

Abstract
Introduction:
The congenital myasthenic syndromes are a heterogeneous group of genetic disorders characterized by an abnormal synaptic transmission at the neuromuscular junction.
Report:
We present a two-year old patient, male, with hypotonia, palpebral ptosis and proximal symmetric weakness with neonatal onset that required several and prolonged hospitalizations for pneumonia and respiratory failure. From two years of age, the parents noticed that the facial and general weakness worsened in the afternoons and with repeated or prolonged physical activity.
The physical examination showed palpebral ptosis, predominantly proximal weakness and fatigability with sustained muscular effort. The electromyogram showed a 27% decrement of the compound muscle action potential. Genetic study of the patient’s parents revealed compound heterozygosity with transmission of two different mutations in the rapsyn gene from both parents. The patient received pyridostigmine with great improvement, achieving optimal performance in school, sports, and daily life activities.
Conclusions:
Weakness and fatigability with neonatal onset, mainly affecting the muscles with brain stem innervation and the decrement greater than 10 percent in the Compound Muscular Action Potential in the electromyographic studies, should make us suspect in a congenital myasthenic syndrome. We review the literature and key clinical points to stablish opportune diagnosis and effective treatment in some of these syndromes.
Keywords: Congenital Myasthenic Syndromes, Neuromuscular Junction Diseases, Myasthenia Gravis, Muscle Proteins, Missense Mutation
Introduction
Congenital myasthenic syndromes are a heterogeneous group of genetic disorders caused by an abnormal transmission in the neuromuscular junction [1]. Most of them are the result of molecular defects in the acetylcholine receptor of the muscle but can be due to different genetic mutations [1],[2]. Congenital myasthenic syndromes must be differentiated from other disorders such as neonatal myasthenia (in which mothers with myasthenia gravis transmit antibodies anti-acetylcholine receptor through the maternal placenta); and from acquired myasthenia gravis (in which the physiopathologic mechanism is an autoimmune phenomenon). The case presented below illustrates the importance of the clinical assessment of the phenomenon of fatigability to establish the suspicion and avoid the delay in diagnosis that can threaten the patient’s life. It should be noted that some congenital myasthenic syndromes have effective treatments to improve the quality of life and prognosis of these patients [3],[4].
Case report
Anamnesis
We present a four-year-old patient with an uneventful prenatal history, born by an elective C-section without any complications, presenting with muscle hypotonia since birth and global muscle weakness characterized by absence of facial expression and limb movements, severe suction-swallowing problems that making it impossible direct breastfeeding, and hypoventilation associated with an episode of pneumonia and sepsis requiring mechanical ventilation for 45 days. He was discharged from neonatal intensive care unit at two months old with the indication of physical therapy, orofacial therapy, and using a nasogastric tube for feeding.
Suction improved slowly until eight months, time in which he could feed directly. He was able to hold his head by eight-months-old, sit at 10 months and walk short distances by two years. Since his early months, parents noticed palpebral ptosis and facial weakness that worsened by the afternoons, in the same way, at age two, when he started walking, parents noticed greater difficulty in the afternoons. Regarding the development of language he started babbling at eight months and at two years and five months he was able to speak short phrases. The social development was normal.
At seven months he had an episode of aspiration pneumonia requiring invasive mechanical ventilation for 12 days and was discharged after one month. At 11 and 15 months he was hospitalized for pneumonia requiring mechanical ventilation for 10 days and four days respectively. He left the hospital with a medical diagnosis of recurrent pneumonia and muscular hypotonia. From the second hospitalization he used inhaled β−2 agonists and parents noticed he had improvement with this treatment.
At 23 months, orchidopexy was performed by bilateral cryptorchidism. The father of 32 years, the mother of 30 years and the brother of two months did not present weakness or other health problems.
Physical exam
Vital functions: heart rate: 96 per minute, respiratory rate: 18 per minute. Weight, height and cephalic circumference were adequate for the age. He looked in good general condition, thin build, collaborator with the interrogation and examination. Immediately when he entered the consulting room, we noticed his myopathic and unexpressive facial features with bilateral palpebral ptosis, lengthened face, arched eyebrows, medium-facial hypoplasia, open mouth with everted lower lip, ogival palate, diastema and pointed chin (Fig. 1).
Figure 1.
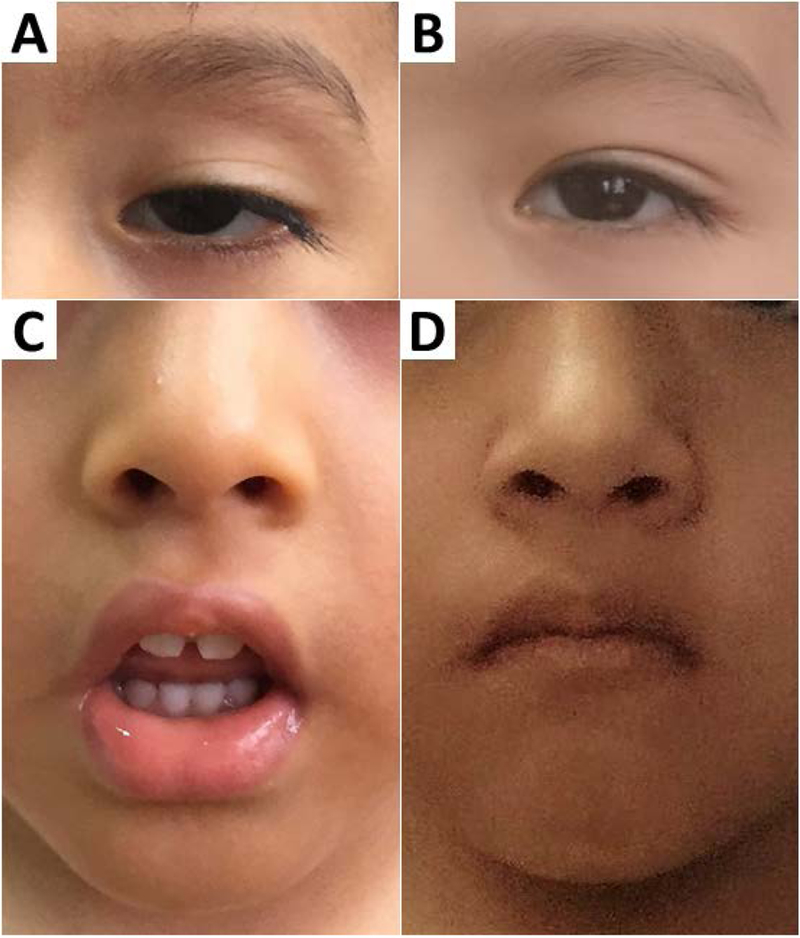
Patient’s facies
(A, C) before treatment with a myophatic unexpressive facies, lengthened face, palpebral ptosis, bilateral epicanthus, medium-facial hypoplasia, semi-opened mouth, everted under lip, ogival palate, diastema, and pointy chin.
(B, D): we can see notorious improvement of the palpebral ptosis, and facial expression in general, after treatment. Source: Patient photos taken by the authors, before and after treatment with pyridostigmine.
In the neurological exam, we found adequate memory, language comprehension according to his age, and mild dysarthria. He had a proximal muscular weakness (Fig. 2a), with a positive Gowers sign, generalized hypotonia, and normal tactile, thermoalgesic and proprioceptive sensibility. Osteotendinous reflexes were normal (Fig. 2b) but a progressive worsening in the bicipital reflex was noted after a 2-minute repetitive stimulation. We found mild muscle hypotrophy. The rest of the exam was within normal limits. Finally, we found fatigability in the palpebral opening provoked by sustained upward gaze for two minutes towards a permanent elevated stimulus (animated child video).
Figure 2.
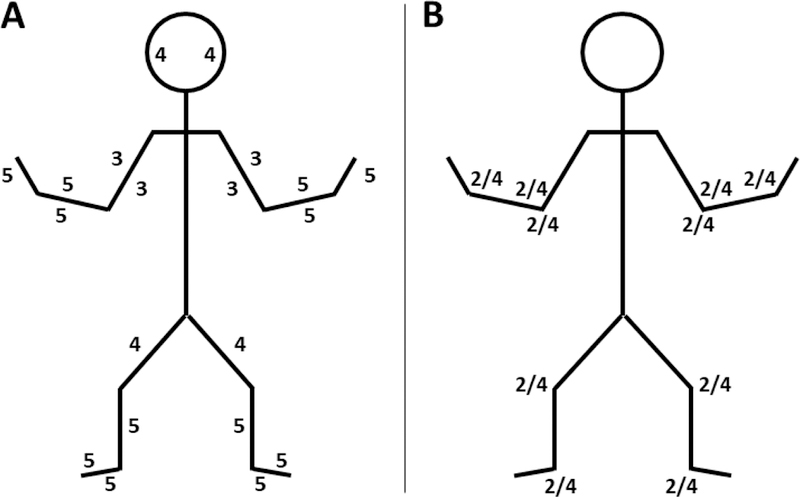
Strength and osteotendinous reflexes valuation.
(A) We found a symmetric proximal weakness
(B) Osteotendinous reflexes were normal at the beginning but exhaustion of the biceps reflex was found with repeated percussion for two minutes. Source: prepared by the authors based on the patient’s clinical findings.
Immediately the clinical suspicion was made of a congenital myasthenic syndrome, based on the myopathic facies, the proximal weakness that was worsening during the day, and the fact of that all symptoms began in the neonatal age. We considered as differential diagnosis limb-girdle muscular dystrophy and some congenital myopathies. We ordered antibodies anti-Acetylcholine and muscle-specific tyrosine kinase, creatine phosphokinase, arterial gases, electrolytes, lactate, pyruvate, and liver enzymes, all of which were normal. The electromyography showed a 27% decrement in the compound muscle action potential in response to repetitive stimulation in the facial nerve. Treatment was initiated with pyridostigmine 7 mg/kg/day, and physical therapy was indicated.
At two-weeks clinical control the parents commented immediate improvement with the use of pyridostigmine in the way of walking (even achieving to run), the palpebral opening, the articulation of the word and the sitting posture. The examination showed improvement of strength and muscle tone, disappearing the sign of Gowers. The treatment with pyridostigmine at seven milligrams per kilo per day in five daily doses is maintained until today.
An MRI of the brain was performed, the first report of which indicated “agenesis of the corpus callosum”. However, a more detailed observation by radiologists and neurologists corrected this report, since the corpus callosum and the whole brain were completely normal.
Genetic study
We sent samples of the patient, both parents, and younger brother to the Mayo Clinic Neuromuscular Research Laboratory in Rochester, Minnesota for the genetic investigation of congenital myasthenic syndromes by a complete exome sequencing (parents-offspring trios). The study shown that the proband (II.1) has a compound heterozygosity because he is carrying two punctual mutations in exon 2 of the rapsyn gene. The first consists of a transversion-type mutation that replaces a cytosine with an adenine at position 264 of deoxyribonucleic acid (DNA), generating a lysine instead of asparagine, in the 88 position of protein (p.N88K). The second mutation is other transversion that substitutes a cytosine with adenine in the 218 position of DNA, creating an aspartate instead of an Alanine in the 73rd position of protein (p.A73D). This makes the patient is a compound heterozygote in the RAPSN gene. It was found that the father (I.1) is a carrier of the missense RAPSN N88K mutation in heterozygosis, which has previously been described as a pathogenic variant. Both the mother (I.2) and the younger brother (II.2) are carriers of the A73D mutation in heterozygosis (Fig. 3).
Figure 3.
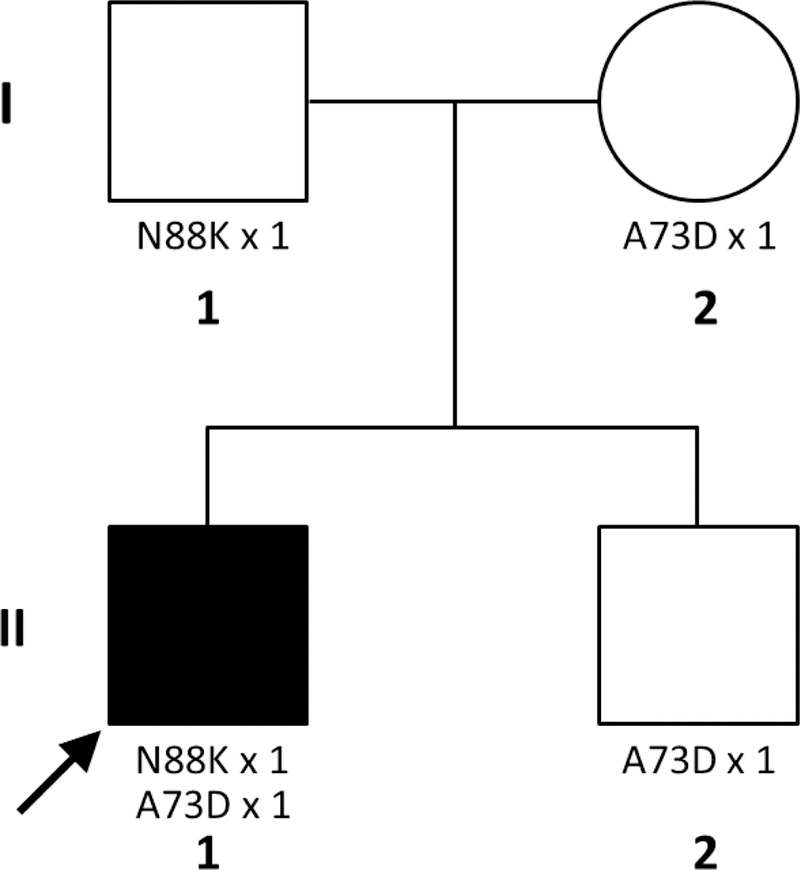
Figure 3 Hederogram of the phenomenon of compound heterozygosis for transmission of two different mutations from both parents on the same gene.
The individual I-1 is heterozygous for the N88K mutation.
I-2 is heterozygous for the A73D mutation.
Proband II-1 is compound heterozygote as it received the two mutations in the rapsyn gene, one from each parent.
Source: prepared by the authors based on the results of the genetic study
This second mutation is not previously described in the genetic bases consulted (SNP databases and Clinvar), but the analysis of this mutation with the effect prediction software programs PolyPhen-2 and Provean / Sift, classifies it as a pathogenic variant (cause of disease).
Discussion
The congenital myasthenic syndromes are a heterogeneous group of genetic diseases caused by an abnormal synaptic transmission in the neuromuscular junction[1],[2]. The prevalence of congenital myasthenic syndromes is estimated in 2.8 – 15.5 per one million, even though it could be underestimated [3],[5].
Most of these diseases are due to molecular defects in the muscular nicotinic receptors for acetylcholine, but can also be caused by mutations in the presynaptic proteins or in the proteins associated to the synaptic basal lamina, defects in the development or maintenance of the neuromuscular junction or defects in the glycosylation of proteins [1],[2].
The symptom that characterizes the congenital myasthenic syndromes is the fatigability of the skeletal muscle, with onset since birth or in infancy. Rarely, the symptoms may appear in the second or third decade of life. It is essential to mention that the severity and history of these diseases are very variable, ranging from mild symptoms to severe progressive weakness. In some of the congenital myasthenic syndromes, the symptoms may be mild, with occasional exacerbations of weakness or even respiratory failure, triggered by fever, infections, or stress. When the onset is in the neonatal period, it is frequently associated with respiratory failure, sudden episodes of apnea, difficulties in feeding, and a weak cry. It can be associated with reduced respiratory effort, stridor, and arthrogryposis. When the onset is later in life, the weakness is exacerbated by physical activity like running or climbing up the stairs, and it is characteristic that this weakness worsens during the day. Other forms of presentation include ptosis and fixed or fluctuating ophthalmoplegia. The cardiac and smooth muscle are not involved in this group of diseases [3],[5],[6]. In the reviewed literature, we have not found any association with cryptorchidism, which may be an isolated clinical problem without any relationship with the genetic disturbances of our patient. It has not been described in the medical literature, differences in the frequency or in the clinical expression of congenital myasthenic syndromes in terms of gender.
We believe that in our patient, the myopathic facies, the palpebral ptosis, and cryptorchidism, notorious at birth, contributed to the generic labeling of “dysmorphic syndrome” that led to search for a malformation of the nervous system. Subsequently, a magnetic resonance image of poor quality and probably an uncaring appreciation or incomplete visualizations, led to the erroneous diagnosis of “agenesis of the corpus callosum” that also constituted a distractor.
In the evolution, during the successive attentions in the emergency due to respiratory disease, more attention had to be paid to the myopathic facies and to the phenomenon of fatigability with the passing of the day. Swallowing problems, recurrent aspiration, recurrent pneumonias and the need for prolonged mechanical ventilation are always suggestive of neuromuscular diseases.
The repetitive nerve stimulation studies the variation in the amplitude of the compound muscle action potential, in response to multiple electrical stimuli over the muscle. Usually, the diminished amplitude between the first and fourth muscle action potential should not be over 8%. When this diminishment is over 10%, we should suspect a Myasthenia Gravis, a Lambert Eaton syndrome, or a congenital myasthenic syndrome [1].
When a congenital myasthenic syndrome is suspected, it is essential to investigate the specific diagnosis through genetic panel’s studies, as it was made in our patient. The utility of the genetic panels remains in the existence of several genes associated with similar clinical presentations, so these studies lower costs. On the other hand, to identify the causal mutation allows us, in some cases, the administration of adequate treatment and to establish the prognosis and to offer adequate genetic counseling to the family [6].
The phenotype described in our patient is coincident as that described in patients with genetic mutations in the RAPSN gene (receptor-associated protein of the synapse) which codifies the rapsyn protein that connects and stabilizes the acetylcholine receptor to the postsynaptic membrane and the subsynaptic cytoskeleton [7],[8]. It has been described that the mutations to this gene cause a postsynaptic congenital myasthenic syndrome characterized by a deficiency of AChR in the neuromuscular plaque [7],[8],[9],[10].
Most of the patients present an early onset clinical picture (usually in the neonatal stage) with myasthenic symptoms as bilateral palpebral ptosis, facial and other truncal muscles weakness, as well as weakness in the upper and lower extremities. The phenomenon of fatigability that worsens during the day is characteristic and should always be investigated during the interrogation. It is common to find in the history of these patients, episodes of severe weakness provoked by stress, fever or infections, usually accompanied by respiratory failure with a requirement of mechanical ventilation (all of which presented in our patient). Other typical manifestations are hypotonia, episodic apneas, and problems to suck or swallow. The clinical course may vary from mild to severe [3],[7],[8],[10].
There are about 200 pathogenic variants identified in the RAPSN gene. The missense mutation c.264C>A (p.N88K) has been identified in many individuals as a pathogenic variant in the coding region in at least one allele, finding evidence of an Indo-European origin; although not all the individuals that present it have the same haplotype [3],[7]. Cases of homozygosity, compound heterozygosity and deletions have been described. Patients with pathogenic variants in the promoter region have also been identified [1],[8],[10],[11].
The A73D mutation we found in our patient, his mother, and his younger brother has not been described before. According to the effect predictive software programs, the bases for genetic variations for polymorphisms of unique nucleotides, and the databases for genomic variations and its relation with human health analyzed, this mutation is identified as a pathogenic variant (meaning cause of disease). In association in our patient with the N88K mutation coming from the father, results in the occurrence of a disease that is expressed in the patient with the clinical manifestations of a congenital myasthenic syndrome. This new A73D pathogenic mutation should be tracked in the population starting with complementary genetic studies in other members of the extended family and looking for variations in the clinical presentation.
Patients with the mutation in N88K RAPSN present a positive response to medication with acetylcholinesterase inhibitors [3],[7],[8],[12]. Our patient had a great response to pyridostigmine. In two studies of a prolonged follow up in patients with congenital myasthenic syndrome due to a rapsyn deficiency, patients had a positive response to pyridostigmine. In patients with a partial response, it is described some improvement with 3,4-diaminopyridine, which can generate an additional improvement and allows to reduce the doses of pyridostigmine. It has also been described improvement with the use of salbutamol, also seen in our patient [1],[5],[8],[12]. The improvement described with the use of these drugs happens in all patients with mutations of the rapsyn gen, independently of the genetic mutation and the kind of inheritance. This is due to a delay in the hydrolysis of acetylcholine, facilitating interaction with the receptors and synaptic transmission.
The long-term prognosis is good. In general, the clinical course is stable except for the intermittent weakness that can be triggered by minor infections or fever and can occur throughout life despite medication. Respiratory crises decrease in both frequency and severity and are very rare after six years of age. On the contrary, exacerbations characterized by cervical weakness and warts, bulbar symptoms and ptosis continue to be observed until adulthood [12]. Long-term follow-up of these patients is necessary to refine the prognosis related to the presented mutations.
Conclusions
As conclusions, we should suspect a congenital myasthenic syndrome in every patient with a history of muscle fatigue, weakness that worsens during the day, mostly affecting the facial, oculomotor, swallowing and respiratory muscles, with onset since birth or early infancy, and an electromyographic response with a decrement of the compound muscle action potential over 10%. Once the suspicion is established, it is essential to look for the specific diagnosis through genetic panels that allows us not only to initiate the most appropriate treatment but to determine a prognosis and the proper genetic counseling to the family.
Main messages.
The congenital myasthenic syndromes are a group of heterogeneous genetic disorders caused by an abnormal transmission in the neuromuscular plate.
The weakness of neonatal onset, the phenomenon of fatigability with the sustained effort and the greater than 10% decrement in the Compound Muscular Action Potential are the key elements to establish the diagnosis.
Acknowledgments
Financing
Work in Dr. Engel’s laboratory was supported by NIH Grant NS6277. The other authors declare no external sources of funding for the preparation of this report.
Footnotes
Declaration of conflicts of interest
The authors have completed the ICMJE conflict of interest declaration form, and declare that they have no conflicts of interest. The forms can be requested by contacting the responsible author or the editorial direction of the Journal.
Informed consent
The informed consent requested by Medwave for the publication of this case report including the images showed has been signed by the patient’s father. The authors declare that the patient’s privacy was respected according to the CIOMS rules, regarding the privacy of the data collected. A copy of the informed consent was forwarded to the editorial direction of the journal.
References
- 1.Engel AG, Shen XM, Selcen D, Sine SM. Congenital Myasthenic Syndromes: Pathogenesis, Diagnosis, and Treatment. Lancet Neurol. 2015; 14(4): 420–34.DOI: 10.1016/S1474-4422(14)70201-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bodamer OA. Neuromuscular Junction Disorders in Newborns and Infants. In: UptoDate. 2018. Available: https://www.uptodate.com/contents/neuromuscular-junction-disorders-in-newborns-and-infants.
- 3.Abicht A, Müller JS, Lochmüller H, Engel AG. Congenital Myasthenic Syndromes. In: University of Washington; 2003. Available: https://www.ncbi.nlm.nih.gov/books/NBK1168/. [Google Scholar]
- 4.Rodriguez Cruz PM, Palace J, Beeson D. Congenital Myasthenic Syndromes and the Neuromuscular Junction. Curr Opin Neurol. 2014; 27(5): 566–75.DOI: 10.1097/WCO.0000000000000134 [DOI] [PubMed] [Google Scholar]
- 5.Hantai D, Nicole S, Eymard B. Congenital Myasthenic Syndromes: An Update. Curr Opin Neurol. 2013; 26(5): 561–8.DOI: 10.1097/WCO.0b013e328364dc0f [DOI] [PubMed] [Google Scholar]
- 6.Souza PV, Batistella GN, Lino VC, Pinto WB, Annes M, Oliveira AS. Clinical and Genetic Basis of Congenital Myasthenic Syndromes. Arq Neuropsiquiatr. 2016; 74(9): 750–60.DOI: 10.1590/0004-282X20160106 [DOI] [PubMed] [Google Scholar]
- 7.Muller JS, Mildner G, Muller-Felber W, Schara U, Krampfl K, Petersen B, et al. Rapsyn N88k Is a Frequent Cause of Congenital Myasthenic Syndromes in European Patients. Neurology. 2003; 60(11): 1805–10 [DOI] [PubMed] [Google Scholar]
- 8.Milone M, Shen XM, Selcen D, Ohno K, Brengman J, Iannaccone ST, et al. Myasthenic Syndrome Due to Defects in Rapsyn: Clinical and Molecular Findings in 39 Patients. Neurology. 2009; 73(3): 228–35.DOI: 10.1212/WNL.0b013e3181ae7cbc [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zuber B, Unwin N. Structure and Superorganization of Acetylcholine Receptor-Rapsyn Complexes. Proc Natl Acad Sci U S A. 2013; 110(26): 10622–7.DOI: 10.1073/pnas.1301277110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohno K, Sadeh M, Blatt I, Brengman JM, Engel AG. E-Box Mutations in the RAPSN Promoter Region in Eight Cases with Congenital Myasthenic Syndrome. Hum Mol Genet. 2003; 12(7): 739–48 [DOI] [PubMed] [Google Scholar]
- 11.Cossins J, Burke G, Maxwell S, Spearman H, Man S, Kuks J, et al. Diverse Molecular Mechanisms Involved in Achr Deficiency Due to Rapsyn Mutations. Brain. 2006; 129(Pt 10): 2773–83.DOI: 10.1093/brain/awl219 [DOI] [PubMed] [Google Scholar]
- 12.Natera-de Benito D, Bestue M, Vilchez JJ, Evangelista T, Topf A, Garcia-Ribes A, et al. Long-Term Follow-up in Patients with Congenital Myasthenic Syndrome Due to RAPSN Mutations. Neuromuscul Disord. 2016; 26(2): 153–9.DOI: 10.1016/j.nmd.2015.10.013 [DOI] [PubMed] [Google Scholar]