

Clostridioides difficile toxin B (TcdB) is an intracellular toxin responsible for many of the pathologies of C. difficile infection. The two variant forms of TcdB (TcdB1 and TcdB2) share 92% sequence identity but have reported differences in rates of cell entry, autoprocessing, and overall toxicity. This 2,366-amino-acid, multidomain bacterial toxin glucosylates and inactivates small GTPases in the cytosol of target cells, ultimately leading to cell death.
KEYWORDS: autoprocessing, Clostridioides difficile, Clostridium difficile, TcdB, toxin B, immunization
ABSTRACT
Clostridioides difficile toxin B (TcdB) is an intracellular toxin responsible for many of the pathologies of C. difficile infection. The two variant forms of TcdB (TcdB1 and TcdB2) share 92% sequence identity but have reported differences in rates of cell entry, autoprocessing, and overall toxicity. This 2,366-amino-acid, multidomain bacterial toxin glucosylates and inactivates small GTPases in the cytosol of target cells, ultimately leading to cell death. Successful cell entry and intoxication by TcdB are known to involve various conformational changes in the protein, including a proteolytic autoprocessing event. Previous studies found that amino acids 1753 to 1852 influence the conformational states of the proximal carboxy-terminal domain of TcdB and could contribute to differences between TcdB1 and TcdB2. In the current study, a combination of approaches was used to identify sequences within the region from amino acids 1753 to 1852 that influence the conformational integrity and cytotoxicity of TcdB2. Four deletion mutants with reduced cytotoxicity were identified, while one mutant, TcdB2Δ1769–1787, exhibited no detectable cytotoxicity. TcdB2Δ1769–1787 underwent spontaneous autoprocessing and was unable to interact with CHO-K1 or HeLa cells, suggesting a potential change in the conformation of the mutant protein. Despite the putative alteration in structural stability, vaccination with TcdB2Δ1769–1787 induced a TcdB2-neutralizing antibody response and protected against C. difficile disease in a mouse model. These findings indicate that the 19-amino-acid region spanning residues 1769 to 1787 in TcdB2 is crucial to cytotoxicity and the structural regulation of autoprocessing and that TcdB2Δ1769–1787 is a promising candidate for vaccination.
INTRODUCTION
Clostridioides difficile toxin B (TcdB) is a 2,366-amino-acid intracellular bacterial toxin that undergoes multiple conformational changes during cellular intoxication. The tertiary structure of TcdB includes distinct domains that confer cell binding, cell entry, and enzymatic activities (1, 2). The linear organization of TcdB’s functional regions includes a glucosyltransferase domain (GTD; amino acids 1 to 543), an autoprocessing domain (APD; amino acids 544 to 767), a multifunction translocation domain involved in cell interaction and membrane translocation (TD; amino acids 768 to 1833), and a carboxyl-terminal domain (CTD; amino acids 1834 to 2366) containing combined repetitive oligopeptide (CROP) sequences (3). The cellular intoxication process is coordinated by stepwise conformational changes, and the timing and cellular location of these structural changes must be tightly regulated for TcdB to successfully intoxicate cells (4). Despite this, the specific regions of TcdB which govern the conformational integrity prior to and during cellular intoxication are poorly defined.
The pH of the acidified endosome triggers the first known conformational change in TcdB and results in the exposure of hydrophobic regions and the unfurling of four buried regions that form ion-conducting channels (5, 6). TcdB undergoes a second conformational change after binding inositol-hexakisphosphate (IP6) in the cell, which triggers the structural rearrangement that allows TcdB to adopt a conformer with an active APD (7–9). Following IP6 binding and APD activation, autoprocessing by the APD results in the release of the GTD into the host cell cytosol, where it inactivates small GTPases by glucosylation (10–13). The timing of both of these events is critical for TcdB intoxication of target cells. Indeed, one of the early descriptions of purified TcdB indicated that the toxin was inactivated under extracellular acid pH conditions (14). In a similar manner, premature autoprocessing also inactivates TcdB (4). Thus, TcdB appears to maintain a soluble structure that allows receptor binding and cell entry and coordinates acid pH-induced translocation and activation of the APD at the appropriate time and location during cellular intoxication.
One region of TcdB that appears to influence the conformation and stability of the protein spans amino acids 1753 to 1852, connecting the TD to the CROP-containing CTD. The region from amino acids 1753 to 1852 influences the exposure of carboxy-terminal epitopes in the CROP domain and contributes to solution multimerization of carboxy-terminal fragments of the toxin (15). Peptides derived from the region from amino acids 1753 to 1852 destabilize and inactivate TcdB through interactions with repeating sequences in the CROP domain (16). This stretch of 99 amino acids also affects the overall activity of TcdB; sequence differences present in this region have been seen to influence the efficiency with which TcdB1 and TcdB2, the two major variants of the toxin, enter cells (17). As additional evidence supporting the importance of this region, Zhang et al. found that deletion of residues 1756 to 1852 prevents TcdB1 from delivering the GTD into cells (18). Subsequent findings showed that residues 1756 to 1780 are critical for the membrane translocation of TcdB1 (19). Taken altogether, these data support the idea that the region from amino acids 1753 to 1852 of TcdB is critical for cellular intoxication and likely plays an important role in maintaining the conformation of the toxin while limiting the exposure of neutralizing epitopes in the toxin.
In the current study, we examined the region from amino acids 1753 to 1852 to identify minimal sequences that influence the cytotoxicity of TcdB while preserving the neutralizing epitopes in the protein. A panel of internal deletion mutants was tested, and the data indicate that deletion of the region from amino acids 1769 to 1787 in TcdB2 leads to premature autoprocessing and prevents interaction with multiple cell types while maintaining both immunogenicity and neutralizing epitopes.
RESULTS
Internal deletion mutants disrupt the cytotoxicity of TcdB2.
A series of internal deletion mutants of TcdB2 was constructed, expressed, and purified. The regions targeted for internal deletion (Fig. 1A) were initially selected on the basis of previous studies suggesting that the region from amino acids 1753 to 1852 influences the efficiency of cell entry (17, 18). To ensure that any effects seen were a result of deletions in this specific region, as a control a 10-amino-acid portion of the CROP domain (amino acids 2213 to 2223), which has no reported function, was deleted from the toxin. Following purification, each mutant was analyzed by SDS-PAGE and Coomassie staining (Fig. 1B). Deletion mutants TcdB2Δ1769–1779, TcdB2Δ1769–1787, TcdB2Δ1847–1856, and TcdB2Δ1751–1761 migrated as multiple bands and included bands at approximately 270 kDa and 200 kDa.
FIG 1.

SDS-PAGE analysis and cytotoxicity of TcdB deletion mutants. (A) Domain layout of TcdB showing the locations of all internal deletions made and the amino acid sequences of all deleted regions. (B) Coomassie-stained SDS-PAGE gel (8%) showing each internal deletion mutant. (C) Results of a cytotoxicity assay showing the cellular viability of CHO-K1 cells after 24 h of treatment with TcdB2 and TcdB2 deletion mutants. Data are presented as the mean ± standard deviation for samples examined in triplicate and are representative of those from three independent experiments.
The impact of the various TcdB2 deletion mutants on cell viability was determined using CHO-K1 cells and a tetrazolium salt-based assay, which measures the extent of dehydrogenase activity in cells. The amount of the colorimetric formazan dye produced by cell dehydrogenase activity is in direct proportion to the number of living, metabolically active cells. As shown in Fig. 1C, cell viability did not change following treatment with TcdB2Δ1769–1779 or TcdB2Δ1769–1787. TcdB2Δ1751–1761, TcdB2Δ1847–1856, and TcdB2Δ1847–1866 exhibited 50% effective concentrations (EC50) of between 50 and 100 pM. Treatment with TcdB2Δ1773–1774 and TcdB2Δ2213–2223 or TcdB2 alone resulted in an EC50 of between 10 and 100 fM. Of the mutants tested, only cells treated with TcdB2Δ1769–1787, the deletion region corresponding to TcdB-derived inhibitory peptide PepB2 (16), showed no loss of cell viability at the highest concentration tested. This observation held true even when cells were exposed to TcdB2Δ1769–1787 at a final concentration of 1 μM (data not shown).
The TcdB2 deletion mutant retains glucosylation activity.
Previous work has suggested that the efficiency of glucosylation of TcdA and TcdB is hindered by conformational restrictions within the proteins (16, 20). It was therefore predicted that deletions which remove conformational constraints in TcdB could result in forms of the protein with altered glucosylation activity. As a quantitative test of glucosylation activity, TcdB2Δ1769–1787 and TcdB2 were incubated with the GTPase Rac1 and UDP-glucose, both of which are substrates of TcdB. Glucosylation of Rac1 was then examined by immunoblotting using an antibody that recognizes only substrate which has not been glucosylated. A reduction in the signal therefore correlates with glucosylation of the substrate, Rac1. TcdB2Δ1769–1787 was seen to exhibit glucosylation activity equal to that of wild-type TcdB2 (Fig. 2), suggesting that the reduction in cytotoxicity is not due to a loss of glucosyltransferase activity.
FIG 2.
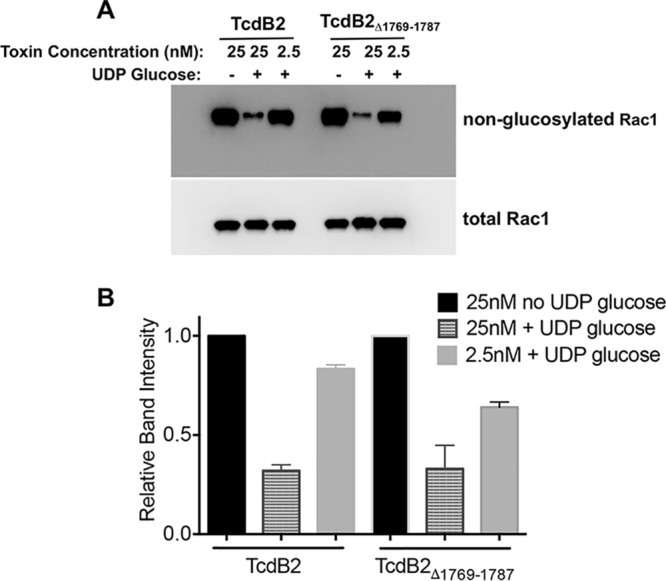
In vitro Rac1 glucosylation assay. (A) Immunoblot analysis of Rac1 glucosylation. TcdB2 and TcdB2Δ1769–1787 (25 and 2.5 nM) were incubated separately with either purified Rac1 and UDP-glucose or purified Rac1 alone. Blots were probed using an antibody specific for nonglucosylated Rac1 and an antibody recognizing total Rac1. A loss of signal was seen upon glucosylation of Rac1 when assessing the blot with the antibody against nonglucosylated Rac1. (B) Densitometry analysis of three independent glucosylation experiment immunoblots. Results are presented as the mean ± standard deviation, with significance being determined by one-way ANOVA on ranks.
The thermal transition profile of TcdB2Δ1769–1787 is similar to that of TcdB2.
Though the results from the glucosylation assay suggested that the structure of TcdB2Δ1769–1787 had not changed in a way that alters enzymatic activity, we performed a second experiment to measure the structural stability of this mutant in comparison to that of TcdB2. The thermal stability across a gradient of increasing temperatures was determined for TcdB2Δ1769–1787 by calculating the thermal denaturation temperature (melting temperature [Tm]), which was then compared to that of TcdB2. Differential scanning fluorimetry was used to measure temperature-induced unfolding and the exposure of hydrophobic domains (21, 22). The fluorescence emission profiles obtained across a temperature gradient of 25°C to 99°C were used to calculate the thermal denaturation temperature (Tm). The emission profiles and calculated denaturation temperatures (Tm) for both proteins are shown in Fig. 3A. Both proteins exhibited similar emission profiles and Tms: TcdB2 at 50.1 ± 0.6°C and TcdB2Δ1769–1787 at 50.9 ± 0.7°C. Thus, deletion of this region did not alter the thermal stability of TcdB2Δ1769–1787.
FIG 3.

Thermal stability, enzymatic activity, and cytotoxicity of TcdB2Δ1769–1787. (A) Differential scanning fluorimetry profile of TcdB2 and TcdB2Δ1769–1787 (0.1 mg/ml) incubated in the presence of SYPRO orange across a temperature gradient (25°C to 99°C). The increase in fluorescence corresponds to protein unfolding and exposure of hydrophobic domains. The first derivative of the profile was used to calculate the melting temperature (Tm). Results are given as the mean ± standard deviation. RFU, relative fluorescence units. (B) Immunoblot analysis of in vitro IP6-induced autoprocessing. TcdB2 and TcdB2Δ1769–1787 were incubated with IP6 (0, 25, 50, and 100 μM) overnight before samples were analyzed by immunoblotting. (C) A Coomassie-stained SDS-PAGE gel (8%) of in vitro autoprocessing samples. (D) Bar graph presenting the results of densitometry analysis of three independent autoprocessing experiments quantifying TcdB (amino acids 1 to 2366) from SDS-PAGE analysis. Results are given as the mean ± standard deviation, with significance being determined by one-way ANOVA on ranks. (E) A Coomassie-stained SDS-PAGE gel (8%) showing TcdB2, TcdB2C698S, TcdB2Δ1769–1787, and TcdB2Δ1769–1787:C698S. (F) Results of a cytotoxicity assay showing the viability of CHO-K1 cells after 24 h of treatment with TcdB2, TcdB2C698S, TcdB2Δ1769–1787, and TcdB2Δ1769–1787:C698S. Data are presented as the mean ± standard deviation.
TcdB2Δ1769–1787 autoprocessing in the absence of exogenous IP6.
Following expression and purification of TcdB2Δ1769–1787, we detected two proteins by SDS-PAGE, one of which appeared to be an autoprocessed form of the full-length mutant. Despite the fact that approximately 50% of the mutant appeared as an unprocessed band on SDS-PAGE gels (Fig. 1B and 3C), cytotoxicity remained undetectable even at concentrations as high as 1 μM (data not shown), suggesting that even the unprocessed fraction of TcdB2Δ1769–1787 lacked cytotoxic activity. In light of this, we next sought to answer two questions. First, is the apparent autoprocessing in the mutant due to endogenous APD activity? And second, is the unprocessed portion of TcdB2Δ1769–1787 capable of IP6-induced autoprocessing?
To address our first question, C698 in TcdB2Δ1769–1787 was converted to S698, a mutation shown to prevent the autoproteolytic cleavage event from occurring even in the presence of IP6 (4). As shown in Fig. 3E, when cysteine protease activity was repressed by mutating C698, TcdB2Δ1769–1787:C698S no longer appeared to spontaneously autoprocess and ran as a single 270-kDa band on SDS-PAGE gels. We next examined the cytotoxicity of TcdB2Δ1769–1787:C698S on CHO-K1 cells and found that preventing autoprocessing with this mutation was not sufficient to restore cytotoxicity (Fig. 3F).
To address our second question, we took advantage of the fact that the protein is purified using a carboxy-terminal His tag. Consequently, the amino-terminal autoprocessed fragment of TcdB2Δ1769–1787, comprised of the GTD (TcdB1–543), is eliminated during purification, as it lacks the His tag and thus will not bind the affinity column. In the mutant, the purification process then results in the isolation of only the full-length mutant and the larger autoprocessing cleavage product. By using an antibody which recognizes the amino-terminal region of TcdB, we were able to assess whether autoprocessing occurred after the addition of IP6 in both TcdB2 and TcdB2Δ1769–1787. As shown in the immunoblot in Fig. 3B, incubating TcdB2Δ1769–1787 with IP6 resulted in additional autoprocessing of the mutant, evidenced by the appearance of the smaller 60-kDa fragment in these samples. Moreover, this appeared to occur at a level similar to that observed in full-length TcdB2 (Fig. 3B to D).
TcdB2Δ1769–1787 exhibits reduced cell association in comparison to TcdB2.
Previous work by Chen et al. indicated that amino acids 1756 to 1780 are necessary for the translocation of TcdB1 across the endocytic vesicle membrane (19). As we did not observe a return of cytotoxicity when autoprocessing was precluded in TcdB2Δ1769–1787:C698S, we wondered if the smaller deletion of residues 1769 to 1787 was having similar effects on the translocation of TcdB2. To first determine if TcdB2Δ1769–1787 associated with cells to the same extent as TcdB2, each protein was applied to both CHO-K1 and HeLa cells. Total cell extracts were then collected and subjected to immunoblot analysis using antibody against the amino-terminal region of TcdB. Recognizing that spontaneous autoprocessing could reduce the total amount of protein capable of interacting with cells, TcdB2Δ1769–1787:C698S and TcdB2C698S were also assessed for interaction with cells. Counter to our prediction that TcdB2Δ1769–1787 would be unable to translocate across the vesicle membrane, no detectable mutant protein interaction was found with these cells, as seen in Fig. 4A to C. In contrast, both TcdB2 and TcdB2C698S interacted with cells (Fig. 4A to C). The lack of detectable cell interactions by both TcdB2Δ1769–1787 and TcdB2Δ1769–1787:C698S suggested that the lack of cell binding in TcdB2Δ1769–1787 was not solely due to spontaneous autoprocessing. While this immunoblot method does not distinguish between surface-bound and internalized toxin, collectively, the data shown indicate that TcdB2Δ1769–1787 is unable to associate with target cells at a level similar to that of TcdB2.
FIG 4.

Cell association of TcdB2Δ1769–1787. (A to C) Immunoblot analysis showing the cell association of TcdB2 and various mutants with CHO-K1 cells following 1 h (A) and 8 h (B) of incubation at 37°C and HeLa cells following 1 h of incubation at 37°C (C). The cells were then washed repeatedly to remove unbound toxin, and the resulting cell lysates were analyzed by immunoblotting. (D to F) Representative fluorescence microscopy images showing CHO-K1 cells treated with Alexa Fluor 488-labeled TcdB2 or Alexa Fluor 488-labeled TcdB2Δ1769–1787 for 1 h (D) and 24 h (E). (F) Representative fluorescence microscopy images showing HeLa cells treated with Alexa Fluor 488-labeled TcdB2 or Alexa Fluor 488-labeled TcdB2Δ1769–1787 for 1 h.
Though both TcdB2 and TcdB2Δ1769–1787 appeared to be equally detected by the anti-TcdB antibody (Fig. 3B), we recognized that it was possible that the TcdB2Δ1769–1787 mutant bound the cell but was rapidly degraded or that the antibody-binding region was altered during interaction with the cell. As a second approach to measure the cell interaction, TcdB2Δ1769–1787 and TcdB2 were labeled with Alexa Fluor 488 (TcdB2Δ1769–1787Alexa Fluor 488 and TcdB2Alexa Fluor 488, respectively) and incubated with both CHO-K1 and HeLa cells before being examined by fluorescence microscopy. Labeled TcdB2 yielded a pronounced signal on treated CHO-K1 cells after 1 and 24 h, while the TcdB2Δ1769–1787Alexa Fluor 488 interaction was below the level of detection (Fig. 4D and E). Consistent with these findings, TcdB2Alexa Fluor 488 was detected on HeLa cells after 1 h, while the TcdB2Δ1769–1787Alexa Fluor 488 interaction remained below the level of detection (Fig. 4F).
TcdB2Δ1769–1787 induces a neutralizing antibody response that correlates with protection from CDI in a murine model.
Previous work using recombinant truncated fragments of TcdB2 has suggested that the region from amino acids 1773 to 1780 of TcdB2 can affect the exposure of epitopes in the proximal CROP region of the toxin (15). This previous observation, along with the autoprocessing data suggesting a conformational change in TcdB2Δ1769–1787, led us to explore whether TcdB2Δ1769–1787 could induce a neutralizing antibody response against TcdB2 and protect against C. difficile infection (CDI).
Mice were vaccinated with TcdB2Δ1769–1787 or TcdB2D270N (which is defective in glucosyltransferase activity) as a control and then boosted at 28 days, as outlined in the schematic in Fig. 5A. At 2 weeks following the booster vaccination, the animals began antibiotic treatment to predispose them to C. difficile infection. Mice were orally gavaged with C. difficile spores to establish infection and then weighed daily for 2 weeks. Animals vaccinated with TcdB2Δ1769–1787 or TcdB2D270N experienced significantly less weight loss than the nonimmunized mice, with their weight loss stabilizing within 48 h (Fig. 5B). The total numbers of C. difficile CFU were measured across groups at days 0, 2, 4, 7, and 10 postgavage, yet there were no significant differences detected between the groups (Fig. 5C). These findings suggest that the vaccinated animals are able to effectively neutralize TcdB, therefore protecting them from the weight loss associated with disease, despite the animals carrying the same burden of C. difficile across infection time points. To specifically test whether or not vaccinated animals produced antibodies capable of neutralizing TcdB2, CHO-K1 cells were treated with TcdB2 either with or without a 1:100 dilution of sera from vaccinated animals and nonimmunized controls. As seen in Fig. 5D, sera from days 14 and 28 showed no effect on cell viability across all groups, while cells treated with sera from day 42 and post-CDI TcdB2Δ1769–1787-vaccinated animals showed significantly increased cell viability compared to cells treated with toxin alone.
FIG 5.

Vaccination with TcdBΔ1769–1787 in a murine model of CDI. (A) Schematic layout of murine experimental schedule. (B) Summary of weight loss data for control and experimentally vaccinated animals. Animals were weighed every 24 h for 14 days following oral gavage with C. difficile spores. The pregavage weight of each individual animal was set as the baseline weight (100%), and subsequent weight loss was calculated as a percentage of the baseline weight. Data are presented as the mean ± standard deviation (n = 5; data are representative of those from two independent experiments). Statistical significance was determined by two-way ANOVA and Dunnett’s posttest, with asterisks indicating significant differences in the percentage of the baseline weight versus the weight of the control. **, P ≤ 0.01 at all time points. (C) Number of fecal CFU during infection. Fecal samples were collected from mice at days 0, 2, 4, 7, and 10 postgavage for analysis. Data are presented as the mean ± standard deviation, with significance being determined by two-way ANOVA and Dunnett’s posttest. (D) Cytotoxicity assay showing the viability of CHO-K1 cells following 24 h of treatment with TcdB2 in the presence or absence of sera from vaccinated mice. Results are given as the mean ± standard deviation, with significance being determined by two-way ANOVA and Dunnett’s posttest. *, P ≤ 0.05; ****, P ≤ 0.0001. Data are representative of those from three independent experiments. d, days.
Endpoint TcdB2-specific antibody titers were determined by enzyme-linked immunosorbent assay (ELISA) at days 14, 28, and 42 postvaccination, as well as post-CDI. Animals vaccinated with TcdB2Δ1769–1787 showed significantly higher TcdB2-specific IgG1 titers at days 14 (P < 0.05), 28 (P < 0.05), and 42 (P < 0.001) and following CDI (P < 0.001) than the nonimmunized controls (Fig. 6A to D). These data show that TcdB2Δ1769–1787 is capable of stimulating an immune response in the murine model and that this response correlates with protection against disease, as measured by weight loss.
FIG 6.

Immune response to vaccination with TcdB2Δ1769–1787. (A to D) Serum was collected at the indicated time points (Fig. 5A), and endpoint TcdB2-specific IgG1 titers were determined by ELISA. Shown are endpoint titers from days 14 (d14) (A), 28 (d28) (B), and 42 (d42) (C) and from a post-CDI collection (D). Each data point represents one individual animal, and bars display the geometric mean ± standard deviation for the group. Statistical significance was determined by ANOVA or one-way ANOVA on ranks followed by Dunnett’s or Dunn’s posttest, with asterisks indicating significant titer increases versus the control. *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001. Data are pooled from two independent experiments.
DISCUSSION
Results from the current study indicate that amino acids 1769 to 1787 have a subtle but critical influence on the conformation of TcdB2. The deletion of this region in TcdB2 led to spontaneous autoprocessing and a reduction in cell binding. Unexpectedly, this mutant protein exhibited a greater than 10,000-fold reduction in cytotoxicity, despite retaining enzymatic activities in vitro. The deletion mutant was still able to undergo IP6-induced autoprocessing at a level similar to that seen for full-length TcdB2. Finally, we found that TcdB2Δ1769–1787 appeared to retain important epitope profiles and induced a strong neutralizing antibody response sufficient to reduce the severity of C. difficile infection in a mouse model.
Autoprocessing in TcdB involves a structural rearrangement that allows the active protease domain to form and subsequently cleave its intramolecular substrate. This conformational change is initiated by the binding of IP6 following TcdB translocation to the host cell cytosol. Though the structural reasons for it are not understood, biochemical evidence suggests that the carboxy-terminal region of the toxin influences autoprocessing. Interestingly, Zhang et al. showed that swapping the CROP domain of TcdB with that of TcdA caused the toxin to become more resistant to IP6-induced autoprocessing (20). Moreover, a deletion mutant of TcdA containing only the first 1,056 amino acids undergoes more efficient and nearly complete autoprocessing compared with that for the full-length toxin (23). In studies using a peptide based on the region from amino acids 1769 to 1787, the peptide destabilized TcdB2 and enhanced autoprocessing in the presence of IP6 (16). In TcdB2Δ1769–1787, we were able to confirm that the spontaneous autoprocessing event involved the APD by showing a loss in autoprocessing when C698 was mutated in the mutant. Shen et al. had previously shown that the APD of TcdB can adopt the active conformer structure in the absence of IP6, but it does so at a rate much lower than that seen in the presence of the molecule (24). Thus, an emerging model is one in which the carboxy-terminal region of TcdB limits conformational flexibility in the APD and prevents premature autoprocessing. It is also important to note that our data do not exclude the possibility that the changes in TcdB2Δ1769–1787 allow the protein to bind the trace amounts of IP6 that are present in the growth medium and thereby trigger autoprocessing. Regardless, the deletion in TcdB2Δ1769–1787 appears to reduce the structural constraints present in TcdB2 that inhibit active APD conformer formation. Molecules that relieve these constraints and cause premature autoprocessing could be used as novel antitoxin drugs.
Examination of the region from amino acids 1769 to 1787 in three-dimensional structures of TcdB1 and TcdA also suggests that disrupting this region could lead to structural instability. Though the complete structures for these toxins have not been reported, structural information from a fragment of TcdB1 (25) includes the region from amino acids 1769 to 1787, as does the structure of TcdA that lacks the CROP domain (7). This region in TcdB1 and TcdA is present at the end of a beta-sandwich structure and transitions into an alpha-helical turn that presumably connects to the beta-solenoid CROP domain. The GTD and APD in the structure of TcdA are proximal to the region from amino acids 1769 to 1787, but the molecular distances could not allow intramolecular contacts. Further work is needed to explain how deletion of the region from amino acids 1769 to 1787 leads to spontaneous autoprocessing, but based on the structural information and in line with the discussion in the previous paragraph, we hypothesize that the region from amino acids 1769 to 1787 influences the structural organization of the CROP domain. Interrupting this structural stability could result in a loss of the CROP domain chaperone effect on the APD, allowing spontaneous autoprocessing. Indeed, in this regard, it is notable that we previously reported that a synthetic peptide of the region from amino acids 1769 to 1787 binds to the CROP region in TcdB2 (16).
Using an antibody-based detection approach, we were able to monitor the TcdB2Δ1769–1787 interaction with target cells. These data suggest that the deletion mutant is unable to interact with TcdB receptors; however, the internal deletion does not coincide with regions known to bind these receptors. Recent work by Chen et al. found that TcdB interacts with frizzled proteins (FZD) through a tricomponent complex that involves the protein receptor, a palmitoleic acid lipid, and the FZD-binding domain of TcdB (TcdB-FBD) (25). The TcdB-FBD sequence that interacted with FZD and the palmitoleic acid falls within a structural region of TcdB covering residues 1434 to 1599; thus, a direct interaction between the region from amino acids 1769 to 1787 would not be expected. Indeed, cocrystal structures of TcdB1285–1804 and FZD reveal no interactions between the region from amino acids 1769 to 1787 and FZD. Accordingly, the absence of cell binding would not appear to be due to a loss of a direct interaction by residues 1769 to 1787 with FZD.
Two other receptors, poliovirus like receptor 3 (PVLR3) (26) and chondroitin sulfate proteoglycan 4 (CSPG4) (27), have been described for TcdB. Data from two studies (27, 28) support the idea that TcdB binds CSPG4 in a region found within amino acids 1831 to 2366, which would be carboxy terminal to the region from amino acids 1769 to 1787. Using a combination of pulldown experiments and antibody-based blocking, Gupta and colleagues further narrowed the region of CSPG4 binding and found that it likely occurs within a region between residues 1824 and 1900 (29). This region is carboxy terminal to amino acids 1769 to 1787 in TcdB. The PVRL3 binding site is less well defined, but it appears to be amino terminal to residue 1834 (26). Thus, it is formally possible that TcdB2Δ1769–1787 is unable to interact with PVRL3. This would not explain why TcdB2Δ1769–1787 is unable to interact with the other two receptors for the toxin. Considering these data in the context of the current findings, reduced binding is unlikely to be due to the loss of a direct interaction of the region from amino acids 1769 to 1787 with all three TcdB receptors. Instead, we hypothesize that the internal deletion structurally alters the proximal binding sites in such a way that precludes binding. The changes that occur in TcdB2Δ1769–1787 allow spontaneous autoprocessing and appear to reduce structural constraints on the process. It is reasonable to conclude that this mutant is also conformationally altered in a way that impedes binding to specific receptors. Finally, work by Chung and colleagues (30) suggests that TcdB2 may not utilize the same receptors as TcdB1, raising the possibility that the region from amino acids 1769 to 1787 could engage a yet-to-be discovered cell surface receptor specific to TcdB2.
Though TcdB2Δ1769–1787 contains a deletion that includes part of the region covering amino acids 1756 to 1780, which was previously found to be essential for membrane translocation (19), a similar defect in TcdB2Δ1769–1787 was not detected. Instead, TcdB2Δ1769–1787 appears to be unable to bind cells. Clearly, both deletions are deleterious to TcdB and reveal regions critical to the toxin’s ability to intoxicate cells. In considering these differences, it is important to note that Chen and colleagues performed their studies using TcdB1, while our studies used TcdB2 (19). These two variants of TcdB share only 77% identity in residues 1753 to 1852, making it possible that the regions are functionally different between the two forms of the toxin.
Despite the apparent structural changes present in the mutant, we found that TcdB2Δ1769–1787 invokes an effective neutralizing antibody response in mice. Previous findings using truncated recombinant fragments of TcdB2 showed that the region from amino acids 1753 to 1852 restricted exposure of neutralizing epitopes in the proximal CROP region of the toxin (15). A similar effect of the region from amino acids 1753 to 1852 in TcdB1 was not observed, suggesting that there was something intrinsic about the region in TcdB2 that was important for this observation. Consequently, we were curious to know if TcdB2Δ1769–1787 could prime a neutralizing antibody response against TcdB2. The data shown in Fig. 5 and 6 indicate that TcdB2Δ1769–1787 primes a neutralizing antibody response against TcdB2 and that this response is protective in a murine model. Thus, it appears that deletion of this region may potentially provide a nontoxic, but effectively immunogenic, candidate for vaccination against C. difficile. In addition, TcdB2Δ1769–1787 has other characteristics that make it an appealing vaccine candidate. For example, the absence of cell binding could supply more protein for professional antigen-presenting cells and antibody recognition. An increased and prolonged availability of antigen to the immune system has been found to be associated with antigen retention in lymph nodes and increased follicular T helper and germinal center B cell numbers (31). Therefore, use of a protein which does not bind cells might increase vaccine potency in comparison to use of a protein which is capable of binding and entering cells. Another beneficial characteristic of TcdB2Δ1769–1787 is that the known functional domains are present, providing a broader array of accessible epitopes. Importantly, TcdB2Δ1769–1787 does not exhibit detectable cytotoxicity; as part of the vaccination protocol, mice were administered 50 μg of protein, which is 2,000 times the calculated 50% lethal dose of TcdB2 (32), and no detrimental effects were observed.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were carried out with approval from the University of Oklahoma Health Sciences Center Institutional Animal Care and Use Committee under protocol number 18-049-HI. The procedures used in this study strictly adhered to the guidelines found in the National Research Council’s Guide for the Care and Use of Laboratory Animals (33).
Statistical analysis.
Results were analyzed using either one-way analysis of variance (ANOVA), two-way ANOVA, or a nonparametric one-way ANOVA on ranks, followed by Bonferroni’s, Dunnett’s, or Dunn’s multiple-comparisons posttest, when appropriate, using the statistical software program Prism.
Cloning, construction of TcdB deletion mutants, and protein purification.
Recombinant TcdB2 was expressed in a Bacillus megaterium system (MoBiTec, Göttingen, Germany) as described previously (15) and affinity purified by Ni2+ chromatography. Mutations in tcdB2 (pC-His1522-tcdB2) were made using a QuikChange II XL site-directed mutagenesis kit (catalog number 200523; Agilent) by addition of a pair of primers with sequences flanking the regions targeted for deletion. Mutants were verified for the appropriate deletion and the absence of off-target mutations by DNA sequencing.
Cell culture.
The hamster epithelial cell line CHO-K1 and the human cervical epithelial cell line HeLa were purchased from the American Type Culture Collection (ATCC). CHO-K1 cells were cultured in F12-K medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. HeLa cells were cultured in Dulbecco modified Eagle medium supplemented with 10% FBS, 100 units/ml penicillin, and 100 μg/ml streptomycin. All cells were grown at 37°C in the presence of 5% CO2.
Glucosyltransferase activity assay.
Glucosyltransferase activity was measured in a cell-free assay. A range of toxin concentrations (2.5 nM to 25 nM) was incubated with 400 nM glutathione S-transferase–Rac1 either with or without 40 μM UDP-glucose. The reactions were carried out at 37°C for 60 min in a buffer comprised of 50 mM HEPES (pH 7.5), 100 mM KCl, 2 mM MgCl2, 1 mM MnCl2, and 100 μg/ml bovine serum albumin (BSA). The reaction was stopped by heating the sample at 95°C for 7 min in SDS-PAGE sample buffer (62.5 mm Tris-HCl [pH 6.8], 2% SDS, 10% glycerol, 5% β-mercaptoethanol, 0.001% bromphenol blue). Twenty-five percent of each reaction mixture was then resolved on an SDS-PAGE gel (12%) before being transferred to a polyvinylidene difluoride (PVDF) membrane for blotting. Glucosylation was detected using an antibody that specifically recognizes nonglucosylated Rac1 (catalog number 610651; BD Biosciences). A second antibody against total Rac1 (catalog number 05-389; Millipore Sigma) was used to ensure that the samples contained similar amounts of Rac1. Primary antibodies were incubated with membranes overnight at 4°C, before the membranes were washed and probed with a horseradish peroxidase (HRP)-conjugated secondary antibody for 15 min. The blots were developed using a chemiluminescent enhancement system (catalog number 1705061; Bio-Rad) and then visualized using a Bio-Rad ChemiDoc MP system.
Autoprocessing assay.
Autoprocessing in TcdB2 and TcdB2Δ1769–1787 was assessed by incubating 5 μg of the respective protein with 25, 50, or 100 μM inositol-hexakisphosphate overnight at 37°C in 20 mM Tris, pH 8. The reaction was stopped by the addition of SDS-PAGE sample buffer, and the sample was heated at 95°C for 7 min, before the sample was resolved on an SDS-PAGE gel (8%). The extent of autoprocessing was assessed by either Coomassie staining or immunoblot analysis. An antibody against the amino terminus of TcdB (catalog number AF6246; R&D Biosystems) was used for the immunoblotting. The blots were developed using a chemiluminescent enhancement system (catalog number 1705061; Bio-Rad) and imaged using the Bio-Rad ChemiDoc MP system. Densitometry analysis was carried out using ImageJ software (34).
ELISA.
Thermo Scientific Pierce 96-well microplates (catalog number PI15041) were coated with TcdB at 10 μg/ml in binding buffer (0.1 M Na2HPO4) overnight at 4°C. The plates were then washed and blocked with 1.0% (wt/vol) BSA in phosphate-buffered saline (PBS)–0.05% (vol/vol) Tween 20 for 2 h at room temperature before washing. Serum was diluted in PBS–0.05% (vol/vol) Tween 20 and applied overnight at 4°C. The plates were then washed on the following day, and HRP-conjugated anti-mouse IgM or IgG1 at 0.2 μg/ml was applied for 1 h at room temperature. Ninety microliters of ABTS [2,2′-azinobis(3-ethylbenzthiazolinesulfonic acid)] developer (catalog number 5120-0035; Seracare) was then added to each well for 3 min. Color development was halted by addition of 110 μl of stop solution (10% [wt/vol] SDS solution). The absorbance was read at 405 nm, and endpoint titers were determined as [(absorbance of the sample) − (average absorbance of the blank wells + 2 standard deviations)], with the endpoint titer being set as the dilution at which the absorbance value fell below 0 after the above-described calculation.
Cell viability assay.
The viability of the cells after toxin exposure was assessed using a tetrazolium salt-based assay (Cell Counting Kit-8 [CCK-8]; catalog number CK04-05; Dojindo Molecular Technologies, Inc.). Cells were seeded in 96-well plates (catalog number 655180; Greiner Bio-One) at a density of 1 × 104 cells per well in 100 μl and incubated overnight at 37°C to allow the cells to grow to approximately 80% confluence. The cells were then treated with toxin at 24 h after plating, with the concentrations ranging from 0.001 to 1,000 pM. The toxins were left on the cells overnight at 37°C, before CCK-8 medium was applied. CHO-K1 cells were incubated with 5% CCK-8 solution in cell culture medium for 4 h before the absorbance was measured at 450 nm. Viability was calculated as [(absorbance of treated cells/absorbance of untreated cells) · 100], using normalized absorbance values.
TcdB cell association assay.
Cells were seeded into 12-well plates (catalog number 3513; Costar) at a density of 5 × 105 cells per well in 1 ml of cell culture medium and incubated overnight at 37°C. The cells were then washed three times with PBS, before 900 μl of fresh medium was applied. The plates were returned to 37°C for 1 h, before 100 μl of 1 μg/ml toxin diluted in cell culture medium, for a final concentration of 100 ng/ml, was applied. The plates were again returned to 37°C for either 1 or 8 h, before being washed three times with 4°C PBS to remove unbound toxin. Following washing, the cells were lysed by addition of 150 μl of lysis buffer (1% SDS, 50 mM Tris, 5 mM EDTA). The plates were placed on ice for 15 min during lysis. The cell lysates were then collected and sonicated before the protein concentration was determined by the Lowry assay. Twenty micrograms of total lysate protein was then resolved by SDS-PAGE (10%) under protein-reducing conditions, before transfer to a PVDF membrane. The membranes were blocked with 5% milk in wash buffer (Tris-buffered saline, 0.1% Tween 20), before being probed overnight with antibodies specific for TcdB (catalog number AF6246; R&D Biosystems) or GAPDH (glyceraldehyde-3-phosphate dehydrogenase; catalog number ab8245; Abcam) as a loading control. The membranes were then washed and incubated with HRP-conjugated secondary antibody at room temperature for 1 h. The blots were developed using a chemiluminescent enhancement system (catalog number 1705061; Bio-Rad), before images were captured using the Bio-Rad ChemiDoc MP system.
TcdB immunization.
Six-week-old female C57BL/6 mice were purchased from The Jackson Laboratory and housed in a pathogen-free facility. Mice were immunized at between the ages of 8 and 10 weeks with a subcutaneous injection divided between both rear flanks. Vaccines consisted of 50 μg of TcdB2D270N or TcdB2Δ1769–1787 in sterile PBS adsorbed to alhydrogel alum (InvivoGen, San Diego, CA) (35). The mice were initially vaccinated on day 0, followed by a booster at day 28. The booster consisted of 25 μg of antigen in sterile PBS with no adjuvant. Retroorbital or submandibular blood collections were done on days 14, 28 (preboost), and 42 and then at 2 weeks postgavage. Samples were allowed to incubate at room temperature for 4 h, before centrifugation at 15,000 × g for 15 min to separate the cells and debris from the serum. The serum was then aliquoted to a new tube and stored at −80°C.
C. difficile challenge.
At between 14 and 20 days following the booster, animals began a 5-day treatment with cefoperazone (catalog number 02199695-CF; MP Bio) in sterile drinking water at a concentration of 0.5 mg/ml. The antibiotic water was switched out every other day, in accordance with the published protocol (36). The mice were then provided untreated drinking water for 2 days before oral gavage. Oral gavage contained approximately 1 × 105 spores of hypervirulent C. difficile strain R20291 in 30 μl of PBS. Following C. difficile gavage, the mice were weighed daily for 14 days to measure disease severity.
Fecal CFU determination.
Fresh fecal samples were collected from mice at days 0, 2, 4, 7, and 10 postgavage for analysis. Fecal pellets were diluted 1:10 (wt/vol) in PBS and incubated anaerobically for 30 min at 37°C. After 30 min, the pellet was disrupted into solution, and serial 1:10 dilutions were made anaerobically. One hundred microliters of each dilution was then transferred to taurocholate-cycloserine-cefoxitin-fructose agar (TCCFA) plates, and the plates were incubated at 37°C for 24 h before the colonies were enumerated. Calculation of the total number of CFU was based on the dilution factor and the initial weight of the feces collected. The dilution plate with the lowest countable number (between 20 and 200 colonies) was used for calculation of the number of CFU per gram of fecal content.
TcdB neutralization assay.
CHO-K1 cells were seeded in 96-well plates (catalog number 655180; Greiner Bio-One) at a density of 7.5 × 103 cells per well in 100 μl and incubated overnight at 37°C. Serum from mice was diluted 1/100 in cell culture medium and mixed with TcdB2 in cell culture medium for a final toxin concentration of 1 pM. This toxin concentration was chosen, as it causes 100% cell rounding and leaves approximately 20% viable cells, as measured by the CCK-8 assay. The serum-toxin mixture was incubated for 1 h at room temperature, before medium was removed from CHO-K1 cells and replaced with either the serum-toxin mixture or toxin alone. Following an overnight incubation at 37°C, a 5% CCK-8 solution was applied to the cells for 4 h before the absorbance was measured at 450 nm. Viability was calculated as [(absorbance of treated cells/absorbance of untreated cells) · 100], using normalized absorbance values.
Differential scanning fluorimetry.
The thermal melting temperature (Tm) of the toxins was measured by combining purified TcdB2 or TcdB2Δ1769–1787 with SYPRO orange fluorescent dye in quadruplicate. The fluorescence emission was monitored over a temperature gradient of 25°C to 99°C, using an Applied Biosystems 7500 real-time PCR system. The reactions were performed in a buffer comprised of 20 mM HEPES (pH 8) and 150 mM NaCl. The Tm value was calculated by graphing the first derivative of the melting curve, with the Tm being equivalent to the midpoint of the transition from the folded to the unfolded state.
Protein labeling with Alexa Fluor 488.
Proteins were fluorescently labeled on primary amines using an Alexa Fluor 488 protein labeling kit from Molecular Probes (catalog number A10235), according to the manufacturer’s directions. Dye incorporation for TcdB2 was approximately 1 mol of dye per mol of protein, while the incorporation for TcdB2Δ1769–1787 was approximately 2.3 mol of dye per mol of protein.
Fluorescence microscopy.
CHO-K1 or HeLa cells were seeded into a 96-well plate (catalog number 655180; Greiner Bio-One) at a density of 7.5 × 103 cells per well in 100 μl and incubated at 37°C overnight to allow adherence. After overnight incubation, cells were treated in triplicate with Alexa Fluor 488-labeled TcdB2 or TcdB2Δ1769–1787 and incubated at 37°C, before cell association was visualized using an Olympus IX51 inverted microscope (Olympus, Waltham, MA). HeLa cells were visualized at 1 h after toxin treatment, while CHO-K1 cells were visualized at 1 and 24 h after toxin treatment. The experiments were repeated three times, and greater than 10 fields per well were examined for fluorescence.
ACKNOWLEDGMENTS
This work was supported by NIH R01AI119048 (to J.D.B.).
We thank Casey Theriot for valuable advice on the cefoperazone mouse model. We acknowledge the assistance of Sheryl Christofferson of the Oklahoma Medical Research Foundation DNA Sequencing Facility for sequencing expertise.
REFERENCES
- 1.Pruitt RN, Chambers MG, Ng KK-S, Ohi MD, Lacy DB. 2010. Structural organization of the functional domains of Clostridium difficile toxins A and B. Proc Natl Acad Sci U S A 107:13467–13472. doi: 10.1073/pnas.1002199107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Albesa-Jové D, Bertrand T, Carpenter EP, Swain GV, Lim J, Zhang J, Haire LF, Vasisht N, Braun V, Lange A, von Eichel-Streiber C, Svergun DI, Fairweather NF, Brown KA. 2010. Four distinct structural domains in Clostridium difficile toxin B visualized using SAXS. J Mol Biol 396:1260–1270. doi: 10.1016/j.jmb.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 3.Pruitt RN, Lacy DB. 2012. Toward a structural understanding of Clostridium difficile toxins A and B. Front Cell Infect Microbiol 2:28. doi: 10.3389/fcimb.2012.00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li S, Shi L, Yang Z, Feng H. 2013. Cytotoxicity of Clostridium difficile toxin B does not require cysteine protease-mediated autocleavage and release of the glucosyltransferase domain into the host cell cytosol. Pathog Dis 67:11–18. doi: 10.1111/2049-632X.12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barth H, Pfeifer G, Hofmann F, Maier E, Benz R, Aktories K. 2001. Low pH-induced formation of ion channels by Clostridium difficile toxin B in target cells. J Biol Chem 276:10670–10676. doi: 10.1074/jbc.M009445200. [DOI] [PubMed] [Google Scholar]
- 6.Qa’Dan M, Spyres LM, Ballard JD. 2000. pH-induced conformational changes in Clostridium difficile toxin B. Infect Immun 68:2470–2474. doi: 10.1128/IAI.68.5.2470-2474.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chumbler NM, Rutherford SA, Zhang Z, Farrow MA, Lisher JP, Farquhar E, Giedroc DP, Spiller BW, Melnyk RA, Lacy DB. 2016. Crystal structure of Clostridium difficile toxin A. Nat Microbiol 1:15002. doi: 10.1038/nmicrobiol.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, Aktories K, Barth H. 2003. Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 278:44535–44541. doi: 10.1074/jbc.M307540200. [DOI] [PubMed] [Google Scholar]
- 9.Reineke J, Tenzer S, Rupnik M, Koschinski A, Hasselmayer O, Schrattenholz A, Schild H, von Eichel-Streiber C. 2007. Autocatalytic cleavage of Clostridium difficile toxin B. Nature 446:415–419. doi: 10.1038/nature05622. [DOI] [PubMed] [Google Scholar]
- 10.Jank T, Aktories K. 2008. Structure and mode of action of clostridial glucosylating toxins: the ABCD model. Trends Microbiol 16:222–229. doi: 10.1016/j.tim.2008.01.011. [DOI] [PubMed] [Google Scholar]
- 11.Just I, Selzer J, Wilm M, von Eichel-Streiber C, Mann M, Aktories K. 1995. Glucosylation of Rho proteins by Clostridium difficile toxin B. Nature 375:500–503. doi: 10.1038/375500a0. [DOI] [PubMed] [Google Scholar]
- 12.Egerer M, Giesemann T, Jank T, Satchell KJF, Aktories K. 2007. Auto-catalytic cleavage of Clostridium difficile toxins A and B depends on cysteine protease activity. J Biol Chem 282:25314–25321. doi: 10.1074/jbc.M703062200. [DOI] [PubMed] [Google Scholar]
- 13.Just I, Wilm M, Selzer J, Rex G, von Eichel-Streiber C, Mann M, Aktories K. 1995. The enterotoxin from Clostridium difficile (ToxA) monoglucosylates the Rho proteins. J Biol Chem 270:13932–13936. doi: 10.1074/jbc.270.23.13932. [DOI] [PubMed] [Google Scholar]
- 14.Sullivan NM, Pellett S, Wilkins TD. 1982. Purification and characterization of toxins A and B of Clostridium difficile. Infect Immun 35:1032–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larabee JL, Krumholz A, Hunt JJ, Lanis JM, Ballard JD. 2015. Exposure of neutralizing epitopes in the carboxyl-terminal domain of TcdB is altered by a proximal hypervariable region. J Biol Chem 290:6975–6985. doi: 10.1074/jbc.M114.612184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larabee JL, Bland SJ, Hunt JJ, Ballard JD. 2017. Intrinsic toxin-derived peptides destabilize and inactivate Clostridium difficile TcdB. mBio 8:e00503-17. doi: 10.1128/mBio.00503-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hunt JJ, Larabee JL, Ballard JD. 2017. Amino acid differences in the 1753-to-1851 region of TcdB influence variations in TcdB1 and TcdB2 cell entry. mSphere 2:e00268-17. doi: 10.1128/mSphere.00268-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang Y, Shi L, Li S, Yang Z, Standley C, Yang Z, ZhuGe R, Savidge T, Wang X, Feng H. 2013. A segment of 97 amino acids within the translocation domain of Clostridium difficile toxin B is essential for toxicity. PLoS One 8:e58634. doi: 10.1371/journal.pone.0058634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen S, Wang H, Gu H, Sun C, Li S, Feng H, Wang J. 2016. Identification of an essential region for translocation of Clostridium difficile toxin B. Toxins (Basel) 8:E241. doi: 10.3390/toxins8080241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Hamza T, Gao S, Feng H. 2015. Masking autoprocessing of Clostridium difficile toxin A by the C-terminus combined repetitive oligo peptides. Biochem Biophys Res Commun 459:259–263. doi: 10.1016/j.bbrc.2015.02.095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sorrell FJ, Greenwood GK, Birchall K, Chen B. 2010. Development of a differential scanning fluorimetry based high throughput screening assay for the discovery of affinity binders against an anthrax protein. J Pharm Biomed Anal 52:802–808. doi: 10.1016/j.jpba.2010.02.024. [DOI] [PubMed] [Google Scholar]
- 22.Vollrath F, Hawkins N, Porter D, Holland C, Boulet-Audet M. 2014. Differential scanning fluorimetry provides high throughput data on silk protein transitions. Sci Rep 4:5625. doi: 10.1038/srep05625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Olling A, Hüls C, Goy S, Müller M, Krooss S, Rudolf I, Tatge H, Gerhard R. 2014. The combined repetitive oligopeptides of Clostridium difficile toxin A counteract premature cleavage of the glucosyl-transferase domain by stabilizing protein conformation. Toxins (Basel) 6:2162–2176. doi: 10.3390/toxins6072162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen A, Lupardus PJ, Gersch MM, Puri AW, Albrow VE, Garcia KC, Bogyo M. 2011. Defining an allosteric circuit in the cysteine protease domain of Clostridium difficile toxins. Nat Struct Mol Biol 18:364–371. doi: 10.1038/nsmb.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen P, Tao L, Wang T, Zhang J, He A, Lam K-H, Liu Z, He X, Perry K, Dong M, Jin R. 2018. Structural basis for recognition of frizzled proteins by Clostridium difficile toxin B. Science 360:664–669. doi: 10.1126/science.aar1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaFrance ME, Farrow MA, Chandrasekaran R, Sheng J, Rubin DH, Lacy DB. 2015. Identification of an epithelial cell receptor responsible for Clostridium difficile TcdB-induced cytotoxicity. Proc Natl Acad Sci U S A 112:7073–7078. doi: 10.1073/pnas.1500791112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yuan P, Zhang H, Cai C, Zhu S, Zhou Y, Yang X, He R, Li C, Guo S, Li S, Huang T, Perez-Cordon G, Feng H, Wei W. 2015. Chondroitin sulfate proteoglycan 4 functions as the cellular receptor for Clostridium difficile toxin B. Cell Res 25:157–168. doi: 10.1038/cr.2014.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tao L, Zhang J, Meraner P, Tovaglieri A, Wu X, Gerhard R, Zhang X, Stallcup WB, Miao J, He X, Hurdle JG, Breault DT, Brass AL, Dong M. 2016. Frizzled proteins are colonic epithelial receptors for C. difficile toxin B. Nature 538:350–355. doi: 10.1038/nature19799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gupta P, Zhang Z, Sugiman-Marangos SN, Tam J, Raman S, Julien J-P, Kroh HK, Lacy DB, Murgolo N, Bekkari K, Therien AG, Hernandez LD, Melnyk RA. 2017. Functional defects in Clostridium difficile TcdB toxin uptake identify CSPG4 receptor-binding determinants. J Biol Chem 292:17290–17301. doi: 10.1074/jbc.M117.806687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung S-Y, Schöttelndreier D, Tatge H, Fühner V, Hust M, Beer L-A, Gerhard R. 2018. The conserved Cys-2232 in Clostridioides difficile toxin B modulates receptor binding. Front Microbiol 9:2314. doi: 10.3389/fmicb.2018.02314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tam HH, Melo MB, Kang M, Pelet JM, Ruda VM, Foley MH, Hu JK, Kumari S, Crampton J, Baldeon AD, Sanders RW, Moore JP, Crotty S, Langer R, Anderson DG, Chakraborty AK, Irvine DJ. 2016. Sustained antigen availability during germinal center initiation enhances antibody responses to vaccination. Proc Natl Acad Sci U S A 113:E6639–E6648. doi: 10.1073/pnas.1606050113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lanis JM, Heinlen LD, James JA, Ballard JD. 2013. Clostridium difficile 027/BI/NAP1 encodes a hypertoxic and antigenically variable form of TcdB. PLoS Pathog 9:e1003523. doi: 10.1371/journal.ppat.1003523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.National Research Council. 2011. Guide for the care and use of laboratory animals, 8th ed National Academies Press, Washington, DC. [Google Scholar]
- 34.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devera TS, Lang GA, Lanis JM, Rampuria P, Gilmore CL, James JA, Ballard JD, Lang ML. 2016. Memory B cells encode neutralizing antibody specific for toxin B from the Clostridium difficile strains VPI 10463 and NAP1/BI/027 but with superior neutralization of VPI 10463 toxin B. Infect Immun 84:194–204. doi: 10.1128/IAI.00011-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Theriot CM, Koumpouras CC, Carlson PE, Bergin II, Aronoff DM, Young VB. 2011. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2:326–334. doi: 10.4161/gmic.19142. [DOI] [PMC free article] [PubMed] [Google Scholar]