

Abstract
N-Acyl sulfamoyladenosines (acyl-AMS) have been used extensively to inhibit adenylate-forming enzymes that are involved in a wide range of biological processes. These acyl-AMS inhibitors are nonhydrolyzable mimics of the cognate acyl adenylate intermediates that are bound tightly by adenylate-forming enzymes. However, the anionic acyl sulfamate moiety presents a pharmacological liability that may be detrimental to cell permeability and pharmacokinetic profiles. We have previously developed the acyl sulfamate OSB-AMS (1) as a potent inhibitor of the adenylate-forming enzyme MenE, an o-succinylbenzoate-CoA (OSB-CoA) synthetase that is required for bacterial menaquinone biosynthesis. Herein, we report the use of computational docking to develop novel, non-acyl sulfamate inhibitors of MenE. A m-phenyl ether-linked analogue (5) was found to be the most potent inhibitor (IC50 = 8 μM; Kd = 244 nM), and its X-ray co-crystal structure was determined to characterize its binding mode in comparison to the computational prediction. This work provides a framework for the development of potent non-acyl sulfamate inhibitors of other adenylate-forming enzymes in the future.
Graphical Abstract

The spread of drug-resistant pathogens such as multi-drug-resistant and extensively drug-resistant Mycobacterium tuberculosis and methicillin-resistant Staphylococcus aureus (MRSA) is a major threat to human health and places a significant burden on our healthcare system.1–5 Thus, the development of novel antibiotics that circumvent existing resistance mechanisms is urgently needed. The biological redox cofactor menaquinone is the sole electron carrier in the electron transport chain of Gram-positive bacteria, all bacteria growing anaerobically, and mycobacteria.6,7 Although mammals use menaquinone (vitamin K2) as a cofactor in many enzymes such as γ-glutamyl carboxylase, this quinone is acquired from the diet or from gut flora rather than through de novo biosynthesis.8–11 Thus, inhibitors that target the bacterial menaquinone biosynthesis pathway are a promising avenue for future antibiotics to address drug-resistant pathogens.12
Menaquinone is derived from chorismate through a series of at least nine distinct enzyme-catalyzed transformations (Figure 1).7,13,14 Inhibitors have been developed against most of the enzymes in the pathway, including MenD,15,16 MenC,17 MenE,18 MenB,19,20 and MenA.21–24 Many of these compounds have significant antimicrobial activity, consistent with genetic studies that have demonstrated the essentiality of men genes in bacteria such as Bacillus subtilis and M. tuberculosis.25–28
Figure 1.
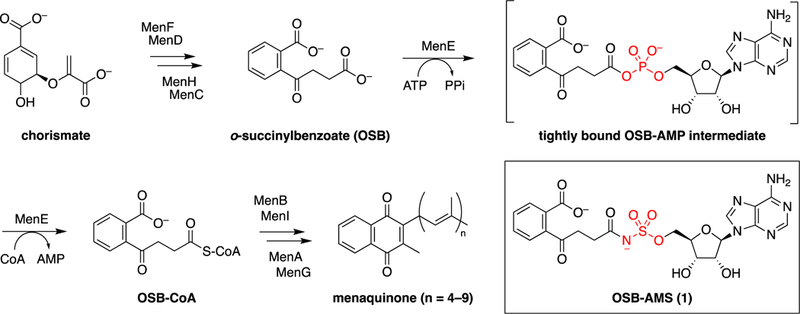
Classical menaquinone biosynthesis pathway and structure of MenE inhibitor OSB-AMS (1). At least nine enzymes catalyze the formation of menaquinone from chorismate. The fifth enzyme, MenE, is an acyl-CoA synthetase that catalyzes the ATP-dependent ligation of CoA to o-succinylbenzoate (OSB) via an OSB-AMP intermediate. OSB-AMS (1) is a stable analogue of OSB-AMP in which the acyl phosphate mixed anhydride is replaced with an acyl sulfamate.
MenE, the acyl-CoA synthetase in the menaquinone biosynthetic pathway, has been the primary focus of our efforts in this field. MenE, a member of the ANL (acyl-CoA synthetase, nonribosomal peptide synthetase adenylation domain, luciferase) family,29 carries out a two-step reaction involving the initial activation of o-succinylbenzoate (OSB) by adenylation to form a tightly bound OSB-adenosine monophosphate (AMP) intermediate, followed by a subsequent thioesterification with CoA to form OSB-CoA (Figure 1).30,31 Our previous efforts to inhibit MenE have focused on the use of acyl-AMS [5′-O-(N-acylsulfamoyl) adenosine] analogues,32–35 inspired by natural products such as ascamycin.36,37 Using this approach, we developed OSB-AMS (1), a tight-binding, low-nanomolar inhibitor of the S. aureus, M. tuberculosis, and Escherichia coli MenE enzymes.33 While effective in biochemical assays, OSB-AMS has significant pharmacological liabilities that limit its further preclinical development. Both the carboxylate and the acyl sulfamate are deprotonated at physiologic pH, and this is the likely source of its poor cellular permeability.38,39 Negative charge is also associated with high levels of plasma protein binding and rapid renal clearance.40,41 Finally, some acyl sulfamates may be metabolically unstable in vivo.42 The free carboxylate of OSB is necessary for potent binding to MenE,34 but we have recently been able to replace the OSB motif with a difluoroindanediol that is neutral at physiologic pH.34,35 Thus, our current efforts have focused on replacing the acyl sulfamate moiety to avoid the pharmacological liabilities associated with this linker.
Previous attempts to identify alternative bioisosteres of the native acyl phosphate motif have met with limited success, with most substitutions resulting in significant loss of biochemical activity against the target adenylate-forming enzyzme.43–50 Herein, we report the design of a virtual library of OSB-AMS linker analogues and the use of computational docking to prioritize compounds for synthesis. Several novel linker analogues were discovered, the most promising of which was further characterized through X-ray crystallography to elucidate its mechanism of binding for comparison to the computational prediction.
MATERIALS AND METHODS
Computational Docking: Protein Preparation.
The OSB-AMS (1)·MenE (R195K) co-crystal structure [Protein Data Bank (PDB) entry 5C5H] was processed using the Protein Preparation Wizard in the Schrödinger Suite (version 2017.2). Bond orders were assigned, explicit hydrogens added, and nonbridging waters >5 Å from the ligand deleted. The protonation and tautomeric states of the protein–ligand complex were generated using Epik at pH 7.4. Hydrogen bond assignment and optimization were performed with PROPKA to sample the hydrogen bonding and orientation of water molecules. Nonbridging waters (fewer than two hydrogen bonds) were removed. Geometric refinement was performed using OPLS_3 force field restrained minimization to a heavy atom convergence of 0.3 Å.
Ligand Preparation.
Ligand preparation was performed using LigPrep in the Schrödinger Suite (version 2017.2). Lowest-energy conformers were obtained using OPLS_3 force field optimization. Ionization and tautomeric states were generated using Epik at pH 7.4.
Grid Generation.
Using the Schrödinger Suite (version 2017.2) receptor grid generator, the receptor-binding site was defined as the area around the co-crystallized ligand with a cube grid with a side length of 10 Å. Nonpolar parts of the receptor were softened using van der Waals radius scaling (factor of 1.0 with a partial cutoff of 0.25). No constraints were defined, and rotations allowed for all hydroxyl groups in the defined binding pocket.
Docking Using Soft Receptor.
Using Schrödinger Glide (version 7.2), ligands were docked to MenE at Glide XP docking precision. Flexible ligand sampling was used, and Epik state penalties were applied to docking scores. Postdocking minimization was performed for all poses.
Inhibitor Synthesis.
See the Supporting Information for complete experimental protocols and analytical data for all new compounds.
Site-Directed Mutagenesis.
The K437A mutation was introduced using high-fidelity Phusion polymerase (New England BioLabs) with the forward primer 5′-AACGGCGG-TATTGCGATTTCACG-3′ and the T7 reverse primer.
Purification of Wild-Type and K437A E. coli MenE.
E. coli MenE (UniProtKB entry P37353) was purified as described previously.33 Briefly, BL21(DE3) pLysS cells were transformed with a pET15b plasmid containing E. coli MenE with an N-terminal His6 tag, then the cells were grown overnight in 10 mL of LB medium containing 200 μg/mL ampicillin. Next, 1 L of LB media containing 200 μg/mL ampicillin was inoculated with the 10 mL overnight culture and the cells were grown until the OD600 reached 0.6. Protein expression was induced by addition of 1 mM isopropyl β-D-1-thiogalactopyranoside, and the culture was shaken overnight at 20 °C. Cells were harvested by centrifugation at 5000 rpm for 10 min at 4 °C. The cell pellet was resuspended in 30 mL of His-binding buffer [20 mM Tris·HCl, 100 mM NaCl, and 5 mM imidazole (pH 8.0)], and the bacteria were disrupted by sonication. Cell debris was removed by centrifugation at 40000 rpm for 60 min at 4 °C, and the clear supernatant was loaded onto a His-bind column (1.5 cm × 15 cm) containing 4 mL of His-bind resin (Novagen) that had been charged with 10 mL of charge buffer (Ni2+). The column was washed with washing buffer containing 60 mM imidazole, and the protein was eluted from the column with elution buffer containing 500 mM imidazole. Fractions containing protein were loaded onto a size-exclusion column (Superdex 75, GE Healthcare) and eluted using 20 mM Tris·HCl buffer (pH 8.0) containing 100 mM NaCl to remove imidazole. The purified protein was >97% pure as determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and stored at −80 °C in storage buffer consisting of 20 mM Tris·HCl (pH 8.0) containing 100 mM NaCl.
Biochemical Assays.
Enzymatic inhibition was assessed using a MenE–MenB coupled assay in which the MenE-catalyzed reaction is the rate-limiting step.32–35 The reaction was initiated by addition of 50 nM wild-type E. coli MenE to a 100 μL reaction system containing 60 μM OSB, 240 μM ATP, 240 μM CoA, 2.5 μM M. tuberculosis MenB, and varying concentrations of the inhibitor in a buffer consisting of 20 mM NaHPO4 (pH 7.4), 150 mM NaCl, and 1 mM MgCl2. The production of 1,4-dihydroxy-2-naphthoyl-CoA was monitored at 392 nm (ε392 = 4000 M−1 cm−1).
Isothermal Titration Calorimetry (ITC).
ITC was performed using a VP-ITC instrument at 22 °C. Inhibitor (1 mM) and E. coli MenE (wt or K437A mutant) (30 μM) were prepared in 20 mM NaHPO4 buffer (pH 7.4) containing 150 mM NaCl and 1 mM MgCl2. The inhibitor was titrated from the injection syringe into the sample cell containing 1.8 mL of a solution of MenE. The data were fit to a single-binding site model with the Origin software package.
X-ray Crystallography.
Wild-type E. coli MenE was prepared as previously described.33 Co-crystallization of analogue 5 with MenE was achieved using the hanging drop diffusion technique, in which 2 μL of the complex solution [10 mg/mL E. coli MenE and 600 μM analogue 5 dissolved in 20 mM Tris·HCl buffer (pH 8.0) containing 100 mM NaCl] was mixed with 2 μL of the reservoir solution [0.1 M Tris·HCl, 0.2 M MgCl2, and 20% PEG (pH 6.5)] and equilibrated against 300 μL of the reservoir solution. Crystals formed at 293.15 K after 4–5 days. A single crystal was selected for diffraction by flash-freezing directly from the crystallization solution. Diffraction data were collected at 100 K using beamline 17-ID-1 (AMX) at National Synchrotron Light Source II at Brookhaven National Laboratory. Data were integrated using XDS51 and scaled using Aimless.52 The structure was determined by molecular replacement with MolRep,53 using our previously determined E. coli MenE (R195K) structure (PDB entry 5C5H) as a search model. The model was refined through successive rounds of manual model building using COOT54 and restrained refinement using REFMAC5.55 Electron density for analogue 5 bound in the active site was clearly visible and was added directly to the difference Fourier map after refinement converged. Ligand restraints were generated using the PRODRG server.56 The data collection and refinement statistics are listed in Table S2. Coordinates have been deposited as PDB entry 6NJ0.
To compare the binding poses of co-crystallized OSB-AMS (1) (PDB entry 5C5H) and docked and co-crystallized m-phenyl ether analogue 5, the N-terminal domains (residues 1–351) of the structures were aligned using PyMOL (align command, default parameters).57
Antibacterial Assays.
Minimum inhibitory concentrations (MICs) were determined using visual growth inspection of cells grown in transparent 96-well plates. E. coli (MG1655), MRSA (ATCC BAA-1762), and B. subtilis (ATCC 6057) were grown to mid log phase (OD600 = 0.6–0.8) in cation-adjusted Miller Hinton (CAMH) medium at 37 °C in an orbital shaker. A final inoculum of 100 μL of 106 colony-forming units/mL of cells was treated with the inhibitor with final concentrations ranging from 0.2 to 100 μg/mL. The MIC was defined as the minimum concentration at which a well showed no obvious growth by visual inspection.
RESULTS
Design and Computational Docking of OSB-AMS Linker Analogues.
We began by designing a 78-member virtual library of OSB-AMS analogues in which the acyl sulfamate motif was replaced by a variety of other potential linkers (Table S1).58 As this is the region of the molecule directly relevant to MenE catalytic activity, there are many crucial interactions between the inhibitor and the enzyme, making substitution at this position difficult. Thus, the virtual library design focused on decreasing polarity while attempting to retain as many of these key interactions as possible as well as the geometry and distance between OSB and adenosine observed in our co-crystal structure of OSB-AMS with E. coli MenE (R195K mutant)34 (Figure 2a).
Figure 2.
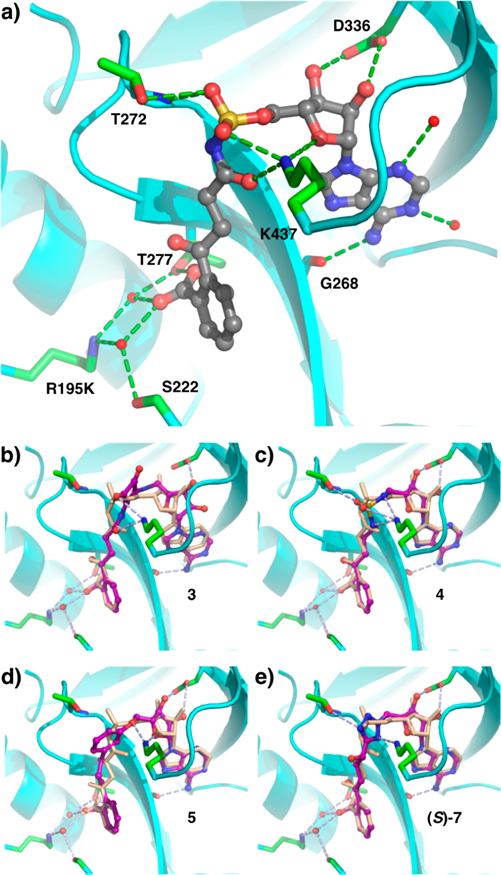
Structures of OSB-AMS (1) and linker analogues bound to E. coli MenE (R195K). (a) X-ray co-crystal structure of OSB-AMS (1, gray) bound in the active site (cyan) of MenE with key binding interactions (green) (PDB entry 5C5H). Computationally docked structures of linker analogues (purple) (b) acyl squaramide 3, (c) alkyl sulfamide 4, (d) m-phenyl ether 5, and (e) α-hydroxytetrazole (S)-7, overlaid with OSB-AMS (1, beige) from the co-crystal structure, with predicted active site interactions (light blue).
The library members were docked with the E. coli MenE (R195K) crystal structure (Schrödinger Glide) (Table 1 and Table S1).58 Briefly, the co-crystal structure (PDB entry 5C5H)34 was minimized (Protein Preparation Wizard), and then OSB-AMS was removed. Analogues were energy minimized (LigPrep), and probable tautomeric and protonation states predicted (Epik) and then docked into the binding pocket using a soft receptor model (Glide XP). Analogues were ranked by docking score, and those with scores above −10 kcal/mol removed from further consideration. Synthetic targets 2–5 were then selected from the list on the basis of combined considerations of docking score, reasonable docking pose that retained key interactions in the binding pocket (Figure 2b–d), likelihood of improving overall physiochemical properties of the scaffold (e.g., elimination of negative charge), synthetic accessibility, and ease of functionalization for further optimization. Linkers that had previously been reported to be ineffective against another ANL family enzyme were also eliminated from consideration.44–46 p-Phenyl ether analogue 6, which had a poor docking score, was selected for synthesis as a negative control.
Table 1.
Computational Docking Scores for OSB-AMS (1), Selected Virtual Library Members (2–6), and Additional Analogues (7–9)
 | ||
|---|---|---|
| analogue | linker | docking score (kcal/mol) |
| 1 |  |
−13.18 |
| 2 |  |
−13.98 |
| 3 |  |
−13.76 |
| 4 |  |
−14.20 |
| 5 |  |
−14.03 |
| 6 |  |
>−10 |
| 7 |  |
−13.36
(S) −12.79 (R) |
| 8 |  |
−12.80 |
| 9 |  |
−13.28 |
Synthesis of Selected OSB-AMS Linker Analogues.
Synthesis of acyl tetrazole analogue 2 began with lithiation of aryl bromide 10, prepared as previously described,34 quenching with dimethyl carbonate, and acid-catalyzed opening of the methyl ketal to afford primary alcohol intermediate 11 (Figure 3). Dess–Martin oxidation and conversion of the resulting aldehyde to the corresponding cyanohydrin 12 set the stage for nitrile 1,3-dipolar cycloaddition to construct the tetrazole motif. Attempted cycloadditions with azide at this stage led to undesired side products stemming from spirocyclization of the free hydroxyl onto the aryl ketone and lactonization of the methyl ester. Accordingly, reprotection as the cyclic methyl ketal 13 allowed efficient cycloaddition to afford the desired tetrazole intermediate 14. Reopening of the cyclic ketal and oxidation of the hydroxyl group afforded linear acyl tetrazole intermediate 15. Coupling to the protected adenosine scaffold 16, prepared as previously described,32 was achieved by Mitsunobu reaction. However, upon attempted deprotection of the penultimate intermediate 17, the acyl tetrazole moiety proved to be hydrolytically unstable in the presence of even relatively mild basic and neutral conditions.
Figure 3.
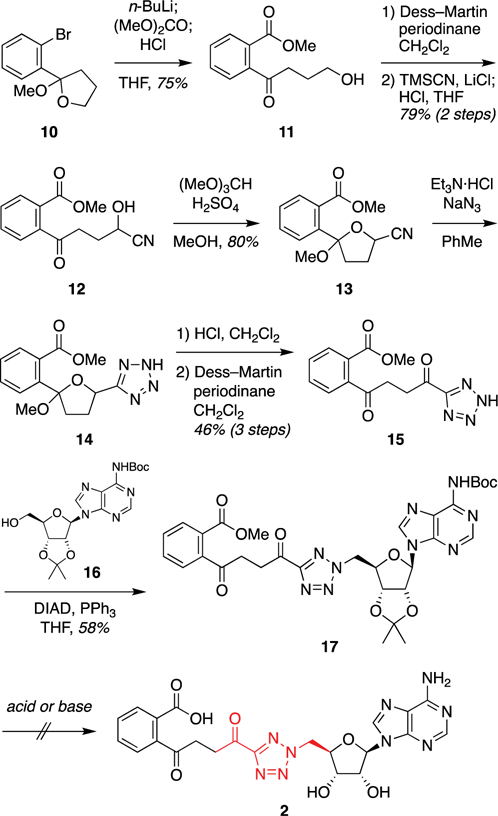
Synthetic approach to acyl tetrazole analogue 2. Abbreviations: Boc, tert-butoxycarbonyl; DIAD, diisopropyl azodicarboxylate; THF, tetrahydrofuran; TMS, trimethylsilyl.
We envisioned that this hydrolytic instability could be avoided using the corresponding α-hydroxytetrazole analogue 7 (Figure 4). Both diastereomers were retrospectively evaluated in our docking model and had promising docking scores [(S)-7, −13.36 kcal/mol; (R)-7, −12.79 kcal/mol (Table 1)] and poses (Figure 2e). Thus, we diverted attention to the synthesis of these analogues. Cyclic ketal-protected tetrazole 14 was coupled to protected adenosine 16 via Mitsunobu reaction to afford disubstituted tetrazole 18. Global deprotection under acidic conditions led to the formation of spirolactone 19, but this was readily saponified to afford the desired α-hydroxytetrazole 7 as a 1:1 mixture of diastereomers.
Figure 4.
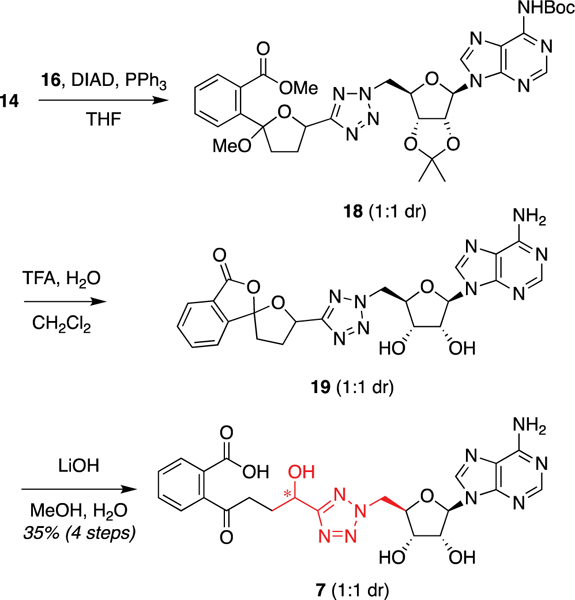
Synthesis of α-hydroxytetrazole analogue 7. Abbreviations: Boc, tert-butoxycarbonyl; DIAD, diisopropyl azodicarboxylate; TFA, trifluoroacetic acid; THF, tetrahydrofuran.
Next, synthesis of acyl squaramide analogue 3 was achieved by initial acylation of protected 5′-aminodeoxyadenosine 20, prepared as previously described,59 with dimethyl squarate followed by quenching with ammonia to form squaramide 21 (Figure 5). N-Acylation with the aromatic benzyl ester of OSB (22) followed by hydrogenolytic debenzylation and desilylation afforded squaramide analogue 3.
Figure 5.
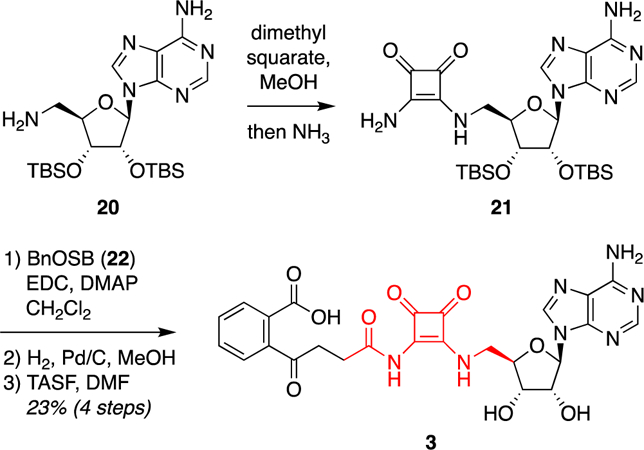
Synthesis of acyl squaramide analogue 3. Abbreviations: BnOSB (22), 4-{2-[(benzyloxy)carbonyl]phenyl}–4-oxobutanoic acid; DMAP, 4-(dimethylamino)pyridine; DMF, dimethylformamide; EDC, N-ethyl-N′-[3-(dimethylamino)propyl]carbodiimide hydrochloride; TASF, tris(dimethylamino)sulfur trimethylsilyl difluoride; TBS, tert-butyldimethylsilyl..
Alkyl sulfamide analogue 4 was synthesized via Mitsunobu coupling of reduced OSB intermediate 23 and protected AMSN scaffold 24, prepared as previously described,32 followed by two-step deprotection (Figure 6).
Figure 6.

Synthesis of alkyl sulfamide analogue 4. Abbreviations: Boc, tert-butoxycarbonyl; Cbz, carbobenzyloxy; DIAD, diisopropyl azodicarboxylate; TFA, trifluoroacetic acid.
To access m-phenyl ether analogue 5, dimethyl phthalate (26) was converted to vinyl ketone 28 in five highly scalable steps (Figure 7). Heck coupling with aryl bromide 29 provided phenol 30, which underwent Mitsunobu coupling to protected adenosine scaffold 16 to afford phenyl ether 31. The side-chain olefin was reduced using Stryker’s reagent, the TBS group removed, and the primary alcohol oxidized to the corresponding carboxylic acid with TPAP/NMO. Global deprotection with TFA provided m-phenyl ether analogue 5.
Figure 7.
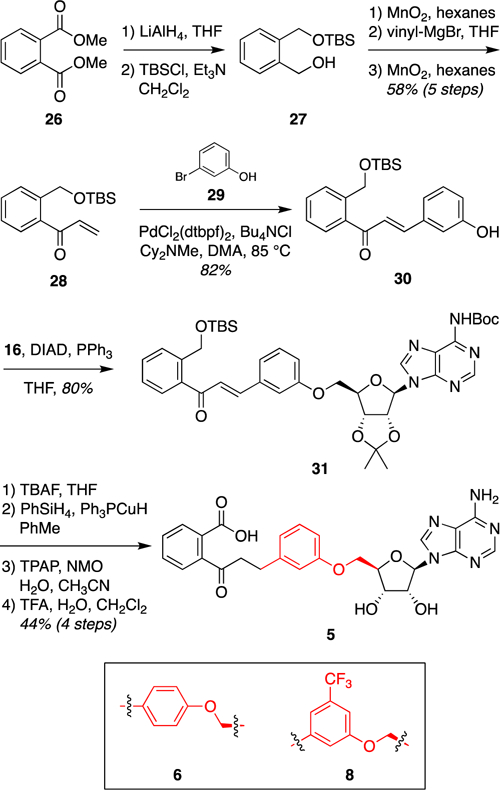
Synthesis of m-phenyl ether analogue 5. p-Phenyl ether analogue 6 and trifluoromethyl-substituted analogue 8 were synthesized by analogous routes from the corresponding Heck coupling partners (cf., 29).58 Abbreviations: Boc, tert-butoxycarbonyl; Cy = cyclohexyl; DIAD, diisopropyl azodicarboxylate; DMA, dimethylacetamide; dtbpf, 1,1′-bis(di-tert-butylphosphino)ferrocene; NMO, N-methylmorpholine N-oxide; TBAF, tetrabutylammonium fluoride; TBS, tert-butyldimethylsilyl; TFA, trifluoroacetic acid; THF, tetrahydrofuran; TPAP, tetrapropylammonium perruthenate.
On the basis of our structural analysis of m-phenyl ether analogue 5 (Figure 1d), we also designed a 3-trifluoromethyl-substituted analogue 8 [docking score of −12.80 (Table 1)] to probe the steric constraints of the linker region as well as the electronics of a possible cation–π interaction between the aryl ring and a conserved lysine K437. This analogue was accessed via the analogous route from the corresponding aryl bromide Heck coupling partner, 3-bromo-5-trifluoromethylphenol (Figure 7).58 In addition, a p-phenyl ether analogue 6 was synthesized as a negative control [docking score of greater than −10 kcal/mol (Table 1)] from the corresponding 4-bromophenol Heck partner.58
We also designed a m-3-pyridyl ether analogue 9 [docking score of −13.28 (Table 1)] to probe the electronics of the possible cation–π interaction between the aryl ring and K437 without the potential steric clashes introduced by the trifluoromethyl group of analogue 8. This required development of an alternative synthetic route to avoid pyridine oxidation during late-stage Ley oxidation, which also proved to be more concise (Figure 8). Thus, initial Suzuki coupling of commercially available aryl boronic acid 32 and vinyl bromide 33 afforded styrene 34. Ozonolysis and one-pot iodination–elimination gave vinyl ketone 35. Heck coupling with pyridyl bromide 36 provided phenol intermediate 37. Coupling to protected adenosine 16 under Mitsunobu conditions gave pyridyl ether 38. Finally, olefin reduction and two-step deprotection provided m-3-pyridyl ether analogue 9.
Figure 8.
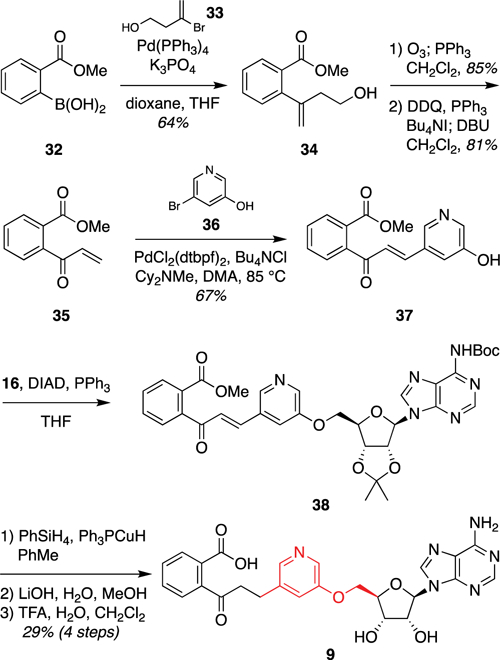
Synthesis of m-3-pyridyl ether analogue 9. Abbreviations: Boc, tert-butoxycarbonyl; Cy, cyclohexyl; DBU, 1,8-diaza-bicyclo[5.4.0]undec-7-ene; DDQ, 2,3-dichloro-5,6-dicyano-1,4-benzoquinone; DIAD, diisopropyl azodicarboxylate; DMA, dimethylacetamide; dtbpf, 1,1′-bis(di-tert-butylphosphino)ferrocene; TFA, trifluoroacetic acid; THF tetrahydrofuran.
Biochemical and Antibacterial Evaluation.
With linker analogues 3–9 in hand, we next evaluated their inhibition of E. coli MenE using our previously reported MenE–MenB coupled assay.32 Consistent with predictions from the docking experiments, all but one of the analogues inhibited MenE, albeit with modest IC50 values compared to that of OSB-AMS (1) (Table 2). Only acyl squaramide analogue 3 was inactive, contrary to prediction. The m-phenyl ether analogue 5 was the most potent inhibitor, with an IC50 of 8.1 ± 0.9 μM, while the corresponding 3-trifluoromethyl-substituted analogue 8 and m-3-pyridyl analogue 9 exhibited slightly weaker inhibition. In contrast, the p-phenyl ether analogue 6 showed no inhibition of MenE, consistent with its docking score of greater than −10 kcal/mol. Taken together, these results demonstrated that computational docking could be used effectively to prioritize OSB-AMS analogues for synthesis and biochemical evaluation against MenE.
Table 2.
Enzyme Inhibition and Docking Scores of Synthesized Compounds
 | |||
|---|---|---|---|
| analogue | linker | IC50a (µM) | docking score (kcal/mol) |
| 1 |  |
0.024 ± 0.003 | −13.78 |
| 3 |  |
>200 | −13.76 |
| 4 |  |
35 ± 3.2 | −14.20 |
| 5 |  |
8.1 ± 0.9b | −14.03 |
| 6 |  |
>200c | >−10.00 |
| 7 |  |
53.5 ± 2.3 | −13.36
(S) −12.79 (R) |
| 8 |  |
26.5 ± 2.3 | −12.80 |
| 9 |  |
11.5 ± 0.8d | −13.28 |
IC50 values were measured using a MenB–MenE coupled assay.32 All measurements were determined in triplicate.
Kd = 244 ± 11 nM as determined by ITC.
No binding detected by ITC.
Kd = 505 ± 14 nM as determined by ITC.
We next investigated the binding affinities of the two most potent inhibitors 5 and 9 with E. coli MenE using ITC.58 The measured Kd values of m-phenyl ether analogue 5 (244 ± 11 nM) and m-3-pyridyl ether analogue 9 (505 ± 14 nM) were generally consistent with IC50 values determined in the biochemical assay. In contrast, p-phenyl ether analogue 6, which did not inhibit MenE, also showed no binding in the ITC assay (Figure S1).
Next, all of the synthesized analogues were evaluated for antibacterial activity against E. coli, MRSA, and B. subtilis. Unfortunately, none of the compounds exhibited an MIC of <100 μg/mL, compared to the MIC of 31.25 μg/mL observed for OSB-AMS (1) against MRSA (ATCC BAA1762).34 We posit that this lack of antimicrobial activity is due to the decreased biochemical potency of these linker analogues in comparison to that of OSB-AMS (1), which may be improved in the future through further optimization of these newly discovered chemotypes.
X-ray Co-Crystal Structure of m-Phenyl Ether Analogue 5 Bound to E. coli MenE.
To understand the binding of m-phenyl ether analogue 5 and to provide guidance for further inhibitor design, the co-crystal structure of 5 bound to wild-type E. coli MenE was determined at 1.8 Å resolution (PDB entry 6NJ0).58 The structure was determined by molecular replacement using our previously reported structure of OSB-AMS (1)-bound E. coli MenE (R195K) (PDB entry 5C5H)34 as a search model. Data collection and refinement statistics are listed in Table S2.
Comparison of the structures of m-phenyl ether analogue 5 bound to wild-type MenE and of OSB-AMS (1) bound to MenE (R195K) revealed differences in the orientation of the small C-terminal domain relative to the large N-terminal domain (Figure 9a). In the structure with m-phenyl ether analogue 5, the C-terminal domain is rotated by 22° (determined using DynDom60) about a hinge residue D352, away from the active site, making it “slightly open” relative to the “closed” conformation seen in the structure with OSB-AMS (1). Notably, apo structures of S. aureus MenE have been reported previously, and the C-terminal domains are rotated much more dramatically, by 144° and 151°, in a fully “open” conformation (PDB entry 3IPL chains A and B).61 Both “open” and “closed” states have been observed previously for other members of the ANL family,29,62 with fully open states typically observed only in unliganded structures.63–68 In the closed conformations, a highly conserved lysine residue (K437 in E. coli MenE)69 serves as a linchpin that coordinates the acyl adenylate intermediate via its acyl group carbonyl, phosphate pro-S oxygen, ribose 4′-oxygen, and/or ribose 5′-oxygen.34,64,65,70 In the OSB-AMS (1)·MenE (R195K) complex, K437 is observed in the active site and binds OSB-AMS (1) through these interactions. In contrast, in the complex of m-phenyl ether analogue 5 bound to wild-type MenE, the loop region from residue 434 to 438 that includes K437 is disordered. We speculate that the loss of the K437–ligand interaction may result in a decreased force holding the C-terminal domain in the closed conformation, resulting in a slight rotation of the C-terminal domain away from the active site. In addition, without the interaction of K437 with the ligand, the loop region becomes highly dynamic and disordered.61
Figure 9.
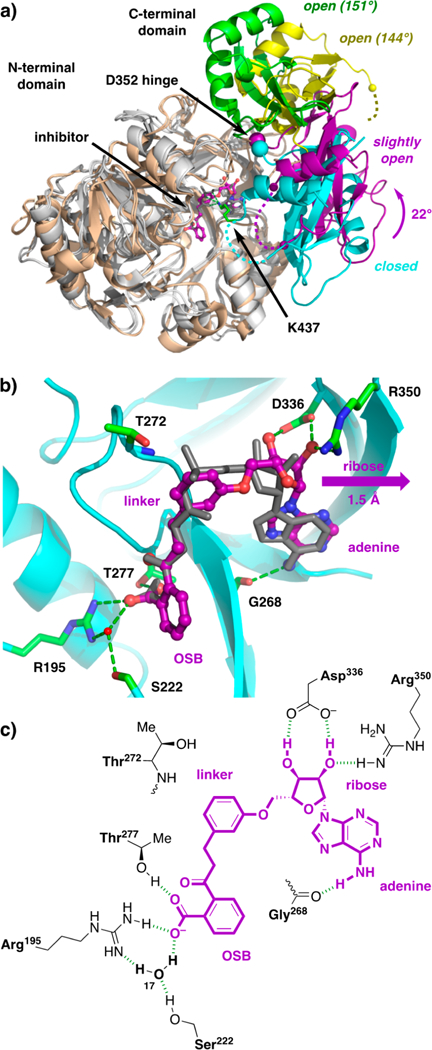
X-ray co-crystal structure of m-phenyl ether analogue 5 bound to wild-type E. coli MenE. (a) Overlay of structures of E. coli wild-type MenE with m-phenyl ether analogue 5 (gray and purple) bound, E. coli MenE (R195K) with OSB-AMS (1) bound (gray and cyan, PDB entry 5C5H), and S. aureus MenE apo structures (wheat and green or yellow, PDB entry 3IPL chains A and B). Alignment of the large N-terminal domains [E. coli residues 1–351, gray, root-mean-square deviation (rmsd) of 0.49 Å; S. aureus residues 1–396, wheat, rmsd of 1.64 Å relative to 5C5H] reveals a 22° rotation of the small C-terminal domain about the E. coli hinge residue D352 in the structure with analogue 5 (purple) compared to that with OSB-AMS (1) (cyan), while larger 144° and 151° rotations of the C-terminal domains are observed in the S. aureus apo structures (green and yellow). The E. coli linchpin residue K437 (green) is observed in the structure with OSB-AMS (1) but is disordered in the structure with m-phenyl ether analogue 5. (b) Active site of wild-type MenE (cyan) with m-phenyl ether analogue 5 (purple) bound, revealing binding interactions (green) and a 1.5 Å shift of the ribose motif compared to OSB-AMS (1) (gray). (c) Schematic of putative active site interactions (green) of m-phenyl ether analogue 5 (purple) with wild-type MenE (black).
Structures of B. subtilus MenE in apo form and in complex with AMP, ATP, and OSB-AMP have also been reported previously (PDB entries 5BUQ, 5BUS, 5BUR, and 5GTD, respectively).71,72 In the liganded structures, the C-terminal domain is also rotated 24° relative to that in the E. coli MenE (R195K)·OSB-AMS (1) complex, but about a distinct axis of rotation that allows the ligands to retain interactions with the corresponding linchpin residue (K471 in B. subtilis MenE). Notably, this region is again disordered in the corresponding apo structure.
Active Site Interactions between m-Phenyl Ether Analogue 5 and E. coli MenE.
The difference in the relative orientation of the C-terminal domain does not appear to impact the overall orientation of m-phenyl ether analogue 5 in the MenE active site compared to that of OSB-AMS (1) (Figure 9b). However, the geometric constraints of the phenyl ether linker force the ribose moiety ≈1.5 Å deeper into the adenine region of the binding pocket.
Enzyme–inhibitor interactions are highlighted in panels b and c of Figure 9. Residues R195, S222, T277, G268 (carbonyl), D336, and R350 form hydrogen-bonding interactions with m-phenyl ether analogue 5, and these residues are conserved in E. coli, S. aureus, and M. tuberculosis MenE.34 In B. subtilis MenE, R195 is replaced by K205, T277 by Q294, and G268 by S285.71 E. coli residues R195, S222, and T277 are clustered around the OSB aromatic carboxylate. Residues R195 and T277 interact directly with the carboxylate, and R195 also engages in a second, water-mediated interaction via water-17, which is coordinated by S222. We had previously proposed that a direct interaction with R195 should be present on the basis of our analysis of the OSB-AMS (1)·MenE (R195K) crystal structure, in which the OSB carboxylate interacts with K195 via two water-mediated interactions (Figure 2a). In contrast, in the structure of wild-type MenE complexed with 5 reported here, the R195 guanidinium group engages the OSB carboxylate via one direct interaction and one water-mediated interaction. In the linker region, no direct interactions are observed with the m-phenyl ether. Residue T272, which binds to the pro-S oxygen of the sulfamate in OSB-AMS, remains within 4 Å of the aromatic ring of m-phenyl ether analogue 5. However, the linchpin K437, which interacts with multiple atoms in the linker region of OSB-AMS, is disordered and not observed in the structure of m-phenyl ether analogue 5. In the ribose region, D336 interacts with the 2′- and 3′-hydroxyl groups of m-phenyl ether analogue 5 in the same way as OSB-AMS (1).
Binding of m-Phenyl Ether Analogue 5 to a MenE (K437A) Linchpin Mutant.
Crucially, we did not observe the predicted interactions between K437 and the linker region of m-phenyl ether analogue 5, because the loop of residues 434–438 was disordered. While analogue 5 lacks the carbonyl and phosphate pro-S oxygen of the cognate intermediate OSB-AMP, we posited that K437 might still hydrogen bond with the ribose 4′- and/or 5′-oxygens and could also engage in a cation–π interaction with the phenyl ether ring, based on our earlier docking predictions (Figure 2d).
To probe for these interactions experimentally, we compared the binding affinity of m-phenyl ether analogue 5 for wild-type E. coli MenE and the K437A mutant. ITC titration curves (Figure S1) show very similar Kd values of 244 and 249 nM, respectively, consistent with the lack of a productive binding interaction of K437 with m-phenyl ether analogue 5.
Comparison of Docked and X-ray Co-Crystal Structures of m-Phenyl Ether Analogue 5 with MenE.
To evaluate the effectiveness of the docking prediction, we compared the binding positions of m-phenyl ether analogue 5 when docked with MenE (R195K) or co-crystallized with wild-type MenE, relative to that of OSB-AMS (1) from its co-crystal structure with MenE (R195K)34 (Figure S2). Overall, there was good overlap between the positions of the docked and co-crystallized inhibitors (root-mean-square deviation of 1.26 Å), with the most significant differences in the aryl linker (rmsd = 1.56 Å) and ribose (rmsd = 1.79 Å) regions. In the docked structure of m-phenyl ether analogue 5, the adenosine region of the inhibitor overlapped well with that of co-crystallized OSB-AMS (1) (rmsd = 0.63 Å). However, the aryl ether linker of analogue 5 protrudes farther from the adenosine-binding pocket than the acyl sulfamate linker of OSB-AMS (1). Nonetheless, conformational differences in the succinyl chain allow the OSB aromatic rings to overlap well (rmsd = 0.76 Å). In contrast, in the co-crystal structure of m-phenyl ether analogue 5, both the OSB and linker regions overlap well with co-crystallized OSB-AMS (1) (rmsd = 0.35 Å for the OSB aromatic ring), resulting in the ribose motif being pushed deeper into the adenosine-binding pocket (rmsd = 1.51 Å). Notably, this translation of the ribose ring deeper into the adenosine-binding pocket moves the ribose 5′-oxygen by 2.0 Å relative to OSB-AMS (1), out of position to hydrogen bond productively with K437 as positioned in the co-crystal structure with OSB-AMS (1). However, the linker aryl ring of 5 appears to be positioned even more favorably for a cation–π interaction with K437 than in the docked structure, so it is still possible that this interaction may be captured in future analogue designs.
DISCUSSION
Acyl-AMS intermediate analogues have been used widely to inhibit ANL family members and other adenylate-forming enzymes.73–81 Several of these compounds have advanced to in vivo proof-of-concept studies in animal models.42,75,82 However, the acyl sulfamate moiety carries several pharmacological liabilities, including a negative charge that can be associated with decreased penetration of Gram-negative bacteria,38,39 high plasma protein binding,41 rapid renal clearance,40 and potential metabolic instability.42 Thus, replacement of the acyl sulfamate linker with stable, neutral isosteres would represent an important advance, providing a path toward pharmacologically optimized inhibitors of the adenylate-forming enzyme.
We previously reported the synthesis and characterization of OSB-AMS (1), an acyl adenylate analogue that inhibits E. coli MenE with an IC50 of 24 ± 3 nM.34 However, despite this potent biochemical inhibition, OSB-AMS has a modest MIC of 31.25 μg/mL against MRSA. We speculated that the poor antibacterial activity of OSB-AMS could result from the double negative charge carried by this molecule at physiological pH. Having recently discovered a difluoroindanediol analogue of the OSB side chain that removes one of these negative charges,35 we set out to develop a new generation of analogues in which the acyl sulfamate linker was replaced with an uncharged motif. As previous efforts to develop new acyl phosphate bioisosteres have proven to be challenging,43–50 we designed a virtual library of linker analogues and docked these candidates to our previously reported structure of E. coli MenE (R195K).34 We used the docking results to prioritize compounds for synthesis, focusing on four main bioisostere chemotypes: acyl squaramide (3), alkyl sulfamide (4), phenyl ether (5), and α-hydroxytetrazole (7). Three out of four of these analogues inhibited E. coli MenE with IC50 values in the low- to mid-micromolar range, validating the overall effectiveness of docking in prioritization of analogues. However, the docking scores did not accurately predict the rank ordering of inhibitor potency between chemotypes, and we posit that this is because binding interactions with MenE are dominated by the OSB and adenosine motifs, which were invariant across all virtual library members. Thus, the docking simply served to predict whether each linker provided suitable geometry for directing these binding anchors to the appropriate pockets. Consistent with this interpretation, p-phenyl ether analogue 6 was predicted not to provide a suitable binding geometry, and this was confirmed in biochemical evaluation of this analogue. On the other hand, docking scores within the m-aryl ether chemotype (5, 8, and 9) did correlate with the rank ordering of experimentally determined IC50 values. Thus, it remains to be seen if such more quantitative predictions can be made robustly across a larger panel of analogues within a given linker chemotype.
In the co-crystal structure of m-phenyl ether analogue 5 with wild-type E. coli MenE, the C-terminal lobe adopts a “slightly open” conformation relative to the large N-terminal domain. This is in contrast to the “closed” conformation that we observed previously in the co-crystal structure of OSB-AMS (1) with E. coli MenE (R195K) and to the fully “open” conformations observed in the apo structures of S. aureus MenE.61 For OSB-AMS (1), the conserved linchpin residue K437 binds the acyl sulfamate and may stabilize the closed conformation. However, in the case of m-phenyl ether analogue 5, both the co-crystal structure and the similar Kd values observed for wild-type and K437A E. coli MenE suggest that this analogue does not interact with K437, and this may lead to the “slightly open” conformation and disorder of residues 434–438 that comprise the loop that includes K437.
Comparison of the available MenE structures reveals how the C- and N-terminal domains may move relative to each other in the course of binding substrates or inhibitors. We posit that the ligand binds to the protein in an open conformation, and subsequently, the small C-terminal domain rotates toward the active site of the large N-terminal domain, leading to an interaction between the ligand and K437 that stabilizes the closed state. Consistent with this interpretation, within the ANL superfamily, fully “open” conformations have also been reported previously for apo structures of luciferase63 and long chain fatty acyl-CoA synthetase,65 and in both cases, co-crystallization with the cognate adenylate or a mimic thereof yields the “closed” conformation.64,65 It must be noted, however, that Gulick has reported a similar “slightly open” conformation for 4-chlorobenzoate-CoA ligase in apo, 4-chlorobenzoate substrate-bound, and adenylate intermediate-bound forms, with the linchpin K492 side chain disordered in all cases,83,84 and it is unclear why the fully closed conformation was not observed in the adenylate-bound structure. In addition, Guo has reported structures of B. subtilis MenE in apo form and in complex with AMP, ATP, and OSB-AMP, all adopting a similar “closed” conformation in which the linchpin residue (K471 in B. subtilis MenE) interacts with the ligand,71,72 highlighting the fact that not all apo structures of ANL enzymes result in “open” conformations.85,86
Notably, Gulick has identified a distinct, ≈140° rotation about the hinge residue that generates a second, distinct “closed” conformation in co-crystal structures of acetyl-CoA synthetase with the adenylate mimic propyl-AMP and the co-substrate CoA, as well as of 4-chlorobenzoate-CoA synthetase with a product analogue 4-chlorophenacyl-CoA.87,88 This conformation has also been observed recently in the structure of B. subtilis MenE with a stable OSB-CoA analogue.89 This “domain alternation” introduces new residues into the active site to catalyze thioesterification in the second half-reaction. Thus, rotation of the C-terminal domain about the hinge residue may be involved in both substrate binding and catalysis of the first half-reaction as well as CoA binding and catalysis of the second half-reaction.
The structure reported in this work also reveals a direct interaction between E. coli MenE R195 and the OSB carboxylate group, as proposed previously.34 While the acyl sulfamate moiety of OSB-AMS (1) is bound by the linchpin residue K437 as well as T272, the phenyl ether linker of analogue 5 does not interact with these residues. A cation–π interaction with K437 appeared to be possible on the basis of the docking model, but this was not observed in the co-crystal structure or evident in ITC binding assays to a K437A mutant. The loss of these two interactions may account for the ≈2 log weaker IC50 value of m-phenyl ether analogue 5 compared to that of OSB-AMS (1). Conversely, however, the m-phenyl ether linker is uncharged, hydrophobic, and not susceptible to hydrolysis, making it an attractive platform for further optimization to develop more potent, non-acyl sulfamate MenE inhibitors. Combination of these optimized linkers with our difluoroindanediol replacement for the OSB side chain35 could then provide analogues in which both negative charges have been removed, affording improved pharmacological properties.
In conclusion, our studies show that replacement of the acyl sulfamate moiety found in most inhibitors of the adenylate-forming enzyme is feasible and provide a path forward for the development of new MenE inhibitors. This strategy of analogue prioritization using computational docking may also be applicable to other adenylate-forming enzymes that have previously been targeted with acyl sulfamate-based inhibitors.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Stephen Walker (Stony Brook University) for assistance with BSL2 studies and Dr. George Sukenick, Rong Wang, and Dr. Sylvi Rusli (Memorial Sloan Kettering Cancer Center) for expert NMR and mass spectrometry support. Access to the Schrödinger Small-Molecule Drug Discovery Suite was generously provided by the Sanders Innovation & Education Initiative of the Tri-Institutional Therapeutics Discovery Institute.
Funding
This work was supported by the National Institutes of Health (NIH) (R01 GM100477 to D.S.T., R01 GM102864 to P.J.T., R35 GM124898 to J.B.F., T32 GM073546-Gross to C.E.E., and Cancer Center Support Grant P30 CA008748 to C. B. Thompson) and a W. Burghardt Turner Fellowship (to J.S.M.). The National Synchrotron Light Source is supported by National Institutes of Health Grant GM0080 (supplement to a PSI program) and U.S. Department of Energy Contract DE-AC02–98CH10886.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.biochem.9b00003.
Complete experimental procedures and analytical data for all new compounds (PDF)
Accession Codes
MenE, UniProtKB entry P37353.
The authors declare the following competing financial interest(s): D.S.T., P.J.T., C.E.E., and J.S.M. are co-inventors on International Patent Application PCT/US2016/055136; D.S.T., P.J.T., C.E.E., and Y.S. are co-inventors on U.S. Provisional Patent Application 62/802,650.
REFERENCES
- (1).Brown ED, and Wright GD (2016) Antibacterial drug discovery in the resistance era. Nature (London, U. K.) 529, 336–343. [DOI] [PubMed] [Google Scholar]
- (2).Lewis K (2013) Platforms for antibiotic discovery. Nat. Rev. Drug Discovery 12, 371–387. [DOI] [PubMed] [Google Scholar]
- (3).Centers for Disease Control and Prevention (2013) Antibiotic Resistance Threats in the United States, 2013 http://www.cdc.gov/drugresistance/threat-report-2013/.
- (4).World Health Organization (2014) Antimicrobial resistance: Global report on surveillance 2014 http://www.who.int/drugresistance/documents/surveillancereport/en/.
- (5).The Pew Charitable Trusts (2016) A Scientific Roadmap for Antibiotic Discovery https://www.pewtrusts.org/en/research-and-analysis/reports/2016/05/a-scientific-roadmap-for-antibiotic-discovery.
- (6).Meganathan R, and Kwon O (2009) Biosynthesis of menaquinone (vitamin K2) and ubiquinone (Coenzyme Q). EcoSal Plus 3, 173–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Meganathan R (2001) Biosynthesis of menaquinone (vitamin K2) and ubiquinone (coenzyme Q): A perspective on enzymatic mechanisms. Vitam. Horm 61, 173–218. [DOI] [PubMed] [Google Scholar]
- (8).Walther B, Karl JP, Booth SL, and Boyaval P (2013) Menaquinones, bacteria, and the food supply: The relevance of dairy and fermented food products to vitamin K requirements. Adv. Nutr 4, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dowd P, Ham SW, Naganathan S, and Hershline R (1995) The mechanism of action of vitamin K. Annu. Rev. Nutr 15, 419–440. [DOI] [PubMed] [Google Scholar]
- (10).Danziger J (2008) Vitamin K-dependent proteins, warfarin, and vascular calcification. Clin. J. Am. Soc. Nephrol 3, 1504–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Nakagawa K, Hirota Y, Sawada N, Yuge N, Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y, and Okano T (2010) Identification of UBIAD1 as a novel human menaquinone-4 biosynthetic enzyme. Nature 468, 117–121. [DOI] [PubMed] [Google Scholar]
- (12).Paudel A, Hamamoto H, Panthee S, and Sekimizu K (2016) Menaquinone as a potential target of antibacterial agents. Drug Discoveries Ther 10, 123–128. [DOI] [PubMed] [Google Scholar]
- (13).Jiang M, Chen X, Guo ZF, Cao Y, Chen M, and Guo Z (2008) Identification and characterization of (1R,6R)-2-succinyl-6-hydroxy-2,4-cyclohexadiene-1-carboxylate synthase in the menaquinone biosynthesis of Escherichia coli. Biochemistry 47, 3426–3434. [DOI] [PubMed] [Google Scholar]
- (14).Begley TP, Kinsland C, Taylor S, Tandon M, Nicewonger R, Wu M, Chiu H-J, Kelleher N, Campobasso N, and Zhang Y (1998) Cofactor biosynthesis: A mechanistic perspective. In Biosynthesis, pp 93–142, Springer. [Google Scholar]
- (15).Fang M, Toogood RD, Macova A, Ho K, Franzblau SG, McNeil MR, Sanders DA, and Palmer DR (2010) Succinylphosphonate esters are competitive inhibitors of MenD that show active-site discrimination between homologous α-ketoglutarate-decarboxylating enzymes. Biochemistry 49, 2672–2679. [DOI] [PubMed] [Google Scholar]
- (16).Fang M, Macova A, Hanson KL, Kos J, and Palmer DR (2011) Using substrate analogues to probe the kinetic mechanism and active site of Escherichia coli MenD. Biochemistry 50, 8712–8721. [DOI] [PubMed] [Google Scholar]
- (17).Pulaganti M, Banaganapalli B, Mulakayala C, Chitta SK, and Anuradha CM (2014) Molecular modeling and docking studies of o-succinylbenzoate synthase of M. tuberculosis – A potential target for antituberculosis drug design. Appl. Biochem. Biotechnol 172, 1407–1432. [DOI] [PubMed] [Google Scholar]
- (18).Tian Y, Suk DH, Cai F, Crich D, and Mesecar AD (2008) Bacillus anthracis o-succinylbenzoyl-CoA synthetase: Reaction kinetics and a novel inhibitor mimicking its reaction intermediate. Biochemistry 47, 12434–12447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Li X, Liu N, Zhang H, Knudson SE, Li HJ, Lai CT, Simmerling C, Slayden RA, and Tonge PJ (2011) CoA adducts of 4-oxo-4-phenylbut-2-enoates: Inhibitors of MenB from the M. tuberculosis menaquinone biosynthesis pathway. ACS Med. Chem. Lett 2, 818–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Li X, Liu N, Zhang H, Knudson SE, Slayden RA, and Tonge PJ (2010) Synthesis and SAR studies of 1,4-benzoxazine MenB inhibitors: Novel antibacterial agents against Mycobacterium tuberculosis. Bioorg. Med. Chem. Lett 20, 6306–6309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kurosu M, and Crick DC (2009) MenA is a promising drug target for developing novel lead molecules to combat Mycobacterium tuberculosis. Med. Chem 5, 197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Dhiman RK, Mahapatra S, Slayden RA, Boyne ME, Lenaerts A, Hinshaw JC, Angala SK, Chatterjee D, Biswas K, Narayanasamy P, Kurosu M, and Crick DC (2009) Menaquinone synthesis is critical for maintaining mycobacterial viability during exponential growth and recovery from non-replicating persistence. Mol. Microbiol 72, 85–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kurosu M, and Begari E (2010) Vitamin K2 in electron transport system: are enzymes involved in vitamin K2 biosynthesis promising drug targets? Molecules 15, 1531–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Li K, Schurig-Briccio LA, Feng X, Upadhyay A, Pujari V, Lechartier B, Fontes FL, Yang H, Rao G, Zhu W, Gulati A, No JH, Cintra G, Bogue S, Liu YL, Molohon K, Orlean P, Mitchell DA, Freitas-Junior L, Ren F, Sun H, Jiang T, Li Y, Guo RT, Cole ST, Gennis RB, Crick DC, and Oldfield E (2014) Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem 57, 3126–3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Sassetti CM, Boyd DH, and Rubin EJ (2003) Genes required for mycobacterial growth defined by high density mutagenesis. Mol. Microbiol 48, 77–84. [DOI] [PubMed] [Google Scholar]
- (26).Kobayashi K, Ehrlich SD, Albertini A, Amati G, Andersen KK, Arnaud M, Asai K, Ashikaga S, Aymerich S, Bessieres P, Boland F, Brignell SC, Bron S, Bunai K, Chapuis J, Christiansen LC, Danchin A, Debarbouille M, Dervyn E, Deuerling E, Devine K, Devine SK, Dreesen O, Errington J, Fillinger S, Foster SJ, Fujita Y, Galizzi A, Gardan R, Eschevins C, Fukushima T, Haga K, Harwood CR, Hecker M, Hosoya D, Hullo MF, Kakeshita H, Karamata D, Kasahara Y, Kawamura F, Koga K, Koski P, Kuwana R, Imamura D, Ishimaru M, Ishikawa S, Ishio I, Le Coq D, Masson A, Mauel C, Meima R, Mellado RP, Moir A, Moriya S, Nagakawa E, Nanamiya H, Nakai S, Nygaard P, Ogura M, Ohanan T, O’Reilly M, O’Rourke M, Pragai Z, Pooley HM, Rapoport G, Rawlins JP, Rivas LA, Rivolta C, Sadaie A, Sadaie Y, Sarvas M, Sato T, Saxild HH, Scanlan E, Schumann W, Seegers JF, Sekiguchi J, Sekowska A, Seror SJ, Simon M, Stragier P, Studer R, Takamatsu H, Tanaka T, Takeuchi M, Thomaides HB, Vagner V, van Dijl JM, Watabe K, Wipat A, Yamamoto H, Yamamoto M, Yamamoto Y, Yamane K, Yata K, Yoshida K, Yoshikawa H, Zuber U, and Ogasawara N (2003) Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U. S. A 100, 4678–4683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Akerley BJ, Rubin EJ, Novick VL, Amaya K, Judson N, and Mekalanos JJ (2002) A genome-scale analysis for identification of genes required for growth or survival of Haemophilus influenzae. Proc. Natl. Acad. Sci. U. S. A 99, 966–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chen WH, Lu G, Chen X, Zhao XM, and Bork P (2017) OGEE v2: An update of the online gene essentiality database with special focus on differentially essential genes in human cancer cell lines. Nucleic Acids Res 45, D940–D944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Gulick AM (2009) Conformational dynamics in the acyl-CoA synthetases, adenylation domains of non-ribosomal peptide synthetases, and firefly luciferase. ACS Chem. Biol 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Meganathan R, and Bentley R (1979) Menaquinone (vitamin K2) biosynthesis: conversion of o-succinylbenzoic acid to 1,4-dihydroxy-2-naphthoic acid by Mycobacterium phlei enzymes. J. Bacteriol 140, 92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kwon O, Bhattacharyya DK, and Meganathan R (1996) Menaquinone (vitamin K2) biosynthesis: Overexpression, purification, and properties of o-succinylbenzoyl-coenzyme A synthetase from Escherichia coli. J. Bacteriol 178, 6778–6781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Lu X, Zhang H, Tonge PJ, and Tan DS (2008) Mechanism-based inhibitors of MenE, an acyl-CoA synthetase involved in bacterial menaquinone biosynthesis. Bioorg. Med. Chem. Lett 18, 5963–5966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lu X, Zhou R, Sharma I, Li X, Kumar G, Swaminathan S, Tonge PJ, and Tan DS (2012) Stable analogues of OSB-AMP: Potent inhibitors of MenE, the o-succinylbenzoate-CoA synthetase from bacterial menaquinone biosynthesis. ChemBioChem 13, 129–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Matarlo JS, Evans CE, Sharma I, Lavaud LJ, Ngo SC, Shek R, Rajashankar KR, French JB, Tan DS, and Tonge PJ (2015) Mechanism of MenE inhibition by acyl-adenylate analogues and discovery of novel antibacterial agents. Biochemistry 54, 6514–6524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Evans CE, Matarlo JS, Tonge PJ, and Tan DS (2016) Stereoselective synthesis, docking, and biological evaluation of difluoroindanediol-based MenE inhibitors as antibiotics. Org. Lett 18, 6384–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Isono K, Uramoto M, Kusakabe H, Miyata N, Koyama T, Ubukata M, Sethi SK, and McCloskey JA (1984) Ascamycin and dealanylascamycin, nucleoside antibiotics from Streptomyces sp. J. Antibiot 37, 670–672. [DOI] [PubMed] [Google Scholar]
- (37).Ueda H, Shoku Y, Hayashi N, Mitsunaga J, In Y, Doi M, Inoue M, and Ishida T (1991) X-ray crystallographic conformational study of 5′-O-[N-(L-alanyl)-sulfamoyl]adenosine, a substrate analog for alanyl-tRNA synthetase. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol 1080, 126–134. [DOI] [PubMed] [Google Scholar]
- (38).Yoshimura F, and Nikaido H (1985) Diffusion of β-lactam antibiotics through the porin channels of Escherichia coli K-12. Antimicrob. Agents Chemother 27, 84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Richter MF, Drown BS, Riley AP, Garcia A, Shirai T, Svec RL, and Hergenrother PJ (2017) Predictive compound accumulation rules yield a broad-spectrum antibiotic. Nature 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Varma MVS, Feng B, Obach RS, Troutman MD, Chupka J, Miller HR, and El-Kattan A (2009) Physicochemical determinants of human renal clearance. J. Med. Chem 52, 4844–4852. [DOI] [PubMed] [Google Scholar]
- (41).Charifson PS, and Walters WP (2014) Acidic and basic drugs in medicinal chemistry: A perspective. J. Med. Chem 57, 9701–9717. [DOI] [PubMed] [Google Scholar]
- (42).Tiwari D, Park SW, Essawy MM, Dawadi S, Mason A, Nandakumar M, Zimmerman M, Mina M, Ho HP, Engelhart CA, Ioerger T, Sacchettini JC, Rhee K, Ehrt S, Aldrich CC, Dartois V, and Schnappinger D (2018) Targeting protein biotinylation enhances tuberculosis chemotherapy. Sci. Transl. Med 10, eaal1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Soares da Costa TP, Tieu W, Yap MY, Pendini NR, Polyak SW, Sejer Pedersen D, Morona R, Turnidge JD, Wallace JC, Wilce MCJ, Booker GW, and Abell AD (2012) Selective inhibition of biotin protein ligase from Staphylococcus aureus. J. Biol. Chem 287, 17823–17832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Somu RV, Boshoff H, Qiao CH, Bennett EM, Barry CE, and Aldrich CC (2006) Rationally designed nucleoside antibiotics that inhibit siderophore biosynthesis of Mycobacterium tuberculosis. J. Med. Chem 49, 31–34. [DOI] [PubMed] [Google Scholar]
- (45).Vannada J, Bennett EM, Wilson DJ, Boshoff HI, Barry CE, and Aldrich CC (2006) Design, synthesis, and biological evaluation of β-ketosulfonamide adenylation inhibitors as potential antitubercular agents. Org. Lett 8, 4707–4710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Qiao CH, Gupte A, Boshoff HI, Wilson DJ, Bennett EM, Somu RV, Barry CE, and Aldrich CC (2007) 5 ‘-O-[(N-Acyl)sulfamoyl]adenosines as antitubercular agents that inhibit MbtA: An adenylation enzyme required for siderophore biosynthesis of the mycobactins. J. Med. Chem 50, 6080–6094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Engelhart CA, and Aldrich CC (2013) Synthesis of chromone, quinolone, and benzoxazinone sulfonamide nucleosides as conformationally constrained inhibitors of adenylating enzymes required for siderophore biosynthesis. J. Org. Chem 78, 7470–7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Dawadi S, Boshoff HIM, Park SW, Schnappinger D, and Aldrich CC (2018) Conformationally constrained cinnolinone nucleoside analogues as siderophore biosynthesis inhibitors for tuberculosis. ACS Med. Chem. Lett 9, 386–391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Callahan BP, Lomino JV, and Wolfenden R (2006) Nanomolar inhibition of the enterobactin biosynthesis enzyme, EntE: Synthesis, substituent effects, and additivity. Bioorg. Med. Chem. Lett 16, 3802–3805. [DOI] [PubMed] [Google Scholar]
- (50).Bisseret P, Thielges S, Bourg S, Miethke M, Marahiel MA, and Eustache J (2007) Synthesis of a 2-indolylphosphonamide derivative with inhibitory activity against yersiniabactin biosynthesis. Tetrahedron Lett 48, 6080–6083. [Google Scholar]
- (51).Kabsch W (2010) Xds. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Evans PR, and Murshudov GN (2013) How good are my data and what is the resolution? Acta Crystallogr., Sect. D: Biol. Crystallogr 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Vagin A, and Teplyakov A (2010) Molecular replacement with MOLREP. Acta Crystallogr., Sect. D: Biol. Crystallogr 66, 22–25. [DOI] [PubMed] [Google Scholar]
- (54).Emsley P, and Cowtan K (2004) Coot: Model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- (55).Murshudov GN, Vagin AA, and Dodson EJ (1997) Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr., Sect. D: Biol. Crystallogr 53, 240–255. [DOI] [PubMed] [Google Scholar]
- (56).Schüttelkopf AW, and Van Aalten DM (2004) PRODRG: A tool for high-throughput crystallography of protein–ligand complexes. Acta Crystallogr., Sect. D: Biol. Crystallogr 60, 1355–1363. [DOI] [PubMed] [Google Scholar]
- (57).PyMOL Molecular Graphics System, version 1.8 (2017) Schrödinger, LLC. [Google Scholar]
- (58). See the Supporting Information for complete details.
- (59).Ferreras JA, Ryu JS, Di Lello F, Tan DS, and Quadri LEN (2005) Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol 1, 29–32. [DOI] [PubMed] [Google Scholar]
- (60).Hayward S, and Berendsen HJC (1998) Systematic analysis of domain motions in proteins from conformational change: New results on citrate synthase and T4 lysozyme. Proteins: Struct., Funct., Genet 30, 144–154. [PubMed] [Google Scholar]
- (61).Patskovsky Y, Toro R, Dickey M, Sauder JM, Chang S, Burley SK, and Almo SC (2009) Crystal structure of o-succinylbenzoic acid-CoA ligase from Staphylococcus aureus subsp. aureus Mu50. Protein Data Bank DOI: 10.2210/pdb3IPL/pdb. [DOI]
- (62).Gulick AM ANL Super Family Solved Structures http://www.acsu.buffalo.edu/~amgulick/RANLChart.html (Accessed on March 6, 2019).
- (63).Conti E, Franks NP, and Brick P (1996) Crystal structure of firefly luciferase throws light on a superfamily of adenylate-forming enzymes. Structure 4, 287–298. [DOI] [PubMed] [Google Scholar]
- (64).Nakatsu T, Ichiyama S, Hiratake J, Saldanha A, Kobashi N, Sakata K, and Kato H (2006) Structural basis for the spectral difference in luciferase bioluminescence. Nature 440, 372–376. [DOI] [PubMed] [Google Scholar]
- (65).Hisanaga Y, Ago H, Nakagawa N, Hamada K, Ida K, Yamamoto M, Hori T, Arii Y, Sugahara M, Kuramitsu S, Yokoyama S, and Miyano M (2004) Structural basis of the substrate-specific two-step catalysis of long chain fatty acyl-CoA synthetase dimer. J. Biol. Chem 279, 31717–31726. [DOI] [PubMed] [Google Scholar]
- (66).Andersson CS, Lundgren CAK, Magnusdottir A, Ge C, Wieslander A, Molina DM, and Hoegbom M (2012) The Mycobacterium tuberculosis very-long-chain fatty acyl-CoA synthetase: Structural basis for housing lipid substrates longer than the enzyme. Structure 20, 1062–1070. [DOI] [PubMed] [Google Scholar]
- (67).Vergnolle O, Xu H, Tufariello JM, Favrot L, Malek AA, Jacobs WR, and Blanchard JS (2016) Post-translational acetylation of MbtA modulates mycobacterial siderophore biosynthesis. J. Biol. Chem 291, 22315–22326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Liu Z, Ioerger TR, Wang F, and Sacchettini JC (2013) Structures of Mycobacterium tuberculosis FadD10 protein reveal a new type of adenylate-forming enzyme. J. Biol. Chem 288, 18473–18483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Stachelhaus T, Mootz HD, and Marahiel MA (1999) The specificity-conferring code of adenylation domains in nonribosomal peptide synthetases. Chem. Biol 6, 493–505. [DOI] [PubMed] [Google Scholar]
- (70).Conti E, Stachelhaus T, Marahiel MA, and Brick P (1997) Structural basis for the activation of phenylalanine in the non-ribosomal biosynthesis of gramicidin S. EMBO J 16, 4174–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Chen Y, Sun Y, Song H, and Guo Z (2015) Structural basis for the ATP-dependent configuration of adenylation active site in Bacillus subtilis o-succinylbenzoyl-CoA synthetase. J. Biol. Chem 290, 23971–23983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chen Y, Jiang Y, and Guo Z (2016) Mechanistic insights from the crystal structure of Bacillus subtilis o-succinylbenzoyl-CoA synthetase complexed with the adenylate intermediate. Biochemistry 55, 6685–6695. [DOI] [PubMed] [Google Scholar]
- (73).Finking R, Neumueller A, Solsbacher J, Konz D, Kretzschmar G, Schweitzer M, Krumm T, and Marahiel MA (2003) Aminoacyl adenylate substrate analogues for the inhibition of adenylation domains of nonribosomal peptide synthetases. Chem-BioChem 4, 903–906. [DOI] [PubMed] [Google Scholar]
- (74).Branchini BR, Murtiashaw MH, Carmody JN, Mygatt EE, and Southworth TL (2005) Synthesis of an N-acyl sulfamate analog of luciferyl-AMP: A stable and potent inhibitor of firefly luciferase. Bioorg. Med. Chem. Lett 15, 3860–3864. [DOI] [PubMed] [Google Scholar]
- (75).Kim S, Lee SW, Choi EC, and Choi SY (2003) Aminoacyl-tRNA synthetases and their inhibitors as a novel family of antibiotics. Appl. Microbiol. Biotechnol 61, 278–288. [DOI] [PubMed] [Google Scholar]
- (76).Koroniak L, Ciustea M, Gutierrez JA, and Richards NG (2003) Synthesis and characterization of an N-acylsulfonamide inhibitor of human asparagine synthetase. Org. Lett 5, 2033. [DOI] [PubMed] [Google Scholar]
- (77).Brown PH, Cronan JE, Grotli M, and Beckett D (2004) The biotin repressor: Modulation of allostery by corepressor analogs. J. Mol. Biol 337, 857–869. [DOI] [PubMed] [Google Scholar]
- (78).Tuck KL, Saldanha SA, Birch LM, Smith AG, and Abell C (2006) The design and synthesis of inhibitors of pantothenate synthetase. Org. Biomol. Chem 4, 3598–3610. [DOI] [PubMed] [Google Scholar]
- (79).Lu X, Olsen SK, Capili AD, Cisar JS, Lima CD, and Tan DS (2010) Designed semisynthetic protein inhibitors of Ub/Ubl E1 activating enzymes. J. Am. Chem. Soc 132, 1748–1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Olsen SK, Capili AD, Lu X, Tan DS, and Lima CD (2010) Active site remodelling accompanies thioester bond formation in the SUMO E1. Nature 463, 906–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL, Ma J, Loke HK, Lingaraj T, Wu D, Hamman KB, Spelman JJ, Cullis CA, Langston SP, Vyskocil S, Sells TB, Mallender WD, Visiers I, Li P, Claiborne CF, Rolfe M, Bolen JB, and Dick LR (2010) Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol. Cell 37, 102–111. [DOI] [PubMed] [Google Scholar]
- (82).Lun S, Guo H, Adamson J, Cisar JS, Davis TD, Chavadi SS, Warren JD, Quadri LEN, Tan DS, and Bishai WR (2013) Pharmacokinetic and in vivo efficacy studies of the mycobactin biosynthesis inhibitor salicyl-AMS in mice. Antimicrob. Agents Chemother 57, 5138–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Gulick AM, Lu X, and Dunaway-Mariano D (2004) Crystal structure of 4-chlorobenzoate:CoA ligase/synthetase in the unliganded and aryl substrate-bound states. Biochemistry 43, 8670–8679. [DOI] [PubMed] [Google Scholar]
- (84).Reger AS, Wu R, Dunaway-Mariano D, and Gulick AM (2008) Structural characterization of a 140° domain movement in the two-step reaction catalyzed by 4-chlorobenzoate:CoA ligase. Biochemistry 47, 8016–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).May JJ, Kessler N, Marahiel MA, and Stubbs MT (2002) Crystal structure of DhbE, an archetype for aryl acid activating domains of modular nonribosomal peptide synthetases. Proc. Natl. Acad. Sci. U. S. A 99, 12120–12125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (86).Hu Y, Gai Y, Yin L, Wang X, Feng C, Feng L, Li D, Jiang X-N, and Wang D-C (2010) Crystal structures of a Populus tomentosa 4-coumarate:CoA ligase shed light on its enzymatic mechanisms. Plant Cell 22, 3093–3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (87).Gulick AM, Starai VJ, Horswill AR, Homick KM, and Escalante-Semerena JC (2003) The 1.75 A crystal structure of acetyl-CoA synthetase bound to adenosine-5’-propylphosphate and coenzyme A. Biochemistry 42, 2866–2873. [DOI] [PubMed] [Google Scholar]
- (88).Reger AS, Carney JM, and Gulick AM (2007) Biochemical and crystallographic analysis of substrate binding and conformational changes in acetyl-CoA synthetase. Biochemistry 46, 6536–6546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Chen Y, Li TL, Lin X, Li X, Li XD, and Guo Z (2017) Crystal structure of the thioesterification conformation of Bacillus subtilis o-succinylbenzoyl-CoA synthetase reveals a distinct substrate-binding mode. J. Biol. Chem 292, 12296–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.