

Abstract
In asthma, airway smooth muscle (ASM) contraction and the subsequent decrease in airflow involve a poorly understood set of mechanical and biochemical events. Organ-level and molecular-scale models of the airway are frequently based on purely mechanical or biochemical considerations and do not account for physiological mechanochemical couplings. Here, we present a microphysiological model of the airway that allows for the quantitative analysis of the interactions between mechanical and biochemical signals triggered by compressive stress on epithelial cells. We show that a mechanical stimulus mimicking a bronchospastic challenge triggers the marked contraction and delayed relaxation of ASM, and that this is mediated by the discordant expression of cyclooxygenase genes in epithelial cells and regulated by the mechanosensor and transcriptional co-activator YAP (Yes-associated protein). A mathematical model of the intercellular feedback interactions recapitulates aspects of obstructive disease of the airways, including pathognomonic features of severe, difficult-to-treat asthma. The microphysiological model could be used to investigate the mechanisms of asthma pathogenesis and to develop therapeutic strategies that disrupt the positive feedback loop that leads to persistent airway constriction.
The bronchial airways can be seen as complex multicellular biological networks capable of distinct self-organized states, driven by mechanisms that are still incompletely understood. One such behavior is bronchospasm, a sudden shortening of the smooth muscle in the walls of the bronchioles that represents a common correlate of obstructive lung diseases, including asthma. In asthma, the constricted airway status can be prolonged, with symptoms frequently persisting even when the triggering stimuli are no longer present. This long-term activation of a specific state is suggestive of underlying ‘switch-like’ mechanisms stabilizing the pathological condition (1). Switch-like activation in biological and other systems is often a product of feedback mechanisms stabilizing distinct states. The switch-like response and the related phenomenon of bistability in airway constriction has been proposed to exist at different biological levels ranging from the emergent phenomena within a bronchial tree (2–5), to molecular interactions in mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) signaling (6). At the intermediate scale of the airway, bistability has been proposed by several models based on mechanosensitive feedback mechanisms that involve the interplay between airway contraction and airway wall mechanics (7–9). Here, we provide evidence that there is an additional chemosensitive feedback element based on the interaction of the epithelium and airway smooth muscle (ASM) via paracrine signaling and mechanosensing.
When bronchospasm is triggered through environmental insults, the ASM compresses the airway and causes mucosal folding (10, 11). During this process, the mucosa forms deep crevasses, exposing the epithelium to considerable compressive stresses (12). Several studies showed that mechanical stress in various forms can regulate the release of epithelium-derived factors, including ATP on a relatively short time scale (seconds) (13) and eicosanoids on longer time scales (minutes to hours) (14–16). ATP, a spasmogen, acts through purinergic receptors on ASM cells (17, 18), while eicosanoids can act at other receptors to evoke both relaxation and contraction (19–22). This points to a potential for complex intercellular feedback interactions between the epithelium and ASM, mediated by paracrine signaling and mechanotransduction over a wide range of time scales. When the positive component of this feedback dominates so that the shortened ASM can be maintained exclusively by the feedback, a switch-like response can lead to irreversibility (23, 24), i.e., trapping of the system in a stable aberrant state even after removal of external stimuli that trigger the bronchospasm.
Another common mechanical input in this process is the periodic stretching characteristic of tidal breathing. While deep inspiration (forced deep breathing) is known to antagonize bronchospasm (25), breathing at tidal volume is not expected to have a significant broncholytic effect. Prior studies in intact airways showed that pressure fluctuations or cyclic stretching that simulate tidal breathing did not reverse bronchospasm (26–28), and in some cases even deep inspiration failed to reverse it (26). Breathing reverses bronchospasm most effectively when the severity of bronchoconstriction is small and the depth of breathing large (i.e., deep inspiration) (25). Also, more severely constricted airways are stiffer, hence exhibit less strain under tidal stress, implicating a reduced mechanical effect of tidal breathing under bronchospasm conditions (25). Finally, it should be noted that while ASM is expected to stretch during tidal breathing, for airway pressures below 10 cmH2O (for breathing at tidal volume the typical pressure is ~3 cmH2O (29)), airway expansion is primarily due to epithelial unfolding, not the stretching of the epithelium (which can occur at higher pressures, such as under deep inspiration) (30). As more explicitly explored later in this study, the effect of cyclic stretch due to breathing is indeed relatively minor, while stretching due to deep inspiration can have a pronounced effect.
Experimental models are needed to investigate these putative intercellular interaction mechanisms and their functional consequences. Although several tractable in vitro airway models have been investigated (31–33), they were not designed to examine these proposed intercellular interaction mechanisms either functionally, or in terms of the underlying molecular mechanisms. While ex vivo models such as lung slices closely approximate the in vivo context (28, 34, 35), it is difficult to control experimental conditions in these systems, e.g., to quantitatively evaluate mechanical properties of the airways, isolate cellular secretions, and test putative mechanisms in a well-controlled and quantitative manner. Similar difficulties are encountered with myograph readings from airway rings. A highly controlled in vitro environment that, at the same time, recapitulates important aspects of the interplay of chemical and mechanical transduction would provide more precise information needed for a mechanistic understanding of the complex intercellular signaling and the emergent property of bronchospasm. In this study, we present such a tractable in vitro environment and perform mechanistic studies in this new experimental model, leading to a proposal of a new mechanism of bronchospasm emergence and progression. This mechanism is based on mechanochemical intercellular interactions, occurring on different time scales. This mechanistic model accounts for the fast switch-like onset of bronchospasm, due to the secretion of spasmogenic factors by compressed epithelial airways cells, which act on ASM cells promoting their further contraction, thus constituting a putative feedback loop. We also provide evidence for a delayed negative feedback, which can trigger relaxation of the bronchospasm, accounting for multiple experimental observations.
Results
A microphysiological airway model supports positive feedback between smooth-muscle shortening and compressive stress on epithelium.
To overcome the aforementioned gaps in current models, we developed a ‘bronchial-chip’ airway model, which reconstitutes 3D co-cultures of early-passage primary human ASM (HASM) and normal human bronchial epithelial (NHBE) cells that are fully-differentiated in an air-liquid interface. This bronchial-chip platform also allows the application of an apical-to-basal compressive stress mimicking severe bronchospasm using a validated experimental technique that reduces the lateral intercellular space of the pseudostratified NHBE layer, representative of compressed airways in vivo (36, 37). The platform is also designed to be compatible with optical magnetic twisting cytometry (OMTC), a method for quantitative analysis of in vitro cellular mechanics (Figs. 1A–B and S1A–C) (38–41). Using this microphysiological model of the bronchial airway, we can test the bronchial epithelial cell response to a relevant mechanical stress, while simultaneously assessing the effect of this response on juxtaposed smooth muscle cells (Fig. 1A). More specifically, the platform allows application of compressive stress at physiological levels on pseudostratified NHBE cells through regulated air pressure. Resulting changes in mechanical properties of HASM cells are then quantitatively measured in real-time using OMTC (Fig. 1B). OMTC utilizes peptide-coated ferrimagnetic microbeads that are functionalized to interact with the actin cytoskeleton through cell surface integrin receptors (beads on HASM layer inside the platform shown in Fig. 1C). Using this experimental setup, we observed that an application of 30 cmH2O compressive stress on the NHBE layer, a stress level observed during severe bronchospasm (12, 42), culminated in a fast and robust stiffening of HASM cells, with the cell stiffness increasing more than two-fold within two minutes (Fig. 1D). This finding suggests that mechanical stress on epithelial cells can trigger their rapid biochemical communication with and subsequent contraction of HASM due to release of epithelium-derived spasmogens.
Fig. 1. Bronchial-chip results indicate positive feedback between smooth-muscle contraction and compressive stress on epithelium.
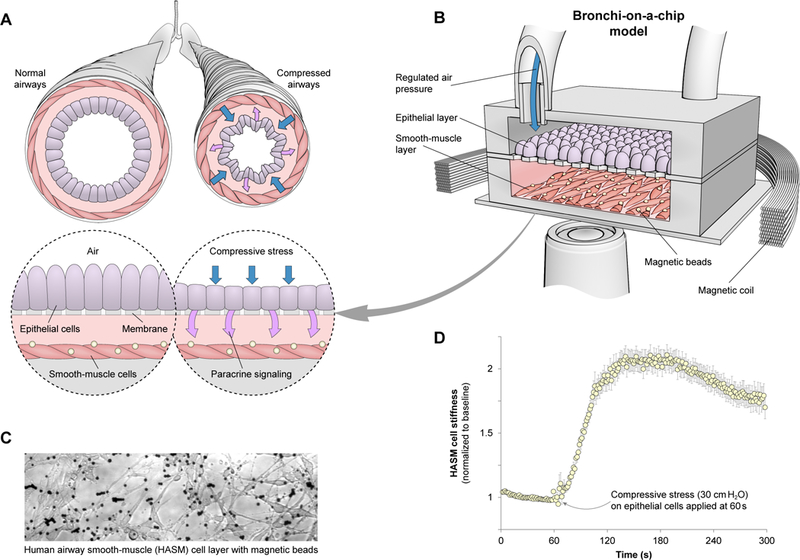
(A) Contraction of airway smooth muscle leads to mechanical stress in airway epithelium, which may result in paracrine signaling. (B) To quantitatively analyze this interaction, a model system using a bronchial-chip platform was developed. Regulated air pressure applies compressive stress at physiological levels on normal human bronchial epithelial (NHBE) cells. Resulting changes in human airway smooth-muscle (HASM) mechanical properties are quantitatively measured using optical magnetic-twisting cytometry. (C) HASM layer inside bronchial-chip, with magnetic beads coated with a peptide binding to cell surface integrin receptors, hence allowing probing of the actin cytoskeleton. (D) 30 cmH2O compressive stress applied after t = 60 seconds on the NHBE layer leads to HASM contraction, whereby cell stiffness changes more than two-fold within two minutes. Mean ± SEM for n = 334 cells from three different chips tested.
Longer-term intercellular interaction through eicosanoid production.
The bronchial-chip platform results indicate release of spasmogenic factors by NHBE cells and ensuing HASM contraction after only a few minutes of mechanical stress. To test for the potential presence of a more protracted cell-cell interaction, we used a setup that allows analysis of the effects of compressive stress on NHBE cells (Figs. 2A and S1D). While this setup does not allow measurements of short-term interactions between NHBE and HASM cells that the bronchial-chip permits, it enables the assaying of much larger cell numbers required for mechanistic analysis. The setup is based on a six-well platform with pseudostratified primary NHBE cells in an air-liquid interface (37, 43). We found that the infranatant (the media collected below cells in the air-liquid interface; Fig. S1E) from NHBE cells exposed to 3 hours of continuous compressive stress (30 cmH2O) also caused HASM cell contraction (Fig. 2B). Recently we have shown that cyclooxygenase plays a significant role in mechanotransduction (44). We therefore further investigated whether the epithelial factors causing HASM contraction included cyclooxygenase products. Pre-incubation of the NHBE cells with celecoxib, a selective inhibitor of cyclooxygenase type 2 (COX2), suppressed the longer-term effects of compressive stress on epithelial cells (Fig. 2C). The infranatant mediated HASM contraction was also observed when media conditioned by epithelial cells was used, i.e., even if NHBE cells were not subjected to compressive stress (Fig. 2C; 0h). This result suggested that basal epithelial secretions can have a tonic effect on HASM cells. In combination, these results pointed towards a critical role for cyclooxygenase, and suggested that prostanoids might constitute a significant portion of the epithelium-derived factors mediating short or long-term HASM contraction in response to compressive stress. This result was supported by observation of substantial reduction of longer-term HASM contraction following treatment of the cells with different eicosanoid receptor antagonists along with epithelial infranatant (Fig. 2D). Overall, these results suggest that a significant contribution to HASM contraction under these physiological circumstances comes from the effect of eicosanoids secreted by NHBE cells.
Fig. 2. Longer-term intercellular interaction through eicosanoid production.

(A) Setup that allows analysis of the effects of compressive stress on epithelial cells in a more controlled environment and with more cell numbers. The setup is based on a six-well platform with pseudostratified NHBE cells in an air-liquid interface. (B) Infranatant from compressive-stress-treated NHBE cells causes strong contraction in HASM cells. Mean ± SEM for n cells from three independent experiments (n = 490 for Cntrl; n = 482 for 3h; Cntrl = untreated medium). (C) COX2 inhibitor celecoxib suppresses the effects of compressive stress on NHBE cells, as HASM cell contraction in response to 0h and 3h-pressure epithelial samples are at similar levels. Mean ± SEM for n cells from three independent experiments (left-to-right n = 202, 111, 102, 218, 209). *P<0.05 indicates significance to all other conditions according to one-way ANOVA with Bonferroni’s post-hoc correction. (D) Response of drug-treated HASM cells to 3h-pressure epithelial infranatant. The drugs antagonize different eicosanoid receptors that are involved in contraction initiation. AL = AL8810, PGF2α receptor (FP) antagonist. ICI = ICI192605, thromboxane receptor (TP) antagonist. Zaf = Zafirlukast, CysLT1 receptor antagonist. Mean ± SEM for n cells from three independent experiments (left-to-right n = 277, 206, 191, 179). *P<0.05 indicates significance to control according to one-way ANOVA with Bonferroni’s post-hoc correction. Differences between different drug treatments are not significant.
Eicosanoid production is regulated by modulation of cyclooxygenase isozymes by compressive stress.
To further test the role of cyclooxygenase products, we analyzed how compressive stress regulates PTGS1 and PTGS2, the genes coding for the two cyclooxygenase isozymes. Following the application of compressive stress we observed a divergent regulation of these two genes in NHBE cells, with PTGS1 (coding for COX1) downregulated, and PTGS2 (coding for COX2) upregulated to levels that depended on the duration of the compressive stress (Fig. 3A). To test the effect of this discordant regulation of cyclooxygenase genes on prostanoid production, we measured the PGE2 content of epithelial infranatants using an ELISA test (Fig. 3B), observing increasing levels of PGE2 concomitant with COX2 upregulation. Furthermore, PGE2 has been shown to regulate COX2 through the EP2/4 receptors (45), constituting a putative feedback loop. This feedback regulation would predict reduced COX2 mRNA expression levels in response to pharmacological COX2 inhibition, which we indeed observed experimentally (Fig. S2A). The tonic effect of the basal NHBE secretion on HASM cells suggested that COX2 can be upregulated under basal conditions. Consistent with this assumption, we found an increased level of secreted PGE2 when the medium, in which NHBE cells were cultured overnight was not changed before the cells were exposed to compressive stress (‘stabilized’, Fig. 3B, left panel). The levels of COX2 expression also showed elevation under these ‘stabilized’ conditions, although the detected difference vs. the fresh medium conditions was not significant. Overall, these results strongly suggested that COX2 expression and function was regulated by the compressive stress in epithelial cells.
Fig. 3. Eicosanoid production is regulated by modulation of cyclooxygenase isozymes by compressive stress.

(A) Compressive stress differentially regulates COX1 and COX2 mRNA expression in NHBE cells. (B) PGE2 content of epithelial infranatant. (A-B) Mean ± SD for n = 3 biologically independent samples. *P<0.05 indicates significance to control according to t-test with Benjamini-Hochberg correction. Controls are samples with no pressure applied. Before pressure experiments, overnight media is washed away, and replaced with fresh media. Stabilized = overnight media remains in the setup during the pressure experiments.
Compressive stress is relayed to cyclooxygenase through the mechanosensor YAP.
We further explored the mechanism of differential regulation of PTGS mRNA levels by compressive stress. Yes-associated protein (YAP) has been implicated as an important mechanosensitive transcriptional co-regulator that can be activated by tension in the actomyosin cytoskeleton and by Rho GTPase (46, 47). We therefore tested HASM contraction in cells treated with infranatant collected from NHBE cells exposed to the compressive stress with or without simultaneous exposure to verteporfin, a pharmacological YAP inhibitor. YAP inhibition abrogated the effects of compressive stress, as HASM cell contraction showed no significant difference in response to infranatants from verteporfin-treated NHBE samples not exposed to or exposed to 3h-compressive stress conditions (Fig. 4A; 0h+Vp vs. 3h+Vp). The role of YAP was further tested by measuring the effect of verteporfin on PTGS1 and PTGS2 mRNA expression in NHBE cells. YAP inhibition downregulated COX2, and abrogated downregulation of COX1 and upregulation of COX2 by compressive stress (Fig. 4B), consistent with the results in Fig. 4A. Both YAP and COX2 inhibition led to a lower PGE2 content of the infranatants, suggesting a functional role for these regulators in mediating the effects of compressive stress (Fig. 4C). Further support for YAP acting as a mechanosensor of compressive stress emerged from the analysis of mRNA expression levels of YAP1 and its target connective tissue growth factor (CTGF) in epithelial cells (Fig. 4D). We observed that CTGF was upregulated by compressive stress, and this upregulation was abrogated by YAP inhibition with verteporfin. On the other hand, celecoxib reduced YAP1 expression, but did not suppress the effect of compressive stress on CTGF. We reasoned therefore that cyclooxygenase is downstream of YAP. We verified this through immunoblotting analysis of COX1 and COX2 expression in epithelial cells, following shRNA-mediated knockdown of YAP (Fig. 4E). These results strongly support YAP-mediated mechanotransduction in NHBE cells as the mechanism of response to the compressive stress leading to prostanoid secretion.
Fig. 4. Compressive stress is relayed to cyclooxygenase through the mechanosensor YAP.

(A) HASM cell contraction in response to infranatant from NHBE cells exposed to compressive stress and/or treated with the YAP inhibitor verteporfin. Mean ± SEM for n cells from three independent experiments (left-to-right n = 111, 102, 250, 249). ***P = 4.6e-11 according to t-test. (B) Effects of celecoxib and verteporfin on COX1 and COX2 mRNA expression in NHBE cells. (C) PGE2 content of epithelial infranatant in response to celecoxib and verteporfin treatment. Cases for which celecoxib inhibited PGE2 production to below the limit of detection (LOD) of the assay were assigned the LOD value (dashed line). (D) Expression of YAP1 mRNA and its target CTGF in NHBE cells. (B-D) Mean ± SD for n = 3 biologically independent samples. *P<0.05 indicates significance to control according to t-test with Benjamini-Hochberg correction. Controls are samples with no pressure applied. (E) Western blots of COX1 and COX2 expression in NHBE cells transduced with shYAP or a control virus. (Full scans of the western blots are provided in Fig. S7.)
Interaction between epithelial and smooth-muscle cells are subject to time resolved positive and negative feedback mechanisms.
The results presented above suggest that compressive stress on NHBE cells can lead to secretion of factors that can control or modulate the contraction and relaxation of HASM cells. We set out to further explore this effect, using HASM cells derived from both non-asthmatics and asthmatic patients. In particular, we were interested in whether NHBE-derived PGE2 might modulate HASM contraction that can be initially triggered by an environmental input. In agreement with our recent study showing asthmatic and non-asthmatic HASM cells possessing different mechanophenotypes (48), we observed stronger contraction in HASM cells derived from the lungs of asthmatic donors in response to methacholine, a synthetic muscarinic receptor agonist used to diagnose bronchial hyperresponsiveness (Fig. 5A). Previously, PGE2 was reported to cause bronchial dilation in non-asthmatic human subjects, while causing either bronchial dilation or constriction in asthmatic patients (21). In our system, we found that subsequent application of PGE2 relaxed both asthmatic and non-asthmatic HASM cells to approximately baseline levels found prior to methacholine application (Fig. 5B). Furthermore, we found that the relaxation effect of PGE2 displayed similar dose response both in ASM derived from fatal asthma and non-asthma subjects (Fig. S3A–C). Given this strong and consistent effect of PGE2, we then examined the expression and abundance of prostaglandin E receptors 1–4 (EP1–4) in normal and asthmatic HASM cells used in the experiments, and the involvement of these receptors in the PGE2 effects. We found a significant increase in mRNA expression of EP2 and increased EP2 and EP4 proteins in asthmatic cells vs. non-asthmatic cells (Fig. S3D–E), which could contribute to the differences in the mechanophenotypes of these cells (49). Importantly, inhibiting the function of EP2 and EP4 receptors revealed that the effect of PGE2 (100 nM) was dependent on both EP2 and EP4 in normal, and only on EP2 in asthmatic cells (Fig. S3F).
Fig. 5. Positive and negative feedback between NHBE and HASM.

(A) Stiffness change of non-asthmatic and asthmatic HASM cells in response to methacholine. Mean ± SEM for n cells from three independent experiments (n = 453 for Non-asthma; n = 486 for Asthma). (B) Stiffness change of HASM cells in response to PGE2 (following methacholine). Mean ± SEM for n cells from three independent experiments (n = 290 for Non-asthma; n = 332 for Asthma). (C) Contractile response of non-asthmatic and asthmatic HASM cells to infranatant samples from NHBE cells treated with different pressure durations. Mean ± SEM for n cells from three independent experiments (left-to-right n = 219, 434, 353, 338 for Non-asthma; n = 248, 439, 425, 470 for Asthma). *P<0.05 indicates significance to all other conditions according to one-way ANOVA with Bonferroni’s post-hoc correction. (D) Epithelial products lead to ASM cell relaxation over extended time periods. HASM cells were incubated with NHBE infranatant samples overnight (18 hours) and then exposed to PGE2. Mean ± SEM for n cells from three independent experiments (left-to-right n = 496, 439, 369). ***P<0.001 according to Wilcoxon rank-sum test (P = 9.1e-15 for Cntrl; P = 4.5e-8 for 0h; P = 8.7e-4 for 6h). (E) Compressive stress on NHBE cells saturates contraction of HASM cells in the bronchial-chip platform. First a 30 cmH2O compressive stress is applied on NHBE cells, leading to strong contraction in HASM cells. After pressure is turned off, 10 μM methacholine is introduced into the chip, followed by 1 μM PGE2, and finally 30 cmH2O compressive stress is reapplied. Mean ± SEM for n cells from three different chips tested (left-to-right n = 334, 335, 327, 282). *P<0.05 and ***P<0.001 according to Wilcoxon rank-sum test (P = 5.4e-18 for Pressure; P = 0.048 for Methacholine).
Our results suggested that PGE2 can be secreted by NHBE cells and can have a relaxation effect on HASM cells. This result was in apparent contradiction to the strongly contractile effect observed in experiments shown in Fig. 1. However, this contradiction can be resolved if the secretion from NHBE cells can have a contractile effect at shorter time scales, and relaxing effect at longer time scales (necessary for YAP-dependent COX2 upregulation and PGE2 synthesis). We indeed observed that, as the duration of compressive stress increased over 6 h, the compressive effect of the secreted factors initially increased and then decreased in both types of HASM cells (3h vs. 6h; Fig. 5C). Furthermore, when we exposed HASM cells to NHBE infranatants overnight, we observed a reduction in the baseline stiffness (muscle tone) (Fig. 5D) in agreement with recent studies (50). This result indicated that, following compressive stress, epithelial cells release factors that work over longer time scales, ultimately leading to relaxation of HASM cells. To further explore this possibility, we again used the bronchial chip platform. We again found that over shorter timescales, application of compressive stress on the NHBE layer in the bronchial-chips led to strong contraction in the HASM layer (Fig. 5E; n = 3 bronchial-chips). Turning off the stress after this relatively short pulse did not lead to relaxation of the baseline in HASM cells, and further treatment of the HASM cells with methacholine only slightly increased cell contraction. These results suggested that factors released by NHBE cells over the short compressive stress duration can persist in the intercellular space and can lead to strong compressive effect on HASM. However, as the compressive stress on NHBE cells was only transient, it did not lead to longer term effects resulting in secretion of the relaxants. Interestingly, treatment of the HASM cells with saturating concentrations of PGE2 subsequent to the brief compressive stress exposure coupled with methacholine did not lead to relaxation either (in contrast to results in Figs. 5B and S3A–C), indicating that HASM cells become progressively more sensitized to the effect of PGE2 over time. The contractile effect of compressive stress could not be further augmented through re-application of compressive stress, indicating that the early contractile factors are secreted rapidly over the first few minutes of NHBE reaction to the mechanical stimulation.
Mechanochemical feedback between epithelium and smooth muscle may underlie different phenotypes of airway contraction.
Collectively, our results paint a complex picture of putative mutual regulation between HASM and NHBE cells. Stimulation of HASM cells by spasmogens can lead to their contraction, which in turn can result in compressive stress on NHBE cells. This stress can lead to secretion of factors that can modulate HASM contractility, constituting a mechanochemical feedback. Over the initial few minutes, release of factors that are poised for secretion can lead to a strong initial contractile effect on HASM cells. Later effects of NHBE cells in the presence of continued contractile stress from HASM cells can lead to the time dependent combination of further positive feedback (contractile factors) followed by a slower negative feedback loop (secretion of relaxants, such as PGE2) (Fig. 6A; with more mechanistic detail provided in Fig. 6B). On the relatively shorter time scale (0–3 h) an initial external stimulus (e.g., histamine release due to an allergic reaction, or acetylcholine release due to autonomic nervous system response to an irritant) can lead to a rapid contraction of HASM cells, which promotes compressive stress on epithelial cells. As a result, as indicated, epithelial cells secrete spasmogens both in an acute manner and in a YAP and COX2 specific delayed fashion. This secretion can also be partially regulated in a cell-autonomous feedback fashion, through a COX2-mediated up-regulation of YAP and COX2 expression. The initial positive feedback interactions resulting from the heterotypic epithelial-ASM cell-cell interaction mechanism, with spasmogens stimulating adjacent ASM cells, can thus result in further compressive stress on epithelial cells, leading to prolongation in time and space of spasmogen secretion. Therefore, the positive feedback interactions can latch the ASM into a state characteristic of persistent contraction and airflow obstruction. On a longer time scale (>3–6 h), YAP and COX2 mediated secretion of relaxants, particularly PGE2, can ultimately lead to antagonism of the pro-contractile state. This interaction thus constitutes a delayed negative feedback leading to full or partial recovery from the initial contractile state.
Fig. 6. Mechanochemical feedback interactions can underlie distinct modes of bronchospasm.

Mechanochemical feedback interactions can underlie distinct modes of bronchospasm. (A) Schematic of airway feedback model: ASM contraction is driven by an external stimulus. Once ASM contract and apply compressive stress on the epithelium, a positive and a slower negative feedback through prostanoid production is activated. (B) Molecular mechanisms studied or identified in this study, alongside the other established relevant signaling pathways. Mechanical stress can lead to the release of ATP by the epithelium, which can cause calcium influx through autocrine signaling, which then leads to the liberation of AA (arachidonic acid), the substrate for COX1 and COX2. This cascade eventually leads to the release of prostaglandins that affect ASM mechanics. Our study shows that bronchospasm stress levels can activate the mechanosensor YAP, which then upregulates COX2 and downregulates COX1, leading to the release of prostanoids that can cause both relaxation and contraction. (C) Determining model parameters from experimental data: Fitting the model to (i) ASM contraction (mean ± SEM for n = 453 cells from three independent experiments), (ii) relative PGE2 production levels (mean ± SEM for n = 3 biologically independent samples), and (iii) ASM contraction in response to epithelial products for different pressure durations (mean ± SEM for n = 370 cells from three independent experiments for each pressure duration) yield Tc, T2, and T1 respectively (95% confidence intervals in parentheses). (iv) Changes in relative spasmogen and relaxant levels based on model and determined time constants when compressive stress is applied for 6 hours on epithelium and then turned off. (D) Distinct ASM contraction profiles for different ASM responsiveness values in response to an external stimulus. (E) Existence of a refractory period where a second stimulus has little effect on ASM contraction. (F) A ‘second wind’ phenomenon, where ASM in a contraction state that slowly oscillates (despite a non-oscillatory stimulus) can paradoxically relax in response to a second stimulus.
To validate and quantify this conceptual model, we expressed the positive and negative feedback interactions suggested by our findings in the form of a mathematical model (Methods section) that was then fitted to experimental data to determine model parameters (Figs. 6C and S4). We exposed the model to a transient simulated input (stimulus) representing an initial trigger (such as exposure to an allergen, airway irritant, cold air, or exercise). The model predicted that due to the faster onset of spasmogens in response to compressive stress, epithelial products are expected to have a contractile effect on ASM at shorter time scales, and a relaxant effect at longer time scales (Fig. 6C–iv). In an agreement with the conceptual model, we found that the action of spasmogens was comparatively fast, occurring on the 1–5 min. time scales, whereas the secretion and action of relaxants was considerably delayed, with the time constant of approximately 3 h. This quantified model predicted that, if the sensitivity of ASM cells to the effect of the stimulus increased, as would be the case in e.g., asthmatic cells (Fig. 5A), the amplitude and duration of ASM contraction were gradually augmented, displaying a range of qualitatively distinct dynamic responses (Fig. 6D). In particular, for relatively low values of responsiveness, the ASM contraction was predicted to closely follow the stimulation in duration, with variable amplitude of the response, proportional to the degree of responsiveness. However, as cell responsiveness further increased, the amplitude of contraction underwent a switch to higher levels, indicative of the onset of the positive feedback, but ultimately subsided with a progressively increasing duration, now exceeding the duration of the initial stimulus. Finally, as the degree of responsiveness was further elevated, the overall response became non-adaptive, with a high level of contraction persisting along with oscillatory fluctuations (expected from the slower negative feedback regulation (51)). Thus, the model predicted that with a high enough responsiveness to a stimulus, which may be characteristic of the asthmatic condition, the epithelial-ASM interaction can maintain the cells in a persistently contracted state, even in the presence of the delayed negative feedback loop, which might suggest the mechanism underlying persistent bronchospasm associated with asthmatic mechanophenotypes.
The model also made several other important predictions. If instead of a single stimulus, two stimuli separated in time were presented to the cells, there were two key unexpected findings. First, under the conditions of intermediate ASM responsiveness, there was a refractory period after the initial stimulation, during which the relaxed cells, when stimulated again, did not undergo a sizable contraction (Fig. 6E). A closer look reveals that this was because the second stimulus was applied during a downturn of contraction when the relative relaxant amount was higher, reducing the effectiveness of the second stimulus (Fig. S5). Second, under the conditions of high ASM responsiveness leading to a persistent but slow oscillatory contraction state following a first non-oscillatory stimulus, a second stimulus could paradoxically relax the simulated airways (Fig. 6F). Furthermore, the relaxation effect became stronger for a stronger second stimulus. This occurred because the second stimulus was “out-of-phase” with the spasmogen oscillations triggered by the first stimulus, but “in-phase” with relaxant oscillations due to their slower dynamics (Fig. S5). Therefore, while the second stimulus enhanced both spasmogen and relaxant levels, a relatively larger increase in relaxant levels was sufficient to completely dampen the oscillations.
Finally, we used the model to explore the effects of cyclic stretch emerging from tidal breathing, distinct from the compressive stress explored up to this point. Because the effect of cyclic stretch on HASM cells has been analyzed in prior studies (see e.g. (25–28)), we first used the bronchial-chip platform to investigate the effects of tidal breathing on NHBE cells experimentally. More specifically, we modeled breathing by applying an oscillating pressure at the frequency and amplitude mimicking the natural setting. For this, we used a compliant membrane that would deflect under pressure so that the epithelial cells would experience a cyclic stretch of ~10% at maximal pressure (Fig. S6A). Application of an oscillating pressure of 3 cmH2O (at 0.5 Hz), corresponding to peak pressures encountered during breathing at tidal volume (29), exerted tension on the NHBE cells resulting in a cellular-area increase of 17% at maximal stretch (Movie S1, Fig. S6B). This strain value compares well to physiological values, as the maximum strain in airways is expected to be in the range of 5% to 15% during tidal breathing (see, e.g., (25)). However, this oscillating pressure did not result in a significant effect on HASM contraction in the bronchial-chips when applied over the course of 15 minutes (Fig. S6C). While we did not observe a significant effect of cyclic stretch applied on the epithelial layer, cyclic stretch of ASM experienced during inspiration is known to reduce its responsiveness, and in some cases antagonize bronchospasm (25–28). To account for this potential effect, we incorporated the reduction of ASM responsiveness into our model using experimental data on human airways from prior studies (25). Our model results show that while breathing at tidal volume has a relatively small effect on bronchospasm, deep inspiration indeed affects it significantly unless the bronchospasm is severe (Fig. S6D), all in agreement with prior studies (25–28).
Discussion
Human tissues are complex self-organized systems composed of cells, which engage in complex chemical and mechanical interactions. Occasionally, these interactions can deviate from the physiologically normal balance, leading to diverse pathological consequences. In this study, using primary human cells cultured in a new experimental platform mimicking the complexity of the bronchial tissue, we show that compressive stress on airway epithelial cells can elicit contraction followed by a delayed partial relaxation of HASM cells. This finding suggests an interplay of positive and negative mechanochemical feedback interactions that might affect the onset and progression of bronchospasm in normal and asthmatic airways.
Our quantitative results indicate that for severe bronchospasm, the positive mechanochemical feedback interaction between smooth muscle and epithelial cells could be strong enough to promote a sustained constricted state and the associated bronchospasm even after the removal of external stimuli. Relatively “fixed”, or recurrent, airflow obstruction is characteristic of both moderate persistent and severe asthma, leading to significant morbidity and mortality (52, 53). The data provided in this report show how adaptive and maladaptive mechanisms can emerge due to interactions between the epithelium and smooth muscle, critically dependent on the secretome associated with epithelial compression. A particularly unexpected finding was the high amplitude and fast rate of the initial HASM contraction response following the application of physiologically relevant mechanical stress on adjacent epithelial cells.
Similar rapid responses have been observed in recent studies using ex vivo lung-tissue slices from rats, where local injury to a single epithelial cell triggered an instantaneous Ca2+ wave throughout the epithelium and induced ASM contraction within several seconds that lasted for tens of seconds (54). It was observed that epithelial injury triggered the release of ATP, and airway contraction was completely blocked by selective inhibition of COX2 (55). While, in these studies, the ASM contraction was triggered by an injury to the epithelium, our study suggests that mechanically stimulating the epithelium can be sufficient to obtain similar results. Indeed, earlier studies using guinea pig trachea in vitro showed that gentle mechanical irritation of the mucosal surface triggers the release of both PGE2 and PGF2α, and that the effect of the mechanical stimulus can be blocked by cyclooxygenase inhibition (56). Prostaglandin release was also induced by contracting the trachea with histamine or acetylcholine (56). Similarly, application of a tensile stress on guinea pig trachea in vitro triggered airway contraction (57). This stretch-induced contraction was maintained only in tissues with an intact epithelium, and was absent in tissues with the epithelium removed, or when prostaglandin synthesis was inhibited (57). It is important to note that, using our microphysiological airway model, we observed a similar sustained contraction state of ASM cells in the presence of an epithelium during the early stages following the onset of the mechanical stress. Despite a difference in the type of mechanical stimuli and the experimental models used, our simpler models could account for these earlier findings. They also allowed further analysis of the role of cyclooxygenase in the initial contractile response of ASM to epithelial factors released following mechanical stress. Finally, our models showed that the short term contractile response is tempered by longer term relaxation effects underscoring the complex dynamics of the process.
In addition to elucidating the phenotypic effects of the compressive stress on airway epithelial cells, the tractability of the experimental system and modeling results allowed us to investigate the underpinnings of the ensuing signaling processes. In particular, our analysis suggests that YAP-associated mechanosensing and YAP-mediated regulation of COX2 expression constitute a likely mechanism of how ASM contraction and relaxation are modulated by the mechanical stress on the epithelium. The prostanoid secretion as a result of COX2 regulation can also potentially affect the epithelial cells themselves, as suggested by our experiments, showing reduced COX2 mRNA expression levels in response to pharmacological COX2 inhibition. These mechanochemical signaling processes can lead to feedback interactions, acting through distinct receptor types in normal vs. asthmatic cells and resulting in complex effects on bronchospasm discussed in detail below. The associated mechanisms, and their quantitative analysis also may provide an opportunity for target identification in controlling bronchospasm in various clinical contexts.
Our results indicate that this mechanical input on the epithelium can lead to diverse effects on ASM contraction, occurring on different time scales. In addition to longer term contraction and relaxation effects (unfolding over several hours), we also observed a much more acute ASM stress (within a few minutes). The mechanism of this more immediate mechanosensing response is less clear. Various mechanosensitive ion channels, known to be expressed in epithelial cells, could potentially mediate this acute response, including transient receptor potential channel (TRP) (58), epithelial sodium channel (59), cystic fibrosis transmembrane conductance regulator (60), and Piezo1 (61, 62). In airway epithelial cells, mechanical transduction through TRP and signaling through the Rho-family small GTPases can mediate direct ATP release through the large transmembrane channel Pannexin1 (63), which also allows efflux of other small molecules including prostaglandins. In other cell types, mechanical loading can lead to Ca2+ dependent rapid vesicular release of ATP, which can then mediate prostaglandin release through autocrine signaling (64). Therefore, besides epithelial injury, spasmogen release due to mechanical activation of ion channels could be a potential mechanism that triggers rapid ASM contraction in response to compressive stress on the epithelium. Taken together, our analysis along with prior data point to the need for more thorough mechanistic studies of epithelium-ASM interactions at different time scales. We believe that the microphysiological airway model presented in this report provides the means for such investigations.
Our results, when expressed in the form of a mathematical model, suggest that the spatial and temporal nature of the ASM response to one or more spasmogens can determine the quantitative and qualitative features of airway constriction. The effect is not simply additive, indicating a degree of complexity not previously appreciated. For example, our results reveal that with increasing ASM responsiveness the duration of constriction can dramatically increase, beyond the duration of the spasmogenic stimulus, becoming potentially irreversible. Unexpectedly, our results also suggest that delivery of a secondary spasmogenic stimulus, depending on its timing, can attenuate bronchospasm. This paradoxical result is directly related to the fact that epithelial-ASM cell interactions can involve both positive and negative long-term interactions, separated in time. Therefore, the secondary stimulus can potentially ‘resonate’ with the relaxant effect of endothelial secretions, leading to abrupt relaxation of the otherwise potentially irreversible process. Interestingly, there are several studies reporting paradoxical relaxation of constricted bronchi that parallel our model results. Studies with freshly isolated feline, equine, and porcine bronchial strips indicate that reapplication of a spasmogen on already contracted airways may exert a bronchi-widening (broncholytic) effect. In isolated feline bronchial strips that were contracted with carbachol, an acetylcholine receptor agonist, a paradoxical relaxation was observed in response to application of the spasmogens histamine, PGF2α, and bradykinin (65). Similar paradoxical broncholytic responses to spasmogenic stimuli were observed in equine (66) and porcine (67) bronchial strips that were initially contracted by spasmogens. Another model prediction, the refractory period related to the action of relaxant, is also frequently reported in the exercise induced asthma, frequently observed in longer distance runners and other athletes. It can potentially account for the positive effects of ‘warm-up’ or initial exertion reported in these settings (68). The model can help elucidate various other phenomena reported in asthma or asthma-like conditions. For instance, aspirin-induced asthma appears to be accompanied by a reduced abundance of PGE2 (69, 70). Our findings suggest that nonsteroidal anti-inflammatory drug (NSAID)-mediated suppression of activity of COX2 can further down-modulate PGE2 synthesis, thus severely decreasing the delayed relaxing effect of epithelial cells on the adjacent ASM cells. This effect can lead to a higher propensity of sustained bronchospasm that can be triggered by low level stimulation, including physiological levels of compressive stress, due to the positive feedback effects not being balanced by the negative feedback ones. The model also suggests that ASM exposure to beta-blockers, while potentially leading to increased airway-constriction acutely, could also lead to more prolonged beneficial effects involving relaxation effects of secondarily released endogenous bronchodilators, e.g., PGE2. This model prediction can provide explanation for paradoxically beneficial longer term effects of beta-blockers in asthma and is complementary to the previously suggested effects of the modulation of the inflammatory response and mucous metaplasia (71).
Our bronchial-chip platform also provides a blueprint for studies in other dynamic organ systems where epithelium-derived factors, particularly prostanoids, serve as mediators in the regulation of smooth-muscle contraction and relaxation. For example in the gastrointestinal tract, epithelium-derived prostanoids (EDPs) regulate the contraction and relaxation of the esophagus and intestines (72). In the urinary tract, EDPs play a role in the regulation of urinary smooth muscle contraction and micturition, and the relaxation of the urethral sphincter in response to increased pressure in the bladder (72, 73). In the female reproductive tract, EDPs, particularly from the endometrium, participate in a series of reproductive events through modulation of smooth muscle contractility. Analogous to the bronchi, the normal muscle tone of the uterus is under the influence of the endometrium (72, 74), and abnormalities in EDP production may lead to excessive uterine contractions. Our tractable bronchi-on-a-chip platform would translate well to these organ systems, and allow quantitative and mechanistic studies to elucidate dynamical interactions underlying disease processes such as dyspepsia, overactive bladder, and dysmenorrhea.
As any model, our experimental and mathematical analysis has a number of limitations, particularly compared to the complexity of the intact bronchial tissue. While our model juxtaposes epithelial and ASM layers at distances mimicking in vivo conditions, unlike in actual airways, the two layers are not directly mechanically coupled. As a result, ASM contraction due to an external stimulus or due to factors released by epithelial cells has no effect on the stress experienced by the epithelial layer. This reductionist design of the model was necessary to precisely control the mechanical stress on the epithelium, and examine the biochemical effects of this stress on the mechanical properties of ASM cells. We examined the predicted effects of the more direct mechanical coupling through a mathematical model, which led to a number of insights into the development and progression of bronchospasm. These predictions can be further validated in more complex airway models, including lung tissues themselves. As a proxy for ASM shortening, we measured stiffness changes in HASM cells using magnetic beads coupled to surface integrin receptors. However, while validated as a predictor of mechanical responses (see e.g. (75)), and a good method to assess acute mechanical responses of HASM over several hours, for longer time durations bead internalization by the cells can be an issue. Another consequence of using this method vs. directly measuring ASM shortening is that the time constants for ASM contraction we determined with cultured cells in a controlled system ( in Fig. 6A) can be somewhat different than those under in vivo conditions, as changes in muscle tone are expected to be generally faster than muscle shortening/lengthening. Nonetheless, measured via ASM shortening is still expected to be much smaller than the feedback time constants (). And while this may affect the time-course of the bronchospasm dynamics predicted by the model, the effect is expected to be small. Finally, despite using HASM cells from both non-asthmatic and asthmatic donors, to model epithelial behavior we only used NHBE cells. However, under certain conditions epithelial cells from asthmatic donors may respond differently to mechanical perturbations, such as slower multicellular reorganization in response to compression (29), which may have an effect on long-term bronchospasm dynamics. Despite these limitations, we believe that the described bronchospasm model can reveal important new mechanisms underlying physiological and pathological correlates of this important process.
Overall, our analysis points to the ability of sophisticated and yet tractable microfabricated organ-on-a-chip systems to yield important insights into the mechanisms of mechanochemical cell-cell interactions and their effects in physiological and pathological function of various tissues. We expect that such platforms, and the bronchial-chip platform in particular, will be a crucially important complement to the analysis performed in vivo, leading to improved disease models and new approaches to treatment of debilitating diseases.
Material and Methods
Bronchial-chip fabrication.
The upper and lower layer of the microfluidic device were fabricated using standard soft lithography techniques by casting a flexible and optically transparent polydimethylsiloxane (PDMS) polymer on a mold and cross-linking at 80°C for 2 hours. The mold was fabricated using optical lithography with an epoxy-based negative photoresist (SU8) that yields thick and robust structures. The two PDMS layers each had compartments with 5 mm width, 20 mm length, and 300 μm height, and around 100 support pillars with 200 μm diameter distributed across the compartments. The two compartments were separated by an optically-clear polycarbonate membrane (Sterlitech) with 5 μm diameter holes, allowing cells on both sides of the membranes to chemically interact. To construct the complete chip, first a PDMS layer and the polycarbonate membrane were exposed to an oxygen plasma (30 W, 700 mTorr, 30 sec) to chemically activate their surfaces. Immediately after the plasma treatment, the PDMS surface was silanized using 5 μl of an APTES solution (1% 3-aminopropyltriethoxysilane in water) and the polycarbonate membrane was allowed to bond to it. After 2 hours of bonding at room temperature, the process was repeated to bond the second PDMS compartment to the other side of the polycarbonate membrane. The APTES silanization allowed a strong permanent bond between the layers that we were not able to pull apart manually. The device was further bonded to a glass coverslip for support using an oxygen plasma. After the chip was sterilized under intense UV light overnight, the two compartments were coated with collagen type I and subsequently seeded with primary normal human bronchial epithelial (NHBE) cells on the top compartment. Following the maturation of the NHBE cells in an air-liquid interface (ALI), primary human airway smooth muscle (HASM) cells were seeded on the bottom compartment.
Cell culture.
All human cells and tissues were obtained from National Disease Research Interchange (NDRI) or The International Institute for the Advancement of Medicine (IIAM) in accordance with their regulations and complying with all relevant ethical regulations. The male and female donors who were de-identified and without chronic illness or medication use, provided lungs from which trachealis and first generation bronchi was dissected and HASM cells isolated. The isolation, characterization and cultivation of HASM cells were described in Panettieri et al. (76). In accordance with the Rutgers Institutional Review Board and NIH, the HASM cells were derived from de-identified donors and as such these studies were not considered human subject research and were exempt from obtaining consent. NHBE cells were grown in collagen-coated flasks (GIBCO) using serum-free BEGM Bronchial Epithelial Cell Growth Medium with BulletKit Supplements (Lonza) and subsequently trypsinized and seeded onto porous polycarbonate membranes (either inside microfluidic devices or on transwell inserts) at 100,000 cells/cm2. The polycarbonate membranes were coated with collagen type I (30 μg/ml; GIBCO) at 37°C for 18 h before cell seeding. After NHBE cells reached confluence (within 3–5 days), the apical medium was removed and the basal medium was replaced with differentiation medium (PneumaCult-ALI Medium and Supplements, StemCell Technologies). The NHBE cells were maintained at the ALI for 2–3 weeks, until they differentiated into a pseudostratified layer. Excess mucus on the air side was washed away with warm PBS weekly. After NHBE differentiation in the microfluidic chips, primary HASM cells were seeded into the bottom compartment. Approximately 18 h before the live-cell micromechanical experiments using MTC, the culture medium of HASM cells was changed to serum-free minimal media (1:1 vol/vol mixture of DMEM Dulbecco’s modified Eagle’s medium and Ham’s F-12 medium, supplemented with insulin (10 mg/ml), transferrin (5.5 mg/ml), and selenium (6.7 mg/ml)), and cells were maintained at 37°C in humidified air containing 5% CO2. The overnight media was then washed and replaced with fresh minimal media before the experiments. This minimal media procedure was applied to NHBE cells as well before compressive-stress application. For experiments involving celecoxib or verteporfin (Sigma Aldrich), NHBE cells were pre-treated with the drugs before the second washing step, as the presence of celecoxib in the infranatant affected HASM mechanics (Fig. S2B–C), and verteporfin in the infranatant would have phototoxic effects on HASM cells during the live-cell micromechanical measurements. Since verteporfin is a photosensitizer, treatment of NHBE cells was always performed in the dark.
Compressive stress application.
To expose NHBE cells in an ALI to apical compressive stress in the microfluidic device, we connected the top compartment to a custom-built pressure regulator that circulates the ambient air through a sterile 0.2 μm pore filter and compresses it to a level of 30 cmH2O pressure, while the basal side of the cells remained at atmospheric pressure. The pressure regulator (Fig. S1D) consists of a micro pump (AP-2P02A, SmartProducts) and a 30 cmH2O pressure check valve (SmartProducts), and is powered by high-temperature compatible lithium batteries. The check valves were selected from a batch of valves manufactured for 0.45 psi with ±20% tolerance, when displaced water height in a tube was exactly 30 cm. To apply compressive stress to NHBE cells on the apical side in transwell inserts, we built a custom transcellular pressure setup consisting of a six-well plate with rubber plugs that have access ports connected first to sterile filters and then in parallel to the pressure regulator (Figs. 2A and S1D). To reduce evaporation during long pressure-application experiments, the wells are covered by a flexible and breathable PDMS cover. Before each experiment, the rubber plugs were autoclaved and the PDMS covers were sterilized with intense overnight UV exposure. To mimic tidal breathing we applied an oscillating pressure (3 cmH2O at 0.5 Hz), and used elastic PDMS as the material for the perforated membrane the NHBE cells were seeded on, fabricated as described previously (77).
HASM stiffness measurements.
Magnetic twisting cytometry with optical detection was used to quantitatively assess dynamic changes in cell stiffness as an indicator of single-cell contraction or relaxation of HASM cells, as previously described (48, 78). In brief, ferrimagnetic microbeads (4.5 μm in diameter) coated with synthetic Arg-Gly-Asp (RGD) containing peptide (American Peptide Company) bound to the cytoskeleton through cell surface integrin receptors were magnetized horizontally. Subsequently, a vertically aligned homogeneous magnetic field varying sinusoidally in time applied an oscillatory torque (0.75 Hz), leading to both a rotation and a pivoting displacement of the bead (Fig. S1B). Lateral bead displacements in response to the resulting oscillatory torque were detected optically (with spatial resolution of ~5 nm), and the ratio of specific torque to bead displacement was computed as the cell stiffness in units of Pa/nm. For stimulus-induced single-cell contraction or relaxation, changes in HASM stiffness were measured in real-time for the duration of 300 seconds. For each individual cell, baseline stiffness was measured during the first 60 seconds, and after stimulus application, stiffness was measured continuously for the next 240 seconds. Baseline values were calculated as the average stiffness between 10–50 seconds, and response values as the average stiffness between 150–190 seconds.
Lentiviral transduction.
Lentiviral transduction particles (Sigma Aldrich) containing empty (TRC1) or YAP shRNA (TRCN0000107266) lentiviral constructs were used as the Control or shYAP virus. Primary NHBE cells were transduced with equal titers of concentrated virus in complete growth media supplemented with 1 μg/ml polybrene (Sigma Aldrich) for 24 hours. Following transduction, cells were given 24 hours to recover before selection in 0.5 μg/ml puromycin (Sigma Aldrich) for a minimum of 6 days. Minimum effective puromycin concentration was determined using kill curves of untransduced cells (data not shown).
Immunoblotting.
Primary NHBE cells were lysed on ice with a cell scraper and radio-immunoprecipitation assay lysis buffer (Pierce) supplemented with protease inhibitor tablets (Roche) and a phosphatase inhibitor cocktail (Pierce). Extracts were incubated for 45 minutes for complete lysis and centrifuged at 10,000 RPM for 10 minutes at 4°C to pellet cell debris. Supernatant protein concentrations were quantified using a BCA Protein Assay kit (Pierce) and a spectrophotometric plate reader (BioTek). Most protein lysates were separated by running 30–100 μg of lysate on 10% Bis-Tris NuPage gels (Invitrogen) and subsequently transferred to 0.2 μm pore polyvinylidene fluoride (PVDF) membranes (BioRad). Primary antibody (Table S2) incubations were according to manufacturer’s recommendations in 0.1% Tween TBS supplemented with 5% non-fat dry milk or BSA, as recommended. Immunoreactive bands were visualized using the appropriate horseradish peroxidase-conjugated anti-IgG antibodies (Pierce). Bands were detected using enhanced chemiluminescence or prime detection reagent (GE Healthcare) whenever appropriate.
Gene expression analysis.
cDNA was generated using random hexamer primers and SuperScript II Reverse Transcriptase (Applied Biosystems). Real-time PCR was performed using TaqMan Universal PCR Master Mix, fluorogenic probes, and oligonucleotide primers. TaqMan assays were repeated in triplicate samples for each of the target genes. The 2-ΔΔCt method was used to calculate the relative fold change (RFC) of transcripts normalized to a house-keeping gene (GAPDH). Primer-probe sets for the analyzed genes are listed in Table S3.
Prostaglandin detection.
Media from the basal compartments (infranatant) were collected from control samples or after application of 30 cmH2O compressive stress on NHBE cells, and were assayed for the presence of PGE2 using a commercial EIA kit (DetectX Prostaglandin E2 High Sensitivity Immunoassay Kit, Arbor Assays, Ann Arbor, MI) following the manufacturer’s instructions. The absorbance in the samples was measured at 450 nm with a microplate reader (Molecular Devices, Sunnyvale, CA). The concentration of PGE2 was calculated from a standard curve derived using calibrated prostaglandin standards. All samples were assayed in triplicate.
Statistics.
Pairwise comparisons of normalized data was performed using the t-test (Fig. 4A, Fig. S3C), and of non-normalized data using the Wilcoxon rank-sum test (Fig. 5D–E), with significance based on *P<0.05, **P<0.01, and ***P<0.001. To adjust the P values when multiple comparisons were performed, the Benjamini-Hochberg procedure (79) was used for exploratory screening with small sample sizes (Fig. 3A–B, Fig. 4B–D, Fig. S2A), and one-way ANOVA followed by Bonferroni’s post-hoc test for confirmatory analysis with large sample sizes (Fig. 2C–D, Fig. 5C, Fig. S2C, Fig. S3A–B, Fig. S3F). Adjustments to the P values took into account any additional comparisons performed in the supplementary data. All statistical tests performed were two-tailed.
Airway ODE model.
The airway ODE model described below was solved with the ode23s solver of Matlab. Preliminary studies and bifurcation analysis were performed with the Oscill8 Dynamical Systems Toolset. Time constants used in the model were fitted to experimental data (Figs. 3 and 5) using a nonlinear least-squares solver (lsqcurvefit), and confidence intervals of the fits were determined through statistical bootstrapping (bootci) in Matlab. To model the system in Fig. 6A we represented the ASM contraction level by , the external stimulus level by , and the levels for the epithelium-derived spasmogens and relaxants as and , respectively. Basal expression rates for spasmogens and relaxants were ignored for simplicity, as changes in ASM contraction were assumed to be relative to the basal tone. The system then was modeled by the following set of ODE equations.
The time scales describing the dynamics of spasmogens and relaxants are represented by and the time scale for contraction by , where . Maximum expression levels are and ASM responsiveness is represented by . Dependence of spasmogen and relaxant production on contraction, and dependence of contraction on stimuli are approximated with Hill functions:
The parameters are chosen to observe effects of the feedback within a maximum stimulus () and a maximum contraction () of order 1. However, we note that compressive stress on NHBE cells at levels observed during severe bronchospasm is sufficient to cause maximum contraction in HASM cells (Fig. 5E), making the range of behaviors predicted by the model in Fig. 6D plausible. We set , , based on values obtained from experiments (Figs. 6C and S4), values to , and values to . Based on typical response curves (see, e.g., (80–82)) we approximate the Hill coefficients as and . The linear stimulus profile (Fig. 6D–F) was chosen as a proxy for tracing a hysteresis loop under equilibrium conditions. However, we note that the general conclusions of the model are independent of the stimulus shape (Fig. S5). In order to model the effects of breathing, namely the cyclic stretch experienced by ASM that is known to reduce its responsiveness (25–28), we reduced the value of by a factor , a function of cyclic stress () and bronchospasm severity (namely ). Based on experimental data from human airways (25), has a value range 0.91 – 0.94 for tidal breathing ( equivalent to strain in relaxed airways), and 0.10 – 0.62 for deep inspiration ( equivalent to strain in relaxed airways, corresponding to full lung inflation) (25). The exact values of within the given ranges depend on the contraction state of the ASM (), with the lower values corresponding to minimal bronchospasm and the higher values corresponding to maximum bronchoconstriction (close to full closure of airway). Fitting to experimental data (25), we obtain for tidal breathing ( fitted to means), and for deep inspiration ( fitted to means). Our model results incorporating the effects of cyclic stretch (Fig. S6D) agree well with prior studies that show tidal breathing has a minimal effect on bronchospasm, while deep inspiration can have a strong broncholytic effect unless bronchospasm is severe (25–28).
Supplementary Material
Acknowledgements
This work was supported by National Institutes of Health grants U01 CA155758 (A.L), U54 CA209992 (A.L), R01 HL107361 (S.S.A), and P01 HL114471 (R.A.P., S.B.L. and S.S.A.). O.K. was a recipient of the American Heart Association Postdoctoral Fellowship (13POST17140090). This work was also supported by a grant from the American Asthma Foundation (A.L. and S.S.A). S.S.A. was also supported by Discovery Award and Catalyst Award from the Johns Hopkins University.
Footnotes
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information. Source data for the figures in this study are available in figshare with the identifier doi:10.6084/m9.figshare.7639898 (ref. 83).
Competing Interests
O.K, S.S.A., and A.L. have a pending patent related to the work in this manuscript. The remaining authors declare no competing interests.
References
- 1.Bates J, editor. Toward a nonlinear network theory of complex disease International Conference on Complex Systems; Boston; 2006. [Google Scholar]
- 2.Venegas JG, Winkler T, Musch G, Melo MFV, Layfield D, Tgavalekos N, et al. Self-organized patchiness in asthma as a prelude to catastrophic shifts. Nature 2005;434(7034):777–82. [DOI] [PubMed] [Google Scholar]
- 3.Winkler T, Venegas JG. Self-organized patterns of airway narrowing. Journal of Applied Physiology 2011;110(5):1482–6. [DOI] [PubMed] [Google Scholar]
- 4.Suki B, Frey U. Temporal dynamics of recurrent airway symptoms and cellular random walk. Journal of Applied Physiology 2003;95(5):2122–7. [DOI] [PubMed] [Google Scholar]
- 5.Mauroy B, Filoche M, Weibel E, Sapoval B. An optimal bronchial tree may be dangerous. Nature 2004;427(6975):633–6. [DOI] [PubMed] [Google Scholar]
- 6.Alam R, Gorska MM. Mitogen-activated protein kinase signalling and ERK1/2 bistability in asthma. Clinical & Experimental Allergy 2011;41(2):149–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lambert RK, Wilson TA, Hyatt RE, Rodarte JR. A computational model for expiratory flow. Journal of Applied Physiology 1982;52(1):44–56. [DOI] [PubMed] [Google Scholar]
- 8.Anafi RC, Wilson TA. Airway stability and heterogeneity in the constricted lung. Journal of Applied Physiology 2001;91(3):1185–92. [DOI] [PubMed] [Google Scholar]
- 9.Donovan GM, Sneyd J, Tawhai MH. The importance of synergy between deep inspirations and fluidization in reversing airway closure. PLoS One 2012;7(11):e48552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber HL, Koessler KK. The pathology of bronchial asthma. Archives of Internal Medicine 1922;30(6):689–760. [Google Scholar]
- 11.James AL, Paré PD, Hogg JC. The mechanics of airway narrowing in asthma. American Review of Respiratory Disease 1989;139(1):242–6. [DOI] [PubMed] [Google Scholar]
- 12.Wiggs BR, Hrousis CA, Drazen JM, Kamm RD. On the mechanism of mucosal folding in normal and asthmatic airways. Journal of Applied Physiology 1997;83(6):1814–21. [DOI] [PubMed] [Google Scholar]
- 13.Okada SF, Ribeiro CM, Sesma JI, Seminario-Vidal L, Abdullah LH, van Heusden C, et al. Inflammation promotes airway epithelial ATP release via calcium-dependent vesicular pathways. American journal of respiratory cell and molecular biology 2013;49(5):814–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Savla U, Sporn PH, Waters CM. Cyclic stretch of airway epithelium inhibits prostanoid synthesis. American Journal of Physiology-Lung Cellular and Molecular Physiology 1997;273(5):L1013–L9. [DOI] [PubMed] [Google Scholar]
- 15.Arold SP, Malavia N, George SC. Mechanical compression attenuates normal human bronchial epithelial wound healing. Respiratory research 2009;10(1):9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Copland IB, Reynaud D, Pace-Asciak C, Post M. Mechanotransduction of stretch-induced prostanoid release by fetal lung epithelial cells. American Journal of Physiology-Lung Cellular and Molecular Physiology 2006;291(3):L487–L95. [DOI] [PubMed] [Google Scholar]
- 17.Burnstock G Purine-mediated signalling in pain and visceral perception. Trends in pharmacological sciences 2001;22(4):182–8. [DOI] [PubMed] [Google Scholar]
- 18.Ferguson D, Kennedy I, Burton T. ATP is released from rabbit urinary bladder epithelial cells by hydrostatic pressure changes–possible sensory mechanism? The Journal of physiology 1997;505(2):503–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cuthbert M Effect on airways resistance of prostaglandin E1 given by aerosol to healthy and asthmatic volunteers. British medical journal 1969;4(5685):723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sweatman W, Collier H. Effects of prostaglandins on human bronchial muscle 1968. [DOI] [PubMed]
- 21.Mathé AA, Hedqvist P. Effect of Prostaglandins F2α and E2 on Airway Conductance in Healthy Subjects and Asthmatic Patients 1–3. American Review of Respiratory Disease 1975;111(3):313–20. [DOI] [PubMed] [Google Scholar]
- 22.Hanna C, Bach M, Pare P, Schellenberg R. Slow-reacting substances (leukotrienes) contract human airway and pulmonary vascular smooth muscle in vitro. Nature 1981;290:343–4 [DOI] [PubMed] [Google Scholar]
- 23.Xiong W, Ferrell JE. A positive-feedback-based bistable ‘memory module’that governs a cell fate decision. Nature 2003;426(6965):460–5. [DOI] [PubMed] [Google Scholar]
- 24.Tian X-J, Zhang X-P, Liu F, Wang W. Interlinking positive and negative feedback loops creates a tunable motif in gene regulatory networks. Physical Review E 2009;80(1):011926. [DOI] [PubMed] [Google Scholar]
- 25.Lavoie TL, Krishnan R, Siegel HR, Maston ED, Fredberg JJ, Solway J, et al. Dilatation of the constricted human airway by tidal expansion of lung parenchyma. American journal of respiratory and critical care medicine 2012;186(3):225–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.LaPrad AS, Szabo TL, Suki B, Lutchen KR. Tidal stretches do not modulate responsiveness of intact airways in vitro. Journal of Applied Physiology 2010;109(2):295–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.LaPrad AS, West AR, Noble PB, Lutchen KR, Mitchell HW. Maintenance of airway caliber in isolated airways by deep inspiration and tidal strains. Journal of Applied Physiology 2008;105(2):479–85. [DOI] [PubMed] [Google Scholar]
- 28.Noble PB, Jones RL, Needi ET, Cairncross A, Mitchell HW, James AL, et al. Responsiveness of the human airway in vitro during deep inspiration and tidal oscillation. Journal of Applied Physiology 2011;110(6):1510–8. [DOI] [PubMed] [Google Scholar]
- 29.Park J-A, Kim JH, Bi D, Mitchel JA, Qazvini NT, Tantisira K, et al. Unjamming and cell shape in the asthmatic airway epithelium. Nature materials 2015;14(10):1040–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Noble P, Sharma A, McFawn P, Mitchell H. Elastic properties of the bronchial mucosa: epithelial unfolding and stretch in response to airway inflation. Journal of Applied Physiology 2005;99(6):2061–6. [DOI] [PubMed] [Google Scholar]
- 31.Huh D, Matthews BD, Mammoto A, Montoya-Zavala M, Hsin HY, Ingber DE. Reconstituting organ-level lung functions on a chip. Science 2010;328(5986):1662–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.West AR, Zaman N, Cole DJ, Walker MJ, Legant WR, Boudou T, et al. Development and characterization of a 3D multicell microtissue culture model of airway smooth muscle. American Journal of Physiology-Lung Cellular and Molecular Physiology 2012;304(1):L4–L16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benam KH, Villenave R, Lucchesi C, Varone A, Hubeau C, Lee H-H, et al. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nature Methods 2016;13(2):151. [DOI] [PubMed] [Google Scholar]
- 34.Noble PB, McFawn PK, Mitchell HW. Responsiveness of the isolated airway during simulated deep inspirations: effect of airway smooth muscle stiffness and strain. Journal of Applied Physiology 2007;103(3):787–95. [DOI] [PubMed] [Google Scholar]
- 35.Vanhoutte PM. Epithelium-derived relaxing factor (s) and bronchial reactivity. Journal of Allergy and clinical Immunology 1989;83(5):855–61. [DOI] [PubMed] [Google Scholar]
- 36.Park J-A, Tschumperlin DJ. Chronic intermittent mechanical stress increases MUC5AC protein expression. American journal of respiratory cell and molecular biology 2009;41(4):459–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tschumperlin DJ, Dai G, Maly IV, Kikuchi T, Laiho LH, McVittie AK, et al. Mechanotransduction through growth-factor shedding into the extracellular space. Nature 2004;429(6987):83–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science 1993;260(5111):1124–7. [DOI] [PubMed] [Google Scholar]
- 39.Wang N, Tolic-Nørrelykke IM, Chen J, Mijailovich SM, Butler JP, Fredberg JJ, et al. Cell prestress. I. Stiffness and prestress are closely associated in adherent contractile cells. American Journal of Physiology-Cell Physiology 2002;282(3):C606–C16. [DOI] [PubMed] [Google Scholar]
- 40.An SS, Laudadio RE, Lai J, Rogers RA, Fredberg JJ. Stiffness changes in cultured airway smooth muscle cells. American Journal of Physiology-Cell Physiology 2002;283(3):C792–C801. [DOI] [PubMed] [Google Scholar]
- 41.An SS, Fabry B, Trepat X, Wang N, Fredberg JJ. Do biophysical properties of the airway smooth muscle in culture predict airway hyperresponsiveness? American journal of respiratory cell and molecular biology 2006;35(1):55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ressler B, Lee RT, Randell SH, Drazen JM, Kamm RD. Molecular responses of rat tracheal epithelial cells to transmembrane pressure. American Journal of Physiology-Lung Cellular and Molecular Physiology 2000;278(6):L1264–L72. [DOI] [PubMed] [Google Scholar]
- 43.Swartz M, Tschumperlin DJ, Kamm R, Drazen J. Mechanical stress is communicated between different cell types to elicit matrix remodeling. Proceedings of the National Academy of Sciences 2001;98(11):6180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yoon A-R, Stasinopoulos I, Kim JH, Yong HM, Kilic O, Wirtz D, et al. COX-2 dependent regulation of mechanotransduction in human breast cancer cells. Cancer biology & therapy 2015;16(3):430–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obermajer N, Muthuswamy R, Lesnock J, Edwards RP, Kalinski P. Positive feedback between PGE2 and COX2 redirects the differentiation of human dendritic cells toward stable myeloid-derived suppressor cells. Blood 2011;118(20):5498–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M, et al. Role of YAP/TAZ in mechanotransduction. Nature 2011;474(7350):179–83. [DOI] [PubMed] [Google Scholar]
- 47.Halder G, Dupont S, Piccolo S. Transduction of mechanical and cytoskeletal cues by YAP and TAZ. Nature Reviews Molecular Cell Biology 2012;13(9):591–600. [DOI] [PubMed] [Google Scholar]
- 48.An SS, Mitzner W, Tang W-Y, Ahn K, Yoon A-R, Huang J, et al. An inflammation-independent contraction mechanophenotype of airway smooth muscle in asthma. Journal of Allergy and clinical Immunology 2016;138(1):294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tilley SL, Hartney JM, Erikson CJ, Jania C, Nguyen M, Stock J, et al. Receptors and pathways mediating the effects of prostaglandin E2 on airway tone. American Journal of Physiology-Lung Cellular and Molecular Physiology 2003;284(4):L599–L606. [DOI] [PubMed] [Google Scholar]
- 50.O’Sullivan MJ, Gabriel E, Panariti A, Park CY, Ijpma G, Fredberg JJ, et al. Epithelial Cells Induce a Cyclo-Oxygenase-1–Dependent Endogenous Reduction in Airway Smooth Muscle Contractile Phenotype. American journal of respiratory cell and molecular biology 2017;57(6):683–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pfeuty B, Kaneko K. The combination of positive and negative feedback loops confers exquisite flexibility to biochemical switches. Physical biology 2009;6(4):046013. [DOI] [PubMed] [Google Scholar]
- 52.Wenzel SE, Fahy JV, Irvin C, Peters SP, Spector S, Szefler SJ, et al. Proceedings of the ATS workshop on refractory asthma. American journal of respiratory and critical care medicine 2000;162(6):2341–51. [DOI] [PubMed] [Google Scholar]
- 53.Bateman E, Hurd S, Barnes P, Bousquet J, Drazen J, FitzGerald M, et al. Global strategy for asthma management and prevention: GINA executive summary. European Respiratory Journal 2008;31(1):143–78. [DOI] [PubMed] [Google Scholar]
- 54.Zhou J, Alvarez-Elizondo MB, Botvinick E, George SC. Local small airway epithelial injury induces global smooth muscle contraction and airway constriction. Journal of Applied Physiology 2012;112(4):627–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhou J, Alvarez-Elizondo MB, Botvinick E, George SC. Adenosine A1 and Prostaglandin E Receptor 3 Receptors Mediate Global Airway Contraction after Local Epithelial Injury. American journal of respiratory cell and molecular biology 2013;48(3):299–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Orehek J, Douglas JS, Bouhuys A. Contractile responses of the guinea-pig trachea in vitro: modification by prostaglandin synthesis-inhibiting drugs. Journal of Pharmacology and Experimental Therapeutics 1975;194(3):554–64. [PubMed] [Google Scholar]
- 57.Gao Y, Vanhoutte PM. Responsiveness of the guinea pig trachea to stretch: role of the epithelium and cyclooxygenase products. Journal of Applied Physiology 1993;75(5):2112–6. [DOI] [PubMed] [Google Scholar]
- 58.Kullmann FA, Shah MA, Birder LA, de Groat WC. Functional TRP and ASIC-like channels in cultured urothelial cells from the rat. American Journal of Physiology-Renal Physiology 2009;296(4):F892–F901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fronius M, Clauss WG. Mechano-sensitivity of ENaC: may the (shear) force be with you. Pflügers Archiv-European Journal of Physiology 2008;455(5):775–85. [DOI] [PubMed] [Google Scholar]
- 60.Zhang WK, Wang D, Duan Y, Loy MM, Chan HC, Huang P. Mechanosensitive gating of CFTR. Nature cell biology 2010;12(5):507–12. [DOI] [PubMed] [Google Scholar]
- 61.Coste B, Mathur J, Schmidt M, Earley TJ, Ranade S, Petrus MJ, et al. Piezo1 and Piezo2 are essential components of distinct mechanically activated cation channels. Science 2010;330(6000):55–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Storch U, y Schnitzler MM, Gudermann T. G protein-mediated stretch reception. American Journal of Physiology-Heart and Circulatory Physiology 2012;302(6):H1241–H9. [DOI] [PubMed] [Google Scholar]
- 63.Seminario-Vidal L, Okada SF, Sesma JI, Kreda SM, van Heusden CA, Zhu Y, et al. Rho signaling regulates pannexin 1-mediated ATP release from airway epithelia. Journal of Biological Chemistry 2011;286(30):26277–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Genetos DC, Geist DJ, Liu D, Donahue HJ, Duncan RL. Fluid Shear-Induced ATP Secretion Mediates Prostaglandin Release in MC3T3-E1 Osteoblasts. Journal of Bone and Mineral Research 2005;20(1):41–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chand N, Eyre P. Atypical (relaxant) response to histamine in cat bronchus. Agents and actions 1977;7(2):183–90. [DOI] [PubMed] [Google Scholar]
- 66.Chand N, Eyre P. Histamine relaxes constricted trachea and bronchi of horse. Veterinary Research Communications 1977;1(1):85–90. [Google Scholar]
- 67.Chand N, DeRoth L. Actions of histamine and other substances on the airway smooth muscle of swine (in vitro). Veterinary Research Communications 1978;2(1):151–5. [Google Scholar]
- 68.Stickland MK, Rowe BH, Spooner CH, Vandermeer B, Dryden DM. Effect of warm-up exercise on exercise-induced bronchoconstriction. Med Sci Sports Exerc 2012;44(3):383–91. [DOI] [PubMed] [Google Scholar]
- 69.Szczeklik A, Stevenson DD. Aspirin-induced asthma: advances in pathogenesis, diagnosis, and management. Journal of Allergy and clinical Immunology 2003;111(5):913–21. [DOI] [PubMed] [Google Scholar]
- 70.Picado C Aspirin-intolerant asthma: role of cyclo-oxygenase enzymes. Allergy 2002;57(s72):58–60. [DOI] [PubMed] [Google Scholar]
- 71.Hanania NA, Dickey BF, Bond RA. Clinical implications of the intrinsic efficacy of beta-adrenoceptor drugs in asthma: full, partial and inverse agonism. Current opinion in pulmonary medicine 2010;16(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruan YC, Zhou W, Chan HC. Regulation of smooth muscle contraction by the epithelium: role of prostaglandins. Physiology 2011;26(3):156–70. [DOI] [PubMed] [Google Scholar]
- 73.Bultitude M, HILLs N, Shuttleworth K. Clinical and experimental studies on the action of prostaglandins and their synthesis inhibitors on detrusor muscle in vitro and in vivo. BJU International 1976;48(7):631–7. [DOI] [PubMed] [Google Scholar]
- 74.Lundström V, Gréen K, Svanborg K. Endogenous prostaglandins in dysmenorrhea and the effect of prostaglandin synthetase inhibitors (PGSI) on uterine contractility. Acta Obstetricia et Gynecologica Scandinavica 1979;58(S87):51–6. [DOI] [PubMed] [Google Scholar]
- 75.Trepat X, Deng L, An SS, Navajas D, Tschumperlin DJ, Gerthoffer WT, et al. Universal physical responses to stretch in the living cell. Nature 2007;447(7144):592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Panettieri R, Murray R, DePalo L, Yadvish P, Kotlikoff M. A human airway smooth muscle cell line that retains physiological responsiveness. American Journal of Physiology-Cell Physiology 1989;256(2):C329–C35. [DOI] [PubMed] [Google Scholar]
- 77.Kilic O, Pamies D, Lavell E, Schiapparelli P, Feng Y, Hartung T, et al. Brain-on-a-chip model enables analysis of human neuronal differentiation and chemotaxis. Lab on a Chip 2016;16(21):4152–62. [DOI] [PubMed] [Google Scholar]
- 78.Fabry B, Maksym GN, Butler JP, Glogauer M, Navajas D, Fredberg JJ. Scaling the microrheology of living cells. Physical review letters 2001;87(14):148102. [DOI] [PubMed] [Google Scholar]
- 79.Benjamini Y, Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the royal statistical society Series B (Methodological) 1995:289–300.
- 80.Van Koppen C, De Miranda J, Beld AJ, Van Herwaarden C, Lammers J, Van Ginneken C. Beta adrenoceptor binding and induced relaxation in airway smooth muscle from patients with chronic airflow obstruction. Thorax 1989;44(1):28–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Aizawa H, Miyazaki N, Shigematsu N, Tomooka M. A possible role of airway epithelium in modulating hyperresponsiveness. British journal of pharmacology 1988;93(1):139–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Healy ZR, Lee NH, Gao X, Goldring MB, Talalay P, Kensler TW, et al. Divergent responses of chondrocytes and endothelial cells to shear stress: cross-talk among COX-2, the phase 2 response, and apoptosis. Proceedings of the National Academy of Sciences of the United States of America 2005;102(39):14010–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kilic O et al. Dataset for A microphysiological model of the bronchial airways reveals the interplay of mechanical and biochemical signals in bronchospasm. figshare 10.6084/m9.figshare.7639898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.