H, Marques MJ. 2013. EPA protects against muscle damage in the mdx mouse model of Duchenne muscular dystrophy by promoting a shift from the M1 to M2 macrophage phenotype. J Neuroimmunol 264:41–47. doi: 10.1016/j.jneuroim.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 33.Villalta SA, Rinaldi C, Deng B, Liu G, Fedor B, Tidball JG. 2011. Interleukin-10 reduces the pathology of mdx muscular dystrophy by deactivating M1 macrophages and modulating macrophage phenotype. Hum Mol Genet 20:790–805. doi: 10.1093/hmg/ddq523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Villalta A, Deng B, Rinaldi C, Wehling-Henricks M, Tidball J. 2011. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J Immunol 187:5419–5428. doi: 10.4049/jimmunol.1101267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Stevens DL, Bryant A. 2002. The role of clostridial toxins in the pathogenesis of gas gangrene. Clin Infect Dis 35(Suppl 1):S93–S100. doi: 10.1086/341928. [DOI] [PubMed] [Google Scholar]
- 36.Takehara M, Takagishi T, Seike S, Ohtani K, Kobayashi K, Miyamoto K, Shimizu T, Nagahama M. 2016. Clostridium perfringens α-toxin impairs innate immunity via inhibition of neutrophil differentiation. Sci Rep 6:28192. doi: 10.1038/srep28192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ciciliot S, Schiaffino S. 2010. Regeneration of mammalian skeletal muscle: basic mechanisms and clinical implications. Curr Pharm Des 16:906–914. doi: 10.2174/138161210790883453. [DOI] [PubMed] [Google Scholar]
- 38.Zhao W, Lu H, Wang X, Ransohoff RM, Zhou L. 2016. CX3CR1 deficiency delays acute skeletal muscle injury repair by impairing macrophage functions. FASEB J 30:380–393. doi: 10.1096/fj.14-270090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flores-Díaz M, Alape-Girón A. 2003. Role of Clostridium perfringens phospholipase C in the pathogenesis of gas gangrene. Toxicon 42:979–986. doi: 10.1016/j.toxicon.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 40.Gutiérrez JM, Ownby CL, Odell GV. 1984. Skeletal muscle regeneration after myonecrosis induced by crude venom and a myotoxin from the snake Bothrops asper (Fer-de-Lance). Toxicon 22:719–731. doi: 10.1016/0041-0101(84)90155-7. [DOI] [PubMed] [Google Scholar]
- 41.Arce V, Brenes F, Gutiérrez JM. 1991. Degenerative and regenerative changes in murine skeletal muscle after injection of venom from the snake Bothrops asper: a histochemical and immunocytochemical study. Int J Exp Pathol 72:211–226. [PMC free article] [PubMed] [Google Scholar]
- 42.Slater CR, Schiaffino S. 2008. Innervation of regenerating muscle, p 303–333. In Schiaffino S, Partridge T (ed), Skeletal muscle repair and regeneration. Springer, Dordrecht, Netherlands. [Google Scholar]
- 43.Kalhovde JM, Jerkovic R, Sefland I, Cordonnier C, Calabria E, Schiaffino S, Lømo T. 2005. “Fast” and “slow” muscle fibres in hindlimb muscles of adult rats regenerate from intrinsically different satellite cells. J Physiol 562:847–857. doi: 10.1113/jphysiol.2004.073684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barbier J, Popoff M, Molgó J. 2004. Degeneration and regeneration of murine skeletal neuromuscular junctions after intramuscular injection with a sublethal dose of Clostridium sordellii lethal toxin. Infect Immun 72:3120–3128. doi: 10.1128/IAI.72.6.3120-3128.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Serrano A, Muñoz-Cánoves P. 2010. Regulation and dysregulation of fibrosis in skeletal muscle. Exp Cell Res 316:3050–3058. doi: 10.1016/j.yexcr.2010.05.035. [DOI] [PubMed] [Google Scholar]
- 46.Deshmane S, Kremlev S, Amini S, Sawaya B. 2009. Monocyte chemoattractant protein-1 (MCP-1): an overview. J Interferon Cytokine Res 29:313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wynn TA. 2008. Cellular and molecular mechanisms of fibrosis. J Pathol 214:199–210. doi: 10.1002/path.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yoshimura A, Wakabayashi Y, Mori T. 2010. Cellular and molecular basis for the regulation of inflammation by TGF-beta. J Biochem 147:781–792. doi: 10.1093/jb/mvq043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hernández R, Cabalceta C, Saravia-Otten P, Chaves A, Gutiérrez JM, Rucavado A. 2011. Poor regenerative outcome after skeletal muscle necrosis induced by Bothrops asper venom: alterations in microvasculature and nerves. PLoS One 6:e19834. doi: 10.1371/journal.pone.0019834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Piller N, Decosterd I, Suter MR. 2013. Reverse transcription quantitative real-time polymerase chain reaction reference genes in the spared nerve injury model of neuropathic pain: validation and literature search. BMC Res Notes 6:266. doi: 10.1186/1756-0500-6-266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods 25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 52.Palomo J, Mastelic-Gavillet B, Woldt E, Troccaz S, Rodriguez E, Palmer G, Siegrist CA, Gabay C. 2016. IL-36-induced toxicity in neonatal mice involves TNF-alpha production by liver myeloid cells. J Immunol 197:2239–2249. doi: 10.4049/jimmunol.1600700. [DOI] [PubMed] [Google Scholar]
- 53.Lee YS, Yang H, Yang JY, Kim Y, Lee SH, Kim JH, Jang YJ, Vallance BA, Kweon MN. 2015. Interleukin-1 (IL-1) signaling in intestinal stromal cells controls KC/CXCL1 secretion, which correlates with recruitment of IL-22-secreting neutrophils at early stages of Citrobacter rodentium infection. Infect Immun 83:3257–3267. doi: 10.1128/IAI.00670-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thacker M, Clark A, Bishop T, Grist J, Yip P, Moon L, Thompson S, Marchand F, McMahon SB. 2009. CCL2 is a key mediator of microglia activation in neuropathic states. Eur J Pain 13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- 55.Han Y, Li Y, Jia L, Cheng JZ, Qi YF, Zhang HJ, Du J. 2012. Reciprocal interaction between macrophages and T cells stimulates IFN-γ and MCP-1 production in Ang II-induced cardiac inflammation and fibrosis. PLoS One 7:e35506. doi: 10.1371/journal.pone.0035506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi R, Chazaud B. 2007. Inflammatory monocytes recruited after skeletal muscle injury switch into antiinflamatory macrophages to support myogenesis. J Exp Med 204:1057–1069. doi: 10.1084/jem.20070075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barbara G, De Giorgio R, Deng Y, Vallance B, Blennerhassett P, Collins S. 2001. Role of immunologic factors and cyclooxygenase 2 in persistent postinfective enteric muscle dysfunction in mice. Gastroenterology 120:1729–1736. doi: 10.1053/gast.2001.24847. [DOI] [PubMed] [Google Scholar]
- 58.Sachdev U, Cui X, Xu J, Xu J, Tzeng E. 2014. MyD88 and TRIF mediate divergent inflammatory and regenerative responses to skeletal muscle ischemia. Physiol Rep 2:e12006. doi: 10.14814/phy2.12006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kilkenny C, Browne WJ, Cuthill IC, Emerson M, Altman DG. 2010. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol 8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.National Research Council. 2004. International guiding principles for biomedical research involving animals. National Academies Press, Washington, DC. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.