

Abstract
Although the concepts of somatic cell reprogramming and human‐induced pluripotent stem cells (hiPSCs) generation have undergone several analyses to validate the usefulness of these cells in research and clinic, it remains still controversial whether the hiPSCs are equivalent to human embryonic stem cells (hESCs), pointing to the need of further characterization for a more comprehensive understanding of pluripotency. Most of the experimental evidence comes from the transcriptome analysis, while a little is available on protein data, and even less is known about the post‐translational modifications. Here, we report a combined strategy of mass spectrometry and gene expression profiling for proteogenomic analysis of reprogrammed and embryonic stem cells. The data obtained through this integrated, multi‐“omics” approach indicate that a small, but still significant, number of distinct pathways is enriched in reprogrammed versus embryonic stem cells, supporting the view that pluripotency is an extremely complex, multifaceted phenomenon, with peculiarities that are characteristic of each cell type.
Keywords: human embryonic stem cells (hESCs), human induced pluripotent stem cells (hiPSCs), Ingenuity Pathway Analysis (IPA), mass spectrometry, pluripotency, post‐translational modifications, proteome, transcriptome
1. INTRODUCTION
Pluripotency is a property shared by certain types of cells that have the ability to differentiate, either spontaneously or following appropriate stimuli, into any of the three germ layers (ectoderm, mesoderm, and endoderm).1 Embryonic stem cells (ESCs) are the main example of pluripotent cells (PSCs) and represent an extraordinary tool for studying normal development, as well as for deciphering the molecular mechanisms underlying a number of complex diseases.2 Their use in laboratory, however, is accompanied by the need to overcome important ethical considerations that can be circumvented by reprogramming somatic cells into stem cells (termed induced pluripotent stem cells, iPSCs) through the forced expression of pluripotency‐promoting transcription factors.3 This strategy, developed by Takahashi and Yamanaka, represented a milestone in stem cell biology and paved the way for hundreds of studies related to this exciting field.4, 5, 6, 7, 8 Despite the numerous advances in stem cells research, the equivalence between human embryonic stem cells (hESCs) and human‐induced pluripotent stem cells (hiPSCs) still remains controversial. Different studies have shown quite a high similarity between embryonic and reprogrammed pluripotent stem cells,9 while others have instead found significant differences.10, 11 At the mRNA level, for example, early passage iPSCs show specific patterns of expression that are not detected in ESCs; this discrepancy seems to become less evident in late passage iPSCs.12 While the epigenomic and transcriptomic profiles of hiPSCs and hESCs have been widely discussed,13, 14, 15 less is instead known about proteomic patterns16 and post‐translational modifications,17 such as phosphorylation.18 Here, we have applied and integrated a multi‐‘omics’ strategy to dissect, at the transcriptional, translational and post‐translational level, two distinct human PSCs, the H9 embryonic stem cell line (hESCs), and the induced pluripotent stem cell line (hiPSC‐1), obtained by reprogramming of peripheral blood T‐lymphocytes from a healthy volunteer. Two additional hiPSC lines, one generated from peripheral blood T‐lymphocytes and one generated from skin fibroblasts (hiPSC‐2 and hiPSC‐3, respectively) were included in this study to validate the proteogenomic data.
2. MATERIAL AND METHODS
2.1. Generation and culture of induced pluripotent stem cells
A written informed consent for human blood and human skin fibroblasts collection was obtained from three healthy donors. The study was approved and supervised by the Ethics Committee of the ‘Magna Graecia’ University of Catanzaro and the research was carried out according to the World Medical Association Declaration of Helsinki. hiPSCs were generated from two different cell sources: T‐Lymphocytes isolated from peripheral blood mononuclear cells (PBMCs) (hiPSC‐1 and hiPSCs‐2 lines) and skin fibroblasts (hiPSCs‐3). Fibroblasts were isolated and expanded by the outgrowth method in DMEM containing 10% fetal bovine serum (FBS, Thermo Fisher Scientific) and 50 U/mL penicillin, and 50 µg/mL streptomycin. Cells were passaged twice before infection for iPSCs generation. Reprogramming of fibroblasts to pluripotency was carried out by non‐integrating Sendai‐virus‐mediated (CytoTune™‐iPS 2.0 Sendai Reprogramming Kit, Thermo Fisher Scientific) transfection of the four canonical transcription factors (Oct4, Sox2, Klf4 and c‐Myc). 3 × 105 fibroblasts were infected at a multiplicity of infection (MOI) of 5, yielding different iPSCs clones generated in feeder‐independent conditions on Matrigel‐coated dishes (BD Biosciences). For hiPSCs generation from T‐lymphocytes, PBMCs were cultured in AIM‐V medium (Thermo Fisher Scientific) containing 20% FBS, 50 U/mL penicillin, and 50 µg/mL streptomycin (Thermo Fisher Scientific), supplemented with 125 ng/mL Interleukine‐2 (IL‐2) (R&D Systems) and finally seeded onto CD3‐coated dishes (10 µg/mL, BD Biosciences) for T‐lymphocytes activation. 5 × 105 T‐Lymphocytes were infected with Sendai virus (SeV)19 at MOI of 20 in feeder‐independent conditions. hiPSC lines (hiPSC‐1, hiPSC‐2, and hiPSC‐3) and hESCs H9 (the latter purchased from WiCell Research Institute) were cultured on Matrigel‐coated (BD Biosciences) dishes in mTeSR1 medium (STEMCELL Technologies) in a humidified incubator at 37°C at 5% CO2. Cells were passaged using Gentle Cell Dissociation Reagent (STEMCELL Technologies). All cell lines were tested for Mycoplasma before being used in experiments.
2.2. Assessment of pluripotency of generated iPSCs
Prior to pluripotency assessment, all generated hiPSCs lines were tested for SeV‐transgenes loss by reverse transcription polymerase chain reaction (RT‐PCR). Detection of SeV‐transgenes was performed in infected parental cells for presence, uninfected parental cells for absence, and generated hiPSCs for loss of viral transgenes. Pluripotency of generated hiPSCs was assessed for the expression of pluripotency‐associated genes (OCT4, NANOG, SOX2, REX1 and DNMT3B) by quantitative real‐time PCR (qRT‐PCR) and for expression of pluripotency markers (NANOG and OCT4) by immunostaining. Additionally, generated hiPSCs were tested for markers of the three germ layers, NESTIN (ectoderm), BRACHYURY (mesoderm), and SOX17 (endoderm) on whole Embryoid Bodies (EBs) by immunostaining and by qRT‐PCR for endoderm (SOX7 and SOX17), mesoderm (HAND1, ACTA2, BMP4), and ectoderm (BMP4 and PAX6) genes. Moreover, an in‐depth gene expression analysis for pluripotency was performed by PluriTest assay.20 hiPSCs characterization assessment is shown in Figure S1.
2.3. RNA extraction, RT‐PCR and qRT‐PCR
Total RNA was extracted using TRIzol reagent (Thermo Fisher Scientific) and 1 µg RNA was used for retro‐transcription using the High‐capacity cDNA Reverse Transcription Kit (Thermo Fisher Scientific). For SeV detection, half µg of cDNA was used for standard PCR reaction. However, qRT‐PCR was used for gene expression quantification using 1 µL of the RT reaction and the Power SYBR Green master Mix (Applied Biosystems). Gene expression levels were normalized to Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) housekeeping gene. qRT‐PCR was performed by StepOnePlus™ Real‐time PCR System (Applied Biosystems). A list of primers is provided in Table S1.
2.4. Immunohistological analysis and alkaline phosphatase staining
Cells were fixed with 4% (vol/vol) paraformaldehyde (PFA) and subjected to immunostaining using the following primary antibodies: NANOG (1:1000; rabbit polyclonal, Abcam), OCT4 (1:400 mouse monoclonal, STEMCELL Technologies) for pluripotency, and BRACHYURY (1:20 goat polyclonal, R&D systems), SOX17 (1:20 goat polyclonal, R&D systems) and NESTIN (1:1000 mouse monoclonal, STEMCELL Technologies) for the three germ layers detection. After incubation with primary antibodies, cells were incubated with Alexa‐Fluor‐647, ‐594 and ‐488 conjugated secondary antibodies (all from Thermo Fisher Scientific) for 1 hour at 37°C. Nuclei were counterstained using 1 µg/mL Hoechst 33528 (Thermo Fischer Scientific). Microscopy was performed using imaging systems (DMi8), filter cubes and software from Leica microsystems. AP staining was performed using the 1‐Step NBT/BCIP (Thermo Fisher Scientific). Fluorescence quantization was achieved by measurement of the corrected total cell fluorescence (CTCF = Integrated Density – [Area of selected cell × Mean fluorescence of background readings]). Five cells/condition were randomly selected for analysis.
2.5. Western blotting
For western blot analysis the cells were lysed in RIPA buffer (Sigma‐Aldrich) supplemented with Halt™ Protease Inhibitor Cocktail and Halt™ Phosphatase Inhibitor Cocktail (both from Thermo Fischer Scientific). The protein concentration was determined using a standard Bradford assay and separated by electrophoresis on acrylamide/bisacrylamide precast gels Mini‐PROTEAN TGX (Bio‐Rad), followed by transfer to the nitrocellulose membrane. Membranes were incubated with the following primary antibodies: anti: anti‐SQSTM1 (Abcam), anti‐RAB17, anti‐phospho HSPB1 (Ser82) and anti‐phospho SQSTM1 (Thr269/Ser272) (Cell Signalling), and next with horseradish peroxidase conjugated secondary antibody anti‐mouse IgG and anti‐rabbit IgG (Jackson ImmunoResearch). However, ɣ‐TUBULIN‐HRP conjugated (Santa Cruz, Biotechnology) or ACTIN (Santa Cruz Biotechnology) were used as internal loading control. Proteins were detected by Clarity™ Western ECL Blotting Substrates (Bio‐Rad).
2.6. Treatment of hESCs and hiPSC lines with retinoic acid and BMS493
For RAR pathway modulation, cells were treated for 24 hours either with 0.5 µM RA or with 5 µM BMS493 (a pan‐retinoic acid receptor inverse agonist) or with a combination of RA and BMS493, directly diluted in the culture media. For EBs formation assay, cells were dissociated into single cells using StemPro Accutase (Thermo Fisher Scientific) and cultured for 7 days on poly (2‐hydroxyethyl methacrylate) (Sigma‐Aldrich) – coated dishes in mTeSR1 medium supplemented with 10 µM of the Rho‐kinase inhibitor Y‐27632 (Selleckchem) for the first three days. At day 7, floating EBs were transferred on 5 µg/mL Biolaminin 521LN (Biolamina)‐coated plates and cultured in adhesion for additional thirteen days in medium consisting of DMEM/F12 containing 20% knockout serum replacement (KSR, Thermo Fisher Scientific), 1% Glutamax (Thermo Fisher Scientific), 1% non‐essential Amino Acids (Thermo Fisher Scientific), 100 µM 2‐mercaptoethanol and 0.5% penicillin and streptomycin.
2.7. Proteomic and phosphoproteomic analysis: Strategy overview
An integrated strategy that combines enzymatic digestion, isobaric mass tag labelling, a selective affinity technique which uses metal oxide affinity material (MOA) for phosphopeptides enrichment, peptides fractionation by strong cation exchange, and nanoLC coupled with high resolution tandem mass spectrometry was adopted. A total of 8 samples (200 µg each), four biological replicates per each cell line, hESCs and hiPSC‐1 respectively, were prepared for phosphoproteomics and proteomics analyses. The core of the strategy is represented by the use of isobaric tags, allowing for relative quantification and consequent identification of differentially expressed proteins (DEPs) between hESCs and hiPSC‐1 lysates. The total area under the chromatograms, the number of proteins and the number of sequenced peptides obtained from single injections were evaluated to estimate protein amounts per sample (Table S2): about 200 µg per sample was handled. These results were further used to ensure that the subsequent labelling reaction would be performed on a comparable amount of starting material. Moreover, nLC‐MS/MS analyses were also carried out after TMT labelling21 (Table S3): normalized median values per each TMT channel were evaluated. Thus, all the samples were combined by mixing different volumes into a single master sample which was further processed for phosphopeptides enrichment; in particular, 90% of the volume was used for the sample labelled 129N; 95% of the volume was used for the samples labelled 127C, 128N, 128C, 129C, 130N and 126, respectively; the whole volume was instead used for both the samples labelled 127N and 130C. Further purification and fractionation steps were needed before nLC‐MS/MS analysis for proteins relative quantification. Detailed Experimental Procedures for proteomics and phosphoproteomics‐enrichment sample preparation and mass spectrometric analysis of hiPSC‐1 and hESCs are provided in Supplementary material and methods section.
2.8. Microarray procedure
Total RNA was extracted using the Stratagene Absolutely RNA kit and resuspended in RNase‐free water. Spectrophotomeric determination of purified RNA yield was performed using the NanoDrop (Thermo Scientific), while total RNA quality was measured using the BioAnalyzer 2100 (Agilent Technologies). Antisense RNA (aRNA) was synthesized, amplified and purified using the Illumina TotalPrep RNA Amplification Kit (Ambion) following the manufacturer's instructions. For microarray, purified aRNA was hybridized to the Human HT‐12v4 Expression BeadChip Kit (Illumina). Samples were scanned on the iSCAN system (Illumina). The output file was statistically analysed.
2.9. Statistical analysis
For proteomic data, statistical analysis was carried out by using both GraphPad Prism (version 7.00 for Mac, GraphPad Software, La Jolla, California USA, www.graphpad.com) and Excel 2011 (version 14.0.0 for Mac, Microsoft; Redmond, WA) software. However, t‐tests were produced in Excel 2011 while Benjamini–Hochberg corrections for q‐value calculation were run through the Prism. Reporter ion intensities from the reference sample (m/z 126) were used as the denominator in both the phopshopeptide and the protein ratio calculations (two different ID lists). Both the protein and the phosphopeptide ratios from four biological replicates per class (N = 4 hESCs; N = 4 hiPSC‐1) were compared by student t‐test corrected for multiple hypothesis testing using the Benjamini‐Hochberg procedure (q‐value <0.05).22 Protein fold‐changes were determined by dividing protein's median fold‐changes (n = 4 replicates) of the two data sets. For microarray analysis, primary raw intensity data produced by Illumina iSCAN were imported in R statistical environment using limma package23 for background subtraction, quantile normalization and log2 transformation signal values. This procedure also removes the control probes, leaving only the regular ones. Moderated t‐test analysis with Benjamini and Hochberg (BH) multiple testing correction were used to identify differentially expressed genes (DEGs) between hESCs and hiPSC‐1. DEGs were selected by a fold‐change analysis of ≥1.5 and based on a P value cut‐off of ≤0.05. The identified DEGs were annotated in Gene Ontology (GO) and pathway analysis. Ingenuity Pathways Analysis (IPA; Ingenuity Systems, http://www.ingenuity.com website) was used for gene set enrichment and gene network analysis.
3. RESULTS
3.1. Whole proteome identification and classification of DEPs
Proteomics data resulting from nano‐Liquid Chromatography Tandem Mass Spectrometry (nLC‐MS/MS) allowed the identification and quantification of 3807 proteins between hiPSC‐1 and hESCs samples (Table S4). For proteomic and phosphoproteomic data, the same statistical cutoff was adapted and 230 statistically significant proteins (q < 0.05) were selected. Interestingly, by comparing hiPSCs vs hESCs, 13 proteins (CRYZ, CES1, SLC2A3, ALB, LBR, HM13, RAB17, SLC3A2, KRI1, RPRD1B, SLC25A1, SLC7A8, SLC6A6) were found enriched, according to a log2 fold change >0.5, and 25 proteins (PLIN2, IFITM2, PKM, GALNT3, KIAA1524, SQSTM1, STK26, PSMD5, ACOT9, ALDH16A1, CHCHD2, GMPR2, PGK1, SLC15A4, TSPAN3, LDHA, IFI30, P4HA2, TSPAN6, TCEAL4, HIST1H4A, HIST1H3A, DHRS4, MT1X, NLRP2) were down‐regulated according to the same log2 fold change (Table 1). Two proteins, RAB17 and SQSTM1, respectively up‐ and down‐regulated in hiPSC‐1 vs hESCs, were selected for biological validation via Western blot analysis (Figure 1A). Although the trend of expression is confirmed for both proteins, only the expression level of SQSTM1 resulted in statistically significant (P value 0.03). RAB17 is a member of the small GTPase superfamily and it has been linked to the down‐regulation of cell growth and proliferation.24 The protein resulted in up‐regulated hiPSCs vs hESCs in our proteogenomic comparison and, in a previous study, we demonstrated that hESCs have indeed a higher proliferation rate compared to hiPSCs as shown by cell cycle analysis.11 SQSTM1 is a hub molecule involved in several biological pathways, including autophagy that represented a highly conserved cellular process in ES cells supporting self‐renewal and regulating differentiation. Moreover, autophagy is activated during reprogramming of somatic cells to iPSCs.25
Table 1.
Differentially expressed proteins in hiPSC‐1 vs hESCs
| Accession | Gene | Description | hiPSC‐1/hESCs Fc | log2 Fc | q value |
|---|---|---|---|---|---|
| A6NP24 | CRYZ | Quinone oxidoreductase (Fragment) | 2.76 | 1.47 | 0.000 |
| P23141 | CES1 | Liver carboxylesterase 1 | 2.37 | 1.25 | 0.002 |
| P11169 | SLC2A3 | Solute carrier family 2, facilitated glucose transporter member 3 | 1.90 | 0.92 | 0.002 |
| Q14739 | LBR | Lamin‐B receptor | 1.60 | 0.68 | 0.030 |
| Q8TCT9 | HM13 | Minor histocompatibility antigen H13 | 1.60 | 0.68 | 0.006 |
| Q9H0T7 | RAB17 | Ras‐related protein Rab‐17 | 1.60 | 0.67 | 0.014 |
| F5GZS6 | SLC3A2 | 4F2 cell‐surface antigen heavy chain | 1.52 | 0.61 | 0.003 |
| A0A0C4DGB6 | ALB | Serum albumin | 1.50 | 0.58 | 0.006 |
| Q8N9T8 | KRI1 | Protein KRI1 homolog | 1.50 | 0.58 | 0.039 |
| Q9NQG5 | RPRD1B | Regulation of nuclear pre‐mRNA domain‐containing protein 1B | 1.49 | 0.57 | 0.009 |
| P53007 | SLC25A1 | Tricarboxylate transport protein, mitochondrial | 1.44 | 0.53 | 0.009 |
| Q9UHI5 | SLC7A8 | Large neutral amino acids transporter small subunit 2 | 1.43 | 0.52 | 0.016 |
| P31641 | SLC6A6 | Sodium‐ and chloride‐dependent taurine transporter | 1.41 | 0.50 | 0.032 |
| Q99541 | PLIN2 | Perilipin‐2 | 0.71 | −0.50 | 0.009 |
| H7BYV1 | IFITM2 | Interferon‐induced transmembrane protein 2 (Fragment) | 0.70 | −0.51 | 0.009 |
| P14618 | PKM | Pyruvate kinase | 0.70 | −0.52 | 0.043 |
| E7EUL0 | GALNT3 | Polypeptide N‐acetylgalactosaminyltransferase 3 | 0.70 | −0.52 | 0.020 |
| Q8TCG1 | KIAA1524 | Protein CIP2A | 0.69 | −0.54 | 0.019 |
| Q13501 | SQSTM1 | Sequestosome‐1 | 0.68 | −0.55 | 0.030 |
| Q9P289 | STK26 | Serine/threonine‐protein kinase 26 | 0.67 | −0.57 | 0.020 |
| Q16401 | PSMD5 | 26S proteasome non‐ATPase regulatory subunit 5 | 0.67 | −0.57 | 0.023 |
| Q9Y305 | ACOT9 | Acyl‐coenzyme A thioesterase 9, mitochondrial | 0.67 | −0.58 | 0.006 |
| Q8IZ83 | ALDH16A1 | Aldehyde dehydrogenase family 16 member A1 | 0.64 | −0.64 | 0.016 |
| Q9Y6H1 | CHCHD2 | Coiled‐coil‐helix‐coiled‐coil‐helix domain‐containing protein 2 | 0.63 | −0.66 | 0.009 |
| H0YMB3 | GMPR2 | GMP reductase | 0.62 | −0.69 | 0.030 |
| P00558 | PGK1 | Phosphoglycerate kinase 1 | 0.62 | −0.69 | 0.034 |
| Q8N697 | SLC15A4 | Solute carrier family 15 member 4 | 0.61 | −0.70 | 0.004 |
| O60637 | TSPAN3 | Tetraspanin‐3 | 0.61 | −0.71 | 0.007 |
| P00338 | LDHA | L‐lactate dehydrogenase A chain | 0.61 | −0.71 | 0.027 |
| P13284 | IFI30 | Gamma‐interferon‐inducible lysosomal thiol reductase | 0.61 | −0.72 | 0.038 |
| O15460 | P4HA2 | Prolyl 4‐hydroxylase subunit alpha‐2 | 0.60 | −0.73 | 0.039 |
| A0A087WZU5 | TSPAN6 | Tetraspanin‐6 | 0.60 | −0.74 | 0.002 |
| A2RQR6 | TCEAL4 | Transcription elongation factor A (SII)‐like 4 variant 1 | 0.58 | −0.78 | 0.020 |
| P62805 | HIST1H4A | Histone H4 | 0.57 | −0.81 | 0.038 |
| P68431 | HIST1H3A | Histone H3.1 | 0.51 | −0.98 | 0.034 |
| Q9BTZ2 | DHRS4 | Dehydrogenase/reductase SDR family member 4 | 0.49 | −1.02 | 0.004 |
| P80297 | MT1X | Metallothionein‐1X | 0.40 | −1.31 | 0.003 |
| A0A0G2JMG8 | NLRP2 | NACHT, LRR and PYD domains‐containing protein 2 | 0.39 | −1.38 | 0.002 |
Among the selected 230 statistically significant proteins (q < 0.05), 13 (in dark grey) enriched in hiPSC‐1 vs hESCs according to a log2 fold change >0.5. 25 proteins (in light grey) were found down‐regulated in hiPSC‐1 vs hESCs according to the same log2 fold change.
hESCs, human embryonic stem cells; hiPSCs, human induced pluripotent stem cells.
Figure 1.
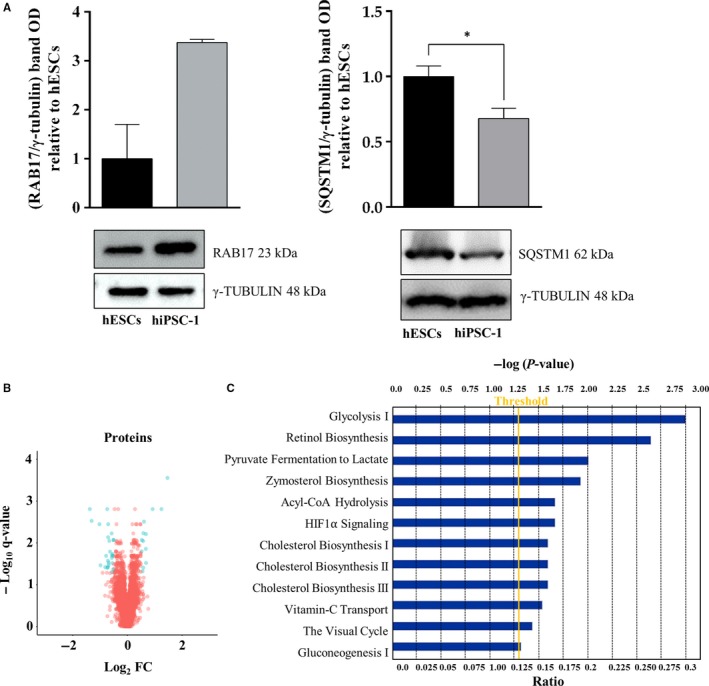
A, Western blot analysis for RAB17 (P value 0.3) and SQSTM1 (P value 0.03), up‐ and down‐regulated in hiPSC‐1 vs hESCs, respectively. Data are mean ± SEM from three independent biological replicates (*P < 0.05, t‐test). B, Volcano Plot of differentially dysregulated proteins (blue dots) between hiPSC‐1 vs hESCs. Unchanged proteins are represented as red dots. The x‐axis specifies the Log2 fold change (Fc) and the y‐axis specifies the q‐value negative logarithm in base 10. C, Ingenuity Pathway Analysis of specific signalling pathways enriched by the differentially expressed proteins. hESCs, human embryonic stem cells; hiPSCs, human induced pluripotent stem cells
Whole proteomic data analysis identified key regulators of pluripotency, including Sox15,26 up‐regulated in hiPSC‐1 vs hESCs, and OCT4, whose expression was similar in reprogrammed and ES cell lines (q‐value 0.43, fold‐change 0.91). Differentially regulated proteins are shown in a Volcano plot (neg log10, q value, vs fold change hiPSC‐1/hESCs) (Figure 1B). IPA of these proteins revealed the enrichment of the following signalling pathways: Glycolysis I, Retinol Biosynthesis, Pyruvate Fermentation to Lactate, Zymosterol Biosynthesis, Acyl‐CoA Hydrolysis, HIF1α Signalling, Cholesterol Biosynthesis, Vitamin‐C Transport, The Visual Cycle, Gluconeogenesis I, listed in Figure 1C. The superpathway of Cholesterol Biosynthesis resulted in enriched hiPSC‐1 vs hESCs, as demonstrated by the presence of two enzymes involved in the cholesterol biosynthesis, namely HMGCR (3‐Hydroxy‐3‐Methylglutaryl‐CoA Reductase) (Table S6) and LBR (Lamin B receptor) (Table S4), respectively down‐ and up‐regulated in hiPSCs.
3.2. Identification and classification of differentially expressed phosphoproteins
Phosphoproteome analysis allowed us to identify and quantify 5958 phophopeptides and 2623 phosphoproteins (Tables S5 and S6). Of these, 69 phophopeptides and 73 phosphoproteins were found statistically significant according to Student's t‐test with the Benjamini‐Hochberg correction (q < 0.05). 19 phosphopeptides and 14 phosphoproteins (SOX15, RPS8, DPF2, WBP4, SLC38A1, ADAR, CASK, PLEKHA6, BSG, SLC3A2, TRAM1, PLEKHG3, NCBP1, OVOL2) were found up‐regulated in hiPSC‐1 (based on log2 hiPSC‐1/hESCs >0.5). SOX15 was found over‐expressed within the hiPSCs‐1 digests according to a q‐value 0.02 and a fold‐change of 2.10. Conversely, 31 phosphopeptides and 20 phosphoproteins (PRKAR2A, BRAP, HMGCR, OSBPL8, CDC6, HSPB1, PPIL1, NPY1R, ANLN, THUMPD1, STX18, MGMT, ANP32B, CDC7, EFNB1, HTR1A, PDAP1, SQSTM1, SEMA4B, SPDL1) were found under‐expressed in hiPSC‐1 samples (based on log2 hiPSC‐1/hESCs <−0.5). Table 2 is relative to differentially expressed phosphoproteins.
Table 2.
Differentially expressed phosphoproteins
| Accession | Gene | Description | hiPSC‐1/hESCs Fc | log2 Fc | q value |
|---|---|---|---|---|---|
| O60248 | SOX15 | Protein SOX‐15 | 2.10 | 1.07 | 0.02 |
| Q5JR95 | RPS8 | 40S ribosomal protein S8 | 1.87 | 0.91 | 0.04 |
| Q92785 | DPF2 | Zinc finger protein Ubi‐D4 | 1.85 | 0.89 | 0.04 |
| O75554 | WBP4 | WW domain‐binding protein 4 | 1.62 | 0.70 | 0.04 |
| F8VX12 | SLC38A1 | Sodium‐coupled neutral amino acid transporter 1 (Fragment) | 1.58 | 0.66 | 0.03 |
| P55265 | ADAR | Double‐stranded RNA‐specific adenine deaminase | 1.54 | 0.62 | 0.00 |
| Q5JS72 | CASK | Peripheral plasma membrane protein CASK | 1.53 | 0.62 | 0.04 |
| Q9Y2H5 | PLEKHA6 | Pleckstrin homology domain‐containing family A member 6 | 1.53 | 0.61 | 0.02 |
| A0A087WUV8 | BSG | Basigin | 1.50 | 0.59 | 0.00 |
| F5GZS6 | SLC3A2 | 4F2 cell‐surface antigen heavy chain | 1.49 | 0.58 | 0.04 |
| G3XAN4 | TRAM1 | Translocating chain‐associated membrane protein 1 | 1.49 | 0.58 | 0.04 |
| A1L390 | PLEKHG3 | Pleckstrin homology domain‐containing family G member 3 | 1.45 | 0.54 | 0.04 |
| F2Z2T1 | NCBP1 | Nuclear cap‐binding protein subunit 1 | 1.44 | 0.52 | 0.04 |
| Q9BRP0 | OVOL2 | Transcription factor Ovo‐like 2 | 1.41 | 0.50 | 0.03 |
| P13861 | PRKAR2A | cAMP‐dependent protein kinase type II‐alpha regulatory subunit | 0.69 | −0.53 | 0.03 |
| J3KNN7 | BRAP | BRCA1‐associated protein | 0.69 | −0.54 | 0.04 |
| P04035 | HMGCR | 3‐hydroxy‐3‐methylglutaryl‐coenzyme A reductase | 0.69 | −0.54 | 0.04 |
| Q9BZF1 | OSBPL8 | Oxysterol‐binding protein‐related protein 8 | 0.68 | −0.55 | 0.04 |
| J3KTI7 | CDC6 | Cell division control protein 6 homolog (Fragment) | 0.67 | −0.57 | 0.04 |
| F8WE04 | HSPB1 | Heat shock protein beta‐1 | 0.67 | −0.58 | 0.04 |
| Q9Y3C6 | PPIL1 | Peptidyl‐prolyl cis‐trans isomerase‐like 1 | 0.65 | −0.62 | 0.01 |
| B4DKL9 | NPY1R | Neuropeptide Y receptor type 1 | 0.64 | −0.65 | 0.01 |
| Q9NQW6 | ANLN | Actin‐binding protein anillin | 0.61 | −0.70 | 0.04 |
| Q9NXG2 | THUMPD1 | THUMP domain‐containing protein 1 | 0.61 | −0.72 | 0.02 |
| D6RC71 | STX18 | Syntaxin‐18 (Fragment) | 0.60 | −0.73 | 0.03 |
| P16455 | MGMT | Methylated‐DNA‐‐protein‐cysteine methyltransferase | 0.60 | −0.74 | 0.04 |
| Q92688 | ANP32B | Acidic leucine‐rich nuclear ph phoprotein 32 family member B | 0.60 | −0.74 | 0.03 |
| O00311 | CDC7 | Cell division cycle 7‐related protein kinase | 0.59 | −0.77 | 0.04 |
| P98172 | EFNB1 | Ephrin‐B1 | 0.56 | −0.84 | 0.04 |
| P08908 | HTR1A | 5‐hydroxytryptamine receptor 1A | 0.53 | −0.92 | 0.02 |
| Q13442 | PDAP1 | 28 kDa heat‐ and acid‐stable phosphoprotein | 0.51 | −0.98 | 0.02 |
| E7EMC7 | SQSTM1 | Sequestosome‐1 | 0.51 | −0.98 | 0.03 |
| Q9NPR2 | SEMA4B | Semaphorin‐4B | 0.41 | −1.29 | 0.00 |
| Q96EA4 | SPDL1 | Protein Spindly | 0.37 | −1.45 | 0.02 |
In dark grey phosphoproteins found up‐regulated in hiPSCs‐1 vs hESCs (based on log2 hiPSC‐1/hESCs >0.5); in light grey phosphoproteins down‐regulated in hiPSC‐1 vs hESCs (based on log2 hiPSC‐1/hESCs <−0.5).
Differentially abundant phosphopetides and phosphoproteins, plotted in Figure 2A and 2, account for the enrichment of Gαi Signalling, Cell Cycle control of chromosomal replication, PCP Pathway, cAMP‐mediated Signalling and G‐Protein‐Coupled receptor Signalling as shown by IPA analysis in Figure 2C. The expression of two phosphoproteins, the phospho‐HSPB1 (Ser82) and the phospho‐SQSTM1 (Thr269/Ser272), both down‐regulated in hiPSC‐1 vs hESCs, was validated via Western Blot analysis in hESCs and three independent hiPSCs lines (hiPSC‐1, hiPSCs‐2, and hiPSCs‐3), confirming the trend of expression obtained with the phoshoproteomic analysis (Figure 2D).
Figure 2.
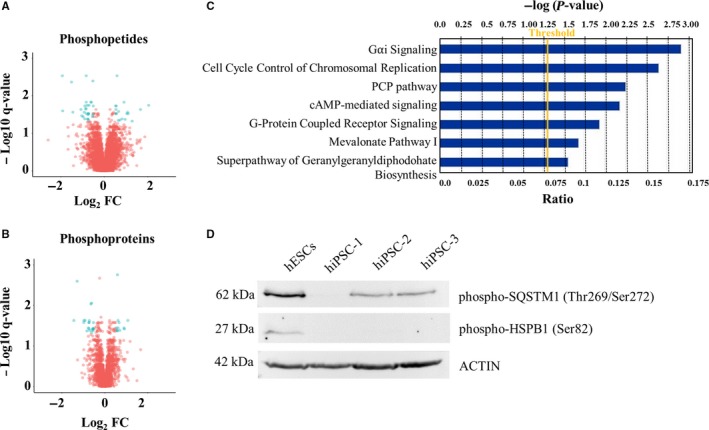
A, Volcano Plot representing differentially abundant phosphopetides between hiPSC‐1 and hESCs. B, Volcano Plot of differentially abundant phosphoproteins between hiPSC‐1 and hESCs. The x‐axis specifies the Log2 fold change (Fc) and the y‐axis specifies the q‐value negative logarithm in base 10. Differentially expressed phosphopeptides and phoshoproteins are represented as blue dots, while the unchanged ones are shown as red dots. C, Ingenuity Pathway Analysis showing signalling pathways enriched by the differentially expressed phosphoproteins. D, Western blot analysis of phospho‐HSPB1 (Ser82) and phospho‐SQSTM1 (Thr269/Ser272), both down‐regulated in hiPSCs (‐1, ‐2, and ‐3) vs hESCs. hESCs, human embryonic stem cells; hiPSCs, human induced pluripotent stem cells
3.3. Transcriptome analysis for the identification and classification of DEGs
Comparative transcriptome analysis of hiPSC‐1 vs hESCs, followed by a functional annotation analysis, highlighted 433 DEGs (Figure 3A), 136 of which were up‐regulated and 297 down‐regulated in hiPSC‐1 vs hESCs (Table S7).
Figure 3.
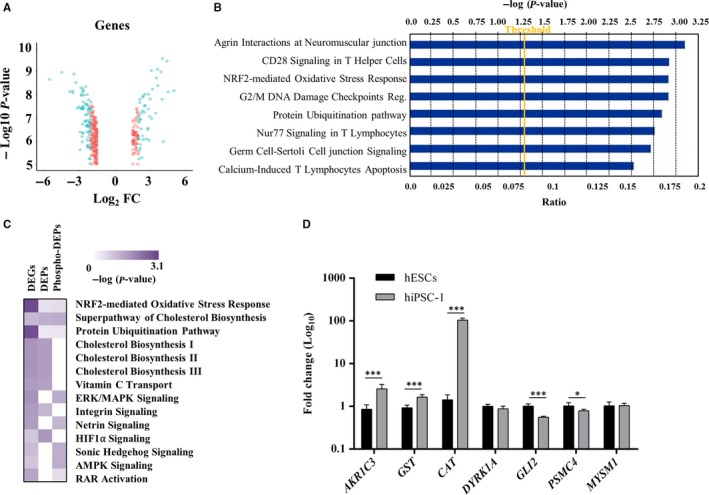
A, Volcano plot representing differentially expressed genes (blue dots) between hiPSC‐1 and hESCs; the unchanged genes are shown as red dots. The x‐axis specifies the Log2 fold change (Fc) and the y‐axis specifies the q‐value negative logarithm in base 10. B, Top canonical signalling pathways uncovered by Ingenuity Pathway Analysis. C, Heat Map representing color‐coded networks derived from the integrative transcriptomic, whole proteomic and phosphoproteomic analysis of hiPSC‐1 and hESCs (the colour intensity scale is proportional to the statistical significance of a specific pathway enriched in each dataset, – log (P‐value). D, Quantitative real‐time polymerase chain reaction analysis of the NRF2‐mediated Oxidative Stress Response (AKR1C3, GST, CAT), Sonic Hedgehog (DYRK1A and GLI2) and Protein Ubiquitination pathways (PSMC4 and MYSM1) ‐ associated genes. Expression values are normalized to GAPDH and relative to hESCs. Data are mean ± SD from three independent experiments (*P < 0.05, ***P < 0.001, t‐test). hESCs, human embryonic stem cells; hiPSCs, human induced pluripotent stem cells
IPA analysis uncovered the top canonical pathways enriched in hiPSC‐1 such as Agrin Interactions at Neuromuscular junction, CD28 Signalling in T Helper cells, NRF2‐mediated Oxidative stress response, G2/M DNA Damage checkpoint regulation, Protein Ubiquitination pathway, Nur77 Signalling in T Lymphocytes, Germ Cell‐Sertoli Cell junction Signalling, and Calcium‐Induced T lymphocytes Apoptosis (Figure 3B).
3.4. Integrative whole proteome, phosphoproteome and transcriptome analysis
For a comprehensive proteogenomic analysis, we performed a ‘cross‐omics’ study using datasets encompassing mRNA, protein and phosphoprotein expression profiles obtained from hESCs and hiPSC‐1. Among the networks commonly shared, we identified the NRF2‐mediated Oxidative Stress Response, Superpathway of Cholesterol Biosynthesis, and Protein Ubiquitination Pathway. The complete list of signalling pathways identified is provided in Table S8. Interestingly, IPA comparison also uncovered a number of pathways enriched only in two out of three datasets given as input. In particular: (a) Cholesterol biosynthesis (I, II, III), Vitamin C Transport, Integrin Signalling and HIF1α Signalling are enriched in transcriptome and whole proteome datasets; (b) ERK/MAPK Signalling, Netrin Signalling, Sonic Hedgehog Signalling, AMPK Signalling and RAR activation are common among transcriptome and phosphoproteome datasets (Figure 3C). For biological validation of the integrative comparison the following genes were analysed by qRT‐PCR: AKR1C3, GST, CAT (NRF2‐associated), PSMC4 and MYSM1 (Protein Ubiquitination Pathway‐associated) (both pathways commonly enriched in all three datasets), DYRK1A and GLI2 (Sonic Hedgehog Signalling‐associated) (shared by transcriptome and phosphoproteome datasets). The qRT‐PCR analysis confirmed the trend of expression of the proteogenomic data: AKR1C3 (Fc 2.56), GST (Fc 1.64), CAT (Fc 104.69) significantly up‐regulated in hiPSC‐1 vs hESCs, GLI2 (Fc 0.55) and PSMC4 (Fc 0.79) significantly down‐regulated in hiPSC‐1 vs hESCs, the expression of DYRK1A, although down‐regulated in hiPSC‐1 (Fc 0.87), is not statistically significant. Finally, the expression level of MYSM1 remains unchanged between the two lines (Figure 3D). Further, we used the cutoff applied in the present manuscript to analyse data from a similar work by Phanstiel et al18 Several pathways, such as those involved in the regulation of cell proliferation such as PI3K/Akt signalling, Sonic Hedgehog, Notch signalling, and in the maintenance of embryonic stem cell self‐renewal and pluripotency such as NRF, and ERK/MAPK signalling resulted commonly enriched (Figure 4A). Although the majority of pathways identified by this cross‐analysis are commonly shared by the two studies, some resulted exclusively enriched in our dataset, such as Retinol Biosynthesis, Cell Cycle Control of Chromosomal Replication, cAMP‐mediated Signalling, and Ephrin Receptor Signalling, FAK Signalling, and RAR Activation pathway. Further, we selected some target genes of these “exclusive” pathways (DHRS4 as target of the Retinol Biosynthesis pathway, MCM7 as target of the Cell Cycle Control of Chromosomal Replication, HTR7 and PLD6 for the cAMP‐mediated signalling, EFNB1, EPHA4 and ROCK1 for the Ephrin Receptor signalling and AKR1C3, EP300, CYP26A1, MAPK10, and TRIM24 for RAR Activation pathway) and validated them by qRT‐PCR analysis in hESCs, and hiPSC‐1, hiPSC‐2, and hiPSC‐3 lines. DHRS4 (average Fc 0.40), EFNB1 (Fc 0.64), EPHA4 (average Fc 0.73), CYP26A1 (average Fc 0.28), MAPK10 (average Fc 0.70) and TRIM24 (average Fc 0.72) resulted down‐regulated in hiPSC vs ESCs. MCM7 (Average Fc 0.9), ROCK1 (Average Fc 0.91) and EP300 (Average Fc 0.80) were not significantly dysregulated, HTR7 (average FC 6.26), PLD6 (average FC 8.14), and AKR1C3 (average FC 2.22) resulted up‐regulated in hiPSCs vs hESCs (Figure 4B). A complete list of Signalling pathways identified is provided in Table S9.
Figure 4.
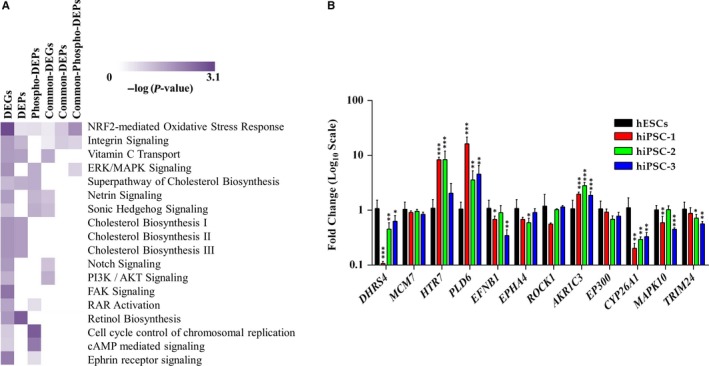
A, Heat Map representing signalling pathways exclusively and commonly enriched in our dataset and in the one coming from Phanstiel et al. B, Quantitative real‐time polymerase chain reaction analysis of some target genes associated with the exclusively enriched pathways. Expression values are normalized to GAPDH and relative to human embryonic stem cells. Data are mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, t‐test)
3.5. Role of the RAR activation pathway and its modulation on the phenotype of human ESCs and human PSCs
Among the pathways identified as differentially regulated in human ESCs and iPSC‐1, we selected the RAR Activation signalling, as retinoids including Vitamin A and its derivatives have been widely associated with embryonic development and differentiation.27, 28 Here, we evaluated the effects on hESCs and hiPSC‐1, ‐2, and ‐3 of two molecules, the all‐trans Retinoic Acid (RA) which functions as positive regulator of the RAR pathway, and BMS493, a powerful Pan‐retinoic acid receptor (pan‐RAR) antagonist that enhances nuclear corepressor (NCoR) interaction with RARs.29 Its binding induces analogous conformational changes in all RAR types (RARα, RARβ and RARγ) inactivating the transcription of target genes. For RAR pathway modulation, cells were treated either with RA or with BMS493 or with a combination of RA and BMS493 together for 24 hours. When exposed to RA, the expression of a direct target of Retinoic Acid, RARβ, increased in all cell lines while the combination of RA and BMS493 induced a reduction in its expression but still higher compared to untreated cells (Figure 5A). The exposure to RA triggered a 6‐fold increased expression of CYP26A1, a target of RA in hESCs and a 22‐ to 40‐fold increased expression in hiPSCs (Figure 5B). Endogenous production of RA is unlikely to take place in ES cell,30 although these cells express RARx, RXRs and Crabp1 required for RA response and for regulation of the expression of its targets such as RARβ and CYP26A1 in a time and dose‐dependent manner. This might explain why the treatment of hESCs and hiPSCs‐2 and ‐3 with BMS493 alone does not affect the expression of RARβ and CYP26A1 compared to cells cultured in Regular Medium. Exposure to either RA or BMS493 or to a combination of both molecules did not induce a significant variation in any of the other genes accounting for the enrichment of the RAR pathway such as AKR1C3, MAPK10, TRIM24 with the exception of EP300 resulted in induced hESCs treated with BMS493 for 24 hours (Figure S2). Further we analysed the effect of RAR modulation on differentiation potential of hESCs and hiPSCs lines. After treatment with RA or BMS493 or both, cells were induced to differentiation by Embryoid Body (EB) formation assay. Even though the overall capability of hESCs and hiPSCs (‐1, ‐2, and 3) to differentiate were maintained, the expression levels of specific germ layer‐associated genes such as NESTIN (ectodermal differentiation), BRACHYURY (mesodermal differentiation), and SOX17 (endodermal differentiation) were differentially affected in response to RA, BMS or both used in combination. While the differentiation ability of hESCs was not significantly affected by the treatment with RA or BMS493 or both in combination as shown by immunofluorescence quantification, the hiPSCs lines (‐1, ‐2, and ‐3) showed an enhanced expression of SOX17 when they were simultaneously exposed to RA and BMS493 (RA+/BMS493+) as shown by immunofluorescence (Figure S3) and its quantification (Figure 5C). qRT‐PCR analysis for NESTIN, MESP1 and SOX17 expression showed a reduction of these markers in hESCs in all the three conditions tested (RA+; BMS493+; RA+/BMS493+); conversely, although the hiPSC lines showed a similar trend when exposed either to RA or BMS493, they completely recovered their endodermal and mesodermal differentiation potential when simultaneously treated with both molecules (RA+/BMS493+), while the ectodermal differentiation capability did not significantly changed (Figure 5D).
Figure 5.

A, Quantitative real‐time polymerase chain reaction (qRT‐PCR) analysis of RARβ. B, qRT‐PCR analysis of CYP26A1 in human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) (hiPSC‐1, ‐2, and ‐3) treated with RA (RA+) or with BMS493 (BMS493+) or with both (RA+/BMS493+); expression values are normalized to GAPDH and relative to regular medium (RM) (mTeSR1 medium). C, Corrected total cell fluorescence of markers specific of each germ layer: Nestin (ectoderm), Brachyury (mesoderm) and Sox17 (endoderm) in EBs derived from hESCs and hiPSCs cell lines (‐1, ‐2, and ‐3) cultured in RM and in medium supplemented for 24 h with 0.5 µM RA, 5 µM BMS493 or with both molecules. Bars represents measurements derived from 5 cells for each condition (mean ± SD). D, qRT‐PCR analysis of NESTIN, MESP1 (mesoderm marker) and SOX17 in EBs derived from hESCs and hiPSC‐1, ‐2, and ‐3 cultured in RM and in medium supplemented for 24 h with 0.5 µM RA, 5 µM BMS493 or with both molecules. Expression values are normalized to GAPDH and relative to EBs derived from untreated hESCs. Data are showed as mean ± SD from three independent experiments (*P < 0.05, **P < 0.01, ***P < 0.001, t‐test)
4. DISCUSSION
Recent breakthroughs in stem cells research allowed to envision a scenario in which a stem cell‐based therapy might become reality. Particularly, the findings that differentiated somatic cells can be reprogrammed back to a pluripotent state and subsequently differentiated towards several cell types showed the feasibility of using the hiPSCs in a clinical setting. Although hiPSCs share a number of characteristics with hESCs, many fundamental questions around their molecular and functional equivalence still remain unanswered. Here we used a combined multi – “omics” strategy for proteogenomic analysis of hESCs and hiPSC, highlighting differences between these cells. IPA analysis of DEPs and transcripts demonstrated the enrichment of signalling pathways mostly associated to pluripotency and cellular metabolism, whose role goes beyond energy production being crucial in cell fate regulation.31 The HIF1α signalling, for instance, enriched in hiPSC‐1 vs hESCs, mediates the effects of oxygen concentration on stem cells proliferation and differentiation by induction of the hypoxia‐associated genes.32 Accordingly, in response to oxygen fluctuations, oxygen signalling controls the balance between pluripotency and differentiation. Highly pluripotent ESCs associate with hypoxia, high levels of glucose consumption and large amount of lactate production.33, 34 The NRF2‐mediated oxidative stress response pathway, commonly enriched in hESCs and hiPSC‐1, has been described as a key regulator of self‐renewal ability of hESCs playing a role in re‐establishment of pluripotency during cellular reprogramming.35, 36 Although PSCs rely more on glycolysis for energy and rapid cell proliferation, the tricarboxylic acid cycle in PSCs provides intermediate metabolites such a citrate and α‐ketoglutarate that are siphoned for lipids and amino acid biosynthesis.37 We found that LDHA and two glycolytic enzymes, phosphoglycerate kinase 1 (PGK1) and the pyruvate kinase (PKM) are down regulated in hiPSC‐1 compared to hESCs, while the mitochondrial carrier SLC25A1, encoding for a trycarboxylate protein that functions as transporter of the mitochondrial citrate into the cytosol, results up‐regulated in hiPSC‐1. Moreover, there are evidences suggesting a co‐existence of both oxidative and glycolytic metabolisms in stem cells.38 Cholesterol contributes to the maintenance of self‐renewal in ESCs by regulation of LIF‐induced signalling, and its derivatives, such as glucocorticoids, regulate cell fate during early development and seem to control cardiac differentiation in mESCs.39 Panopoulus et al40 demonstrated that the level of some unsaturated fatty acids such as arachidonic acid, implicated in the cholesterol homeostasis regulation, differs quantitatively between iPSCs and ESCs.41 Another pathway that resulted enriched in hiPSC‐1 vs hESCs is represented by the anti‐oxidant system as shown by the over‐expression in hiPSC‐1 of the glucose transporter SLC2A3, previously associated with an anti‐oxidant role.42
To further strengthen our findings, we used the cutoff applied in this study to analyse data from Phanstiel et al18 Several signalling pathways were commonly enriched in both studies, while others (Retinol Biosynthesis, Cell cycle Control of Chromosomal Replication, cAMP‐mediated Signalling, Ephrin Receptor Signalling, FAK Signalling, and RAR Activation Signalling) resulted exclusively enriched in our datasets and validated by qRT‐PCR analysis. Among these exclusive pathways, we focused on the RAR Activation Pathway since Retinoic acid and its receptors are classically linked both to embryonic development43, 44 and to pluripotency maintenance.28, 45 Subtle regulation of RA signalling is fundamental for the PSCs in order to choose between self‐renewal and differentiation programmes, as demonstrated by the fact that hESCs and hiPSCs behave differently when they undergo to treatment with Retinoic acid or BMS493. To investigate the role of RAR pathway, we first looked at the expression level of its target genes such as AKR1C3, MAPK10, EP300 and TRIM24 in hESCs and hiPSC‐1, ‐2, and ‐3 cultured in regular medium (mTeSR1), showing an increased expression of AKR1C3 in hiPSCs lines compared to hESCs, while the expression levels of MAPK10, EP300, CYP26A1 and TRIM24 had an opposite trend. When cells were treated with RA the expression of CYP26A1 was dramatically increased in hiPSC lines compared to hESCs. These data suggest that hiPSCs are more sensitive to RA than hESCs. This greater sensitivity of hiPSCs to RA can explain the higher expression of CYP26A1, a member of the cytochrome P450 family that controls the levels of RA by its oxidation to a less biologically active form, aiming to keep the pathway in a balanced activity. This hypothesis is supported by previously published data demonstrating that CYP26A1 ‐/‐ ESCs exhibit an increase of intracellular level of RA accompanied by a reduction of the differentiation capability respect to wild‐type ESCs.46 hESCs and hiPSCs showed a different propensity to enter a specific lineage commitment in response to treatment (RA+ or BMS493+ or RA+/BMS493+). hiPSC lines treated with RA or BMS493 alone show a slight resistance to the endodermal lineage. A complete re‐establishment of hiPSCs differentiation potential is instead observed when cells are simultaneously treated with RA and BMS493, supporting the fact that a synergistic action of both molecules is sufficient to restore the activity of the pathway.
In conclusion, our data indicate that the molecular correspondence between hiPSCs and hESCs is not exactly linear, as demonstrated by the presence of specific pathways representative for each cell line. The discovery of pathways enriched in hiPSC‐1 vs hESCs and the identification of pathways exclusively present in our dataset are in agreement with the view that pluripotency is an extremely complex and multifaceted phenomenon, with peculiarities that are characteristic of each pluripotent cell type, and that hESCs and hiPSCs cannot be considered equivalent from a functional point of view. Our data provide evidence that reprogrammed cells possess a unique molecular signature that can have functional and phenotypic consequences when a given pathway is modulated. Future investigations of the identified pathways and their relative components will provide new insights into the complex mechanisms of pluripotency and self‐renewal of reprogrammed cells and will give the opportunity to understand how molecular variations can impact the phenotypic and functional behaviors of reprogrammed stem cells.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
AUTHOR CONTRIBUTIONS
GC conceived the study; EIP, SS, DT, MTD, GS, MG and GS performed the experiments; analysed the data; EIP, SS, DT, GS analysed the data and performed interpretation; GS and DT conducted the bioinformatic and statistical analysis; EIP, SS, DT, GC and GS wrote this manuscript; GC and GS reviewed and edited the final version of this article; GC provided the funds and materials. All authors have read and approved the final edition of this manuscript.
ETHICAL APPROVAL
The use and inclusion of human samples was approved by the Ethics Committee of the ‘Magna Graecia’ University of Catanzaro and the Azienda Ospedaliero – Universitaria “Mater Domini”. Skin biopsy and whole blood withdrawal were obtained from three individuals without clinical evidences for any specific disease after written informed consent from the donor.
Supporting information
ACKNOWLEDGMENTS
This work was supported by in part by the MIUR grant PON03PE_00009_2 (iCARE) to G.C.
Parrotta EI, Scalise S, Taverna D, et al. Comprehensive proteogenomic analysis of human embryonic and induced pluripotent stem cells. J Cell Mol Med. 2019;23:5440–5453. 10.1111/jcmm.14426
Elvira Immacolata Parrotta, Stefania Scalise, and Domenico Taverna are equally contributed to this work.
Contributor Information
Gianluca Santamaria, Email: gsantamaria@unicz.it.
Giovanni Cuda, Email: cuda@unicz.it.
REFERENCES
- 1. Thomson JA, Itskovitz‐eldor J, Shapiro SS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282(5391):1145‐1147. [DOI] [PubMed] [Google Scholar]
- 2. Fatehullah A, Tan SH, Barker N. Organoids as an in vitro model of human development and disease. Nat. Cell. Biol. 2016;18:246‐254. [DOI] [PubMed] [Google Scholar]
- 3. Takahashi K, Tanabe K, Ohnuki M, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861‐872. [DOI] [PubMed] [Google Scholar]
- 4. Tiscornia G, Vivas EL, Belmonte J. Diseases in a dish: modeling human genetic disorders using induced pluripotent cells. Nat. Med. 2011;17:1570‐1576. [DOI] [PubMed] [Google Scholar]
- 5. Deshmukh RS, Kovács KA, Dinnyés A. Drug discovery models and toxicity testing using embryonic and induced pluripotent stem‐cell‐derived cardiac and neuronal cells. Stem Cells Int. 2012;2012:5440‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Robinton DA, Daley GQ. The promise of induced pluripotent stem cells in research and therapy. Nature. 2012;481(7381):295‐305. 10.1038/nature10761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Birket MJ, Ribeiro MC, Verkerk AO, et al. Expansion and patterning of cardiovascular progenitors derived from human pluripotent stem cells. Nat. Biotechnol. 2015;33:970‐979. [DOI] [PubMed] [Google Scholar]
- 8. Wakeman DR, Hiller BM, Marmion DJ, et al. Cryopreservation Maintains Functionality of Human iPSC Dopamine Neurons and Rescues Parkinsonian Phenotypes In Vivo. Stem Cell Reports. 2017;9:149‐161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Park I‐H, Zhao R, West JA, et al. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008;451:141‐146. [DOI] [PubMed] [Google Scholar]
- 10. Chin MH, Mason MJ, Xie W, et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Parrotta E, De Angelis MT, Scalise S, et al. Two sides of the same coin? Unraveling subtle differences between human embryonic and induced pluripotent stem cells by Raman spectroscopy. Stem Cell Res. Ther. 2017;8:271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kim K, Doi A, Wen B, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285‐290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ghosh Z, Wilson KD, Wu YI, Hu S, Quertermous T, Wu JC. Persistent donor cell gene expression among human induced pluripotent stem cells contributes to differences with human embryonic stem cells. PLoS ONE. 2010;5(2):e8975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Marchetto M, Yeo GW, Kainohana O, Marsala M, Gage FH, Muotri AR. Transcriptional signature and memory retention of human‐induced pluripotent stem cells. PLoS ONE. 2009;4:e7076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Ohi Y, Qin H, Hong C, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nat. Cell Biol. 2011;13:541‐549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kim SY, Kim M‐J, Jung H, et al. Comparative proteomic analysis of human somatic cells, induced pluripotent stem cells, and embryonic stem cells. Stem Cells Dev. 2012;21:1272‐1286. [DOI] [PubMed] [Google Scholar]
- 17. Wang Y‐C, Peterson SE, Loring JF. Protein post‐translational modifications and regulation of pluripotency in human stem cells. Cell Res. 2014;24:143‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Phanstiel DH, Brumbaugh J, Wenger CD, et al. Proteomic and phosphoproteomic comparison of human ES and iPS cells. Nat. Methods. 2011;8:821‐827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seki T, Yuasa S, Fukuda K. Generation of induced pluripotent stem cells from a small amount of human peripheral blood using a combination of activated T cells and Sendai virus. Nat. Protoc. 2012;7:718‐728. [DOI] [PubMed] [Google Scholar]
- 20. Müller F‐J, Schuldt BM, Williams R, et al. A bioinformatic assay for pluripotency in human cells. Nat. Methods. 2011;8:315‐317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Taverna D, Mignogna C, Gabriele C, et al. An optimized procedure for on‐tissue localized protein digestion and quantification using hydrogel discs and isobaric mass tags: analysis of cardiac myxoma. Anal. Bioanal. Chem. 2017;409:2919‐2930. [DOI] [PubMed] [Google Scholar]
- 22. Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Series B Stat. Methodol. 1995;57:289‐300. [Google Scholar]
- 23. Ritchie ME, Phipson B, Wu DI, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47‐e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi J, Zhao P, Li F, et al. Down‐regulation of Rab17 promotes tumourigenic properties of hepatocellular carcinoma cells via Erk pathway. Int. J. Clin. Exp. Pathol. 2015;8:4963‐4971. [PMC free article] [PubMed] [Google Scholar]
- 25. Wang S, Xia P, Ye B, Huang G, Liu J, Fan Z. Transient activation of autophagy via Sox2‐mediated suppression of mTOR is an important early step in reprogramming to pluripotency. Cell Stem Cell. 2013;13:617‐625. [DOI] [PubMed] [Google Scholar]
- 26. Sarkar A, Hochedlinger K. The Sox family of transcription factors: versatile regulators of stem and progenitor cell fate. Cell Stem Cell. 2013;12:15‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Khillan JS. Vitamin A/retinol and maintenance of pluripotency of stem cells. Nutrients. 2014;6:1209‐1222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. De Angelis MT, Parrotta EI, Santamaria G, Cuda G. Short‐term retinoic acid treatment sustains pluripotency and suppresses differentiation of human induced pluripotent stem cells. Cell Death Dis. 2018;9(1):6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Germain P, Gaudon C, Pogenberg V, et al. Article differential action on coregulator interaction defines inverse retinoid agonists and neutral antagonists. Chem. Biol. 2009;16:479‐489. [DOI] [PubMed] [Google Scholar]
- 30. Simandi Z, Balint BL, Poliska S, Ruhl R, Nagy L. Activation of retinoic acid receptor signaling coordinates lineage commitment of spontaneously differentiating mouse embryonic stem cells in embryoid bodies. FEBS Lett. 2010;584:3123‐3130. [DOI] [PubMed] [Google Scholar]
- 31. Mathieu J, Ruohola‐Baker H. Metabolic remodeling during the loss and acquisition of pluripotency. Development. 2017;144(4):541‐551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Keith B, Simon MC. Hypoxia‐Inducible Factors, Stem Cells, and Cancer. Cell. 2007;129(3):465‐472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saito S, Lin Y‐C, Tsai M‐H, et al. Emerging roles of hypoxia‐inducible factors and reactive oxygen species in cancer and pluripotent stem cells. Kaohsiung J. Med. Sci. 2015;31:279‐286. [DOI] [PubMed] [Google Scholar]
- 34. Forristal CE, Christensen DR, Chinnery FE, et al. Environmental oxygen tension regulates the energy metabolism and self‐renewal of human embryonic stem cells. PLoS ONE. 2013;8:5440‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hawkins K, Joy S, Delhove J, et al. NRF2 orchestrates the metabolic shift during induced pluripotent stem cell reprogramming. Cell Rep. 2016;14:1883‐1891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jang J, Wang Y, Kim H‐S, Lalli MA, Kosik KS. Nrf2, a regulator of the proteasome, controls self‐renewal and pluripotency in human embryonic stem cells. Stem Cells. 2014;32:2616‐2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zhang J, Nuebel E, Daley G, Koehler C, Teitell M. Metabolic regulation in pluripotent stem cells during reprogramming and self‐renewal. Cell Stem Cell. 2012;11:589‐595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Teslaa T, Teitell MA. Pluripotent stem cell energy metabolism: an update. EMBO J. 2015;34(2):138‐153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Cabral‐Teixeira J, Martinez‐Fernandez A, Cai W, Terzic A, Mercola M, Willems E. Cholesterol‐derived glucocorticoids control early fate specification in embryonic stem cells. Stem Cell Res. 2015;15:88‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Panopoulos AD, Yanes O, Ruiz S, et al. The metabolome of induced pluripotent stem cells reveals metabolic changes occurring in somatic cell reprogramming. Cell Res. 2012;22:168‐177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Demetz E, Schroll A, Auer K, et al. The arachidonic acid metabolome serves as a conserved regulator of cholesterol metabolism. Cell Metab. 2014;20(5):787‐798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rivas CI, Zúñiga FA, Salas‐Burgos A, Mardones L, Ormazabal V, Vera JC. Vitamin C transporters. J Physiol Biochem. 2008;64(4):357‐375. [DOI] [PubMed] [Google Scholar]
- 43. Gudas LJ. Retinoids and vertebrate development. J Biol Chem. 1994;269(22):15399‐15402. [PubMed] [Google Scholar]
- 44. Mark M, Ghyselinck NB, Chambon P. Function of retinoic acid receptors during embryonic development. Nucl Recept Signal. 2009;7:e002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wang R, Liang JI, Yu H‐M, Liang HE, Shi Y‐J, Yang H‐T. Retinoic acid maintains self‐renewal of murine embryonic stem cells via a feedback mechanism. Differentiation. 2008;76:931‐945. [DOI] [PubMed] [Google Scholar]
- 46. Langton S, Gudas LJ. CYP26A1 knockout embryonic stem cells exhibit reduced differentiation and growth arrest in response to retinoic acid. Dev Biol. 2008;315(2):331‐354. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials