

Abstract
Balanced multi‐ion channel‐blocking drugs have low torsade risk because they block inward currents. The Comprehensive In Vitro Proarrhythmia Assay (CiPA) initiative proposes to use an in silico cardiomyocyte model to determine the presence of balanced block, and absence of heart rate corrected J‐Tpeak (J‐Tpeakc) prolongation would be expected for balanced blockers. This study included three balanced blockers in a 10‐subject‐per‐drug parallel design; lopinavir/ritonavir and verapamil met the primary end point of ΔΔJ‐Tpeakc upper bound < 10 ms, whereas ranolazine did not (upper bounds of 8.8, 6.1, and 12.0 ms, respectively). Chloroquine, a predominant blocker of the potassium channel encoded by the ether‐à‐go‐go related gene (hERG), prolonged ΔΔQTc and ΔΔJ‐Tpeakc by ≥ 10 ms. In a separate crossover design, diltiazem (calcium block) did not shorten dofetilide‐induced ΔQTc prolongation, but shortened ΔJ‐Tpeakc and prolonged ΔTpeak‐Tend. Absence of J‐Tpeakc prolongation seems consistent with balanced block; however, small sample size (10 subjects) may be insufficient to characterize concentration‐response in some cases.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑Drug‐induced hERG block and QTc prolongation are associated with torsade de pointes. However, there are QTc prolonging drugs that have low torsade risk because they have balanced block of inward currents (late sodium or L‐type calcium) in addition to hERG.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑Can concentration‐response analysis of the J‐Tpeakc interval be used in small sample size studies to confirm that balanced ion channel‐blocking drugs do not prolong J‐Tpeakc?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑A lack of J‐Tpeakc prolongation can confirm balanced ion channel block; however, small sample size studies (10 subjects per drug) may be insufficient to characterize concentration‐response relationships in some cases.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑A comprehensive nonclinical CiPA assessment combined with assessment of the J‐Tpeakc interval may be able to inform the intensity of ECG monitoring in phase III trials and drug labeling for mild‐to‐moderate QTc prolonging drugs.
Current regulatory guidelines from the International Council on Harmonisation (ICH) describe nonclinical (ICH S7B1) and clinical strategies (ICH E142) to assess the potential of a drug to prolong cardiac repolarization as well as its clinical proarrhythmic potential. No drugs with unexpected QT prolongation or torsade de pointes (torsade) risk have reached the market after the adoption of ICH S7B and E14 guidelines. However, there are drugs that block the potassium channel encoded by the human ether‐à‐go‐go related gene (hERG) and prolong the heart rate corrected QT (QTc) interval but have low torsade risk (e.g., ranolazine, verapamil, and amiodarone) because they block other inward currents (i.e., late sodium and/or L‐type calcium currents). Block of these inward currents is anti‐arrhythmic by preventing early afterdepolarizations.3, 4, 5, 6, 7, 8, 9 ICH S7B and E14 discuss the importance of the drug's effects on other ion channels in addition to hERG and encourage interested parties to develop models for directly assessing drug‐induced proarrhythmia of QTc prolonging drugs.1, 2 In line with this, the goal of the Comprehensive In Vitro Proarrhythmia Assay (CiPA) is to perform a mechanistic assessment of a drug's torsade risk.10
Under CiPA, the torsade risk of a drug will be determined by an in silico model that integrates the drug's effects on multiple ion channel currents of the human ventricular myocyte. The CiPA in silico model outputs a Torsade Metric Score called qNet,11 which reflects the balance of inward and outward currents throughout the action potential. Multichannel blocking drugs, i.e., drugs that block hERG as well as late sodium and/or L‐type calcium currents, will be considered balanced ion channel blockers (referred to as “balanced blockers”) if qNet predicts the drug to be low risk, and “predominant hERG blockers” otherwise (i.e., drugs that only block hERG or multichannel blockers predicted to be intermediate or high risk). For a balanced blocker, the CiPA initiative proposes to use electrocardiogram (ECG) analysis in early phase I clinical trials to determine if there is evidence of unexpected ion channel effects in humans compared with the preclinical data.12 As described in a review of the CiPA initiative that included the rationale and design of this clinical study,13 prior work identified that the heart rate corrected J‐Tpeak (J‐Tpeakc) interval could be used to differentiate predominant hERG blockers from balanced blockers. Specifically, predominant hERG blockers prolong QTc and J‐Tpeakc, whereas balanced blockers prolong QTc with no J‐Tpeakc prolongation.14, 15, 16, 17, 18
This study was designed in two parts with separate objectives.13 The primary objective of part 1 was to assess whether balanced ion channel‐blocking drugs (ranolazine, verapamil, and lopinavir/ritonavir) do not cause J‐Tpeakc prolongation in a small sample size (10 subjects per drug and placebo) phase‐I type parallel‐study design. One predominant hERG‐blocking drug (chloroquine) was included as a control. Balanced block was determined based on ion channel data available during study design,13, 19 and not based on qNet. Thus, drug categories may be different if based on data acquired with current CiPA protocols20 and qNet. Prolongation of J‐Tpeakc and QTc was defined using a concentration‐response model as an upper bound of the predicted effect at maximum drug plasma concentration (Cmax) ≥ 10 ms. The primary objective of part 2 (crossover study) was to test the hypothesis that diltiazem (calcium channel block) can reduce the QTc prolongation by dofetilide (predominant hERG block) by shortening J‐Tpeakc. Sample size and J‐Tpeakc threshold were determined based on analyses from prior clinical studies.14, 15, 17, 18 and by resampling the data using similar methodology of Ferber et al.21(see online supplement in Vicente et al.13 for details).
RESULTS
Sixty healthy subjects (22 women) with a mean ± SD age of 31.7 ± 8.7 years and body mass index of 25.8 ± 2.7 kg/m2 were randomized with 50 subjects assigned to part 1 (parallel study) and 10 subjects assigned to part 2 (crossover study). No serious or unexpected treatment‐related adverse events were observed. Two subjects were discontinued after receiving the second dose of diltiazem in part 2, and one subject withdrew consent prior to the second day of treatment in part 1. All other subjects completed the study. Supplementary Material S1 includes full baseline characteristics, adverse events frequencies by treatment, and Consolidated Standards of Reporting Trials (CONSORT) flow diagram, as well as detailed results for multiple ECG biomarkers, as described in the statistical analysis plan.13 The clinical data and raw annotated ECG waveforms are publicly available at the PhysioNet22 website (<https://physionet.org/physiobank/database/ecgcipa/>).
Part 1: Parallel study
Administered drugs, their dosing times, and their pharmacokinetic (PK) time profiles are shown in Figure 1. The observed geometric mean Cmax after first dose on days 1 and 3 are shown in Table 1. As planned, subjects were exposed to a lower drug concentration on day 1 and a higher drug concentration on day 3.
Figure 1.
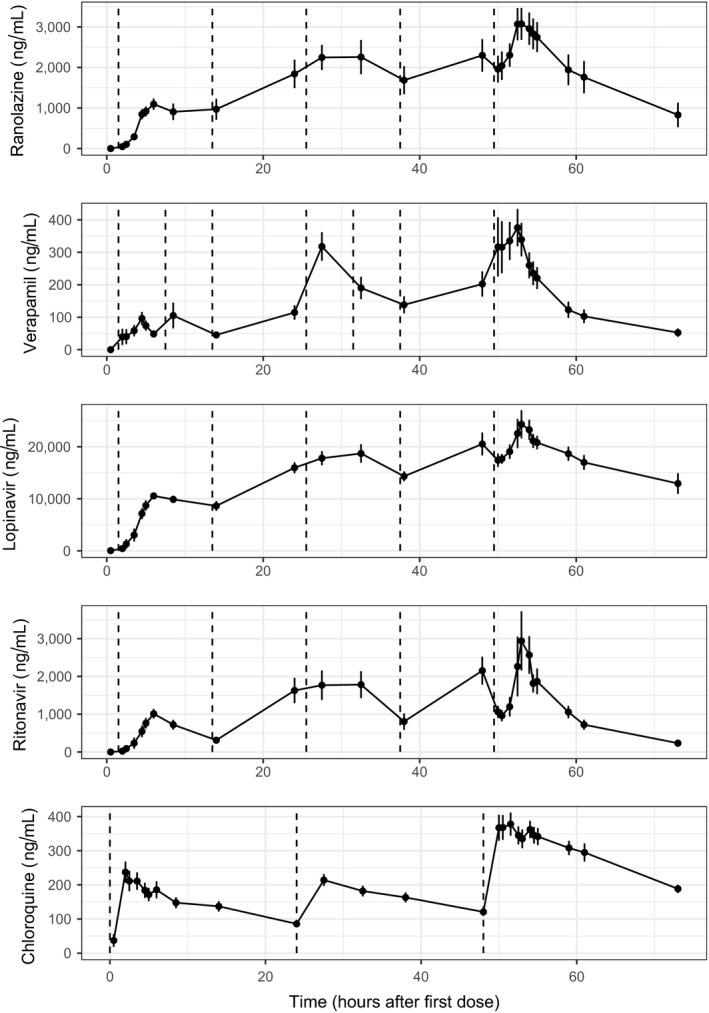
Part 1 pharmacokinetic (PK) time profiles. Mean (dots) and standard error (error bars) of PK plasma concentration profiles for ranolazine, verapamil, lopinavir, ritonavir, and chloroquine per timepoint after first dose on day 1. Dashed vertical lines show the time of active treatment doses for ranolazine (1,500 mg after breakfast and in the evening), verapamil (120 mg immediate release after breakfast and in the afternoon, 240 mg extended release in the evening), lopinavir/ritonavir (lopinavir 800 mg/ritonavir 200 mg after breakfast and in the evening), and chloroquine (1,000 mg day 1, 500 mg day 2, and 1,000 mg day 3, all before breakfast). Oral placebo was administered at dosing timepoints that had no active treatment dosing as well as throughout all the dosing timepoints in the placebo treatment arm (not shown).
Table 1.
Part 1 drug‐induced ECG predicted changes
| Drug | Day | Cmax, ng/mL | ΔΔQTc, ms | ΔΔJ‐Tpeakc, ms | ΔΔTpeak‐Tend, ms | ΔΔPR, ms | ΔΔQRS, ms |
|---|---|---|---|---|---|---|---|
| Ranolazine | 1 | 1,191.2 | 14.1 (7.5 to 20.8) | 2.2 (−4.3 to 8.7) | 11.9 (5.9 to 17.9) | 0.7 (−2.5 to 3.8) | −0.5 (−3.3 to 2.3) |
| 3 | 3,046.3 | 28.2 (18.3 to 38.2) | 1.2 (−9.5 to 12.0) | 25.7 (14.7 to 36.7) | 3.1 (−0.8 to 6.9) | −0.2 (−3.5 to 3.0) | |
| Verapamil | 1 | 101.0 | 8.6 (4.4 to 12.9) | 3.6 (−2.4 to 9.7) | 4.4 (0.6 to 8.2) | 13.2 (7.2 to 19.2) | 0.3 (−2.0 to 2.7) |
| 3 | 398.4 | 13.9 (10.0 to 17.8) | −2.6 (−11.4 to 6.1) | 14.9 (9.1 to 20.6) | 52.2 (27.9 to 76.6) b | 1.8 (−1.7 to 5.4) | |
| Lopinavir | 1 | 10,734.2 | a | 1.2 (−5.4 to 7.8) | a | 14.8 (10.3 to 19.4) | a |
| 3 | 24,351.5 | a | −2.9 (−14.6 to 8.8) | a | 33.5 (22.7 to 44.4) b | a | |
| Chloroquine | 1 | 228.8 | 30.7 (22.6 to 38.9) | a | 13.1 (8.4 to 17.9) | 9.3 (2.8 to 15.8) | 5.5 (3.0 to 8.0) |
| 3 | 404.9 | 50.5 (38.5 to 62.5) | a | 24.9 (17.2 to 32.7) | 14.8 (4.7 to 24.9) | 9.9 (6.1 to 13.6) |
Cmax, peak plasma concentration; ECG, electrocardiogram; J‐Tpeakc, heart rate corrected J‐Tpeak; QTc, heart rate corrected QT. Drug‐induced placebo‐corrected changes from baseline for QTc, J‐Tpeakc, Tpeak‐Tend, PR, and QRS intervals at Cmax of days 1 and 3 for each treatment in part 1.
aValues not reported because poor fit of concentration‐response linear models did not allow for reliable predictions.
bGoodness‐of‐fit plots suggest linear models may overestimate ΔΔPR effects around Cmax (see text and Supplementary Material S1 for comparison with maximum effect (Emax) and sigmoid models).
Figure 2 shows time profiles of placebo‐corrected changes from baseline (ΔΔ) for ΔΔQTc and ΔΔJ‐Tpeakc for each treatment and timepoint (see Supplementary Material S1 for ΔΔTpeak‐Tend plots). Table 1 shows predictions for all ECG biomarkers at low and high concentrations on days 1 and 3, respectively, from concentration‐response models with proper fit (i.e., linear relationship supported by observed data). Goodness‐of‐fit concentration‐response plots are shown in Figure 3 and Supplementary Material S1 .
Figure 2.
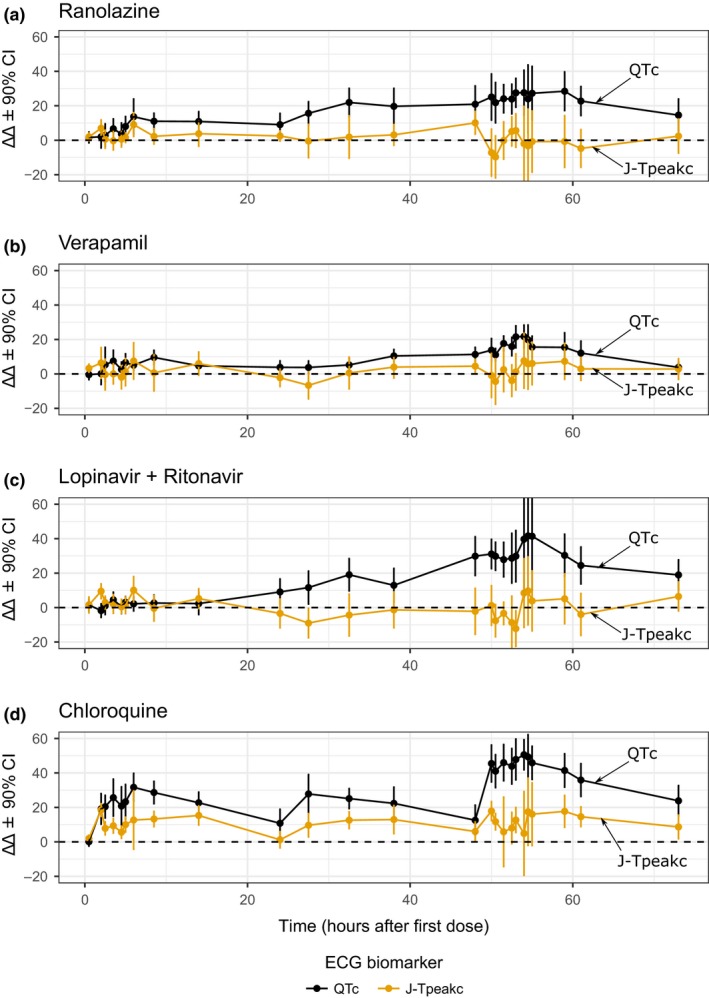
Part 1 pharmacodynamic time profiles. Drug‐induced changes (mean ± 90% confidence interval (CI)) for the placebo‐corrected change from baseline (ΔΔ) of heart rate corrected QT (QTc) (black) and heart rate corrected J‐Tpeak (J‐Tpeakc) (orange) for (a) ranolazine, (b) verapamil, (c) lopinavir/ritonavir, and (d) chloroquine. Horizontal dashed line corresponds with 0 ms. The y‐axis range of each panel has been adjusted to enhance interpretation. See electrocardiogram (ECG) analysis report in the Supplementary Material S1 for plots with full free‐scale y‐axis range including Tpeak‐Tend.
Figure 3.
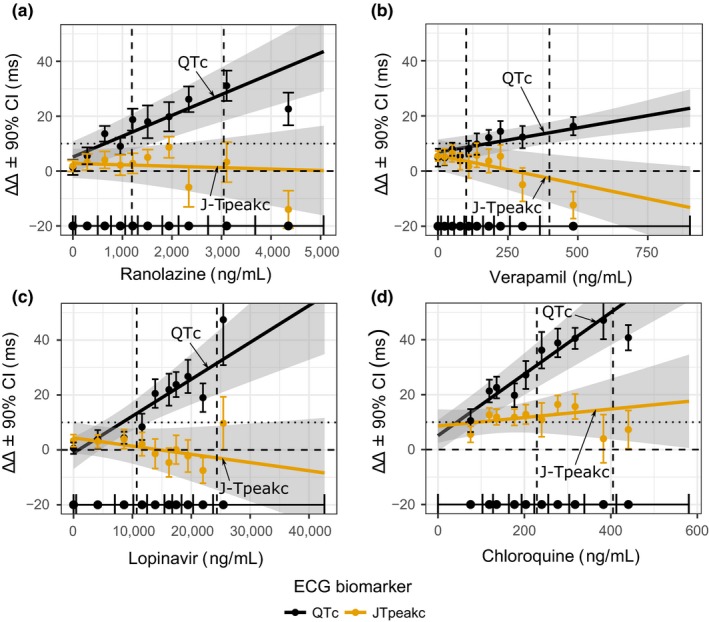
Part 1 concentration‐response plots. Predicted drug‐induced placebo‐corrected changes from baseline (ΔΔ) using concentration‐response models for heart rate corrected QT (QTc) (black) and heart rate corrected J‐Tpeak (J‐Tpeakc) (orange) for (a) ranolazine, (b) verapamil, (c) lopinavir/ritonavir, and (d) chloroquine. The solid line with gray shaded area denotes the model‐predicted mean placebo‐adjusted ΔΔ with 90% confidence interval (CI) as a function of concentration. Horizontal solid line with tick marks show the range of plasma concentrations divided into deciles. Vertical error bars denote the observed means and 90% CI for the ΔΔ within each plasma concentration decile. Vertical dashed lines correspond with population average maximum concentration (Cmax) on days 1 (low Cmax) and 3 (high Cmax). Horizontal dashed and dotted lines correspond with 0 ms and 10 ms ΔΔ, respectively. The y‐axis range of each panel has been adjusted to enhance interpretation. See electrocardiogram (ECG) analysis report in Supplementary Material S1 for plots with full free‐scale y‐axis range as well as other ECG measurements.
Ranolazine, lopinavir/ritonavir, and verapamil.
The prespecified primary end point for the balanced ion channel‐blocking drugs was that they would not prolong J‐Tpeakc defined by an upper bound of the two‐sided 90% confidence interval of ΔΔJ‐Tpeakc < 10 ms at Cmax on day 3. Although not included in the primary end point, QTc prolongation (i.e., upper bound of ΔΔQTc ≥ 10 ms at Cmax on day 3) was also expected because these drugs are known to block hERG.
The three balanced blockers had flat or negative mean ΔΔJ‐Tpeakc slope and mean ΔΔJ‐Tpeakc effects < 5 ms (Figure 3). The prespecified criterion was met with verapamil (mean 90% confidence interval of −2.6 (−11.4 to 6.1) ms) and lopinavir/ritonavir (−2.9 (−14.6 to 8.8) ms), but the criterion was not met for ranolazine (1.2 (−9.5 to 12.0) ms).
Ranolazine prolonged QTc by 14.1 (7.5–20.8) ms on day 1 and 28.2 (18.3–38.2) ms on day 3, whereas verapamil prolonged QTc by 8.6 (4.4–12.9) ms on day 1 and 13.9 (10.0–17.8) ms on day 3 (Figure 3 and Table 1). Lopinavir/ritonavir prolonged QTc by > 20 ms on day 3, but exhibited nonlinear concentration‐dependent QTc prolongation.
Chloroquine.
Chloroquine was included as a nonbalanced (predominant hERG) blocker drug. The prespecified primary end point was that it would prolong ΔΔQTc defined by an upper bound of the two‐sided 90% confidence interval of ΔΔQTc ≥ 10 ms at Cmax on day 1. Although not included in the primary end point, chloroquine was expected to prolong ΔΔJ‐Tpeakc.
Chloroquine met the prespecified criterion prolonging ΔΔQTc by 30.7 (22.6–38.9) ms at Cmax on day 1 (Figure 3). ΔΔJ‐Tpeakc was prolonged as defined by it being ≥ 10 ms throughout the concentration range in the study (Figure 3); however, the goodness‐of‐fit plot showed a poor fit of the concentration‐J‐Tpeakc linear model with a large positive intercept (8.6 ms) and flat slope.
Part 2: Crossover part ‐ Dofetilide vs. dofetilide + diltiazem
The hypothesis for this part of the study was that diltiazem (L‐type calcium channel block) can shorten the QTc prolongation caused by dofetilide (predominant hERG block) by shortening the J‐Tpeakc interval. This was assessed in a crossover study design by subjects receiving dofetilide alone in one period, whereas in a second period having subjects receive diltiazem alone on days 1 and 2 and then diltiazem combined with dofetilide on day 3. Dosing schedule and PK results are shown in Figure 4. Diltiazem observed Cmax on day 3 was 385.2 ng/mL. Dofetilide Cmax concentrations on day 3 were 1.5 and 1.2 ng/mL for dofetilide alone and diltiazem + dofetilide treatment periods, respectively. When dofetilide was administered alone (Figure 5), it prolonged ΔQTc by prolonging both ΔJ‐Tpeakc and ΔTpeak‐Tend. When diltiazem was co‐administered with dofetilide, diltiazem did not shorten ΔQTc (Figure 5 a,d), thus not meeting the primary end point. In the subsequent exploratory analysis, diltiazem shortened ΔJ‐Tpeakc by 11.5 (2.3–20.7) ms (Figure 5 b,e) and increased ΔTpeak‐Tend by 16.2 (4.8–27.7) ms (Figure 5 c,f).
Figure 4.
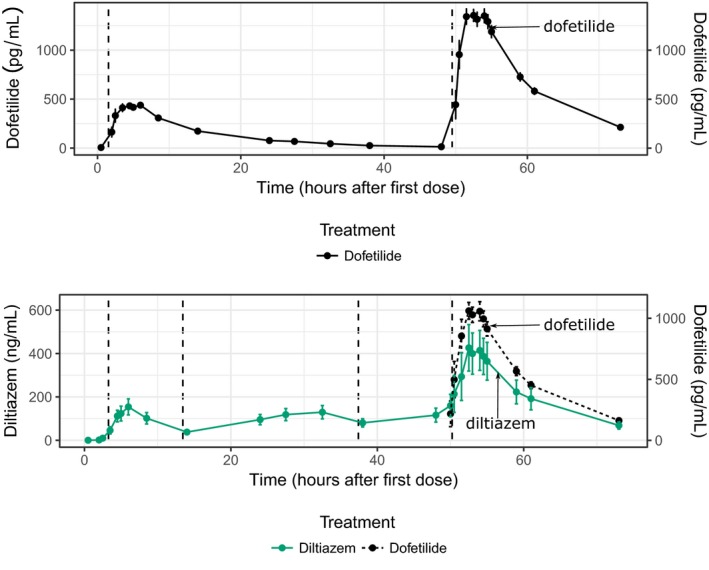
Part 2 pharmacokinetic (PK) time profiles. Mean (dots) and standard error (error bars) of PK plasma concentration profiles per timepoint after first dose on day 1 for dofetilide by itself (top panel) and for diltiazem + dofetilide combination (bottom panel). Bottom panel shows diltiazem concentration (left y‐axis, green solid lines) and dofetilide (right y‐axis, black dashed line). Dosing times were in the morning (after breakfast) and in the evening of days 1 and 2, and after breakfast in the morning of day 3. Dashed vertical lines show the time of active treatment doses for dofetilide (0.125 and 0.375 mg after breakfast on days 1 and 3, respectively, of dofetilide alone period, and 0.25 mg after breakfast on day 3 in the diltiazem + dofetilide period) and diltiazem (120 mg immediate release after breakfast on days 1 and 3, and 240 mg in the evening of days 1 and 2). Oral placebo was administered at dosing timepoints matching part 1 and that had no active treatment dosing (not shown).
Figure 5.
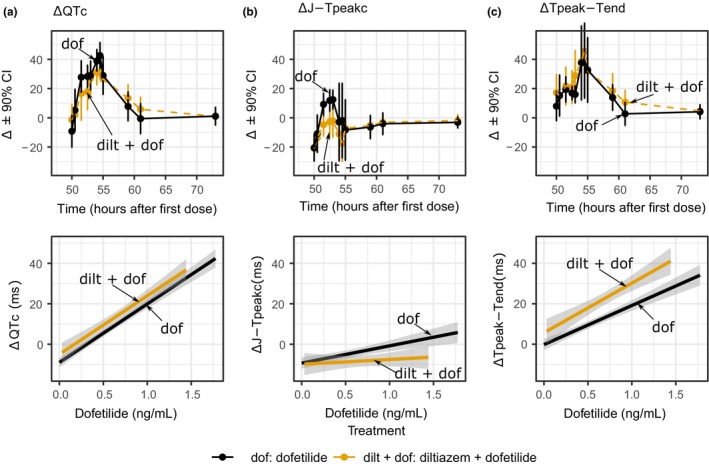
Part 2 pharmacodynamic time‐profiles and concentration‐response linearity plots. Top row panels show drug‐induced changes (mean ± 90% confidence interval (CI)) for the change from baseline (Δ) of (a) heart rate corrected QT (QTc), (b) heart rate corrected J‐Tpeak (J‐Tpeakc), and (c) Tpeak‐Tend on day 3 of dofetilide alone (black) and diltiazem + dofetilide (orange) treatments. The y‐axis range of each panel has been adjusted to enhanced interpretation. Bottom row panels show exploratory plots with linear regression fit through all the data for dofetilide alone (black) and diltiazem + dofetilide (orange) for drug‐induced Δ in (a) QTc, (b) J‐Tpeakc, and (c) Tpeak‐Tend. Note that there is no change in ΔQTc prolongation associated with dofetilide (hERG block) when diltiazem (calcium block) is co‐administered. See Supplementary Material S1 for full time profiles as well as concentration‐response plots of dofetilide (hERG block) vs. dofetilide + diltiazem (hERG + calcium) vs. dofetilide + mexiletine (hERG + late sodium).
PR and QRS effects for all study drugs
Chloroquine, verapamil, lopinavir/ritonavir, and diltiazem prolonged ΔΔPR in a concentration‐dependent fashion (Table 1). Goodness‐of‐fit plots supported a linear concentration‐ΔΔPR relationship for chloroquine, which caused 14.8 (4.7–24.9) ms prolongation at Cmax on day 3. With verapamil, lopinavir/ritonavir, and diltiazem, concentration‐dependent ΔΔPR prolongation was linear at lower concentrations, but goodness‐of‐fit plots (see Supplementary Material S1 ) suggested that it plateaued around 30 ms, which was the effect seen at Cmax on day 3. Ranolazine and dofetilide did not prolong PR.
Chloroquine prolonged QRS in a concentration‐dependent fashion, with ΔΔQRS of 9.9 (6.1–13.6) ms at Cmax on day 3 (Table 1). No QRS prolongation was observed with other drugs.
DISCUSSION
Drug‐induced torsade develops when there is an imbalance of inward and outward repolarization currents that triggers early afterdepolarizations. The CiPA initiative proposes to assess the effects of drugs on multiple ion channel currents (e.g., hERG, L‐type calcium, and late sodium) and integrate them in a mechanistic, in silico model of the human ventricular cardiomyocyte to predict torsade risk. The torsade risk metric (qNet) from the in silico model reflects the balance of inward and outward currents (i.e., how close a drug is to generating an early afterdepolarization, the trigger for torsade).11 CiPA proposes to assess ECG data from early phase I clinical studies to determine if there are unexpected ion channel effects in humans compared with the results from the in vitro assays that inform the in silico model. For a balanced ion channel‐blocking drug (i.e., multichannel blocker that has a low torsade risk qNet), a lack of QTc prolongation would confirm that there are no unexpected ion channel effects. Moreover, if the balanced blocker causes QTc prolongation, a lack of J‐Tpeakc prolongation could confirm presence of balanced ion channel effects. Thus, presence of J‐Tpeakc prolongation would not be expected for balanced blockers.
Part 1
Drug categories were assigned based on ion channel data available during study design, and not on the qNet score from the in silico model.13, 19 The prespecified criterion to test the hypothesis for the balanced ion channel drugs in this study was that the upper bound of the two‐sided 90% confidence interval of the predicted ΔΔJ‐Tpeakc using a concentration‐response model would be < 10 ms at Cmax on day 3. All three balanced ion channel drugs had flat or negative concentration‐response relationships for ΔΔJ‐Tpeakc, and the primary end point was met for verapamil (upper bound 6.1 ms) and for lopinavir/ritonavir (8.8 ms); however, it was missed for ranolazine (12.0 ms). Of note, with ranolazine this occurred when the QTc was prolonged by ~30 ms, whereas on day 1 the mean ΔΔQTc was ~14 ms and ΔΔJ‐Tpeakc upper bound was < 10 ms (2.2 (−4.3 to 8.7) ms). One predominant hERG‐blocking drug (chloroquine) that is associated with torsade23, 24 was included in the parallel part of the study as control. As hypothesized, chloroquine prolonged ΔΔQTc in a concentration‐dependent fashion, meeting the primary end point with ΔΔQTc upper bound at Cmax on day 1 ≥ 10 ms. Chloroquine also prolonged ΔΔJ‐Tpeakc throughout the 3 days of the study as defined by an upper bound ≥ 10 ms, but the prolongation was not linearly related to concentration.
Concentration‐response model observations and limitations.
In the parallel part (part 1) of the study, concentration‐response models for the primary end points had flat or negative J‐Tpeakc slopes for ranolazine, verapamil, and lopinavir/ritonavir, and a positive QTc slope for chloroquine, as expected. For the nonprimary end points, it was observed that the lopinavir‐ΔΔQTc relationship was nonlinear and the chloroquine‐ΔΔJ‐Tpeakc model had a positive intercept with a flat slope (Figure 3). With lopinavir, lower concentrations showed no QTc changes followed by a sharp increase of ~20 ms in mean ΔΔQTc at the higher concentrations on day 3. This nonlinear pattern was also present for ΔΔTpeak‐Tend ( Supplementary Material S1 ), and both are consistent with the time profile (Figure 2).
The positive intercept with a flat slope for the chloroquine‐ΔΔJ‐Tpeakc linear model may have been due to not having enough data to characterize ΔΔJ‐Tpeakc response at low chloroquine concentrations (Figure 3). Positive intercepts have been reported before with small sample sizes (see table 3 in Darpo et al.25), and were observed in some other concentration‐ECG relationships in this study (e.g., ranolazine‐QTc, Supplementary Material S1 ). In addition, multi‐ion channel effects may play a more substantial role at higher chloroquine concentrations leading to a plateau of ΔΔJ‐Tpeakc prolongation. This hypothesis is supported by PR and QRS prolongation caused by chloroquine in this study (Table 1 and Supplementary Material S1 ), which is consistent with prior observations.26, 27 In addition, chloroquine is known to accumulate in heart tissue, so the tissue concentration may have been substantially higher than the plasma concentration (e.g., up to ~150‐fold),28 leading to additional ion channel block. Last, QRS prolongation may be also explained by block of the inward rectifier potassium current by chloroquine.19, 29, 30 Overall, the ECG findings for chloroquine may warrant further investigation.
Last, the variability of ΔΔJ‐Tpeakc measures observed at high ranolazine concentrations was due to heart‐rate increase in two timepoints in two subjects. The observed variability could have been an artifact due to the use of previously developed population based heart‐rate correction.14
Part 2
A prior study demonstrated that lidocaine and mexiletine (late sodium current blockers) shorten QTc prolongation by dofetilide by shortening J‐Tpeakc.17 We hypothesized that diltiazem (L‐type calcium channel blocker) would exert a similar effect. Diltiazem did not affect the slope of the dofetilide concentration‐ΔQTc relationship (28.4 (21.0–35.9) ms per ng/mL with diltiazem and 29.2 (21.2–37.2) ms per ng/mL without), which were similar to those reported in our two prior studies (30.2 (26.7–33.7) ms per ng/mL and 27.9 (24.7–31.1) ms per ng/mL).15, 17 The reason for the lack of QTc shortening with diltiazem is not clear. Multiple nonclinical studies clearly demonstrate that L‐type calcium channel block shortens action potential duration.31, 32, 33 However, prior clinical studies combining diltiazem with quinidine and diltiazem with moxifloxacin also demonstrated a lack of QTc shortening by diltiazem.17, 34 This could be something specific to diltiazem, including potential secondary effects due to an autonomic response from diltiazem's effect on blood pressure or changes in ventricular loading from atrioventricular delay with mechano‐electrical feedback from stretch‐activated ion channels.35, 36, 37, 38
Diltiazem did shorten J‐Tpeakc, but also increased Tpeak‐Tend, which differs from what was observed for late sodium current block combined with predominant hERG block.17 Nevertheless, these findings were considered an exploratory analysis according to the statistical analysis plan.39 Of note, verapamil and lopinavir/ritonavir, which block hERG and L‐type calcium,19 did prolong QTc without prolonging J‐Tpeakc, as expected (Figure 3). Thus, these specific drugs with a combination of L‐type calcium and hERG block exhibited QTc prolongation without J‐Tpeakc prolongation.
Summary of QTc and JTpeakc effects with comparison to prior studies
Figure 6 shows ΔΔQTc and ΔΔJ‐Tpeakc changes predicted from the linear‐mixed effects models for balanced ion channel blockers (ranolazine and verapamil in this study along with ranolazine in a prior study) in comparison to predominant hERG blockers (dofetilide in two studies, quinidine and moxifloxacin). Concentration has been normalized in the x‐axis to facilitate comparison.15, 17 Chloroquine and lopinavir/ritonavir are not shown because poor fit of concentration‐response linear models did not allow for reliable predictions of ΔΔJ‐Tpeakc for chloroquine and ΔΔQTc for lopinavir/ritonavir. At concentrations causing mean ΔΔQTc prolongations above 10 ms, the concentration‐matched mean ΔΔJ‐Tpeakc changes remained close to zero or decreased for balanced blockers but increased proportionally to QTc for the predominant hERG blockers. The normalized concentration‐response relationships are consistent across studies and drugs with balanced blockers prolonging QTc without prolonging J‐Tpeakc, whereas predominant hERG blockers prolong QTc and J‐Tpeakc.16, 18
Figure 6.
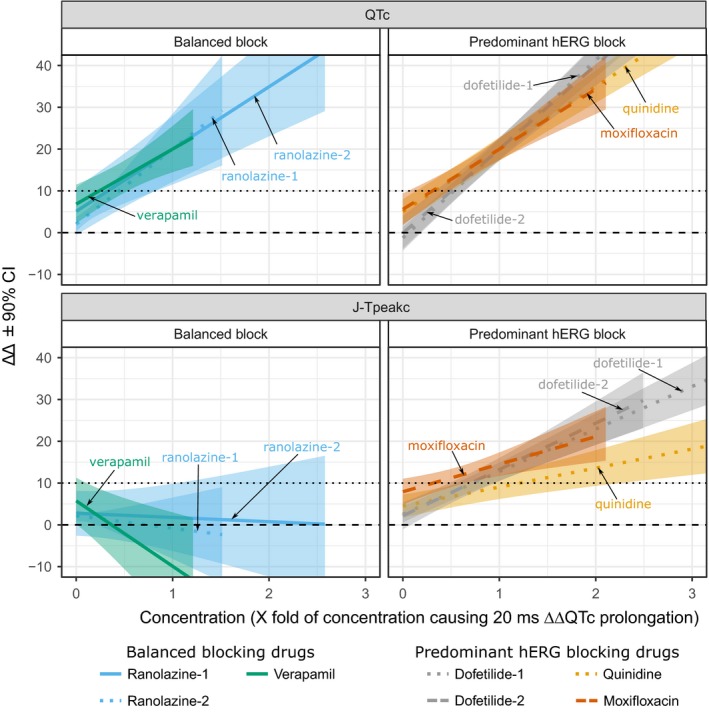
Heart rate corrected QT (ΔΔQTc) and heart rate corrected J‐Tpeak (ΔΔJ‐Tpeakc) concentration‐response for several predominant hERG and balanced ion channel‐blocking drugs. As concentration of drug increases, balanced blockers prolong ΔΔQTc (top left) without prolonging ΔΔJ‐Tpeakc (bottom left), but predominant hERG blockers prolong both ΔΔQTc (top right) and ΔΔJ‐Tpeakc (bottom right). Plots show mean (color lines) and 90% confidence intervals (CIs) (shade areas) predictions for drug‐induced baseline‐corrected and placebo‐corrected changes in QTc (ΔΔQTc, y‐axis, top panels) and ΔΔJ‐Tpeakc (y‐axis, bottom panels) vs. drug concentration (x‐axis) from concentration‐response models. Before plotting, each drug concentration was normalized to the concentration that caused 20 ms ΔΔQTc prolongation in its corresponding study. Balanced blockers shown include ranolazine (hERG + late sodium; blue), and verapamil (hERG + calcium; green). Predominant hERG blockers shown are dofetilide (gray), moxifloxacin (dark orange), and quinidine (light orange). Data from this study for multiple dosing of ranolazine (ranolazine‐2) and verapamil (solid lines) and from two prior studies with single oral dose design (ranolazine‐1, dofetilide‐1, and quinidine from Johannesen et al.15 dashed lines; dofetilide‐2 and moxifloxacin from Johannesen et al.17 dotted lines). Chloroquine and lopinavir/ritonavir not shown because poor fit of concentration‐response linear models did not allow for reliable predictions of ΔΔJ‐Tpeakc (chloroquine) and ΔΔQTc (lopinavir/ritonavir). Black horizontal lines show 0 ms (dashed) and 10 ms (dotted) thresholds for QTc (top panels) and ΔΔJ‐Tpeakc (bottom panels). Ranges of x‐ and y‐axes adjusted to facilitate comparison.
It is important to note that the presence vs. absence of J‐Tpeakc prolongation interpreted in isolation alone does not necessarily portend the presence vs. absence of pro‐arrhythmic risk. The CiPA proposes that the ECG data should be interpreted with the nonclinical ion channel data and in silico Torsade Metric Score. In addition, moxifloxacin is a drug that is a predominant hERG blocker; however, it has weak hERG block at standard clinical exposures and, thus, a substantially lower risk of torsade compared with dofetilide or quinidine.
Potential implications of CiPA
ICH E14 states that it is inconclusive as to whether drugs that prolong QTc are pro‐arrhythmic and that certain factors may modify the risk of QTc prolongation, including prolonging QTc up to a “plateau” value and that pro‐arrhythmic risk might be influenced by other pharmacologic effects (e.g., other ion channel effects in addition to hERG block).2 Although ICH S7B and E14 guidelines state that the presence of QTc prolongation does not mean that a drug will cause arrhythmias and the presence of QTc prolongation should support planning for and interpretation of subsequent clinical studies, the way they have been used in practice has been to sometimes drop compounds or drugs from development, which may not always be appropriate.40
One of the potential impacts of CiPA is to better inform when QTc prolonging drugs require intensive ECG monitoring in phase III trials. ICH E14 describes a nuanced approach for ECG monitoring in late stage trials in the question and answer (Q&A; see figure 1 in Vicente et al.13). Briefly, the results can be divided into categories of whether the mean ΔΔQTc at therapeutic exposures is < 10 ms, 10–20 ms or > 20 ms. In the current ICH E14 Q&A, if the ΔΔQTc at maximal “worst case” therapeutic exposures is < 10 ms, only routine ECG monitoring is recommended in late phase trials. However, if higher exposures are only expected in a limited subset of patients, intensive ECG monitoring may only be required in that specific patient population. If the ΔΔQTc effect is 10–20 ms at therapeutic exposure, intensive ECGs are needed, and if ΔΔQTc is > 20 ms, intensive ECGs and risk mitigation strategies are warranted.
It has been proposed that CiPA could impact clinical development for a drug with ΔΔQTc effect of 10–20 ms at worst‐case therapeutic exposures, in which a combined nonclinical and clinical integrated risk assessment should be taken into account to determine what level of ECG monitoring is appropriate in phase III. If the ΔΔQTc effect is > 20 ms at therapeutic concentrations, it is anticipated that intensive ECG monitoring would still be required; however, the combined nonclinical and clinical data could be taken into account to better inform the type and extent of ECG monitoring and risk mitigation strategies. In addition, a goal of CiPA is to enable updating drug labels to be more informative about pro‐arrhythmic risk, not just QT prolongation.
Conclusions
Results of this study suggest that concentration‐response analysis of QTc and J‐Tpeakc can differentiate QTc prolonging drugs with predominant hERG block from drugs that have balanced ion channel block. However, small sample sizes (10 subjects) may be insufficient to characterize concentration‐response relationships for some multichannel blocking drugs.
Methods
This study (clinicaltrials.gov number NCT03070470) was approved by the US Food and Drug Administration (FDA) Research Involving Human Subjects Committee and the local institutional review board. All subjects gave written informed consent and the study was performed at a phase I clinic (Spaulding Clinical, West Bend, WI). The study rationale and design along with the protocol were previously published.13 The design is briefly summarized below.
Part 1 was a double‐blind, randomized, placebo‐controlled, one‐period parallel design to assess the effects of three balanced blockers (ranolazine, verapamil, and lopinavir/ritonavir combination), one predominant hERG blocker (chloroquine), and one placebo on the QTc and J‐Tpeakc intervals in 50 healthy subjects. This parallel design is similar to early phase I studies and resulted in each study drug being administered to 10 subjects, and placebo to 10 subjects, in 3 consecutive days to achieve low and high drug plasma concentration levels on days 1 and 3, respectively. Administered drugs, their dosing times, and their PK time profiles are shown in Figure 1.
Part 2 was a double‐blind, randomized, two‐period crossover design in 10 subjects to assess whether diltiazem (calcium channel block) can reduce the QTc prolongation from dofetilide (hERG block) by shortening J‐Tpeakc. In the dofetilide alone period, subjects received dofetilide on days 1 and 3. In the diltiazem + dofetilide period, subjects received diltiazem alone on days 1 and 2, and diltiazem + dofetilide on day 3. Dosing times and PK profiles are shown in Figure 4.
ECG assessment
Continuous ECG recordings were performed using the Mortara Surveyor system (Mortara, Milwaukee, WI) sampled at 500 Hz with an amplitude resolution of 2.5 μV. From the continuous recording, three ECGs were extracted before the draw of each PK sample, based on heart rate stability and signal quality41 at 28 timepoints during the treatment periods: −1 hour, −30 minutes, 0 (immediately before first oral dose on day 1), 0.5, 2, 2.5, 3.5, 4.5, 5, 6, 8.5, 14, 24, 27.5, 32.5, 38, 48, 50, 50.5, 51.5, 52.5, 53, 54, 54.5, 55, 59, 61, and 73 hours after time 0 or the first oral dose on day 1. The ECG extractions at −1 hour, −30 minutes, and time 0 (immediately before first oral dose on day 1) were used for the baseline.13 Semi‐automated measurements of the PR, QRS, J‐Tpeak, Tpeak‐Tend, and QT interval were performed using the derived vector magnitude lead in the median beat. Briefly, automatic annotations from CalECG (AMPS, New York, NY) for global P onset, QRS onset and offset, and from a previously developed algorithm42 for Tpeak and Tend were generated. Next, two independent ECG readers blinded to treatment and time adjusted the measurements using high‐resolution images using previously developed software.43
None of the drugs caused an absolute mean ΔΔHR > 10 beats per minute, so the QT interval and J‐Tpeak intervals were corrected for heart rate with prespecified population correction formulas.13 Specifically, the QT interval was corrected for heart rate using Fridericia's correction (QTc = QT/(RR/1,000 ms)1/3, with RR in ms).44 Similarly, the J‐Tpeak interval was corrected for heart rate (J‐Tpeakc = J‐Tpeak/(RR/1,000 ms)0.58, with RR in ms).14 No correction was performed for the Tpeak‐Tend interval, as prior studies have shown a lack of rate dependence at resting heart rates.14, 45
PK sample analysis
Six validated liquid chromatography tandem mass spectrometric methods were used for PK sample analysis. The first method was the determination of dofetilide in K2EDTA human plasma using deuterated dofetilide as the internal standard (IS) using simple methanol‐based protein precipitation. Reversed‐phase ultraperformance liquid chromatographic separation was achieved with an Acquity ultraperformance liquid chromatographic C18 column (2.1 × 100 mm, 1.8 micron). Mass spectrometric detection was set at mass transitions of m/z 442.2→198.2. The second, third, and fourth methods were developed for diltiazem, ranolazine, and verapamil. All three analytes and their deuterated IS were chromatographed on Zorbax SB‐C18 column (50 × 2.1 mm, 3.5 micron) after simple protein precipitation using acetonitrile. Mass spectrometric detections were set at m/z 415.3→178.0, m/z 428.3→279.0, and m/z 455.3→177.1 for diltiazem, ranolazine, and verapamil, respectively. The fifth method, determination of chloroquine, and sixth method, simultaneous determination of lopinavir and ritonavir in K2EDTA human plasma were developed. All three analytes and their deuterated IS were chromatographed on Phenomenex, Kinetex C18 column (50 × 2.1 mm, 1.7 micron). Mass spectrometric detections were set at mass transitions of m/z 320.2→247.2, m/z 629.0→155.2, and m/z 722.0→268.1 for chloroquine, lopinavir, and ritonavir, respectively.
Drug categories
Drug categories (i.e., balanced vs. predominant hERG block) were assigned based on ion channel data available during study design, as previously described,13, 19 and not on the qNet score from the in silico model. Ranolazine and verapamil are balanced blockers (similar potency for hERG and late sodium and for hERG and calcium, respectively). Lopinavir/ritonavir blocks hERG, calcium, and late sodium at similar potencies and was included as a balanced blocker. Chloroquine has substantially more potent hERG block compared with calcium or late sodium and was labeled as a predominant hERG blocker. Drug categories may be different if based on qNet and new ion channel data acquired with current CiPA protocols.20
Statistical analysis
The statistical analysis plan was previously published.13 Concentration‐response modeling was performed following current best practices46 with prespecified models. Linear‐mixed effects models were used to characterize the relationship between drug concentration and ECG changes. The change from baseline (Δ) for each ECG biomarker (e.g., the average ΔQTc, ΔJ‐Tpeakc) by timepoint was the dependent variable, for which baseline was defined as the mean of the three predose ECG readings on day 1 for each period. In part 1, we used linear mixed‐effects models in which ΔECG was the dependent variable (e.g., ΔQTc, ΔJ‐Tpeakc) and drug concentration (set to 0 for placebo), nominal timepoint, treatment (coded 0 for placebo and 1 for active drug) were included as fixed effects, and subject was included as random effect on intercept and drug concentration (i.e., allowing each subject to have his own concentration‐response relationship). In part 2, we used a linear mixed‐effects model in which ΔECG was the dependent variable (e.g., ΔQTc, ΔJ‐Tpeakc), dofetilide, and diltiazem concentrations as well as dofetilide‐diltiazem interaction were included as fixed effects, and subject was included as a random effect on intercept and dofetilide and diltiazem concentrations. All data before withdrawals or discontinuations were included in the analysis. All statistical analyses were performed in R 3.5.0 (R Foundation for Statistical Computing, Vienna, Austria).
Funding
This study was internally funded by the FDA/CDER Safety Research Interest Group.
Conflicts of Interest
P.T.S. has consulting agreements with Biomedical Systems, Charles River, and iCardiac. The other authors report no conflicts of interest.
Author Contributions
D.G.S., J.V., R.Z., L.J., R.O.‐J., J.W.M., C.S., S.K., P.T.S., V.P., M.K.M., J.L., J.F., C.G., and N.S. wrote the manuscript. D.G.S., J.V., R.Z., L.J., J.W.M., P.T.S., V.P., M.K.M., C.G., and N.S. designed the research. D.G.S., J.V., R.Z., L.J., J.W.M., C.S., and S.K. performed the research. J.V., R.O.‐J., R.Z., V.P., M.K.M., L.J., J.F., C.G., and D.G.S. analyzed the data.
Disclaimer
This article reflects the views of the authors and should not be construed to represent the FDA's views or policies. The mention of commercial products, their sources, or their use in connection with material reported herein is not to be construed as either an actual or implied endorsement of such products by the FDA.
Supporting information
Supplementary Material S1. HTML with analysis code.
Acknowledgments
This work was supported by the FDA Safety Research Interest Group and Critical Path Initiative in the Center for Drug Evaluation and Research, the FDA's Office of Women's Health, and by a research fellowship from the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the FDA. We would like to thank staff at Spaulding Clinical Research for executing the clinical study (NCT03070470).
Contributor Information
Jose Vicente, Email: jose.vicenteruiz@fda.hhs.gov.
David G. Strauss, Email: david.strauss@fda.hhs.gov
References
- 1. International Council on Harmonisation . ICH topic S7B the nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals <http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Safety/S7B/Step4/S7B_Guideline.pdf> (2005). [PubMed]
- 2. International Council on Harmonisation . Guideline for industry E14 clinical evaluation of QT/QTc interval prolongation and proarrhythmic potential for non‐antiarrhythmic drugs <https://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Efficacy/E14/E14_Guideline.pdf> (2005). [PubMed]
- 3. January, C.T. & Riddle, J.M. Early afterdepolarizations: mechanism of induction and block. A role for L‐type Ca2+ current. Circ. Res. 64, 977–990 (1989). [DOI] [PubMed] [Google Scholar]
- 4. Duff, H.J. , Roden, D. , Primm, R.K. , Oates, J.A. & Woosley, R.L. Mexiletine in the treatment of resistant ventricular arrhythmias: enhancement of efficacy and reduction of dose‐related side effects by combination with quinidine. Circulation 67, 1124–1128 (1983). [DOI] [PubMed] [Google Scholar]
- 5. Duff, H. , Mitchell, B. , Manyari, D. & Wyse, G. Mexiletine‐quinidine combination: electrophysiologic correlates of a favorable antiarrhythmic interaction in humans. J. Am. Coll. Cardiol. 10, 1149–1156 (1987). [DOI] [PubMed] [Google Scholar]
- 6. Giardina, E.‐G.V. & Wechsler, M.E. Low dose quinidine‐mexiletine combination therapy versus quinidine monotherapy for treatment of ventricular arrhythmias. J. Am. Coll. Cardiol. 15, 1138–1145 (1990). [DOI] [PubMed] [Google Scholar]
- 7. Chézalviel‐Guilbert, F. , Davy, J.‐M. , Poirier, J.‐M. & Weissenburger, J. Mexiletine antagonizes effects of sotalol on QT interval duration and its proarrhythmic effects in a canine model of torsade de pointes. J. Am. Coll. Cardiol. 26, 787–792 (1995). [DOI] [PubMed] [Google Scholar]
- 8. Guo, D. , Zhao, X. , Wu, Y. , Liu, T. , Kowey, P. & Yan, G.‐X. L‐type calcium current reactivation contributes to arrhythmogenesis associated with action potential triangulation. J. Cardiovasc. Electrophysiol. 18, 196–203 (2007). [DOI] [PubMed] [Google Scholar]
- 9. Badri, M. et al Mexiletine prevents recurrent torsades de pointes in acquired long QT syndrome refractory to conventional measures. JACC Clin. Electrophysiol. 1, 315–322 (2015). [DOI] [PubMed] [Google Scholar]
- 10. Sager, P. , Gintant, G. , Turner, R. , Pettit, S. & Stockbridge, N. Rechanneling the cardiac proarrhythmia safety paradigm: a meeting report from the Cardiac Safety Research Consortium. Am. Heart J. 167, 292–300 (2014). [DOI] [PubMed] [Google Scholar]
- 11. Dutta, S. et al Optimization of an in silico cardiac cell model for proarrhythmia risk assessment. Front. Physiol. 8, 616 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vicente, J. , Stockbridge, N. & Strauss, D. Evolving regulatory paradigm for proarrhythmic risk assessment for new drugs. J. Electrocardiol. 49, 837–842 (2016). [DOI] [PubMed] [Google Scholar]
- 13. Vicente, J. et al Mechanistic model‐informed proarrhythmic risk assessment of drugs: review of the “CiPA” initiative and design of a prospective clinical validation study. Clin. Pharmacol. Ther. 103, 54–66 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Johannesen, L. et al Improving the assessment of heart toxicity for all new drugs through translational regulatory science. Clin. Pharmacol. Ther. 95, 501–508 (2014). [DOI] [PubMed] [Google Scholar]
- 15. Johannesen, L. et al Differentiating drug‐induced multichannel block on the electrocardiogram: randomized study of dofetilide, quinidine, ranolazine, and verapamil. Clin. Pharmacol. Ther. 96, 549–558 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Vicente, J. et al Comprehensive T wave morphology assessment in a randomized clinical study of dofetilide, quinidine, ranolazine, and verapamil. J. Am. Heart Assoc. 4, e001615 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johannesen, L. et al Late sodium current block for drug‐induced long QT syndrome: results from a prospective clinical trial. Clin. Pharmacol. Ther. 99, 214–223 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vicente, J. et al Electrocardiographic biomarkers for detection of drug‐induced late sodium current block. PLoS One 11, e0163619 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Crumb, W.J. Jr , Vicente, J. , Johannesen, L. & Strauss, D.G. An evaluation of 30 clinical drugs against the comprehensive in vitro proarrhythmia assay (CiPA) proposed ion channel panel. J. Pharmacol. Toxicol. Methods 81, 251–262 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Comprehensive In Vitro Proarrhythmia Assay (CiPA) Initiative . Recommended cardiac ion channel protocols under CiPA <http://cipaproject.org/wp-content/uploads/sites/24/2018/06/CiPA-protocol-061318.pdf> (2018). Accessed July 13, 2018.
- 21. Ferber, G. , Zhou, M. & Darpo, B. Detection of QTc effects in small studies—implications for replacing the thorough QT study. Ann. Noninvasive Electrocardiol. 20, 368–377 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Goldberger, A.L. et al PhysioBank, PhysioToolkit, and PhysioNet: components of a new research resource for complex physiologic signals. Circulation 101, e215–e220 (2000). [DOI] [PubMed] [Google Scholar]
- 23. Demazière, J. , Fourcade, J.M.N. , Busseuil, C.T.A. , Adeleine, P. , Meyer, S.M. & Saïssy, J.M. The hazards of chloroquine self prescription in West Africa. J. Toxicol. Clin. Toxicol. 33, 369–370 (1995). [DOI] [PubMed] [Google Scholar]
- 24. Haverkamp, W. et al The potential for QT prolongation and proarrhythmia by non‐antiarrhythmic drugs: clinical and regulatory implications. Report on a Policy Conference of the European Society of Cardiology. Eur. Heart J. 21, 1216–1231 (2000). [DOI] [PubMed] [Google Scholar]
- 25. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Pharmacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 26. Westward Pharmaceutical Corp . Chloroquine label <https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/083082s050lbl.pdf> (2009).
- 27. Looareesuwan, S. et al Cardiovascular toxicity and distribution kinetics of intravenous chloroquine. Br. J. Clin. Pharmacol. 22, 31–36 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Browning, D.J. Pharmacology of chloroquine and hydroxychloroquine In Hydroxychloroquine and Chloroquine retinopathy 35–63 (Springer New York, New York, NY, 2014). [Google Scholar]
- 29. Rodríguez‐Menchaca, A.A. et al The molecular basis of chloroquine block of the inward rectifier Kir2.1 channel. Proc. Natl. Acad. Sci. USA 105, 1364–1368 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Nada, A. et al The evaluation and management of drug effects on cardiac conduction (PR and QRS Intervals) in clinical development. Am. Heart J. 165, 489–500 (2013). [DOI] [PubMed] [Google Scholar]
- 31. January, C.T. , Riddle, J.M. & Salata, J.J. A model for early afterdepolarizations: induction with the Ca2+ channel agonist Bay K 8644. Circ. Res. 62, 563–571 (1988). [DOI] [PubMed] [Google Scholar]
- 32. Martin, R.L. , McDermott, J.S. , Salmen, H.J. , Palmatier, J. , Cox, B.F. & Gintant, G.A. The utility of hERG and repolarization assays in evaluating delayed cardiac repolarization: influence of multi‐channel block. J. Cardiovasc. Pharmacol. 43, 369–379 (2004). [DOI] [PubMed] [Google Scholar]
- 33. Blinova, K. et al Comprehensive translational assessment of human‐induced pluripotent stem cell derived cardiomyocytes for evaluating drug‐induced arrhythmias. Toxicol. Sci. 155, 234–247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Laganière, S. et al Pharmacokinetic and pharmacodynamic interactions between diltiazem and quinidine. Clin. Pharmacol. Ther. 60, 255–264 (1996). [DOI] [PubMed] [Google Scholar]
- 35. Taggart, P. & Sutton, P.M.I. Cardiac mechano‐electric feedback in man: clinical relevance. Prog. Biophys. Mol. Biol. 71, 139–154 (1999). [DOI] [PubMed] [Google Scholar]
- 36. Eckardt, L. , Kirchhof, P. , Breithardt, G. & Haverkamp, W. Load‐induced changes in repolarization: evidence from experimental and clinical data. Basic Res. Cardiol. 96, 369–380 (2001). [DOI] [PubMed] [Google Scholar]
- 37. Trayanova, N. , Li, W. , Eason, J. & Kohl, P. Effect of stretch‐activated channels on defibrillation efficacy. Heart Rhythm 1, 67–77 (2004). [DOI] [PubMed] [Google Scholar]
- 38. Trayanova, N. , Constantino, J. & Gurev, V. Models of stretch‐activated ventricular arrhythmias. J. Electrocardiol. 43, 479–485 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. US Food and Drug Administration (FDA) . Multiple endpoints in clinical trials. Guidance for industry. Draft guidance <https://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM536750> (2017).
- 40. Stockbridge, N. , Morganroth, J. , Shah, R. & Garnett, C. Dealing with global safety issues: was the response to QT‐liability of non‐cardiac drugs well coordinated? Drug Saf. 36, 167–182 (2013). [DOI] [PubMed] [Google Scholar]
- 41. Badilini, F. , Vaglio, M. & Sarapa, N. Automatic extraction of ECG strips from continuous 12‐lead holter recordings for QT analysis at prescheduled versus optimized time points. Ann. Noninvasive Electrocardiol. 14(Suppl 1), S22–S29 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johannesen, L. , Vicente, J. , Hosseini, M. & Strauss, D. Automated algorithm for J‐Tpeak and Tpeak‐tend assessment of drug‐induced proarrhythmia risk. PLoS One 11, e0166925 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Vicente, J. , Johannesen, L. , Galeotti, L. & Strauss, D.G. ECGlab: user friendly ECG/VCG analysis tool for research environments. Comput Cardiol (Cinc.) 40, 775–778 (2013). [Google Scholar]
- 44. Fridericia, L.S. Die Systolendauer im Elektrokardiogramm bei normalen Menschen und bei Herzkranken. Acta Med. Scand. 54, 17–50 (1921). [Google Scholar]
- 45. Smetana, P. , Batchvarov, V. , Hnatkova, K. , John Camm, A. & Malik, M. Sex differences in the rate dependence of the T wave descending limb. Cardiovasc. Res. 58, 549–554 (2003). [DOI] [PubMed] [Google Scholar]
- 46. Garnett, C. et al Scientific white paper on concentration‐QTc modeling. J. Pharmacokinet. Pharmacodyn. 45, 383–397 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material S1. HTML with analysis code.